On the Role of the Cathode for the Electro-Oxidation of Perfluorooctanoic Acid

 ,
,  ,
,
Abstract
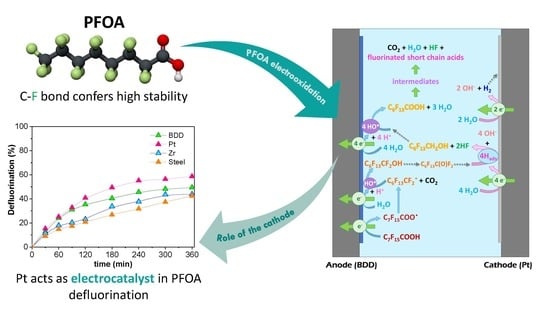
:
1. Introduction
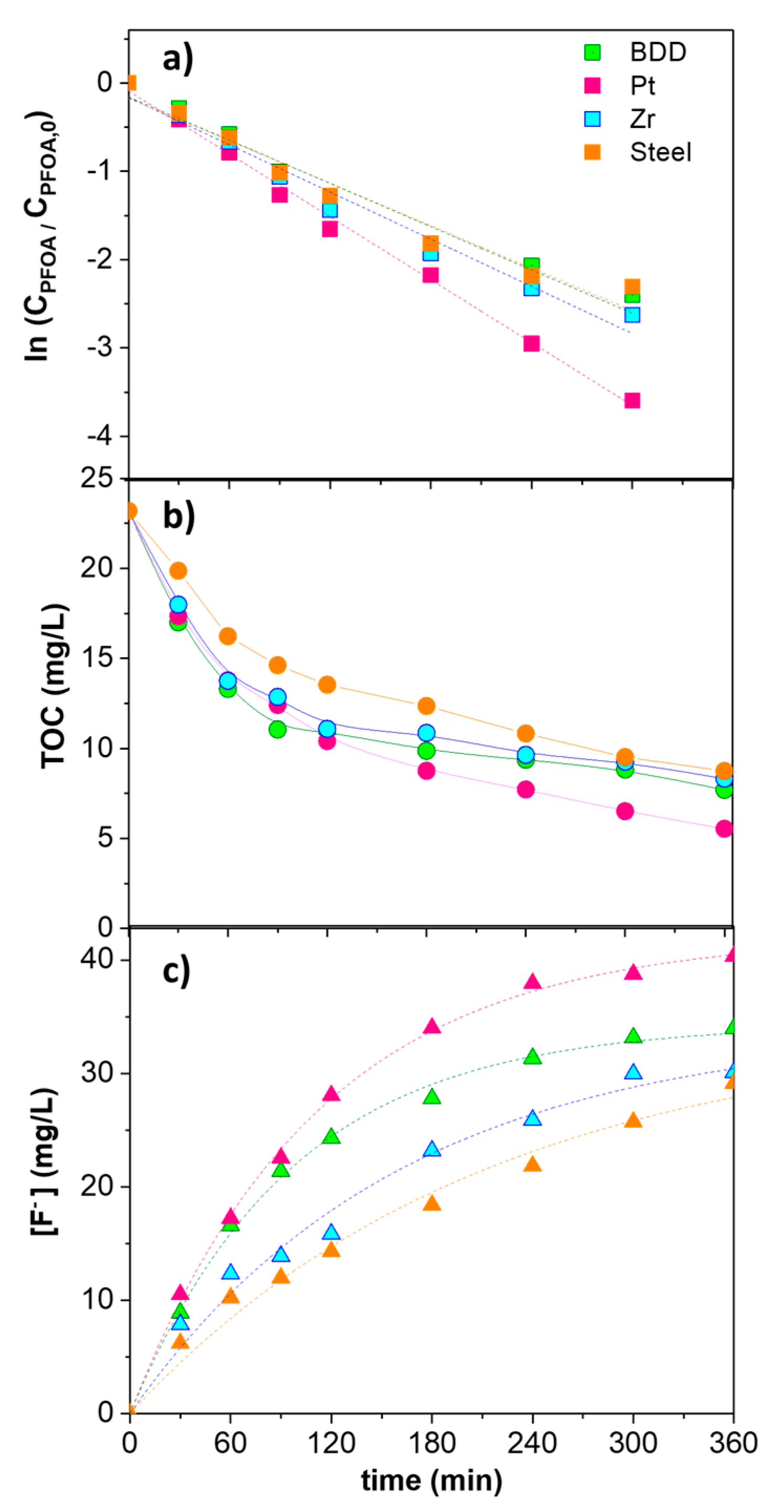
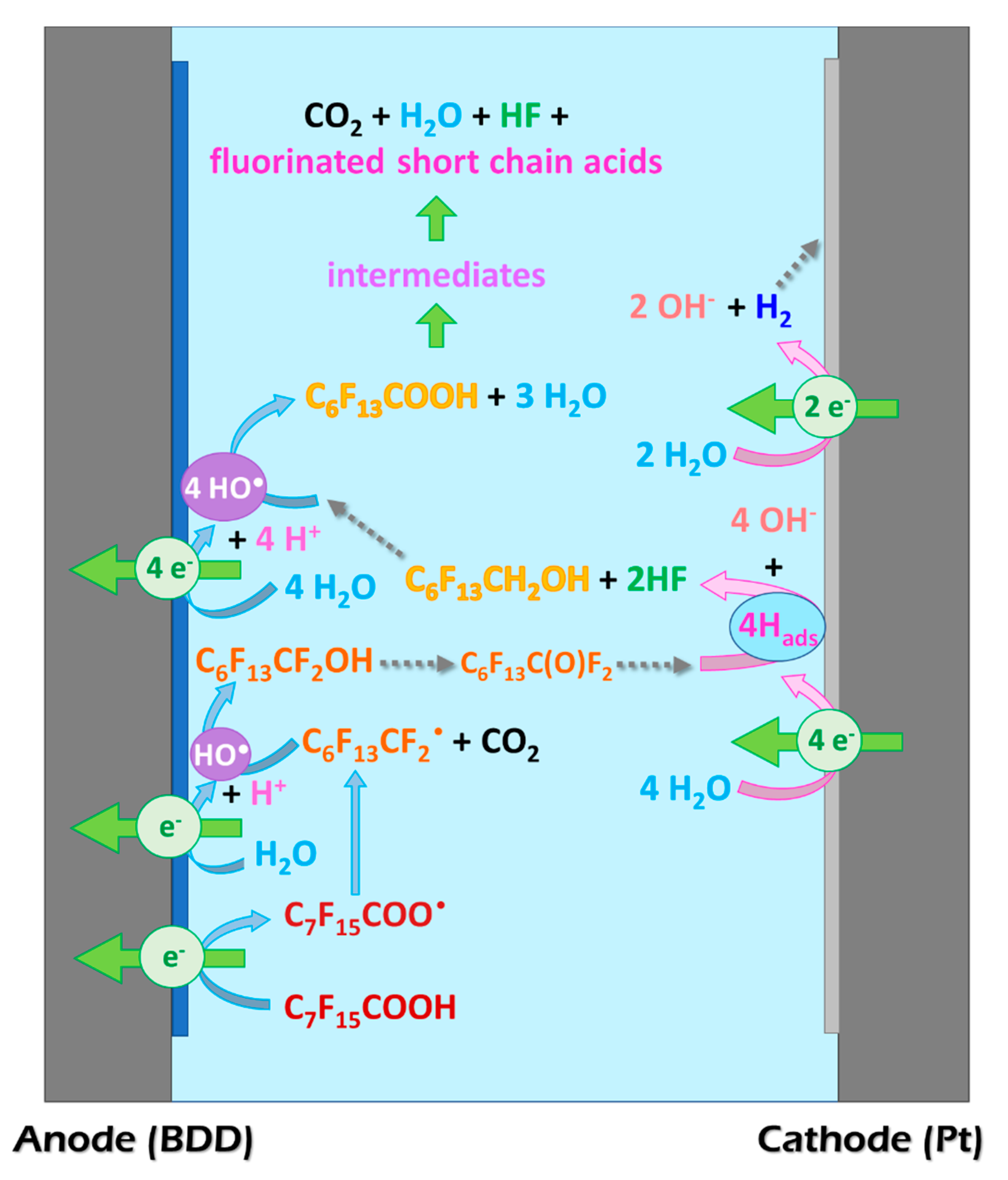
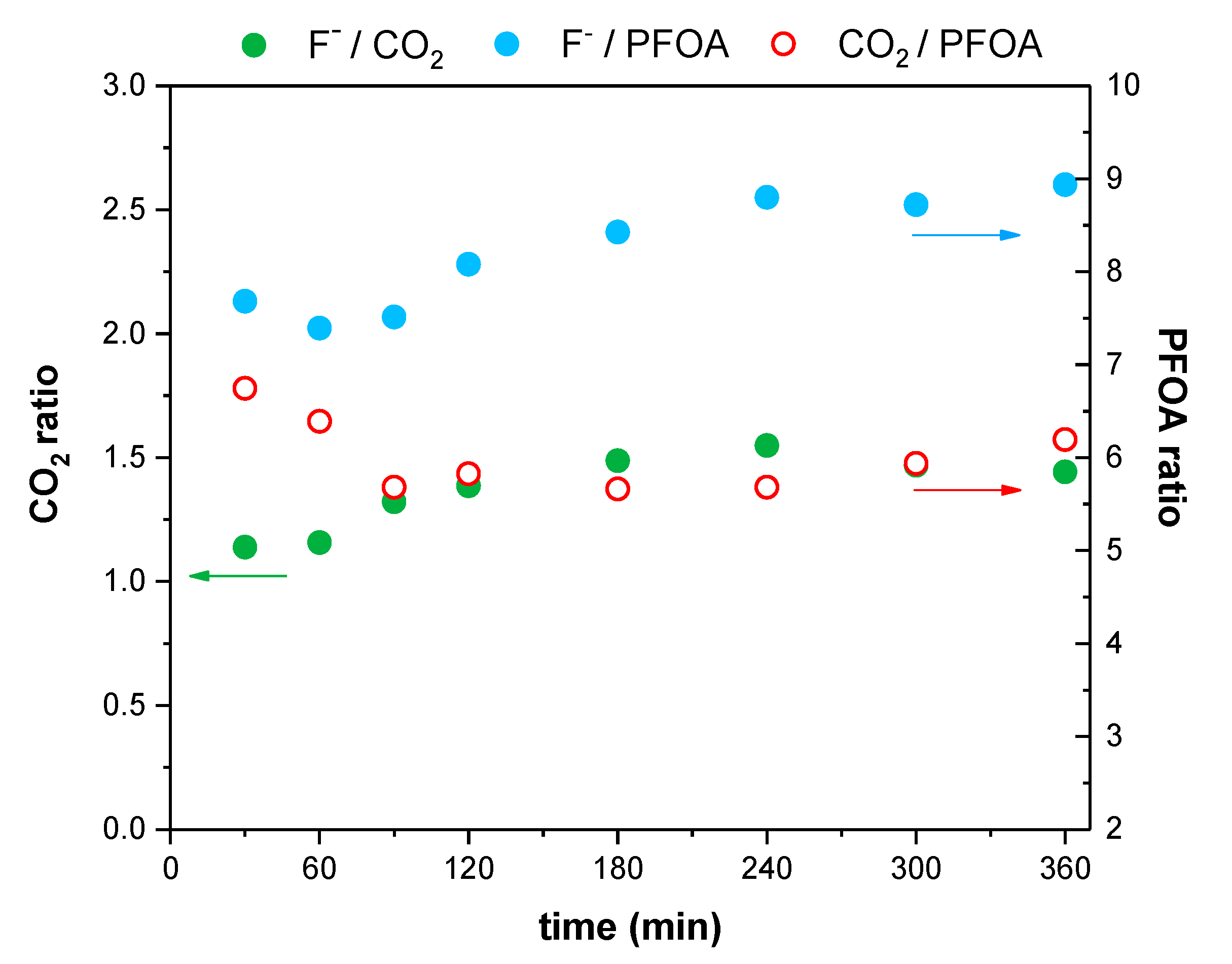
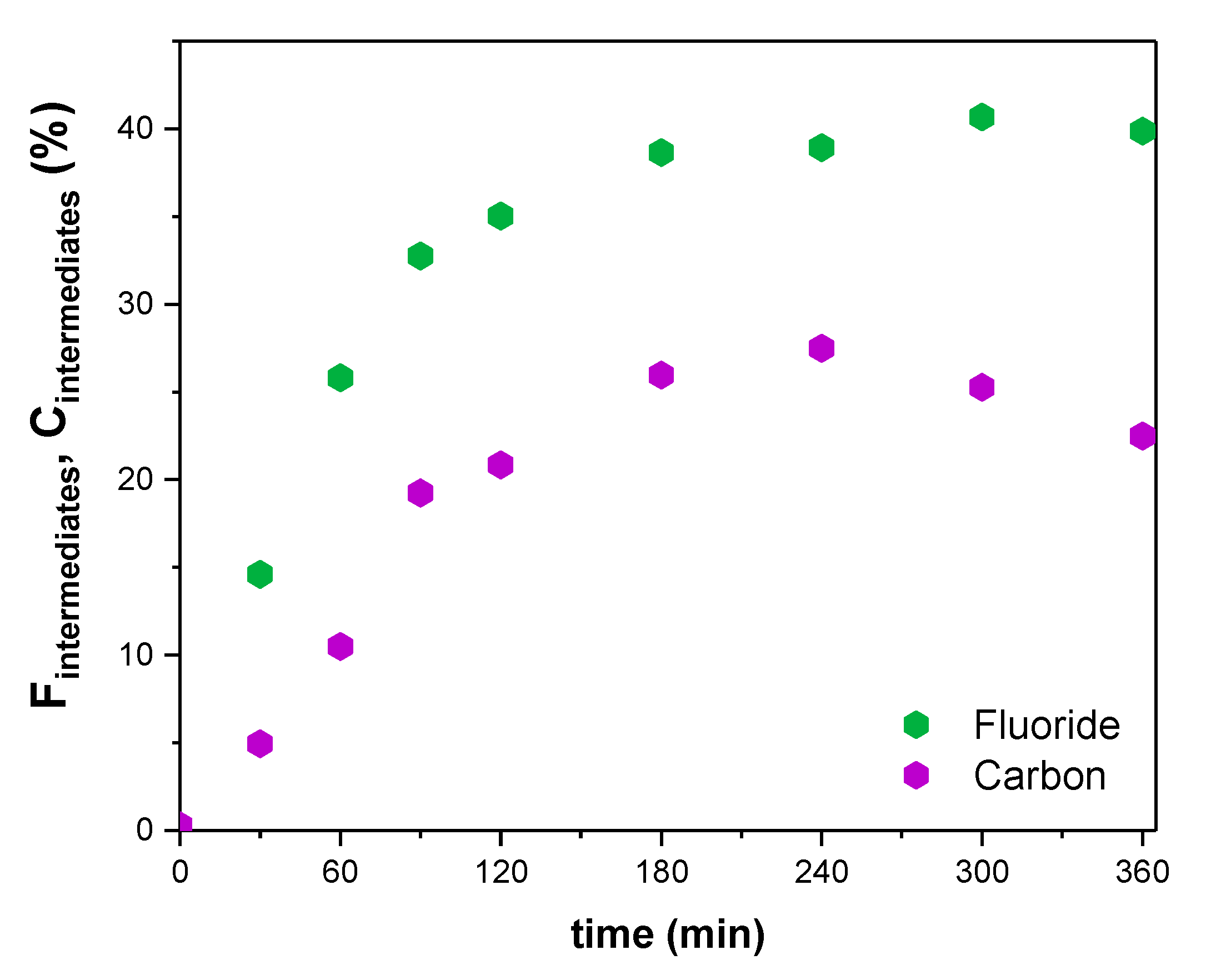
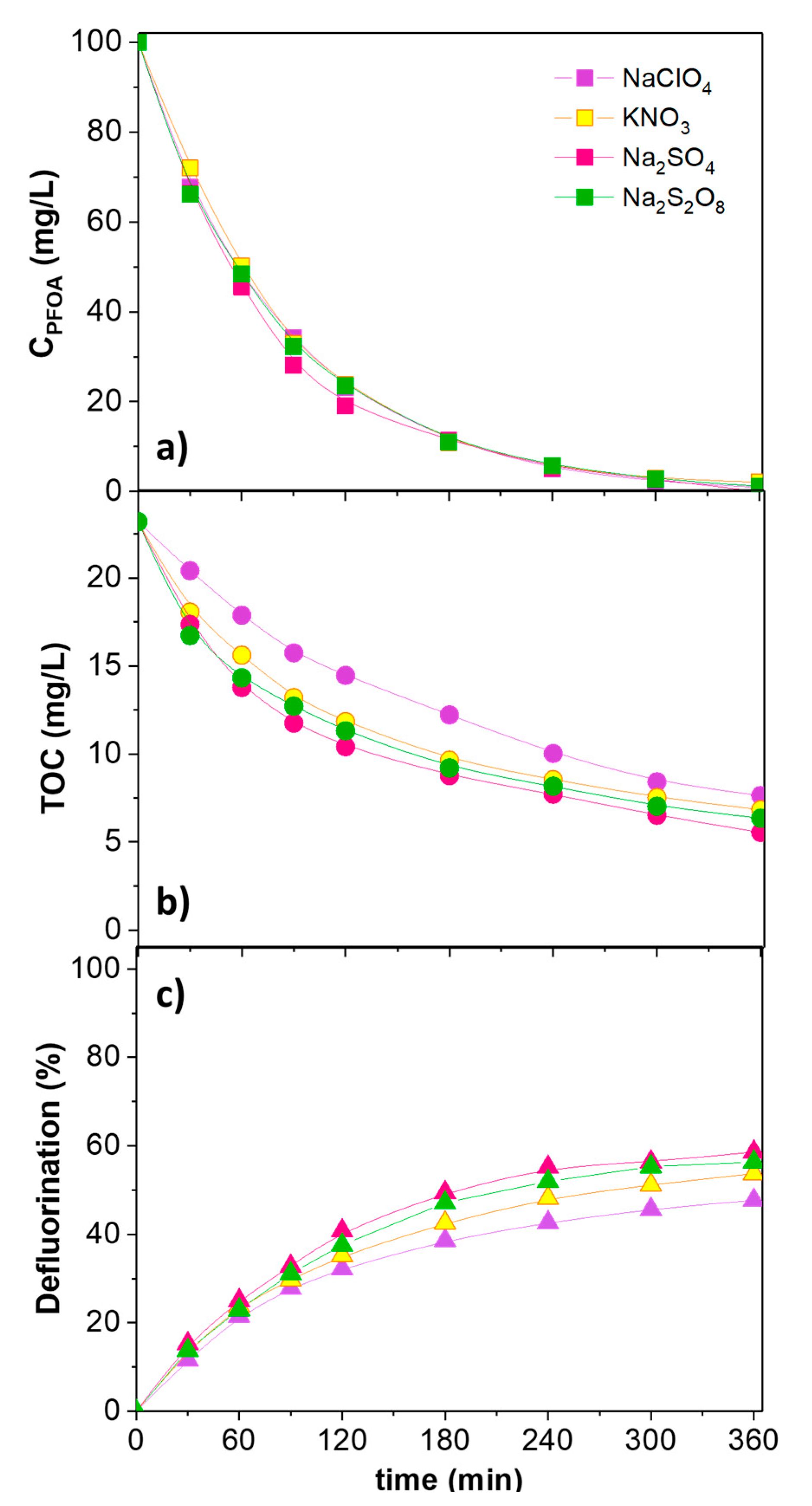
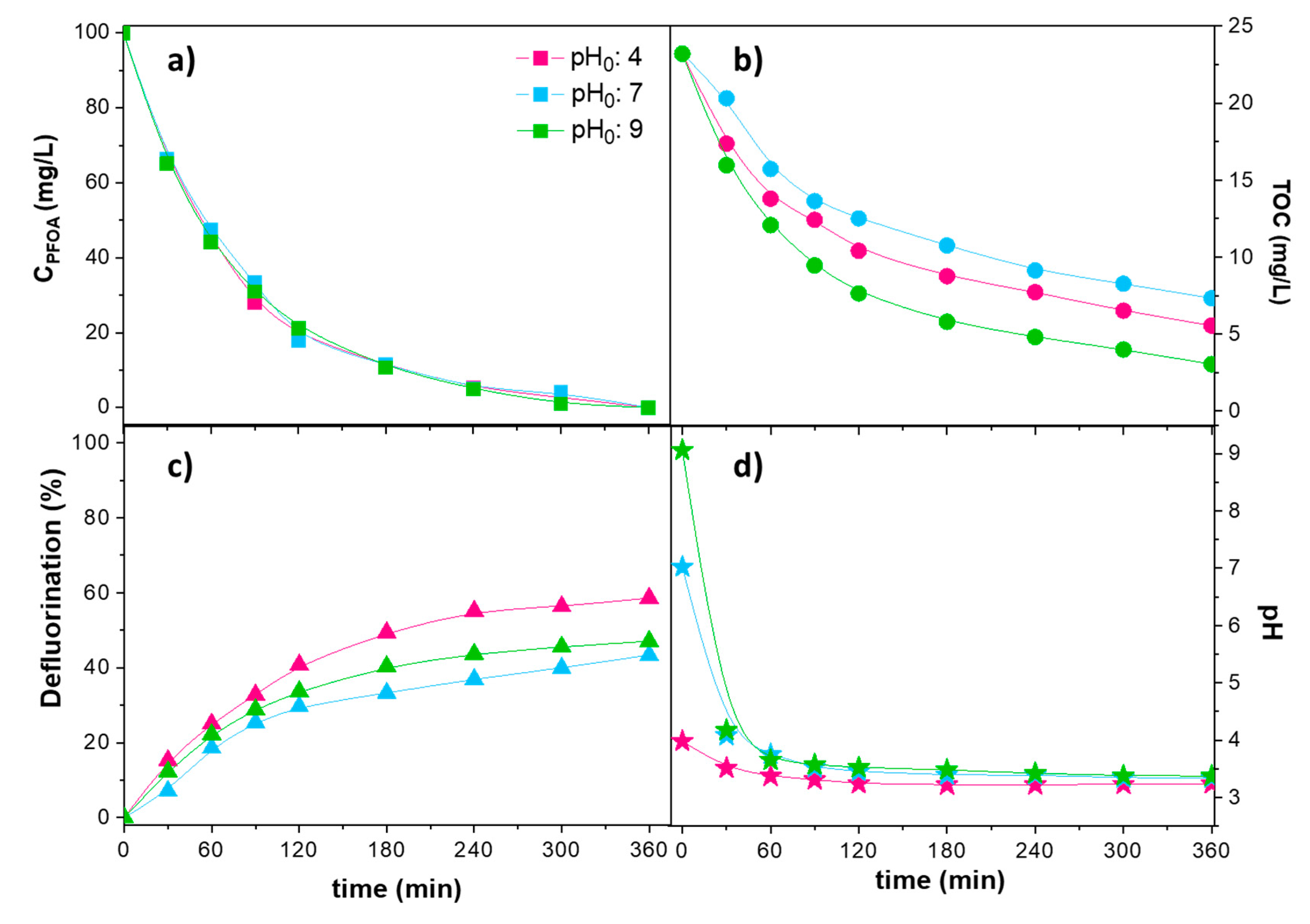
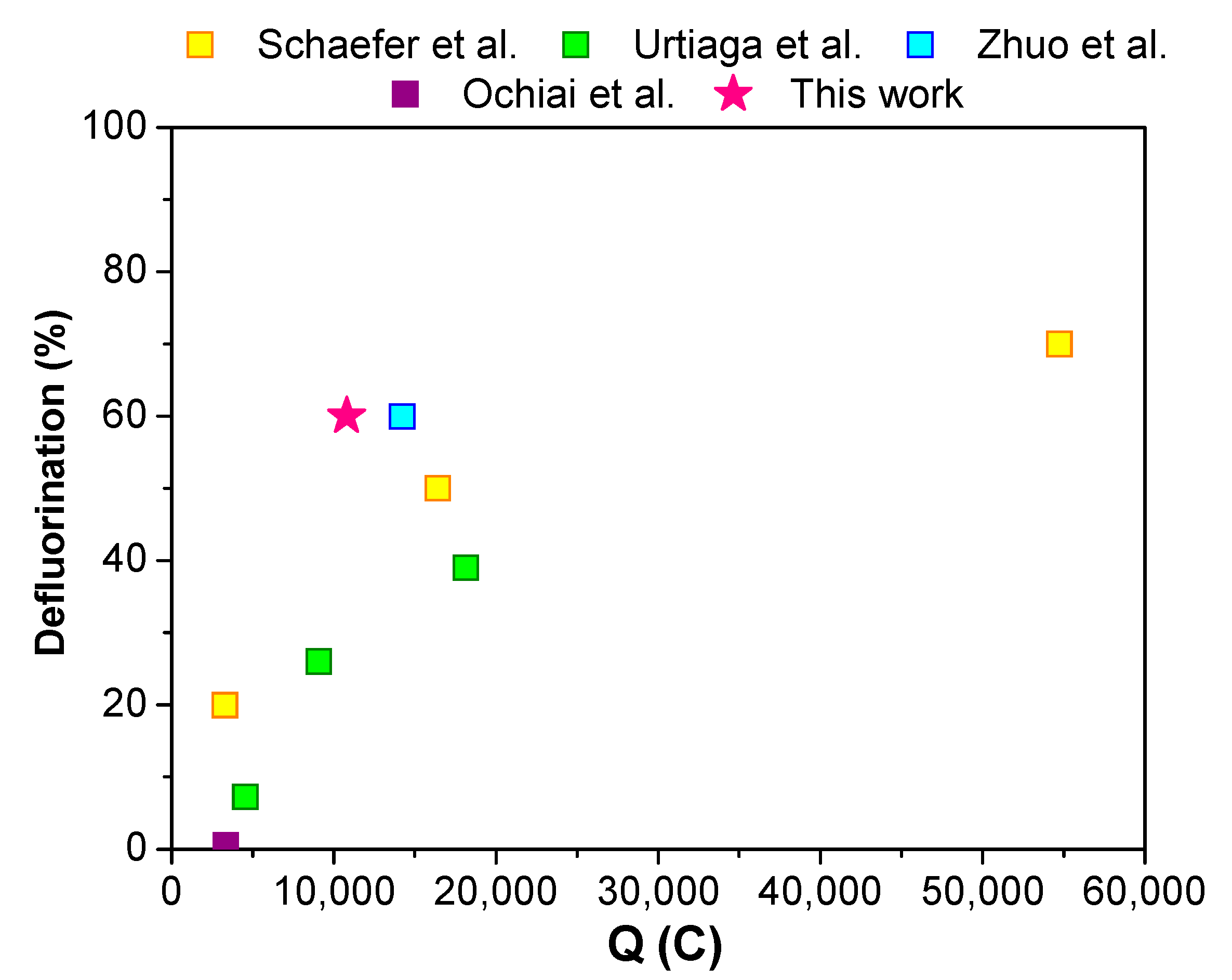
2. Results and Discussion
- (i)
- With adsorbed hydrogen generated by water electro-reduction at the cathode, releasing 2 F− (Equation (6)).As the first carbon in the alkyl chain is now defluorinated, HO• can attack it once again leading to the formation of C6F13COOH. This mechanism is similar to that presented for PFOA photocatalytic degradation by Wang et al. [20] and theoretic quantum calculations and experimental data collected by Trojanowicz et al. [31]. Hence, this step depends strongly on the cathode material.
- (ii)
- With hydroxyl radicals leading to the formation of COF2, as related by Niu et al. [31] and Zhang et al. [28], following Equations (7)–(9):According to George et al. [30] hydrolysis of carbonyl fluoride COF2 in the aqueous phase is extremely fast since its half-life is 0.7 s at T = 273 K.
- (iii)
3. Materials and Methods
3.1. Reactants
3.2. Experimental Set-Up
3.3. Analytical Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gebbink, W.A.; van Leeuwen, S.P.J. Environmental Contamination and Human Exposure to PFASs Near a Fluorochemical Production Plant: Review of Historic and Current PFOA and GenX Contamination in the Netherlands. Environ. Int. 2020, 137, 11. [Google Scholar] [CrossRef] [PubMed]
- Ao, J.J.; Yuan, T.; Xia, H.; Ma, Y.N.; Shen, Z.M.; Shi, R.; Tian, Y.; Zhang, J.; Ding, W.J.; Gao, L.; et al. Characteristic and Human Exposure Risk Assessment of Per- and Polyfluoroalkyl Substances: A study Based on Indoor Dust and Drinking Water in China. Environ. Pollut. 2019, 254, 9. [Google Scholar] [CrossRef] [PubMed]
- Hori, H.; Hayakawa, E.; Einaga, H.; Kutsuna, S.; Koike, K.; Ibusuki, T.; Kiatagawa, H.; Arakawa, R. Decomposition of Environmentally Persistent Perfluorooctanoic Acid in Water by Photochemical Approaches. Environ. Sci. Technol. 2004, 38, 6118–6124. [Google Scholar] [CrossRef] [PubMed]
- Kannan, K.; Koistinen, J.; Beckmen, K.; Evans, T.; Gorzelany, J.F.; Hansen, K.J.; Jones, P.D.; Helle, E.; Nyman, M.; Giesy, J.P. Accumulation of Perfluorooctane Sulfonate in Marine Mammals. Environ. Sci. Technol. 2001, 35, 1593–1598. [Google Scholar] [CrossRef]
- Wang, G.H.; Wang, X.L.; Xing, Z.N.; Lu, J.J.; Chang, Q.G.; Tong, Y.B. Occurrence and Distribution of Perfluorooctane Sulfonate and Perfluorooctanoic Acid in Three Major Rivers of Xinjiang, China. Environ. Sci. Pollut. Res. 2019, 26, 28062–28070. [Google Scholar] [CrossRef]
- Pico, Y.; Blasco, C.; Farre, M.; Barcelo, D. Occurrence of Perfluorinated Compounds in Water and Sediment of L’Albufera Natural Park (Valencia, Spain). Environ. Sci. Pollut. Res. 2012, 19, 946–957. [Google Scholar] [CrossRef]
- Domingo, J.L.; Nadal, M. Human Exposure to Per-And Polyfluoroalkyl Substances (PFAS) through Drinking Water: A Review of the Recent Scientific Literature. Environ. Res. 2019, 177, 10. [Google Scholar] [CrossRef]
- Saeidi, N.; Kopinke, F.D.; Georgi, A. Understanding the Effect of Carbon Surface Chemistry on Adsorption of Perfluorinated Alkyl Substances. Chem. Eng. J. 2020, 381, 11. [Google Scholar] [CrossRef]
- Wang, F.; Shih, K.M. Adsorption of Perfluorooctanesulfonate (PFOS) and Perfluorooctanoate (PFOA) on Alumina: Influence of Solution pH and Cations. Water Res. 2011, 45, 2925–2930. [Google Scholar]
- Maimaiti, A.; Deng, S.B.; Meng, P.P.; Wang, W.; Wang, B.; Huang, J.; Wang, Y.J.; Yu, G. Competitive Adsorption of Perfluoroalkyl Substances on Anion Exchange Resins in Simulated AFFF-Impacted Groundwater. Chem. Eng. J. 2018, 348, 494–502. [Google Scholar] [CrossRef]
- Garcia-Costa, A.L.; Zazo, J.A.; Casas, J.A. Microwave-Assisted Catalytic Wet Peroxide Oxidation: Energy Optimization. Sep. Purif. Technol. 2019, 215, 62–69. [Google Scholar] [CrossRef]
- Garcia-Costa, A.L.; Zazo, J.A.; Rodriguez, J.J.; Casas, J.A. Intensification of Catalytic Wet Peroxide Oxidation With Microwave Radiation: Activity and Stability of Carbon Materials. Sep. Purif. Technol. 2019, 209, 301–306. [Google Scholar] [CrossRef]
- Maruthamuthu, P.; Padmaja, S.; Huie, R.E. Rate Constants for Some Reactions of Free-Radicals with Haloacetates in Aqueous-Solution. Int. J. Chem. Kinet. 1995, 27, 605–612. [Google Scholar] [CrossRef]
- Moriwaki, H.; Takagi, Y.; Tanaka, M.; Tsuruho, K.; Okitsu, K.; Maeda, Y. Sonochemical Decomposition of Perfluorooctane Sulfonate and Perfluorooctanoic Acid. Environ. Sci. Technol. 2005, 39, 3388–3392. [Google Scholar] [CrossRef] [PubMed]
- Javed, H.; Lyu, C.; Sun, R.N.; Zhang, D.N.; Alvarez, P.J.J. Discerning the Inefficacy of Hydroxyl Radicals during Perfluorooctanoic Acid Degradation. Chemosphere 2020, 247, 6. [Google Scholar] [CrossRef]
- Santos, A.; Rodriguez, S.; Pardo, F.; Romero, A. Use of Fenton Reagent Combined with Humic Acids for the Removal of PFOA from Contaminated Water. Sci. Total Environ. 2016, 563, 657–663. [Google Scholar] [CrossRef]
- Chen, M.J.; Lo, S.L.; Lee, Y.C.; Huang, C.C. Photocatalytic Decomposition of Perfluorooctanoic Acid by Transition-Metal Modified Titanium Dioxide. J. Hazard. Mater. 2015, 288, 168–175. [Google Scholar] [CrossRef]
- Chen, M.J.; Lo, S.L.; Lee, Y.C.; Kuo, J.; Wu, C.H. Decomposition of Perfluorooctanoic Acid by Ultraviolet Light Irradiation with Pb-Modified Titanium Dioxide. J. Hazard. Mater. 2016, 303, 111–118. [Google Scholar] [CrossRef]
- Gomez-Ruiz, B.; Ribao, P.; Diban, N.; Rivero, M.J.; Ortiz, I.; Urtiaga, A. Photocatalytic Degradation and Mineralization of Perfluorooctanoic Acid (PFOA) Using a Composite TiO2–rGO Catalyst. J. Hazard. Mater. 2018, 344, 950–957. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.N.; Yang, Q.; Chen, F.; Sun, J.; Luo, K.; Yao, F.B.; Wang, X.L.; Wang, D.B.; Li, X.M.; Zeng, G.M. Photocatalytic Degradation of Perfluorooctanoic Acid and Perfluorooctane Sulfonate in Water: A Critical Review. Chem. Eng. J. 2017, 328, 927–942. [Google Scholar] [CrossRef]
- Schaefer, C.E.; Andaya, C.; Burant, A.; Condee, C.W.; Urtiaga, A.; Strathmann, T.J.; Higgins, C.P. Electrochemical Treatment of Perfluorooctanoic Acid and Perfluorooctane Sulfonate: Insights into Mechanisms and Application to Groundwater Treatment. Chem. Eng. J. 2017, 317, 424–432. [Google Scholar] [CrossRef]
- Urtiaga, A.; Fernandez-Gonzalez, C.; Gomez-Lavin, S.; Ortiz, I. Kinetics of the Electrochemical Mineralization of Perfluorooctanoic Acid on Ultrananocrystalline Boron Doped Conductive Diamond Electrodes. Chemosphere 2015, 129, 20–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuo, Q.F.; Deng, S.B.; Yang, B.; Huang, J.; Wang, B.; Zhang, T.T.; Yu, G. Degradation of Perfluorinated Compounds on a Boron-Doped Diamond Electrode. Electrochim. Acta 2012, 77, 17–22. [Google Scholar] [CrossRef]
- Ochiai, T.; Iizuka, Y.; Nakata, K.; Murakami, T.; Tryk, D.A.; Fujishima, A.; Koide, Y.; Morito, Y. Efficient Electrochemical Decomposition of Perfluorocarboxylic Acids by the Use of a Boron-Doped Diamond Electrode. Diam. Relat. Mater. 2011, 20, 64–67. [Google Scholar] [CrossRef]
- Xiao, H.S.; Lv, B.Y.; Zhao, G.H.; Wang, Y.J.; Li, M.F.; Li, D.M. Hydrothermally Enhanced Electrochemical Oxidation of High Concentration Refractory Perfluorooctanoic Acid. J. Phys. Chem. A 2011, 115, 13836–13841. [Google Scholar] [CrossRef]
- Mais, L.; Mascia, M.; Palmas, S.; Vacca, A. Photoelectrochemical Oxidation of Phenol With Nanostructured TiO2-PANI Electrodes under Solar Light Irradiation. Sep. Purif. Technol. 2019, 208, 153–159. [Google Scholar] [CrossRef]
- Weiss, E.; Groenen-Serrano, K.; Savall, A.; Comninellis, C. A Kinetic Study of the Electrochemical Oxidation of Maleic Acid on Boron Doped Diamond. J. Appl. Electrochem. 2007, 37, 41–47. [Google Scholar] [CrossRef] [Green Version]
- Walsh, F.C. A First Course in Electrochemical Engineering. The Electrochemical Consultancy. 1993, 381. [Google Scholar] [CrossRef]
- Pizarro, A.H.; Molina, C.B.; Fierro, J.L.G.; Rodriguez, J.J. On the Effect of Ce Incorporation on Pillared Clay-Supported Pt and Ir Catalysts for Aqueous-Phase Hydrodechlorination. Appl. Catal. B Environ. 2016, 197, 236–243. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Moores, A.; Liu, J.X.; Ghoshal, S. New Insights into the Degradation Mechanism of Perfluorooctanoic Acid by Persulfate from Density Functional Theory and Experimental Data. Environ. Sci. Technol. 2019, 53, 8672–8681. [Google Scholar] [CrossRef]
- Trojanowicz, M.; Bojanowska-Czajka, A.; Bartosiewicz, I.; Kulisa, K. Advanced Oxidation/Reduction Processes Treatment for Aqueous Perfluorooctanoate (PFOA) and Perfluorooctanesulfonate (PFOS)—A Review of Recent Advances. Chem. Eng. J. 2018, 336, 170–199. [Google Scholar] [CrossRef]
- Niu, J.F.; Lin, H.; Gong, C.; Sun, X.M. Theoretical and Experimental Insights into the Electrochemical Mineralization Mechanism of Perfluorooctanoic Acid. Environ. Sci. Technol. 2013, 47, 14341–14349. [Google Scholar] [CrossRef] [PubMed]
- Stefanova, A.; Ayata, S.; Erem, A.; Ernst, S.; Baltruschat, H. Mechanistic Studies on Boron-Doped Diamond: Oxidation of Small Organic Molecules. Electrochim. Acta 2013, 110, 560–569. [Google Scholar] [CrossRef]
- Zazo, J.A.; Casas, J.A.; Mohedano, A.F.; Rodriguez, J.J. Catalytic Wet Peroxide Oxidation of Phenol with a Fe/active Carbon Catalyst. Appl. Catal. B Environ. 2006, 65, 261–268. [Google Scholar] [CrossRef]
- Serrano, K.; Michaud, P.A.; Comninellis, C.; Savall, A. Electrochemical Preparation of Peroxodisulfuric Acid Using Boron Doped Diamond Thin Film Electrodes. Electrochim. Acta 2002, 48, 431–436. [Google Scholar] [CrossRef] [Green Version]
- Lan, Y.D.; Coetsier, C.; Causserand, C.; Serrano, K.G. On the Role of Salts for the Treatment of Wastewaters Containing Pharmaceuticals by Electrochemical Oxidation Using a Boron Doped Diamond Anode. Electrochim. Acta 2017, 231, 309–318. [Google Scholar] [CrossRef] [Green Version]
- Kolthoff, I.M.; Miller, I.K. The Chemistry of Persulfate.1. The Kinetics and Mechanism of the Decomposition of the Persulfate ion in Aqueous Medium. J. Am. Chem. Soc. 1951, 73, 3055–3059. [Google Scholar] [CrossRef]
- Neta, P.; Madhavan, V.; Zemel, H.; Fessenden, R.W. Rate Constants and Mechanism of Reaction of Sulfate Radical Anion with Aromatic Compounds. J. Am. Chem. Soc. 1977, 99, 163–164. [Google Scholar] [CrossRef]
- Yang, L.; He, L.Y.; Xue, J.M.; Ma, Y.F.; Xie, Z.Y.; Wu, L.; Huang, M.; Zhang, Z.L. Persulfate-Based Degradation of Perfluorooctanoic Acid (PFOA) and Perfluorooctane Sulfonate (PFOS) in Aqueous Solution: Review on Influences, Mechanisms and Prospective. J. Hazard. Mater. 2020, 393, 11. [Google Scholar] [CrossRef]
- Qian, Y.J.; Guo, X.; Zhang, Y.L.; Peng, Y.; Sun, P.Z.; Huang, C.H.; Niu, J.F.; Zhou, X.F.; Crittenden, J.C. Perfluorooctanoic Acid Degradation Using UV-Persulfate Process: Modeling of the Degradation and Chlorate Formation. Environ. Sci. Technol. 2016, 50, 772–781. [Google Scholar] [CrossRef]
- Silveira, J.E.; Garcia-Costa, A.L.; Cardoso, T.O.; Zazo, J.A.; Casas, J.A. Indirect Decolorization of Azo Dye Disperse Blue 3 by Electro-Activated Persulfate. Electrochim. Acta 2017, 258, 927–932. [Google Scholar] [CrossRef]
- Lan, Y.; Coetsier, C.; Causserand, C.; Serrano, K.G. An Experimental and Modelling Study of the Electrochemical Oxidation Of Pharmaceuticals Using a Boron-Doped Diamond Anode. Chem. Eng. J. 2018, 333, 486–494. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electro-Oxidation System | Operating Conditions | Results | Remarks | Ref |
|---|---|---|---|---|
| Anode: BDD Cathode: W Area: 38 cm2 Spacing: 4 mm | [PFOA]0: 15 mg/L 8Electrolyte: 1500 mg/L Na2SO4 or 1500 mg/L Na2SO4 + 167 mg/L NaCl T: 20 °C, j: 3, 15, 50 mA/cm2 V: 250 mL, t: 480 min | xPFOA: 60–100% xF−: 30–80% | No apparent influence of Cl− in the process | [21] |
| Anode: BDD Cathode: W Area: 42 cm2 Spacing: 8 mm | [PFOA]0: 100 mg/L Electrolyte: 1.4–8.4 g/L NaClO4, 5 g/L NaSO4 T: 20 °C, j: 50, 100, 200 mA/cm2 V: not specified, t: 360 min | xPFOA: 93% xTOC: 95% xF−: 38% | In the tested conditions, SO42− did not produce additional oxidants. higher j, higher degradation | [22] |
| Anode: BDD Cathode: BDD Area: 85 cm2 Spacing: 30 mm | [PFOA]0: 50 mg/L Electrolyte: 1.4 g/L NaClO4 pH: 3, 9, 12, T: 32 °C, j: 23.24 mA/cm2 V: 40 mL, t: 120 min | xPFOA: 100% xF−: 58% | Slightly better results obtained at pH0 3 than pH0 9 | [23] |
| Anode: BDD Cathode: Pt Area: 77.4 cm2 Spacing: 10 mm | [PFOA]0: 50 mg/L Electrolyte: 1.2 g/L NaClO4 T: not specified, j: 0.04–1.2 mA/cm2 V: 300 mL, t: 480 min | xPFOA: 85% | F− deposition on the BDD surface. | [24] |
| Anode: BDD Cathode: Pt Area: 5.5 cm2 Spacing: 20 mm | [PFOA]0: 200 mg/L Electrolyte: 7.1 g/L Na2SO4 P: 0.3 MPa, T: 80–120 °C, j: 20 mA/cm2 V: 400 mL, t: 360 min | xPFOA: 95% xTOC: 90% xF−: 90% | High temperature process greatly enhances the PFOA degradation in relation to the room temperature system. | [25] |
| kPFOA·103 (min−1) | r2 | kF−·103 (min−1) | r2 | |
|---|---|---|---|---|
| BDD | 8.92 ± 0.68 | 0.988 | 8.97 ± 0.28 | 0.999 |
| Pt | 11.86 ± 0.32 | 0.994 | 10.36 ± 0.44 | 0.997 |
| Zr | 8.16 ± 0.58 | 0.979 | 6.23 ± 0.86 | 0.982 |
| Steel | 7.98 ± 0.42 | 0.981 | 4.70 ± 0.78 | 0.982 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia-Costa, A.L.; Savall, A.; Zazo, J.A.; Casas, J.A.; Groenen Serrano, K. On the Role of the Cathode for the Electro-Oxidation of Perfluorooctanoic Acid. Catalysts 2020, 10, 902. https://doi.org/10.3390/catal10080902
Garcia-Costa AL, Savall A, Zazo JA, Casas JA, Groenen Serrano K. On the Role of the Cathode for the Electro-Oxidation of Perfluorooctanoic Acid. Catalysts. 2020; 10(8):902. https://doi.org/10.3390/catal10080902
Chicago/Turabian StyleGarcia-Costa, Alicia L., Andre Savall, Juan A. Zazo, Jose A. Casas, and Karine Groenen Serrano. 2020. "On the Role of the Cathode for the Electro-Oxidation of Perfluorooctanoic Acid" Catalysts 10, no. 8: 902. https://doi.org/10.3390/catal10080902
APA StyleGarcia-Costa, A. L., Savall, A., Zazo, J. A., Casas, J. A., & Groenen Serrano, K. (2020). On the Role of the Cathode for the Electro-Oxidation of Perfluorooctanoic Acid. Catalysts, 10(8), 902. https://doi.org/10.3390/catal10080902