Cr-Phthalocyanine Porous Organic Polymer as an Efficient and Selective Catalyst for Mono Carbonylation of Epoxides to Lactones


Abstract
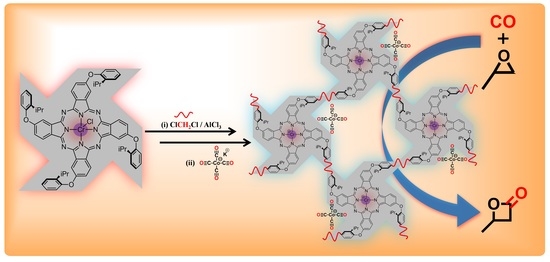
:
1. Introduction
2. Results and Discussion
2.1. Synthesis and Characterization of POP-Pc’Cr(III)Cl
2.2. Synthesis and Characterization of POP-Pc’Cr(III)Co(CO)4
2.3. Carbonylation Activity of POP-Pc’Cr(III)Co(CO)4
3. Experimental Section
3.1. Materials and Methods
3.2. Synthesis of Pc’ Ligand
3.3. Synthesis of Pc’Cr(III)Cl
3.4. Synthesis of POP-Pc’Cr(III)Cl
3.5. Synthesis of [POP-Pc’Cr(III)]][Co(CO)4]
3.6. PO Carbonylation Procedure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dunn, E.W.; Lamb, J.R.; Lapointe, A.M.; Coates, G.W. Carbonylation of Ethylene Oxide to β-Propiolactone: A Facile Route to Poly(3-hydroxypropionate) and Acrylic Acid. ACS Catal. 2016, 6, 8219–8223. [Google Scholar] [CrossRef]
- Rajendiran, S.; Park, G.; Yoon, S. Direct Conversion of Propylene Oxide to 3-Hydroxy Butyric Acid Using a Cobalt Carbonyl Ionic Liquid Catalyst. Catalysts 2017, 7, 228. [Google Scholar] [CrossRef] [Green Version]
- Getzler, Y.D.Y.L.; Kundnani, V.; Lobkovsky, E.B.; Coates, G.W. Catalytic Carbonylation of β-Lactones to Succinic Anhydrides. J. Am. Chem. Soc. 2004, 126, 6842–6843. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.L.; Christenson, J.K.; Wackett, L.P. Biosynthesis and chemical diversity of β-lactone natural products. Nat. Prod. Rep. 2019, 36, 458–475. [Google Scholar] [CrossRef]
- Wang, Y.C.; Tennyson, R.L.; Romo, D. β-lactones: Intermediates for Natural Product Total Synthesis and New Transformations. Heterocycles 2004, 64, 605–658. [Google Scholar] [CrossRef]
- Li, Z.; Yang, J.; Loh, X.J. Polyhydroxyalkanoates: Opening doors for a sustainable future. NPG Asia Mater. 2016, 8, e265. [Google Scholar] [CrossRef]
- Jiang, J.; Rajendiran, S.; Piao, L.; Yoon, S. Base Effects on Carbonylative Polymerization of Propylene Oxide with a [(salph)Cr(THF)2]+[Co(CO)4](-) Catalyst. Top. Catal. 2017, 60, 750–754. [Google Scholar] [CrossRef]
- Rajendiran, S.; Gunasekar, G.H.; Yoon, S. A heterogenized cobaltate catalyst on a bis-imidazolium-based covalent triazine framework for hydroesterification of epoxides. New J. Chem. 2018, 42, 12256–12262. [Google Scholar] [CrossRef]
- Rajendiran, S.; Park, K.; Lee, K.; Yoon, S. Ionic-Liquid-Based Heterogeneous Covalent Triazine Framework Cobalt Catalyst for the Direct Synthesis of Methyl 3-Hydroxybutyrate from Propylene Oxide. Inorg. Chem. 2017, 56, 7270–7277. [Google Scholar] [CrossRef]
- Kramer, J.W.; Rowley, J.M.; Coates, G.W. Ring-Expanding Carbonylation of Epoxides. In Organic Reactions; Denmark, S.E., Ed.; Wiley: Hoboken, NJ, USA, 2015; pp. 1–104. [Google Scholar]
- Getzler, Y.D.Y.L.; Mahadevan, V.; Lobkovsky, E.B.; Coates, G.W. Synthesis of β-Lactones: A Highly Active and Selective Catalyst for Epoxide Carbonylation. J. Am. Chem. Soc. 2002, 124, 1174–1175. [Google Scholar] [CrossRef]
- Kramer, J.W.; Lobkovsky, E.B.; Coates, G.W. Practical β-Lactone Synthesis: Epoxide Carbonylation at 1 atm. Org. Lett. 2006, 8, 3709–3712. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.A.R.; Mahadevan, V.; Getzler, Y.D.Y.L.; Coates, G.W. A Readily Synthesized and Highly Active Epoxide Carbonylation Catalyst Based on a Chromium Porphyrin Framework: Expanding the Range of Available β-Lactones. Org. Lett. 2004, 6, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.A.R.; Lobkovsky, E.B.; Coates, G.W. Chromium (III) Octaethylporphyrinato Tetracarbonylcobaltate: A Highly Active, Selective, and Versatile Catalyst for Epoxide Carbonylation. J. Am. Chem. Soc. 2005, 127, 11426–11435. [Google Scholar] [CrossRef] [PubMed]
- Park, H.D.; Dincă, M.; Román-Leshkov, Y. Heterogeneous Epoxide Carbonylation by Cooperative Ion-Pair Catalysis in Co(CO)4−-Incorporated Cr-MIL-101. ACS Central Sci. 2017, 3, 444–448. [Google Scholar] [CrossRef]
- Rajendiran, S.; Natarajan, P.; Yoon, S. A covalent triazine framework-based heterogenized Al-Co bimetallic catalyst for the ring-expansion carbonylation of epoxide to beta-lactone. RSC Adv. 2017, 7, 4635–4638. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Yoon, S. A Metalated Porous Porphyrin Polymer with [Co(CO)4]− Anion as an Efficient Heterogeneous Catalyst for Ring Expanding Carbonylation. Sci. Rep. 2018, 8, 13243. [Google Scholar] [CrossRef]
- Ganesan, V.; Yoon, S. Hyper-Cross-Linked Porous Porphyrin Aluminum(III) Tetracarbonylcobaltate as a Highly Active Heterogeneous Bimetallic Catalyst for the Ring-Expansion Carbonylation of Epoxides. ACS Appl. Mater. Interfaces 2019, 11, 18609–18616. [Google Scholar] [CrossRef]
- Ganesan, V.; Yoon, S. Direct Heterogenization of Salphen Coordination Complexes to Porous Organic Polymers: Catalysts for Ring-Expansion Carbonylation of Epoxides. Inorg. Chem. 2020, 59, 2881–2889. [Google Scholar] [CrossRef]
- Rajendiran, S.; Ganesan, V.; Yoon, S. Balancing between Heterogeneity and Reactivity in Porphyrin Chromium-Cobaltate Catalyzed Ring Expansion Carbonylation of Epoxide into β-Lactone. Inorg. Chem. 2019, 58, 3283–3289. [Google Scholar] [CrossRef]
- Jiang, J.; Rajendiran, S.; Yoon, S. Double Ring-Expanding Carbonylation Using an In Situ Generated Aluminum Phthalocyanine Cobalt Carbonyl Complex. Asian J. Org. Chem. 2018, 8, 151–154. [Google Scholar] [CrossRef]
- Rowley, J.M.; Lobkovsky, E.B.; Coates, G.W. Catalytic Double Carbonylation of Epoxides to Succinic Anhydrides: Catalyst Discovery, Reaction Scope, and Mechanism. J. Am. Chem. Soc. 2007, 129, 4948–4960. [Google Scholar] [CrossRef] [PubMed]
- Church, T.L.; Getzler, Y.D.; Byrne, C.M.; Coates, G.W. Carbonylation of heterocycles by homogeneous catalysts. Chem. Commun. 2007, 7, 657–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, C.-G.; Bergbreiter, D.E. Highly organic phase soluble polyisobutylene-bound cobalt phthalocyanines as recyclable catalysts for nitroarene reduction. Catal. Commun. 2016, 77, 89–93. [Google Scholar] [CrossRef] [Green Version]
- Ghani, F.; Kristen, J.; Riegler, H. Solubility Properties of Unsubstituted Metal Phthalocyanines in Different Types of Solvents. J. Chem. Eng. Data 2012, 57, 439–449. [Google Scholar] [CrossRef]
- Bulgakov, B.A.; Sulimov, A.V.; Babkin, A.V.; Kepman, A.V.; Malakho, A.P.; Avdeev, V.V. Dual-curing thermosetting monomer containing both propargyl ether and phthalonitrile groups. J. Appl. Polym. Sci. 2017, 134, 44786. [Google Scholar] [CrossRef]
- Lever, A. The Phthalocyanines. In Advances in Inorganic Chemistry and Radiochemistry; Emeléus, H.J., Sharpe, A.G., Eds.; Academic Press: Cambridge, MA, USA, 1965; Volume 7, pp. 27–114. [Google Scholar]
- Lee, W.; Yuk, S.B.; Choi, J.; Jung, D.H.; Choi, S.-H.; Park, J.; Kim, J.P. Synthesis and characterization of solubility enhanced metal-free phthalocyanines for liquid crystal display black matrix of low dielectric constant. Dye. Pigment. 2012, 92, 942–948. [Google Scholar] [CrossRef]
- Padmanaban, S.; Yoon, S. Surface Modification of a MOF-based Catalyst with Lewis Metal Salts for Improved Catalytic Activity in the Fixation of CO2 into Polymers. Catalysts 2019, 9, 892. [Google Scholar] [CrossRef] [Green Version]
- Donohue, M.; Aranovich, G. A new classification of isotherms for Gibbs adsorption of gases on solids. Fluid Phase Equilibria 1999, 158, 557–563. [Google Scholar] [CrossRef]
- He, W.-L.; Wu, C. Incorporation of Fe-phthalocyanines into a porous organic framework for highly efficient photocatalytic oxidation of arylalkanes. Appl. Catal. B: Environ. 2018, 234, 290–295. [Google Scholar] [CrossRef]
- McKeown, N.B.; Makhseed, S.; Budd, P.M. Phthalocyanine-based nanoporous network polymers. Chem. Commun. 2002, 2780–2781. [Google Scholar] [CrossRef]
- Maffei, A.V.; Budd, P.M.; McKeown, N.B. Adsorption Studies of a Microporous Phthalocyanine Network Polymer. Langmuir 2006, 22, 4225–4229. [Google Scholar] [CrossRef] [PubMed]
- Neti, V.S.P.K.; Wang, J.; Deng, S.; Echegoyen, L. High and selective CO2 adsorption by a phthalocyanine nanoporous polymer. J. Mater. Chem. A 2015, 3, 10284–10288. [Google Scholar] [CrossRef]
- Ma, P.; Lv, L.; Zhang, M.; Yuan, Q.; Cao, J.; Zhu, C. Synthesis of catalytically active porous organic polymer from iron phthalocyanine and diimide building blocks. J. Porous Mater. 2015, 22, 1567–1571. [Google Scholar] [CrossRef]
- Xue, Q.; Xu, Z.; Jia, D.; Li, X.; Zhang, M.; Bai, J.; Li, W.; Zhang, W.; Zhou, B.; Wang, J. Solid-Phase Synthesis Porous Organic Polymer as Precursor for Fe/Fe3 C-Embedded Hollow Nanoporous Carbon for Alkaline Oxygen Reduction Reaction. ChemElectroChem 2019, 6, 4491–4496. [Google Scholar] [CrossRef]
- Ganji, P.; Doyle, D.J.; Ibrahim, H. In situ Generation of the Coates Catalyst: A Practical and Versatile Catalytic System for the Carbonylation of meso-Epoxides. Org. Lett. 2011, 42, 3142–3145. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.H.; Song, T.; Seo, U.R.; Park, J.E.; Cho, K.; Lee, S.M.; Kim, H.; Ko, Y.-J.; Chung, Y.K.; Son, S.U. Hollow and microporous catalysts bearing Cr(III)–F porphyrins for room temperature CO2 fixation to cyclic carbonates. J. Mater. Chem. A 2017, 5, 23612–23619. [Google Scholar] [CrossRef]
- Deng, F.-G.; Hu, B.; Sun, W.; Chen, J.; Xia, C. Novel pyridinium based cobalt carbonyl ionic liquids: Synthesis, full characterization, crystal structure and application in catalysis. Dalton Trans. 2007, 4262–4267. [Google Scholar] [CrossRef]
- Edgell, W.F.; Lyford, J. Preparation of sodium cobalt tetracarbonyl. Inorg. Chem. 1970, 9, 1932–1933. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry a | Catalyst | Solvent | Epoxide/Co Ratio b | T (°C) | Time (h) | Yield c (%) | Lactone (%) | Acetone (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | 4 | DME | 200 | 60 | 24 | >99 | >99 | <1 |
| 2 | 4 | THF | 200 | 60 | 24 | 50 | 99 | 1 |
| 3 | 4 | 1,4-dioxane | 200 | 60 | 24 | 52 | >99 | <1 |
| 4 | 4 | Toluene | 200 | 60 | 24 | 75 | >99 | <1 |
| 5 | [Pc’Cr][Co(CO)4] | DME | 200 | 60 | 24 | >99 | 99 | 1 |
| 6 | 4 | DME | 400 | 60 | 24 | 52 | >99 | <1 |
| 7 | 4 | DME | 200 | 60 | 12 | 70 | >99 | <1 |
| 8 | 4 | DME | 200 | 60 | 1 | 22 | >99 | <1 |
| 9 | 4 | DME | 100 | 30 | 24 | 40 | >99 | trace |
| 10 | 3 | DME | 200 | 60 | 24 | 12 d |
| Cycle | Yield (%) | Selectivity (%) |
|---|---|---|
| β-Butyrolactone/Acetone | ||
| 1 | >99 | >99/<1 |
| 2 | 98 | >99/<1 |
| 3 | 85 ± 6 | >99/<1 |
| 4 * | 98 | >99/<1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ganesan, V.; Yoon, S. Cr-Phthalocyanine Porous Organic Polymer as an Efficient and Selective Catalyst for Mono Carbonylation of Epoxides to Lactones. Catalysts 2020, 10, 905. https://doi.org/10.3390/catal10080905
Ganesan V, Yoon S. Cr-Phthalocyanine Porous Organic Polymer as an Efficient and Selective Catalyst for Mono Carbonylation of Epoxides to Lactones. Catalysts. 2020; 10(8):905. https://doi.org/10.3390/catal10080905
Chicago/Turabian StyleGanesan, Vinothkumar, and Sungho Yoon. 2020. "Cr-Phthalocyanine Porous Organic Polymer as an Efficient and Selective Catalyst for Mono Carbonylation of Epoxides to Lactones" Catalysts 10, no. 8: 905. https://doi.org/10.3390/catal10080905
APA StyleGanesan, V., & Yoon, S. (2020). Cr-Phthalocyanine Porous Organic Polymer as an Efficient and Selective Catalyst for Mono Carbonylation of Epoxides to Lactones. Catalysts, 10(8), 905. https://doi.org/10.3390/catal10080905