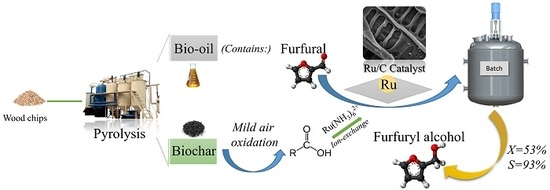
1. Introduction
In an effort to find alternatives to fossil fuel, biomass has been proposed as a source of carbon to produce bio-oil [
1]. The bio-oil, however, has much lower energy than traditional petroleum fuels, due to its high oxygen content (45 to 50 wt.%), low pH value, complex composition, and instability leading to phase separation with time. Bio-oil conversion to liquid fuel thus requires refining processes typically performed on crude-oil, such as aqueous-phase processing, hydrotreating, and thermal cracking [
2,
3].
Sanna et al. [
4] mentioned that despite the expenses of hydrotreating processes, they should be implemented as they provide higher selectivities to desired hydrocarbon products. Elliott [
5] suggested considering hydrodeoxygenation (HDO) as a crucial step of bio-oils treatment, removing the oxygen present in unsaturated oxygen-containing functionalities in the liquid. Following earlier suggestions [
6], he has thus proposed two-step HDO processes for pyrolysis oil, first performing mild hydrogenation under low temperature conditions, e.g., 100 to 140
, to saturate aldehydes and ketones. This step increases the stability of the oil and reduces the rate of coke formation under the severe conditions of HDO [
7]. The second step is then bio-oil hydrotreating at higher temperature (200 to 300
), with the aim of completely deoxygenating the bio-oil. The present work focuses on the first step, namely aldehyde conversion exemplified by furfural hydrogenation.
Investigating the opportunities for bio-based products, Bozell and Petersen [
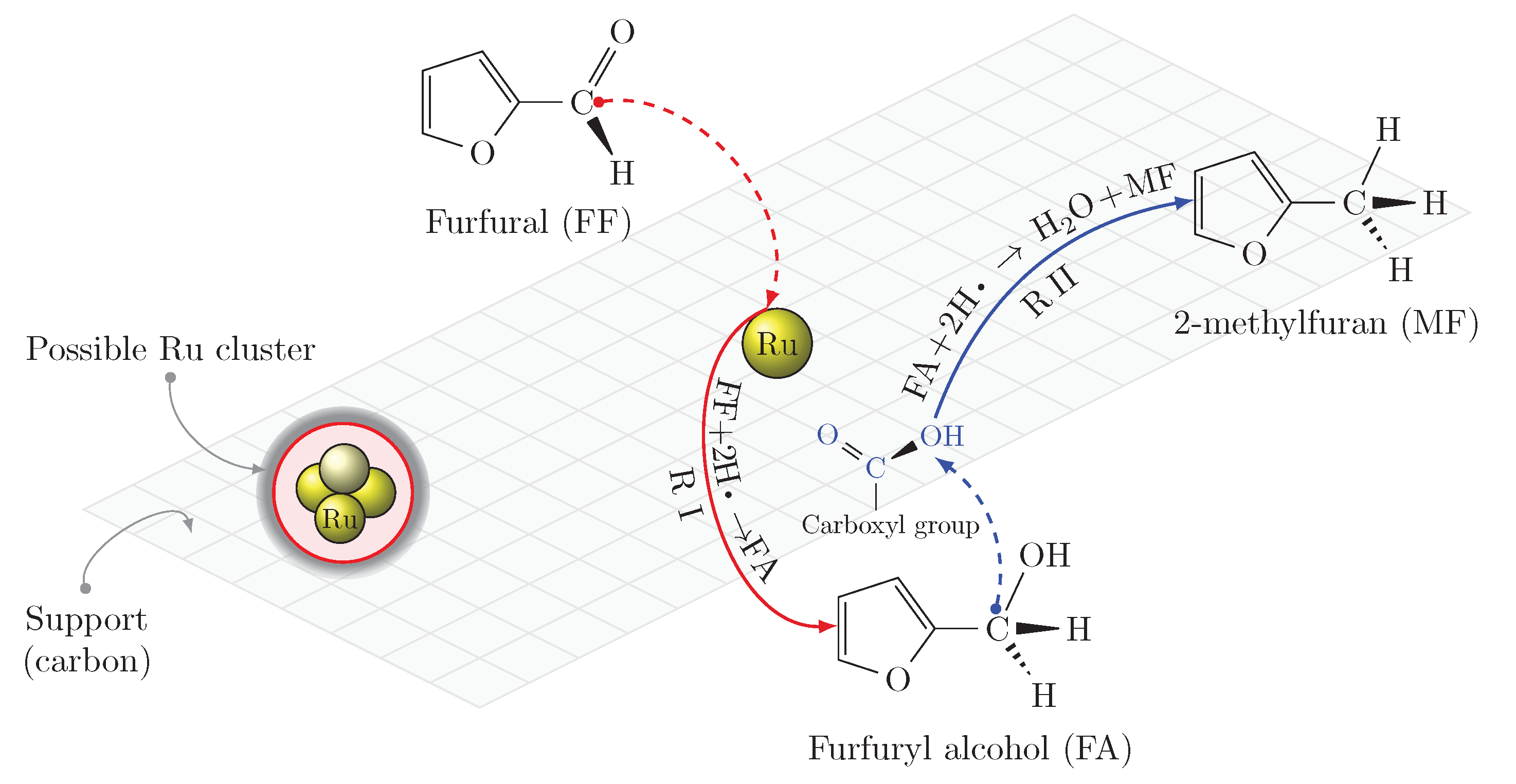
8] have listed furfural as one of the top ten chemicals, possessing nine special criteria and having received significant attention in the literature, to be considered to be an energy platform molecule. Furfuryl alcohol is one of the most interesting furfural derivatives, used in the production of dark thermostatic resins, synthetic fibers, and some dietary supplements, such as vitamin C [
9,
10,
11]. Furfuryl alcohol is the almost exclusive intermediate in the commercial production of levulinic acid, another identified platform molecule [
12].
Hydrogenation of furfural to furfuryl alcohol requires catalysts. As reviewed by Long et al. [
13], it was performed using non-precious metal such as Cu-based catalysts, ferrous metal (Fe, Ni, Co)-based catalysts, solid acid-based catalysts such as
, and precious metals such as Ru, Pt, Pd-based catalysts. Apart from reaction conditions, the type of metal indeed affects product selectivity. For instance, Cu-based catalysts could not open furan rings due to their exclusion from copper surfaces, but they have been reported to strongly react with C=O bonds. Using these catalysts, the reaction should however be performed under severe high temperature and pressure conditions [
14]. According to a review of aqueous-phase hydrogenation of bio-sourced chemicals published by Besson et al. [
15], ruthenium metal particles supported on carbons and oxides have been reported as the most promising catalysts, resulting in a rapid and selective conversion of carbonyl moieties of aldehydes into their corresponding alcohols [
16]. To the best of our knowledge, Kaliaguine’s group was the first to report successful hydrogenation of aldehydes in bio-oils using Ru-based catalysts [
6].
Ru catalysts can be synthesized in supported or unsupported form. The deposition of active metals on a support prevents their sintering, thereby improving the catalytic activity. Among the different types of supports such as silica [
17], alumina [
18], and zeolites [
19], carbonaceous materials have been vastly used, notably owing to their unique characteristics [
20]. These include high surface area, the presence of oxygen-containing surface functional groups, physical stability, and chemical inertness [
21,
22]. Most of biochars show however poor specific surface area and surface density of oxygen-containing functional groups, thus requiring surface modification [
23]. A common method of catalysts preparation is incipient wetness impregnation (IWI), where a porous support is filled with a certain quantity of precursor solution equivalent to its total pore volume. Catalysts preparation via adsorption process is another method [
24,
25,
26,
27,
28,
29,
30,
31,
32,
33,
34]. Adsorption from aqueous solutions is based on ion-exchange (IE), mainly performed when a low metal loading is targeted [
35]. In comparison with IWI, adsorption is used for supports with high density of acidic surface functional groups. It was reported that IE results in higher dispersion than IWI [
24,
36].
In the 2017 review reported by Lee et al. [
37], biochar was mentioned as an excellent catalyst support, but there is still meager research in this particular application. The main problem is still finding an efficient approach toward biochar impregnation with metal particles. A unique pyrolysis biochar with very low specific surface area was produced by Pyrovac Inc. [
38]. In the present work, this biochar surface was modified by mild air/steam oxidation to provide this material with high density of carboxylic surface functional groups, allowing catalysts preparation via ion-exchange. The prepared catalysts were characterized using
physisorption, elemental and ICP analyses, SEM/EDX, TEM,
chemisorption, TPR, and XPS. Catalysts activity was examined in the hydrogenation of furfural under mild temperature conditions using an autoclave batch reactor. The products were quantified using GC analysis. The effects of reaction parameters including temperature,
pressure, mass of catalyst, and agitation time were also investigated.
3. Experimental
3.1. Materials and Methods
Chemicals were all purchased from Sigma-Aldrich
® Co. (Oakville, ON, Canada) and Alfa Aesar (Fisher Scientific™ Inc. (Ottawa, ON, Canada)). Gases were acquired from Praxair Inc. of Canada. Biochar sample designated as “BC” was provided by Pyrovac Inc. (Saint-Lambert-de-Lauzon, Quebec, Canada), and produced using the pyrolysis process described elsewhere [
42].
Oxidation was performed in multiple steps under flowing steam and air at 230
. About 4
of sample was placed in the center of a furnace using stainless steel sample holder. Temperature was raised with heating ramp of 3
/
−1. Boehm titration was used to quantify the concentration of total acidic functional groups (
), as described elsewhere [
38]. The best results in terms of the highest value of
equal to
, were obtained for 4
of sample oxidation under the conditions described below.
At 230 , air was fed into the reactor at a flow rate of −1 for 1 . The temperature detected by a thermocouple on sample surface was suddenly raised and reached 375 after 40 under flowing air, and then dropped off very slowly. After 1 of air injection when the recorded temperature was still of 345 , steam at 230 was fed to the reactor keeping the air flow. This step was considered to control the raise in temperature arising from the biochar heat of combustion. The temperature monitored from sample surface went back to 230 after 30 of steam injection. After 2 of the steam injection, the sample was held in the reactor for an extra hour under dry air, while no change in the final recorded temperature was observed. The oxidized sample was designated as sample “OBC”.
3.2. Catalysts Preparation and Reaction Procedures
A Ru loading of 2.5 wt.% was targeted in the synthesis of catalysts. Around 272 of was added to 1 of distilled water mixed with 10 of (35 wt.%), to prevent the hydrolysis of ruthenium cations. About 4 of samples “BC” or “OBC” were separately added to the prepared solutions, mixed for 24 , and then filtered. The filtrates were analyzed by ICP, and the solids were dried overnight at 75 . The catalyst particle size was about 105 (a sieve no. 140). Samples “BC” and “OBC” loaded with ruthenium were designated as “Ru-BC” and “Ru-OBC”, respectively.
The catalytic activity was investigated in the hydrogenation of furfural using a high-pressure autoclave reactor, connected to a pressurized pure hydrogen cylinder. Reaction mixture was prepared by vigorously mixing 1
of FF in 50
deionized water. Prior to each reaction test, the catalyst was first reduced at 450
(heating ramp of 7
−1) for 2
, using a 20
−1 flow of pure hydrogen. The reduced catalyst was cooled down to room temperature under flowing hydrogen, and immediately transferred to the reaction mixture. The reactor was sealed, and slowly purged with pure hydrogen for 30
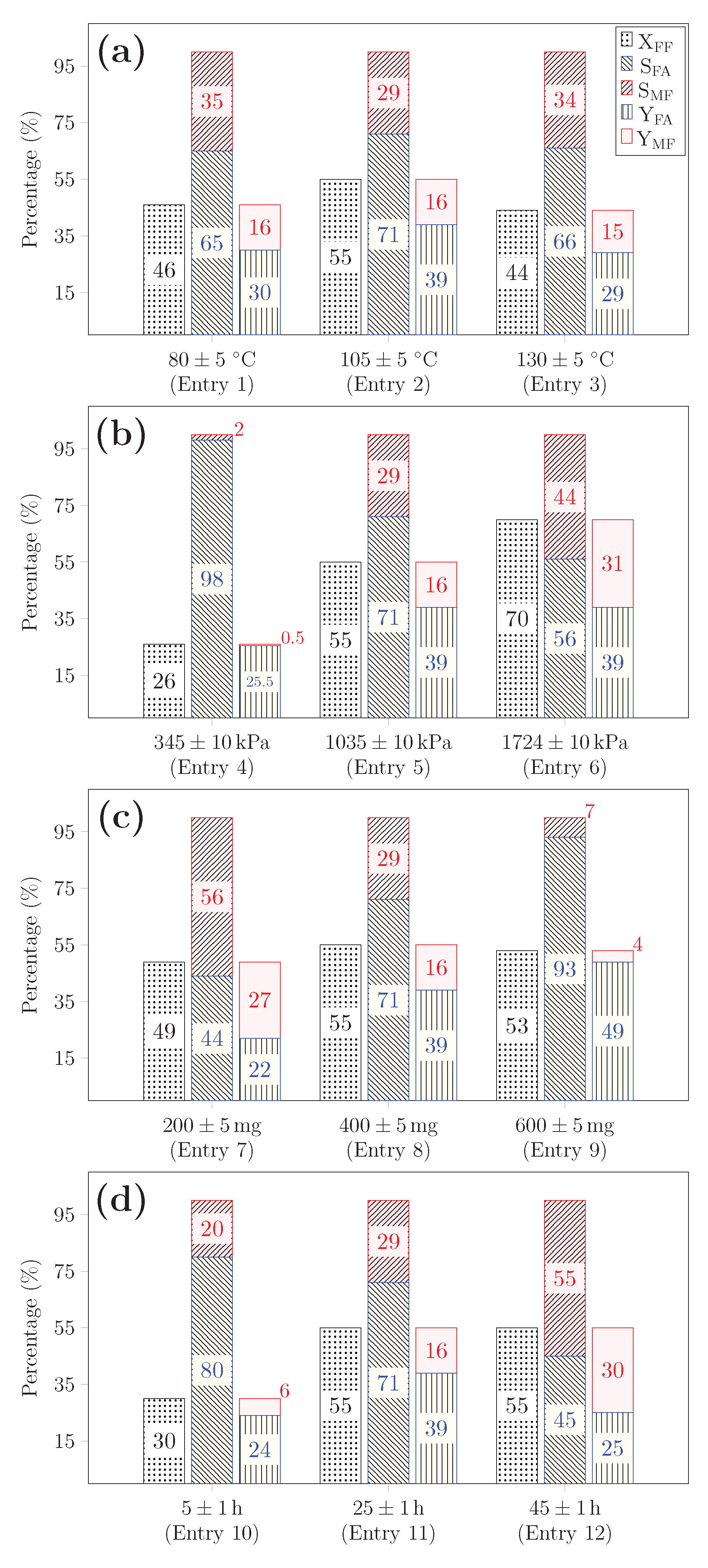
. It was then pressurized to the desired pressure, followed by rising temperature to a targeted value. The mixture was continuously agitated. To stop the reaction, the reactor was first placed in a bath of iced water, then slowly depressurized. The reaction mixtures were separated from catalysts by filtration. Furfural conversion (
X), furfuryl alcohol selectivity (
) and yield (
) were established using Equations (
1)–(
3):
and
are the initial and the total consumed moles of furfural, respectively.
is the number of moles of furfuryl alcohol.
3.3. Analyses
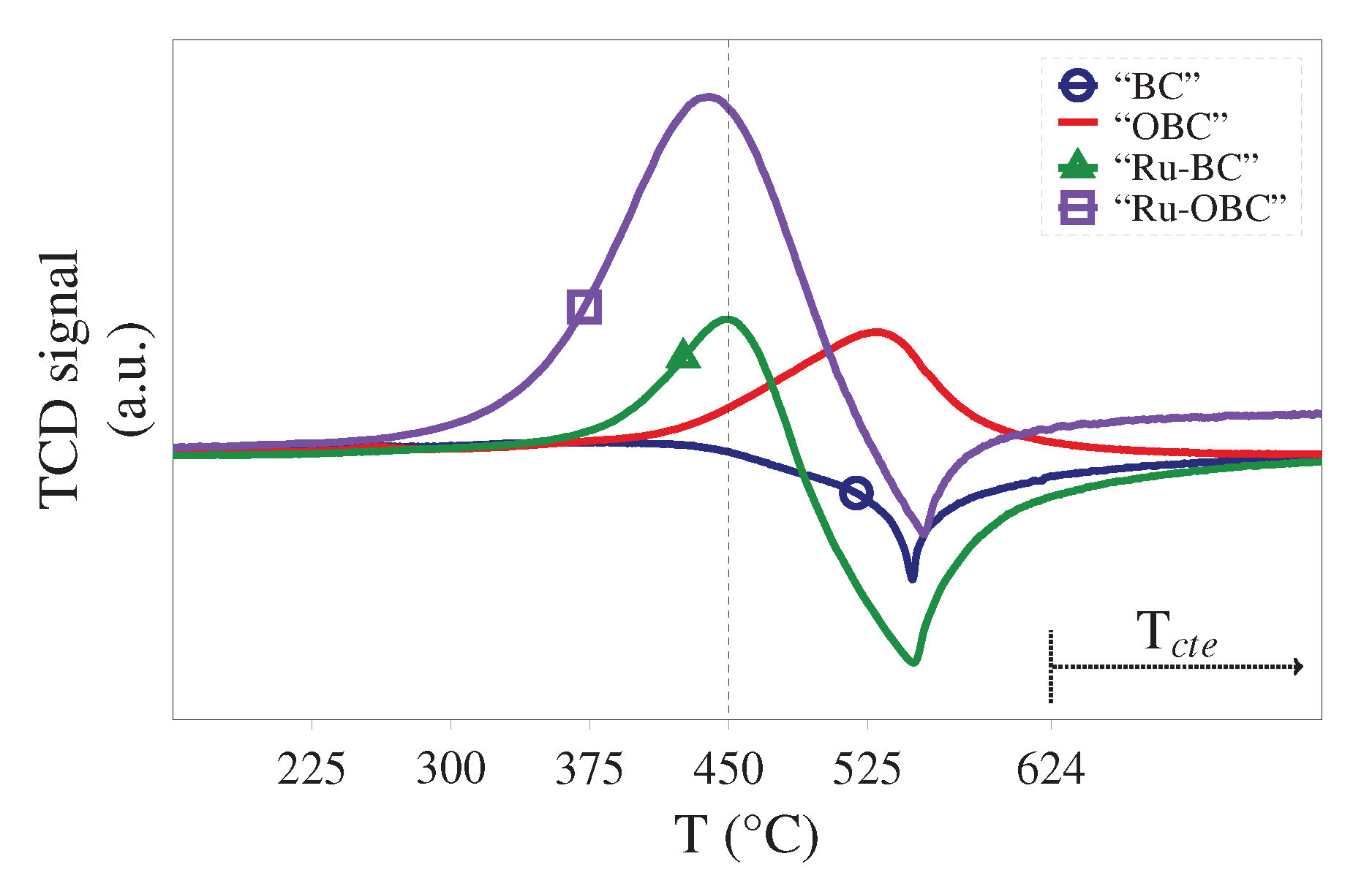
Surface area and pore size distribution of catalyst samples were obtained from nitrogen adsorption-desorption isotherms analysis using a Quantachrome NOVA 2000 surface area analyzer. Sample degassing was performed at 300 for 48 under vacuum. Adsorption-desorption isotherms were analyzed using the Quantachrome Autosorb 1 software (version 1.55). Specific surface area was obtained using BET equation. Pore size distribution was established using BJH method and the Density Functional Theory (DFT) (two approaches of Non-Linear (NLDFT) and Quenched Solid (QSDFT)).
Elemental analysis was obtained by CHN and Inductively Coupled Plasma-Optical Emission Spectrometry (ICP-OES) analyses, using a LECO CHN 628 series elemental analyzer (LECO Inc., St. Joseph, MI, USA) and an Agilent 5110 SVDV spectrometer (Agilent Inc., Santa Clara, CA, USA), respectively.
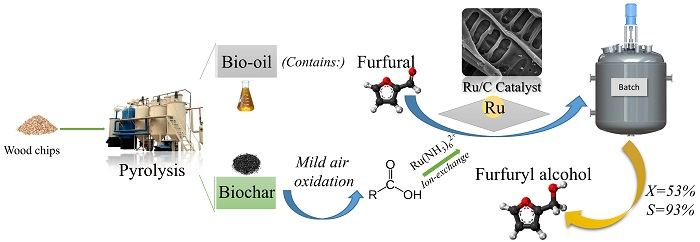
Samples were analyzed by Temperature-Programmed Reduction (TPR) using an (ASDI, RXM-100 model) analyzer. The results were collected using the ASDI software (version 4.12). About 100 of sample was placed in a quartz U-shaped tube reactor. Temperature was raised from room temperature to 450 at a heating ramp of 7 −1 and kept under pure flowing argon for 2 . The sample was then cooled down to room temperature under flowing argon. TCD signal was recorded upon heating the sample from room temperature to 625 under flowing 5 vol.% in Ar. Reaching 625 , the sample was held for an extra hour. Moisture leaving the sample surface was trapped prior to the TCD, in a long loop immersed in a bath of dry ice/ethanol.
Ruthenium dispersion was measured by pulse
-chemisorption using an AutoChem II chemisorption analyzer of Micromeritics Inc. To ascertain the metallic form of ruthenium, 100
sample was first heated to 450
with heating ramp of 8
−1 and kept for 2
under flowing pure hydrogen at a flow rate of 40
−1. It was then cooled down to room temperature after switching
to pure Ar. The first sorption of 5 vol.%
in Ar was performed at 75
as suggested by Shen et al. [
70], using a 100
loop. Then, the sample was again heated to 350
to remove reversibly adsorbed hydrogen atoms, cooled down to 75
, and followed by a second sorption in the same conditions. The stoichiometric ratio of H/Ru was considered equal to 1. The difference in the volume of adsorbed
between the first and the second sorptions indicates the amount of strongly adsorbed hydrogen. Dispersion and particle average size calculations were made from this amount, using Refs. [
45,
71].
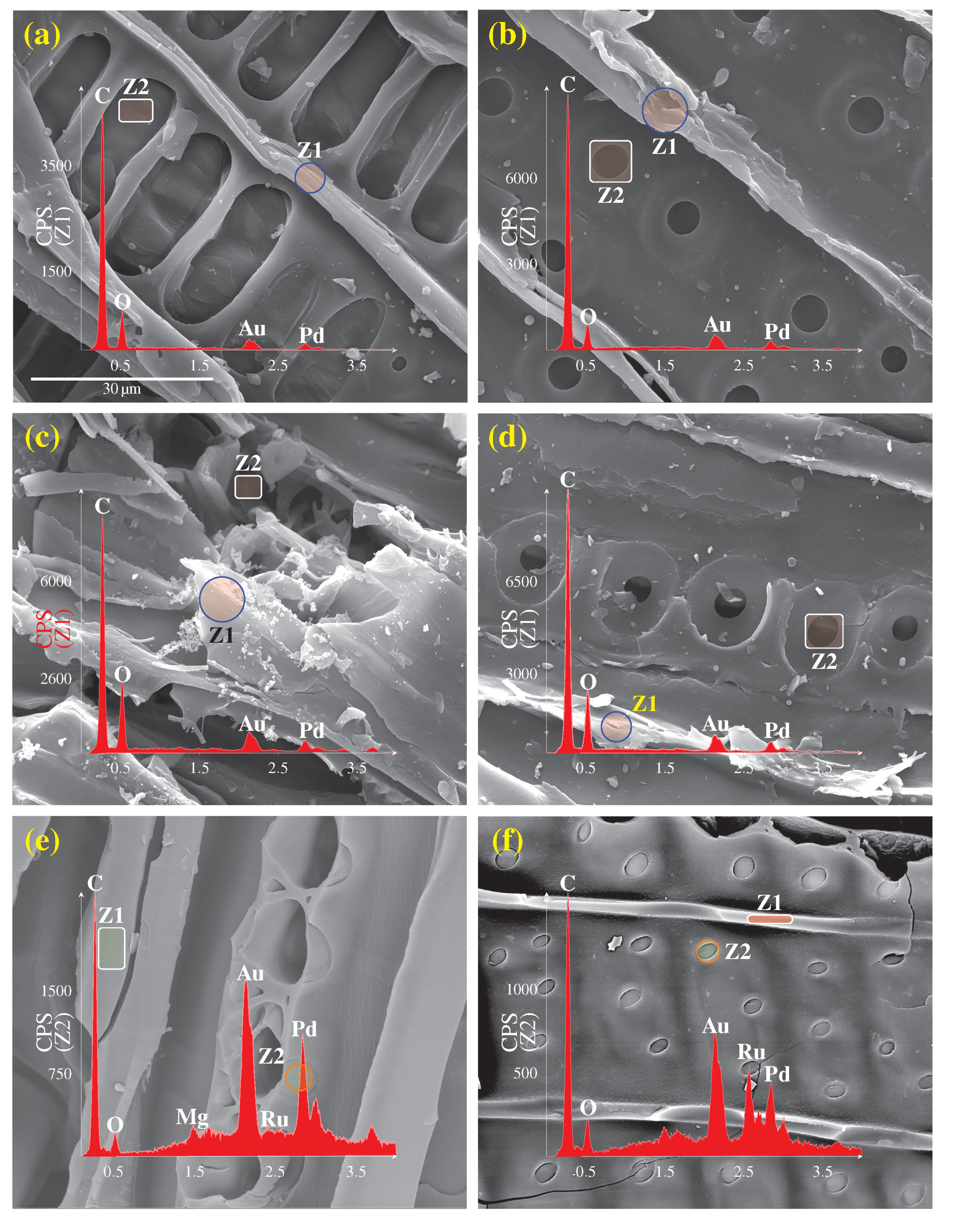
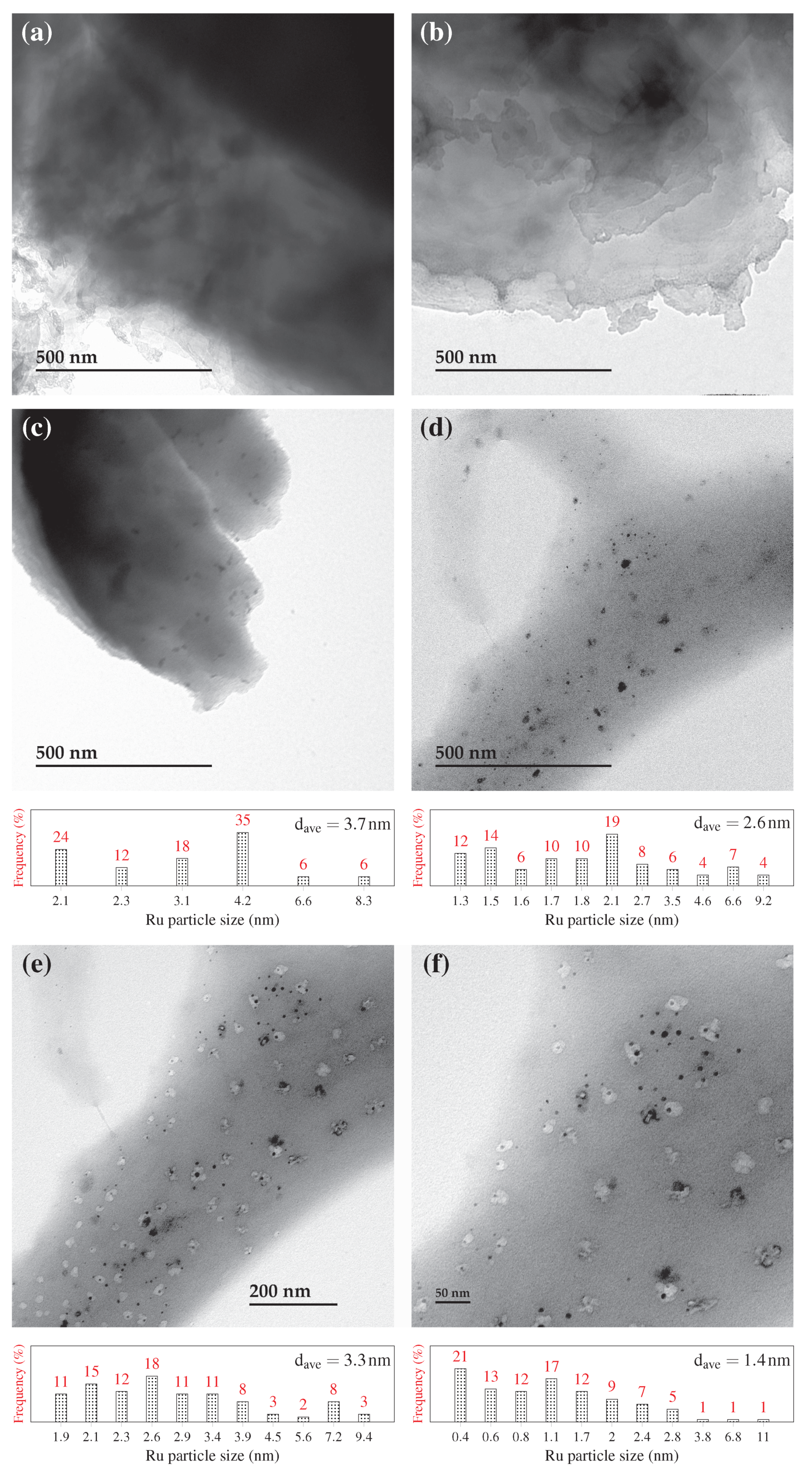
The morphology of samples was investigated using a Scanning Electron Microscope (SEM) equipped with Energy Dispersive X-Ray (EDX) and a Transmission Electron Microscope (TEM) (JSM-840A and JEM-1230 JEOL series microscopes, respectively). For SEM analysis, the dried specimen was mounted on a copper sample holder, then sputtered with palladium and gold in a vacuum chamber. For samples loaded with Ru, the specimen surface was first examined with SEM electron back-scattering. The EDX signals were thus recorded from places of interest using SEM secondary images. For TEM analysis, dried specimen in form of fine powder was first suspended in methanol. The solution was then deposited on a nickel grid support, followed by drying at room temperature. TEM images were processed using MATLAB
® software (MathWorks Inc., version R2018a), to determine the particle size distribution of Ru, assuming spherical particles. Ruthenium dispersion (
) was determined from TEM photographs using Equation (
4) [
72]:
and
are the volume and the surface occupied by a Ru atom equal to
and
, respectively. The average Ru particle diameter was obtained from
using TEM images, where
is the frequency of a particle with diameter
.
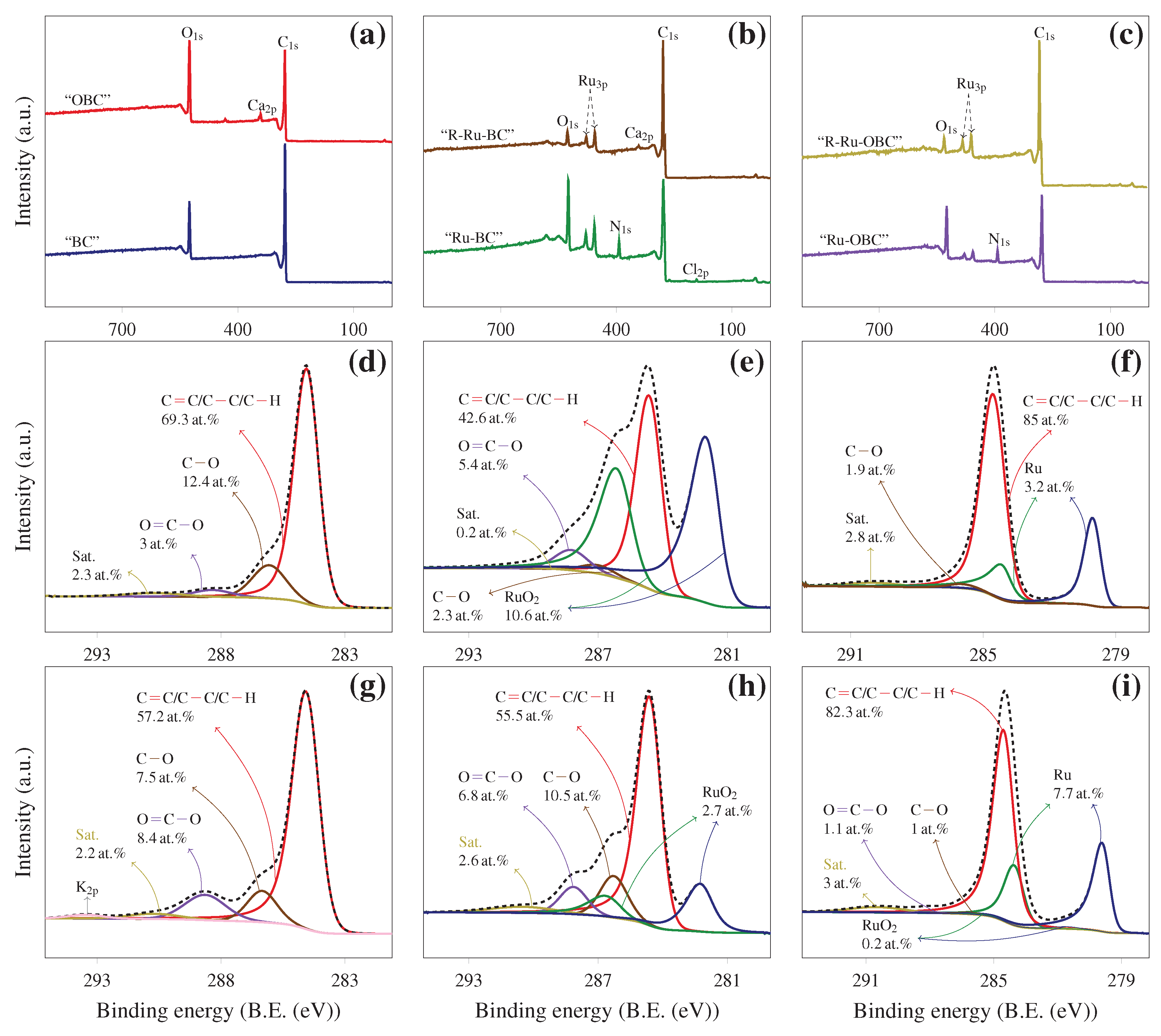
The surface chemistry of sample specimen was investigated by quantitative X-ray Photoelectron Spectroscopy (XPS) analysis using an ESCA spectrometer (Shimadzu Kratos AXIS-Ultra instrument, Shimadzu Inc., Wharfside Manchester, UK). The deconvolution of XPS spectra was obtained by setting binding energy scales to eV for graphitic carbon , using Gaussian-Lorentzian (GL) curve fitting. The XPS results were then treated using the CasaXPS software (version 2.3.15). Samples “Ru-BC” and “Ru-OBC”, were analyzed before (as made) and after reduction. The reduced samples were respectively designated as “R-Ru-BC” and “R-Ru-OBC”. Reduction was performed prior to the XPS analysis under conditions suggested by TPR analysis namely at 450 for 2 with a heating ramp of 7 −1 under pure flowing hydrogen.
In the catalytic tests, the concentrations of the reactant and products were obtained in −1 according to calibration curves obtained from standard solutions of detected components, using a Varian CP-3800 Gas Chromatograph (GC).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}