The Role of CO2 as a Mild Oxidant in Oxidation and Dehydrogenation over Catalysts: A Review


Abstract
:
1. Introduction
2. Effect of CO2 in Oxidation
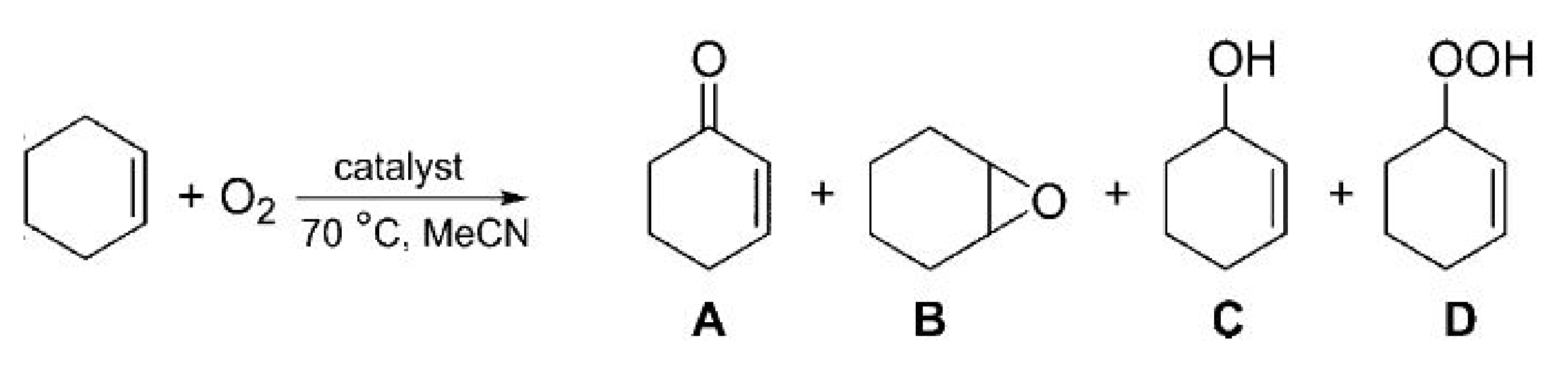
2.1. Influence of CO2 on Oxidation of Cyclohexene
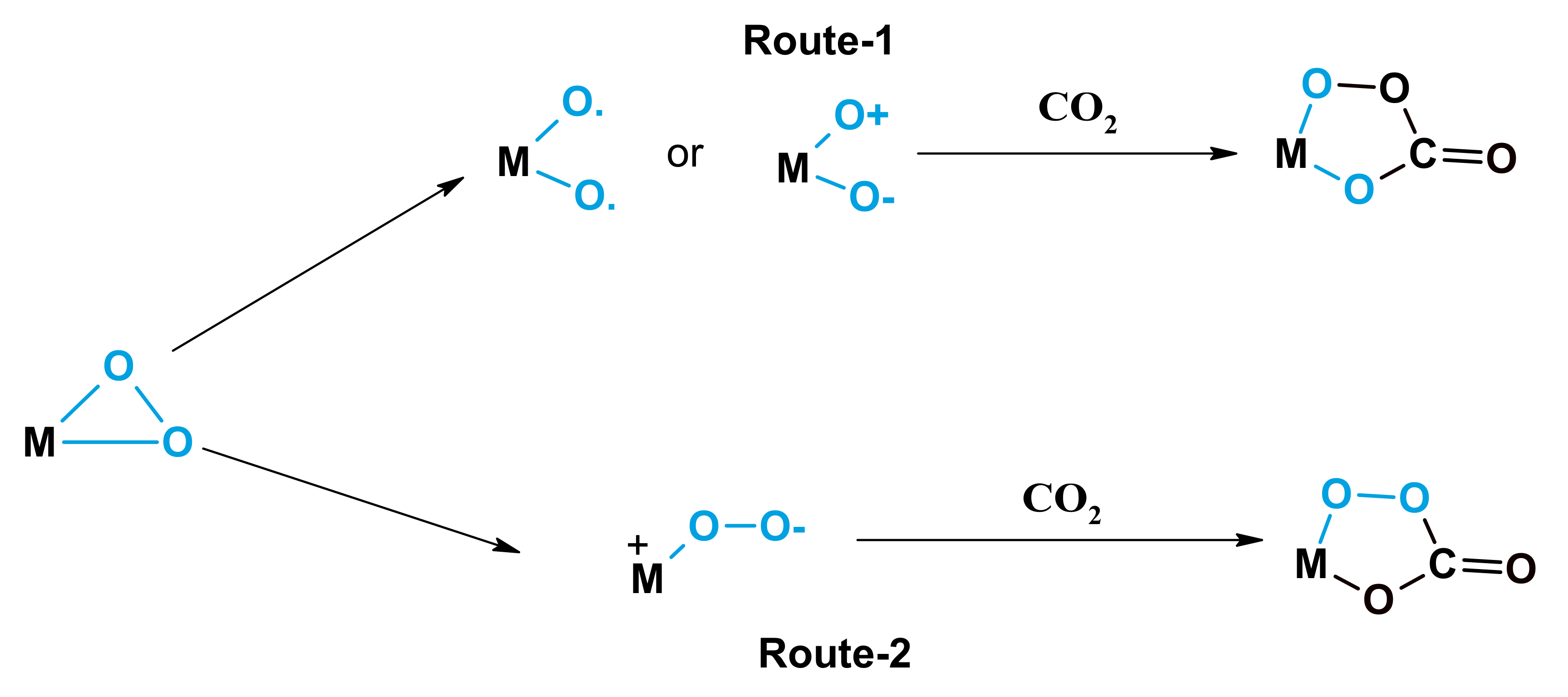
2.2. Promotional Effect of CO2 on Oxidation of Cyclic Olefins
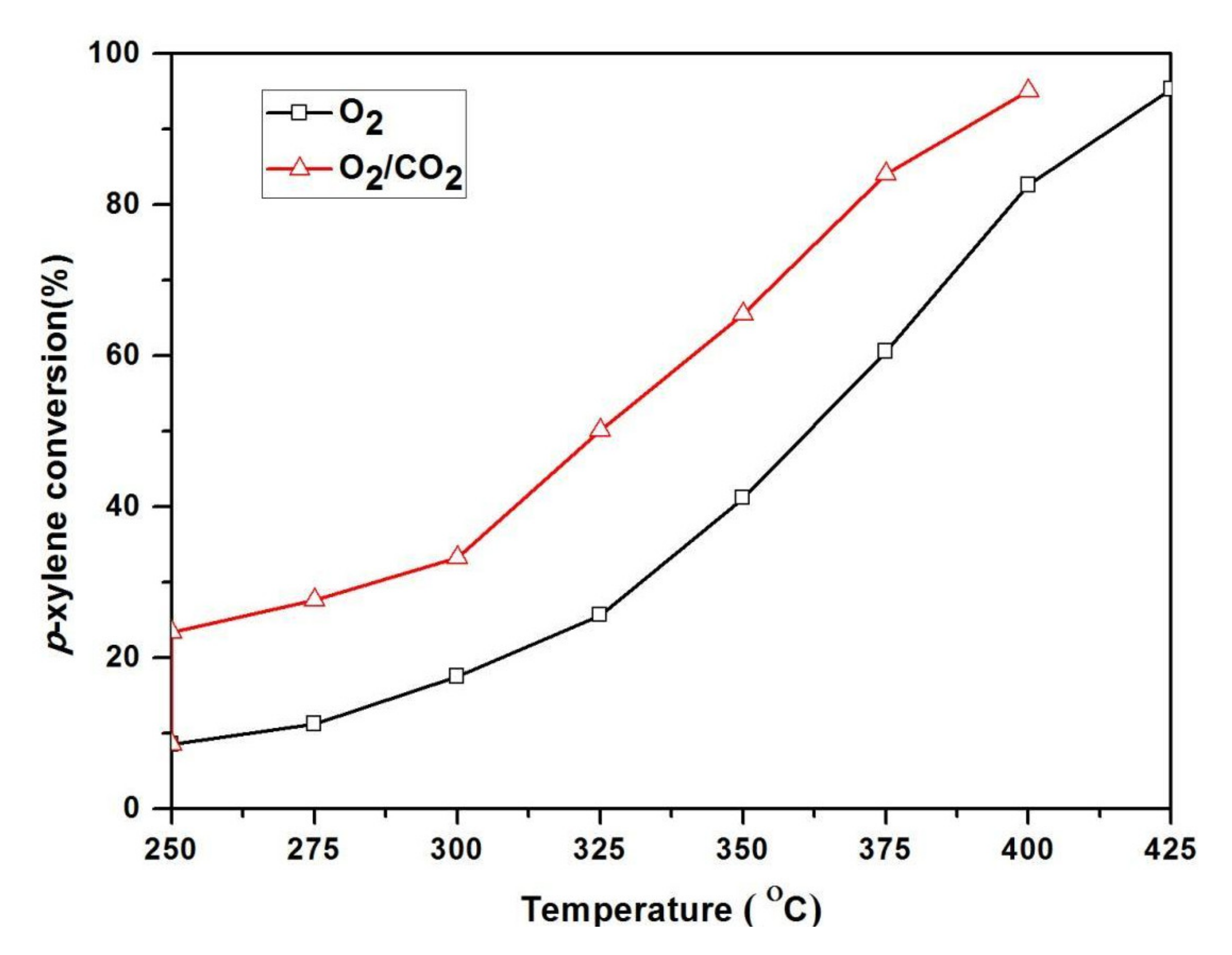
2.3. Influence of CO2 on Oxidation of p-Xylene
2.4. Oxidation of p-Toluic Acid and p-Methyl-Anisole
3. Performance of CO2 in Oxidative Dehydrogenation
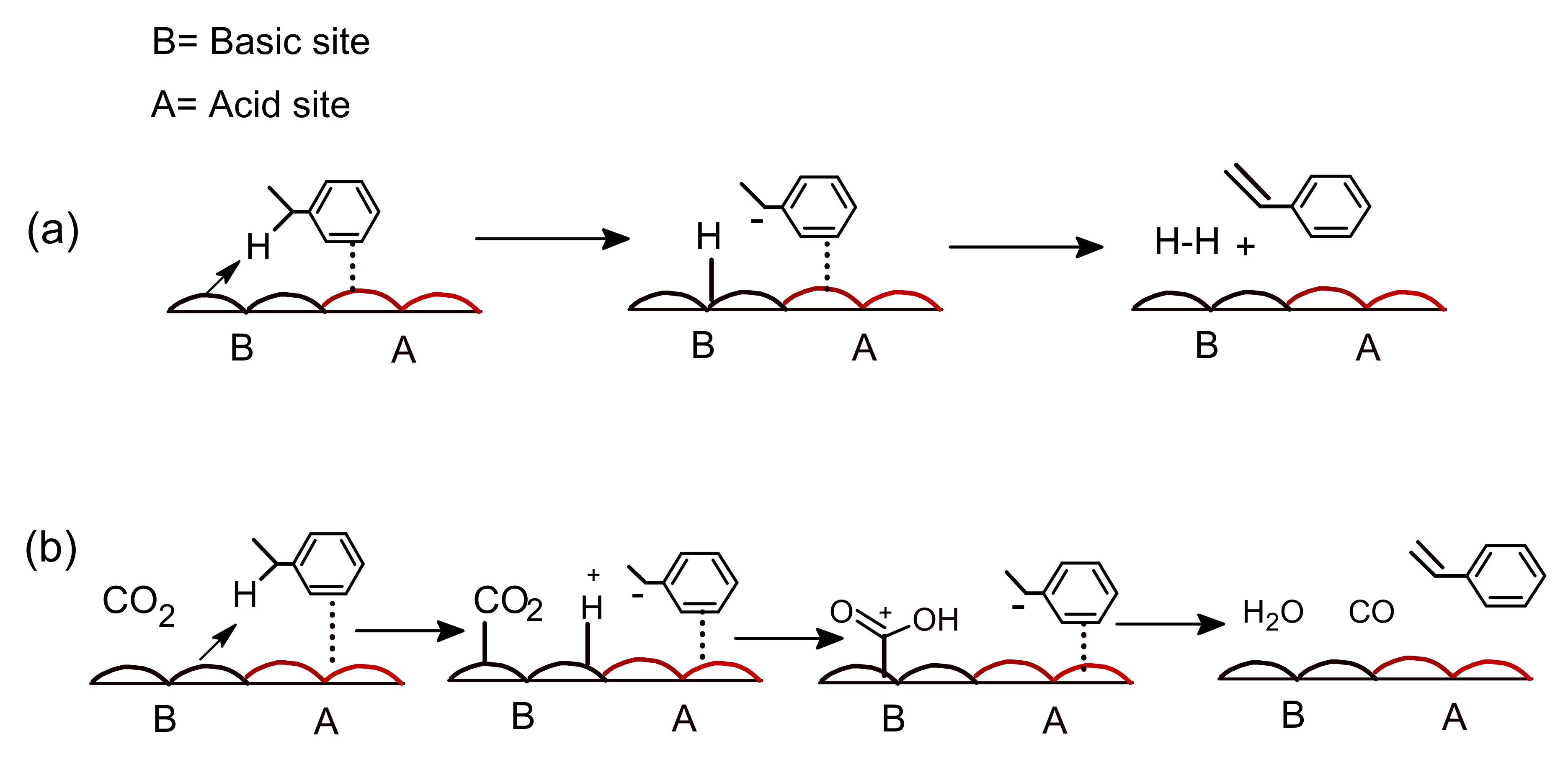
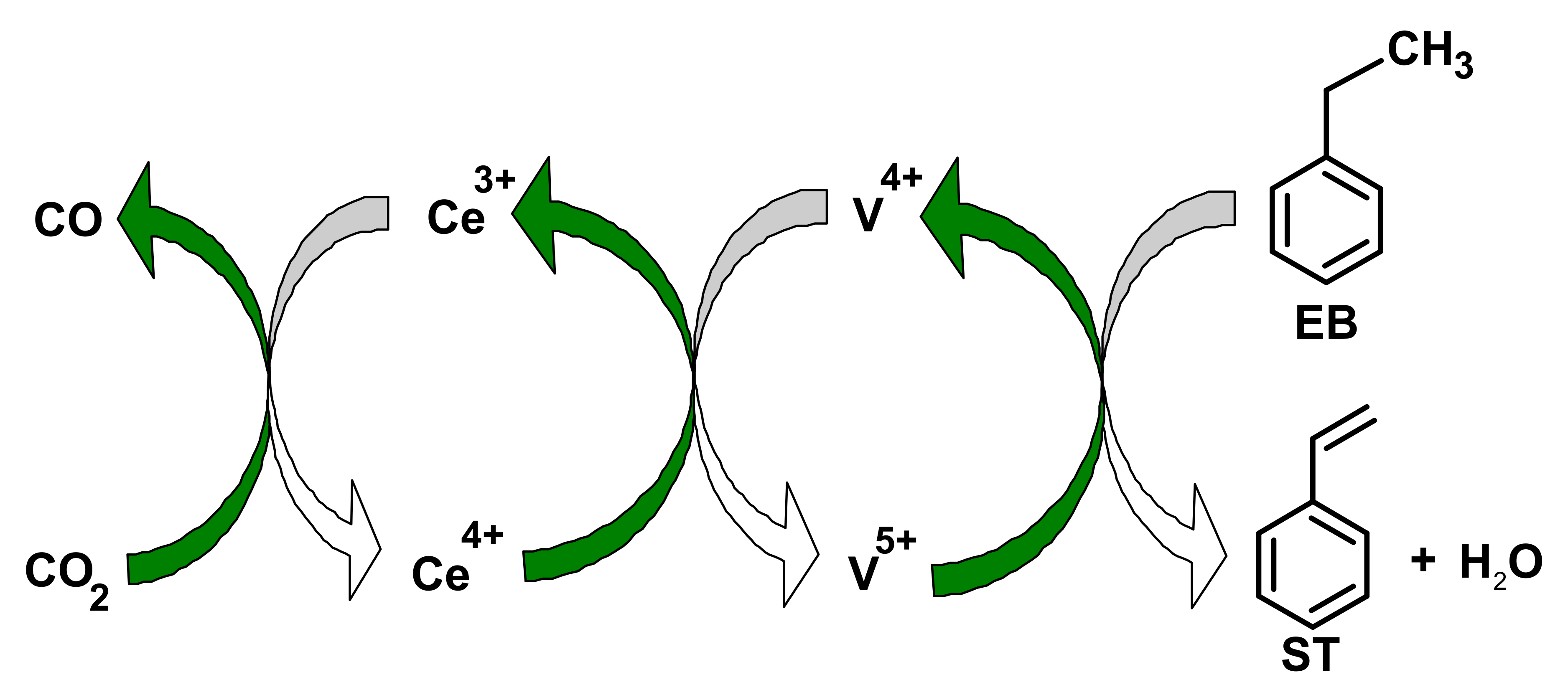
3.1. Influence of CO2 on Dehydrogenation of Ethyl Benzene
3.2. Performance of CO2 on Dehydrogenation of Ethane
3.3. Influence of CO2 on the Alkylation of Toluene Side-Chain
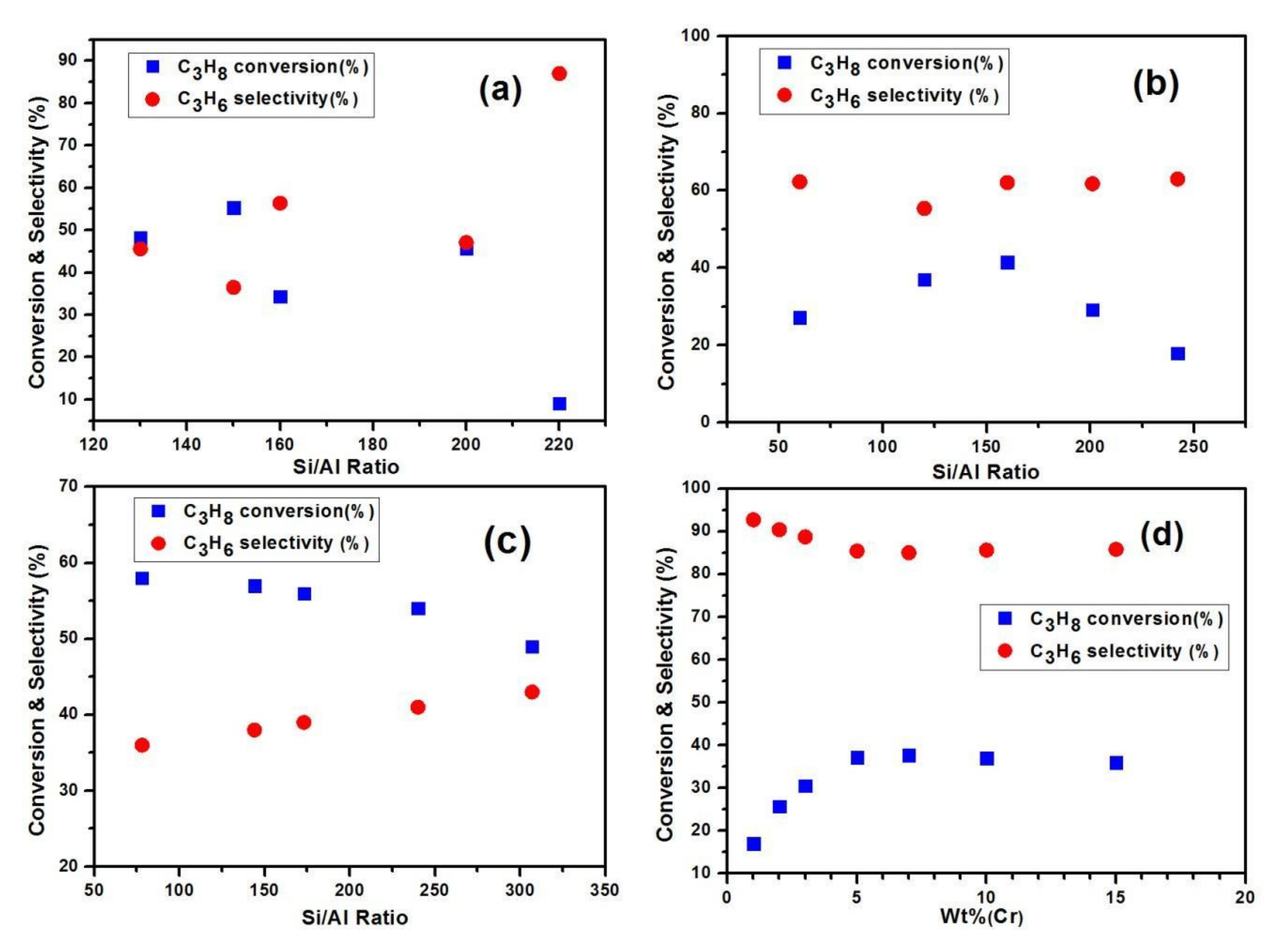
3.4. Role of CO2 on Dehydrogenation of Propane

4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kar, S.; Goeppert, A.; Prakash, G.K.S. Integrated CO2 Capture and Conversion to Formate and Methanol: Connecting Two Threads. Acc. Chem. Res. 2019, 52, 2892–2903. [Google Scholar] [CrossRef] [PubMed]
- Smol, J.P. Climate Change: A Planet in flux. Nature 2012, 483, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Ross, M.B.; De Luna, P.; Li, Y.; Dinh, C.-T.; Kim, D.; Yang, P.; Sargent, E.H. Designing materials for electrochemical carbon dioxide recycling. Nat. Catal. 2019, 2, 648–658. [Google Scholar] [CrossRef] [Green Version]
- De Luna, P.; Hahnet, C.; Higgins, D.; Jaffer, S.A.; Jaramillo, T.F.; Sargent, E.H. What would it take for renewably powered electrosynthesis to displace petrochemical processes? Science 2019, 364. [Google Scholar] [CrossRef] [Green Version]
- Nugent, P.; Belmabkhout, Y.; Burd, S.D.; Cairns, A.J.; Luebke, R.; Forrest, K.; Pham, T.; Ma, S.; Space, B.; Wojtas, L.; et al. Porous materials with optimal adsorption thermodynamics and kinetics for CO2 separation. Nature 2013, 495, 80–84. [Google Scholar] [CrossRef]
- Rehman, A.; Park, S.-J. From chitosan to urea-modified carbons: Tailoring the ultra-microporosity for enhanced CO2 adsorption. Carbon 2020, 159, 625–637. [Google Scholar] [CrossRef]
- Lee, S.H.; Sullivan, I.; Larson, D.M.; Liu, G.; Toma, F.M.; Xiang, C.; Drisdell, W.S. Correlating Oxidation State and Surface Area to Activity from Operando Studies of Copper CO Electroreduction Catalysts in a Gas Fed Device. ACS Catal. 2020, 10, 8000–8011. [Google Scholar] [CrossRef]
- Han, L.; Zhou, W.; Xiang, C. High-Rate Electrochemical Reduction of Carbon Monoxide to Ethylene Using Cu-Nanoparticles Based Gas Diffusion Electrodes. ACS Energy Lett. 2018, 3, 855–860. [Google Scholar] [CrossRef] [Green Version]
- Heo, Y.J.; Park, S.-J. Facile Synthesis of MgO-Modified Carbon Adsorbents with Microwave-Assisted Methods: Effect of MgO Particles and Porosities on CO2 Capture. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Qi, S.-C.; Wu, J.-K.; Lu, J.; Yu, G.-X.; Zhu, R.R.; Liu, Y.; Liu, X.-Q.; Sun, L.-B. Underlying mechanism of CO2 adsorption onto conjugated azacyclo-copolymers: N-doped adsorbents capture CO2 chiefly through acid-base interaction? J. Mater. Chem. A 2019, 7, 17842–17853. [Google Scholar] [CrossRef]
- Qi, S.C.; Liu, Y.; Peng, A.Z.; Xue, D.M.; Liu, X.; Liu, X.Q.; Sun, L.B. Fabrication of porous carbons from mesitylene for highly efficient CO2 capture: A rational choice improving the carbon loop. Chem. Eng. J. 2019, 361, 945–952. [Google Scholar] [CrossRef]
- Rehman, A.; Park, S.-J. Tunable nitrogen-doped microporous carbons: Delineating the role of optimum pore size for enhanced CO2 adsorption. Chem. Eng. J. 2019, 362, 731–742. [Google Scholar] [CrossRef]
- Jessop, P.G.; Ikariya, T.; Noyori, R. Homogeneous catalysis in supercritical fluids. Science 1995, 269, 1065–1069. [Google Scholar] [CrossRef]
- Musie, G.; Wei, M.; Subramaniam, B.; Busch, D.H. Catalytic oxidations in carbon dioxide-based reaction media, including novel CO2-expanded phases. Coord. Chem. Rev. 2001, 219–221, 789–820. [Google Scholar] [CrossRef]
- Heldebrant, D.J.; Jessop, P.G.; Thomas, C.A.; Eckert, C.A.; Liotta, C.L. The reaction of 1,8-diazabicyclo [5.4.0]undec-7-ene (DBU) with carbon dioxide. J. Org. Chem. 2005, 70, 5335–5338. [Google Scholar] [CrossRef]
- Chang, J.S.; Vislovskiy, V.P.; Park, M.S.; Hong, D.Y.; Yoo, J.S.; Park, S.E. Utilization of carbon dioxide as soft oxidant in the dehydrogenation of ethyl benzene over supported vanadium-antimony oxide catalysts. Green Chem. 2003, 5, 587–590. [Google Scholar] [CrossRef]
- Park, M.S.; Chang, J.S.; Kim, D.S.; Park, S.-E. Oxidative dehydrogenation of ethyl benzene with carbon dioxide over zeolite-supported iron oxide catalysts. Res. Chem. Intermed. 2002, 28, 461–469. [Google Scholar] [CrossRef]
- Sakakura, T.; Choi, J.C.; Yasuda, H. Transformation of carbon dioxide. Chem. Rev. 2007, 107, 2365–2387. [Google Scholar] [CrossRef]
- Reddy, B.M.; Lakshmanan, P.; Loridant, S.; Yamada, Y.; Kobayashi, T.; López-Cartes, C.; Rojas, T.C.; Fernández, A. Structural characterization and oxidative dehydrogenation Activity of v2O5/CexZr1-xO2/SiO2 catalysts. J. Phys. Chem. B 2006, 110, 9140–9147. [Google Scholar] [CrossRef]
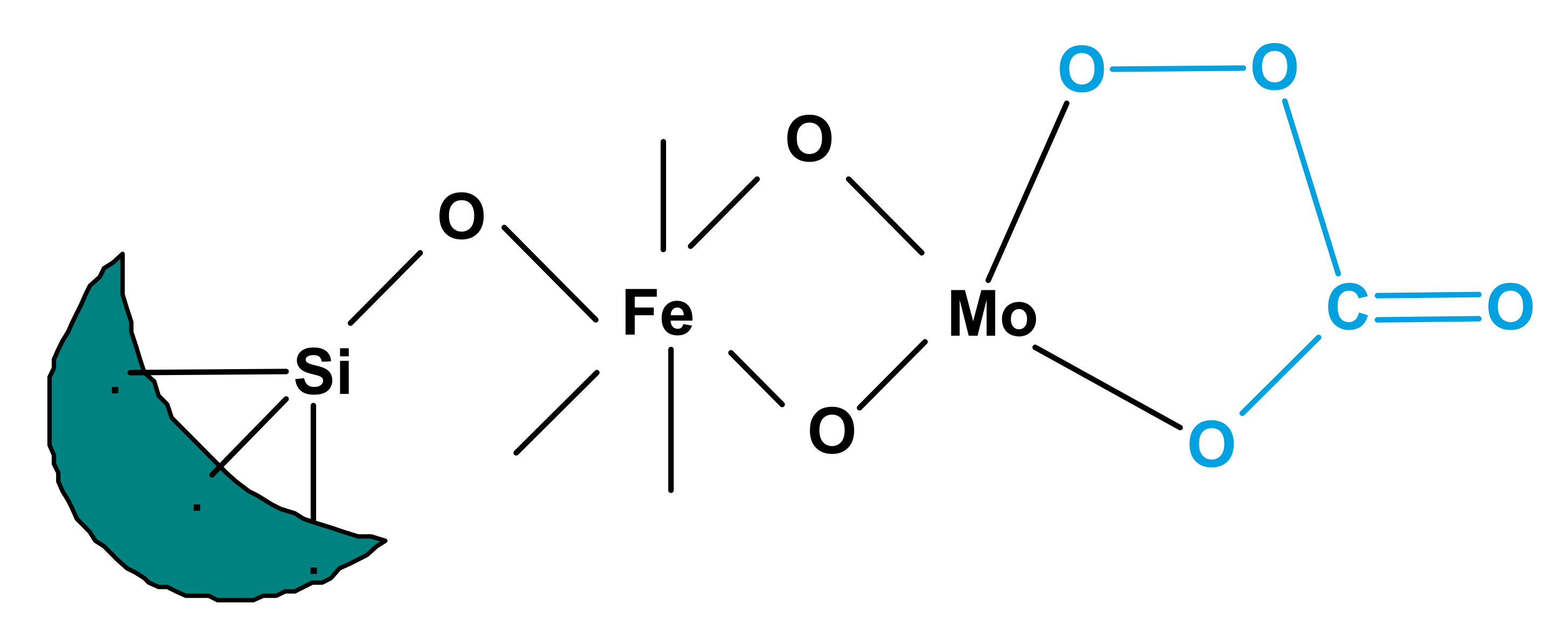
- Yoo, J.S.; Lin, P.S.; Elfline, S.D. Gas-phase oxygen oxidations of alkyl aromatics over CVD Fe/Mo/borosilicate molecular sieve. II. The role of carbon dioxide as a co-oxidant. Appl. Catal. A Gen. 1993, 106, 259–273. [Google Scholar] [CrossRef]
- Sun, A.; Qin, Z.; Wang, J. Reaction coupling of ethylbenzene dehydrogenation with water-gas shift. Appl. Catal. A Gen. 2002, 234, 179–189. [Google Scholar] [CrossRef]
- Ansari, M.B.; Park, S.E. Carbon dioxide utilization as a soft oxidant and promoter in catalysis. Energy Environ. Sci. 2012, 5, 9419–9437. [Google Scholar] [CrossRef]
- Abanades, S.; Le Gal, A. CO2 splitting by thermo-chemical looping based on ZrxCe1-xO2 oxygen carriers for synthetic fuel generation. Fuel 2012, 102, 180–186. [Google Scholar] [CrossRef]
- Wang, S.; Wang, X. Imidazolium ionic liquids, imidazolylidene heterocyclic carbenes, and zeolitic imidazolate frameworks for CO2 capture and photochemical reduction. Angew. Chemie Int. Ed. 2016, 55, 2308–2320. [Google Scholar] [CrossRef] [PubMed]
- Nikulshina, V.; Hirsch, D.; Mazzotti, M.; Steinfeld, A. CO2 capture from air and co-production of H2 via the Ca(OH)2-CaCO3 cycle using concentrated solar power-Thermodynamic analysis. Energy 2006, 31, 1715–1725. [Google Scholar] [CrossRef]
- Spinner, N.S.; Vega, J.A.; Mustain, W.E. Recent progress in the electrochemical conversion and utilization of CO2. Catal. Sci. Technol. 2012, 2, 19–28. [Google Scholar] [CrossRef]
- Singh, G.; Lakhi, K.S.; Ramadass, K.; Sathish, C.I.; Vinu, A. High-Performance Biomass-Derived Activated Porous Biocarbons for Combined Pre- and Post-Combustion CO2 Capture. ACS Sustain. Chem. Eng. 2019, 7, 7412–7420. [Google Scholar] [CrossRef]
- Grice, K.A. Carbon dioxide reduction with homogenous early transition metal complexes: Opportunities and challenges for developing CO2 catalysis. Coord. Chem. Rev. 2017, 336, 78–95. [Google Scholar] [CrossRef]
- Bartholomew, C.H. Catalyst Deactivation 1991; Elsevier: Amsterdam, The Netherlands, 1991; pp. 96–112. [Google Scholar]
- Denekamp, I.M.; Antens, M.; Slot, T.K.; Rothenberg, G. Selective Catalytic Oxidation of Cyclohexene with Molecular Oxygen: Radical Versus Nonradical Pathways. ChemCatChem 2018, 10, 1035–1041. [Google Scholar] [CrossRef]
- Ansari, M.B.; Min, B.H.; Mo, Y.H.; Park, S.-E. CO2 activation and promotional effect in the oxidation of cyclic olefins over mesoporous carbon nitrides. Green Chem. 2011, 13, 1416–1421. [Google Scholar] [CrossRef]
- Aresta, M.; Tommasi, I.; Quaranta, E.; Fragale, C.; Tranquille, M.; Galan, F.; Fouassier, M. Mechanism of formation of peroxycarbonates RhOOC(O)O(Cl)(P)3 and Their Reactivity as Oxygen Transfer Agents Mimicking Monooxygenases. The First Evidence of CO2 Insertion into the O-O Bond of Rh(η2-O2) Complexes. Inorg. Chem. 1996, 35, 4254–4260. [Google Scholar] [CrossRef] [PubMed]
- Iwata, R.; Ido, T.; Fujisawa, Y.; Yamazaki, S. On-line interconversion of [15O]O2 and [15O]CO2 via metal oxide by isotopic exchange. Int. J. Radiat. Appl. Instrum. Part A. 1988, 39, 1207–1211. [Google Scholar] [CrossRef]
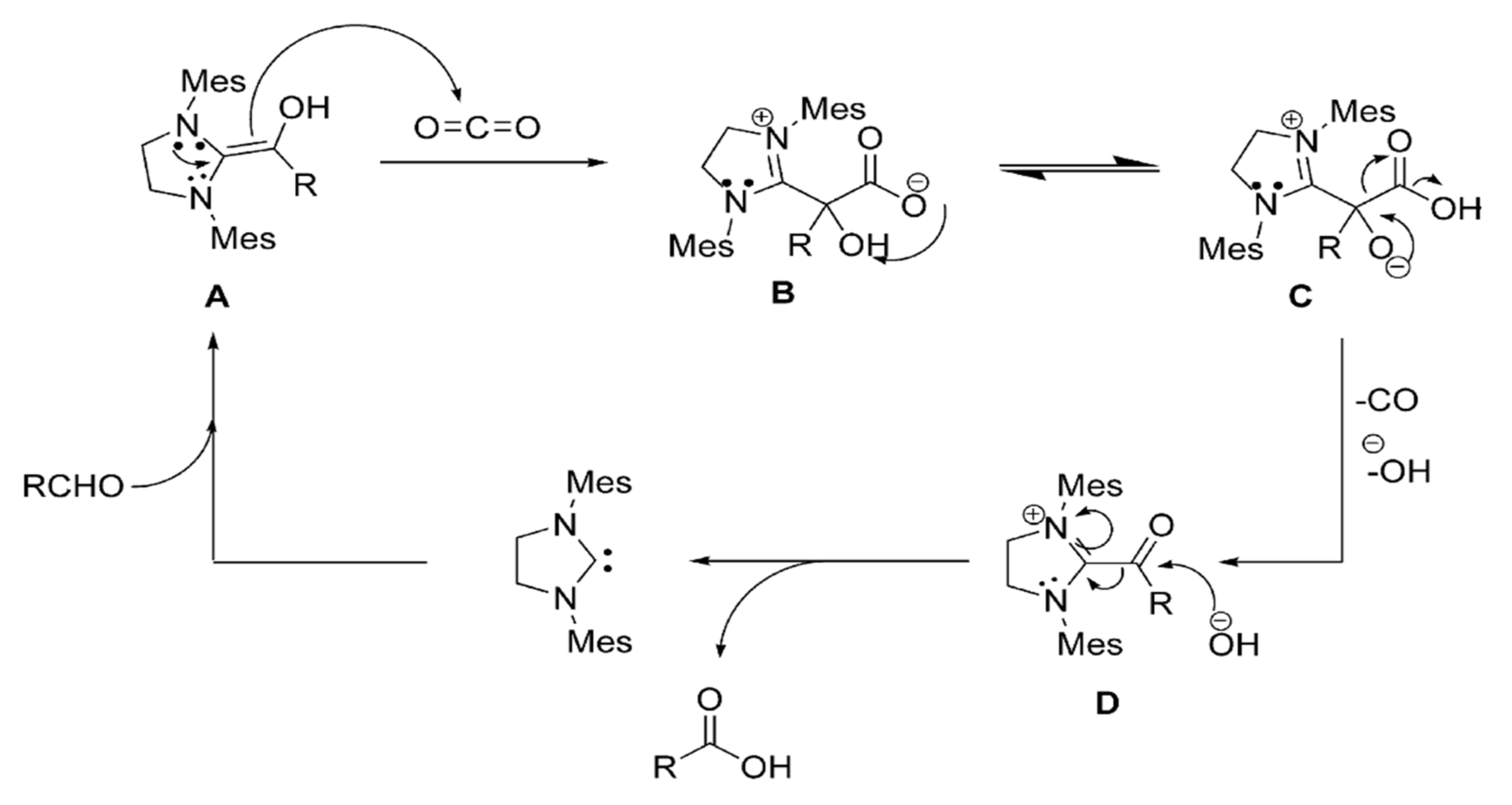
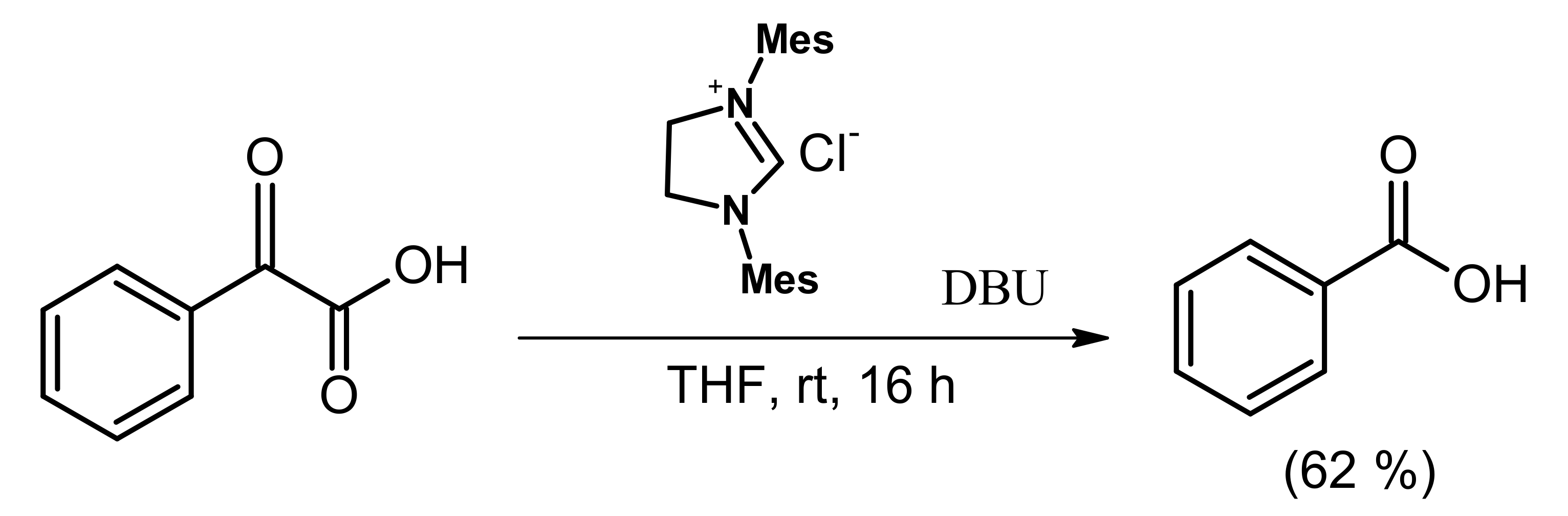
- Nair, V.; Varghese, V.; Paul, R.R.; Jose, A.; Sinu, C.R.; Menon, R.S. NHC catalyzed transformation of aromatic aldehydes to acids by carbon dioxide: An unexpected reaction. Org. Lett. 2010, 12, 2653–2655. [Google Scholar] [CrossRef]
- Chiang, P.C.; Bode, J.W. On the role of CO2 in NHC-catalyzed oxidation of aldehydes. Org. Lett. 2011, 13, 2422–2425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, L.; Zhang, Y. Unexpected CO2 splitting reactions to form CO with N-heterocyclic carbenes as organocatalysts and aromatic aldehydes as oxygen acceptors. J. Am. Chem. Soc. 2010, 132, 914–915. [Google Scholar] [CrossRef]
- Lane, B.S.; Vogt, M.; De Rose, V.J.; Kevin, B. Manganese-Catalyzed Epoxidations of Alkenes in Bicarbonate Solutions. J. Am. Chem. Soc. 2002, 124, 11946–11954. [Google Scholar] [CrossRef]
- Park, S.-E.; Yoo, J.S. Studies in Surface Science and Catalysis 153; Elsevier: Amsterdam, The Netherlands, 2004; pp. 303–314. [Google Scholar]
- Cavani, F.; Trifiro, F. Alternative Processes for the Production of Sponge Iron. Appl. Catal. A Gen. 1995, 133, 219–239. [Google Scholar] [CrossRef]
- Li, X.; Li, W. Effect of TiO2 loading on the activity of V/TiO2-Al2O3 in the catalytic oxidehydrogenation of ethylbenzene with carbon dioxide. Front. Chem. Eng. China 2010, 4, 142–146. [Google Scholar] [CrossRef]
- Nowicka, E.; Reece, C.; Althahban, S.M.; Mohammed, K.M.H.; Kondrat, S.A.; Morgan, D.J.; He, Q.; Willock, D.J.; Golunski, S.; Kiely, C.J.; et al. Elucidating the Role of CO2 in the Soft Oxidative Dehydrogenation of Propane over Ceria-Based Catalysts. ACS Catal. 2018, 8, 3454–3468. [Google Scholar] [CrossRef] [Green Version]
- Kovacevic, M.; Agarwal, S.; Mojet, B.L.; Van Ommen, J.G.; Lefferts, L. The effects of morphology of cerium oxide catalysts for dehydrogenation of ethylbenzene to styrene. Appl. Catal. A Gen. 2015, 505, 354–364. [Google Scholar] [CrossRef]
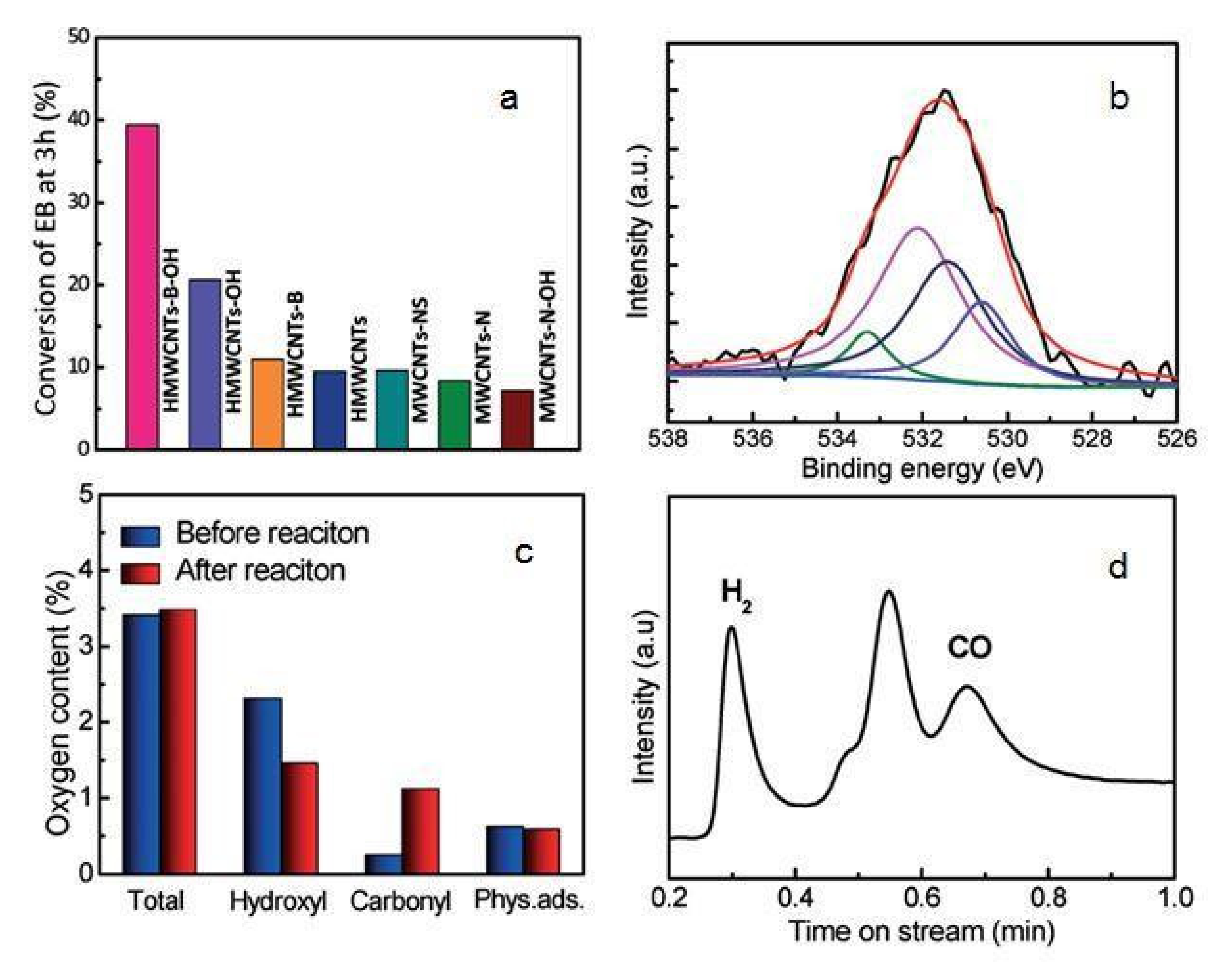
- Rao, R.; Zhang, Q.; Liu, H.; Yang, H.; Ling, Q.; Yang, M.; Zhang, A.; Chen, W. Enhanced catalytic performance of CeO2 confined inside carbon nanotubes for dehydrogenation of ethylbenzene in the presence of CO2. J. Mol. Catal. A Chem. 2012, 363–364, 283–290. [Google Scholar] [CrossRef]
- Rao, R.; Yang, M.; Ling, Q.; Li, C.; Zhang, Q.; Yang, H.; Zhang, A. A novel route of enhancing oxidative catalytic activity: Hydroxylation of MWCNTs induced by sectional defects. Catal. Sci. Technol. 2014, 4, 665–671. [Google Scholar] [CrossRef]
- Sakurai, Y.; Suzaki, T.; Ikenaga, N.O.; Suzuki, T. Dehydrogenation of ethylbenzene with an activated carbon-supported vanadium catalyst. Appl. Catal. A Gen. 2000, 192, 281–288. [Google Scholar] [CrossRef]
- Fan, H.; Feng, J.; Li, X.; Guo, Y.; Li, W.; Xie, K. Ethylbenzene dehydrogenation to styrene with CO2 over V2O5(001): A periodic density functional theory study. Chem. Eng. Sci. 2015, 135, 403–411. [Google Scholar] [CrossRef]
- Rao, K.N.; Reddy, B.M.; Abhishek, B.; Seo, Y.H.; Jiang, N.; Park, S.E. Effect of ceria on the structure and catalytic activity of V2O5/TiO2-ZrO2 for oxidehydrogenation of ethylbenzene to styrene utilizing CO2 as soft oxidant. Appl. Catal. B Environ. 2009, 91, 649–656. [Google Scholar] [CrossRef]
- Liu, Z.W.; Wang, C.; Fan, W.B.; Liu, Z.T.; Hao, Q.Q.; Long, X.; Lu, J.; Wang, J.G.; Qin, Z.F.; Su, D.S. V2O5/Ce0.6Zr0.4O2-Al2O3 as an efficient catalyst for the oxidative dehydrogenation of ethylbenzene with carbon dioxide. ChemSusChem 2011, 4, 341–345. [Google Scholar] [CrossRef]
- Burri, A.; Jiang, N.; Ji, M.; Park, S.-E.; Khalid, Y. Oxidative dehydrogenation of ethylbenzene to styrene with CO2 over V2O5-Sb2O5-CeO2/TiO2-ZrO2 catalysts. Top. Catal. 2013, 56, 1724–1730. [Google Scholar] [CrossRef]
- Liu, B.S.; Rui, G.; Chang, R.Z.; Au, C.T. Dehydrogenation of ethylbenzene to styrene over LaVOx/SBA-15 catalysts in the presence of carbon dioxide. Appl. Catal. A Gen. 2008, 335, 88–94. [Google Scholar] [CrossRef]
- Li, Z.; Su, K.; Cheng, B.; Shen, D.; Zhou, Y. Effects of VOx /AlMCM-41 surface structure on ethyl benzene oxydehydrogenation in the presence of CO2. Catal. Lett. 2010, 135, 135–140. [Google Scholar] [CrossRef]
- Jiang, N.; Burri, A.; Park, S.-E. Ethylbenzene to styrene over ZrO2-based mixed metal oxide catalysts with CO2 as soft oxidant. Chin. J. Catal. 2016, 37, 3–15. [Google Scholar] [CrossRef]
- Reddy, B.M.; Khan, A. Recent advances on TiO2-ZrO2 mixed oxides as catalysts and catalyst supports. Catal. Rev. Sci. Eng. 2005, 47, 257–296. [Google Scholar] [CrossRef]
- Jiang, N.; Han, D.S.; Park, S.-E. Direct synthesis of mesoporous silicalite-1 supported TiO2-ZrO2 for the dehydrogenation of EB to styrene with CO2. Catal. Today 2009, 141, 344–348. [Google Scholar] [CrossRef]
- Manríquez, M.E.; López, T.; Gómez, R.; Navarrete, J. Preparation of TiO2-ZrO2 mixed oxides with controlled acid-basic properties. J. Mol. Catal. A Chem. 2004, 220, 229–237. [Google Scholar] [CrossRef]
- Zangeneh, F.T.; Sahebdelfar, S.; Ravanchi, M.T. Conversion of carbon dioxide to valuable petrochemicals: An approach to clean development mechanism. J. Nat. Gas Chem. 2011, 20, 219–231. [Google Scholar] [CrossRef]
- Balasamy, R.J.; Tope, B.B.; Khurshid, A.; Al-Ali, A.A.S.; Atanda, L.A.; Sagata, K.; Asamoto, M.; Yahiro, H.; Nomura, K.; Sano, T.; et al. Ethylbenzene dehydrogenation over FeOx/(Mg,Zn)(Al)O catalysts derived from hydrotalcites: Role of MgO as basic sites. Appl. Catal. A Gen. 2011, 398, 113–122. [Google Scholar] [CrossRef]
- Braga, T.P.; Pinheiro, A.N.; Teixeira, C.V.; Valentini, A. Dehydrogenation of ethylbenzene in the presence of CO2 using a catalyst synthesized by polymeric precursor method. Appl. Catal. A Gen. 2009, 366, 193–200. [Google Scholar] [CrossRef]
- Pochamoni, R.; Narani, A.; Varkolu, M.; Dhar Gudimella, M.; Prasad Potharaju, S.S.; Burri, D.R.; Rao Kamaraju, S.R. Studies on ethylbenzene dehydrogenation with CO2 as soft oxidant over Co3O4/COK-12 catalysts. J. Chem. Sci. 2015, 127, 701–709. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; Chen, H.; Li, J.; Wang, Q.; Wang, Y.; Ma, S.; Zhu, Z. Acid-based co-catalysis for oxidative dehydrogenation of ethylbenzene to styrene with CO2 over X zeolite modified by alkali metal cation exchange. RSC Adv. 2015, 5, 75787–75793. [Google Scholar] [CrossRef]
- Periyasamy, K.; Aswathy, V.T.; Ashok Kumar, V.; Manikandan, M.; Shukla, R.; Tyagi, A.K.; Raja, T. An efficient robust fluorite CeZrO4-δ oxide catalyst for the eco-benign synthesis of styrene. RSC Adv. 2015, 5, 3619–3626. [Google Scholar] [CrossRef]
- Mukherjee, D.; Park, S.-E.; Reddy, B.M. CO2 as a soft oxidant for oxidative dehydrogenation reaction: An eco benign process for industry. J. CO2 Util. 2016, 16, 301–312. [Google Scholar] [CrossRef]
- Sato, S.; Ohhara, M.; Sodesawa, T.; Nozaki, F. Combination of ethylbenzene dehydrogenation and carbon dioxide shift-reaction over a sodium oxide/alumina catalyst. Appl. Catal. 1988, 37, 207–215. [Google Scholar] [CrossRef]
- Chen, S.; Qin, Z.; Wang, G.; Dong, M.; Wang, J. Promoting effect of carbon dioxide on the dehydrogenation of ethylbenzene over silica-supported vanadium catalysts. Fuel 2013, 109, 43–48. [Google Scholar] [CrossRef]
- Burri, D.R.; Choi, K.M.; Han, S.C.; Burri, A.; Park, S.-E. Dehydrogenation of ethylbenzene to styrene with CO2 over TiO2-ZrO2 bifunctional catalyst. Bull. Korean Chem. Soc. 2007, 28, 53–58. [Google Scholar]
- Burri, D.R.; Choi, K.M.; Han, S.C.; Burri, A.; Park, S.-E. Selective conversion of ethylbenzene into styrene over K2O/TiO2-ZrO2 catalysts: Unified effects of K2O and CO2. J. Mol. Catal. A Chem. 2007, 269, 58–63. [Google Scholar] [CrossRef]
- Burri, A.; Jiang, N.; Park, S.-E. High surface area TiO2-ZrO2 prepared by caustic solution treatment, and its catalytic efficiency in the oxidehydrogenation of para-ethyl toluene by CO2. Catal. Sci. Technol. 2012, 2, 514–520. [Google Scholar] [CrossRef]
- Burri, D.R.; Choi, K.M.; Han, D.S.; Sujandi; Jiang, N.; Burri, A.; Park, S.-E. Oxidative dehydrogenation of ethylbenzene to styrene with CO2 over SnO2-ZrO2 mixed oxide nanocomposite catalysts. Catal. Today 2008, 131, 173–178. [Google Scholar] [CrossRef]
- Burri, D.R.; Choi, K.M.; Han, D.S.; Koo, J.B.; Park, S.-E. CO2 utilization as an oxidant in the dehydrogenation of ethylbenzene to styrene over MnO2-ZrO2 catalysts. Catal. Today 2006, 115, 242–247. [Google Scholar] [CrossRef]
- Rahmani, F.; Haghighi, M.; Amini, M. The beneficial utilization of natural zeolite in preparation of Cr/clinoptilolite nanocatalyst used in CO2-oxidative dehydrogenation of ethane to ethylene. J. Ind. Eng. Chem. 2015, 31, 142–155. [Google Scholar] [CrossRef]
- Mimura, N.; Takahara, I.; Inaba, M.; Okamoto, M.; Murata, K. High-performance Cr/H-ZSM-5 catalysts for oxidative dehydrogenation of ethane to ethylene with CO2 as an oxidant. Catal. Commun. 2002, 3, 257–262. [Google Scholar] [CrossRef]
- Liu, L.; Li, H.; Zhang, Y. A comparative study on catalytic performances of chromium incorporated and supported mesoporous MSU-x catalysts for the oxidehydrogenation of ethane to ethylene with carbon dioxide. Catal. Today 2006, 115, 235–241. [Google Scholar] [CrossRef]
- Shi, X.; Ji, S.; Wang, K. Oxidative dehydrogenation of ethane to ethylene with carbon dioxide over Cr-Ce/SBA-15 catalysts. Catal. Lett. 2008, 125, 331–339. [Google Scholar] [CrossRef]
- Deng, S.; Li, S.; Li, H.; Zhang, Y. Oxidative dehydrogenation of ethane to ethylene with CO2 over Fe-Cr/ZrO2 catalysts. Ind. Eng. Chem. Res. 2009, 48, 7561–7566. [Google Scholar] [CrossRef]
- Shi, X.; Ji, S.; Li, C. Oxidative dehydrogenation of ethane with CO2 over novel Cr/SBA-15/Al2O3/FeCrAl monolithic catalysts. Energy Fuels 2008, 22, 3631–3638. [Google Scholar] [CrossRef]
- Tedeeva, M.A.; Kustov, A.L.; Pribytkov, P.V.; Leonov, A.V.; Dunaev, S.F. Dehydrogenation of Propane with CO2 on Supported CrOx/SiO2 Catalysts. Russ. J. Phys. Chem. A 2018, 92, 2403–2407. [Google Scholar] [CrossRef]
- Wang, S.; Murata, K.; Hayakawa, T.; Hamakawa, S.; Suzuki, K. Dehydrogenation of ethane with carbon dioxide over supported chromium oxide catalysts. Appl. Catal. A Gen. 2000, 196, 1–8. [Google Scholar] [CrossRef]
- Weckhuysen, B.M.; Schoonheydt, R.A.; Jehng, J.M.; Wachs, I.E.; Cho, S.J.; Ryoo, R.; Kijlstra, S.; Poels, E. Combined DRS-RS-EXAFS-XANES-TPR study of supported chromium catalysts. J. Chem. Soc. Faraday Trans. 1995, 91, 3245–3253. [Google Scholar] [CrossRef] [Green Version]
- Mimura, N.; Okamoto, M.; Yamashita, H.; Oyama, S.T.; Murata, K. Oxidative dehydrogenation of ethane over Cr/ZSM-5 catalysts using CO2 as an oxidant. J. Phys. Chem. B 2006, 110, 21764–21770. [Google Scholar] [CrossRef]
- Heracleous, E.; Lemonidou, A.A. Ni-Nb-O mixed oxides as highly active and selective catalysts for ethene production via ethane oxidative dehydrogenation. Part I: Characterization and catalytic performance. J. Catal. 2006, 237, 162–174. [Google Scholar] [CrossRef]
- Heracleous, E.; Lemonidou, A.A. Ni-Nb-O mixed oxides as highly active and selective catalysts for ethene production via ethane oxidative dehydrogenation. Part II: Mechanistic aspects and kinetic modeling. J. Catal. 2006, 237, 175–189. [Google Scholar]
- Heracleous, E.; Delimitis, A.; Nalbandian, L.; Lemonidou, A.A. HRTEM characterization of the nanostructural features formed in highly active Ni-Nb-O catalysts for ethane ODH. Appl. Catal. A Gen. 2007, 325, 220–226. [Google Scholar] [CrossRef]
- Koirala, R.; Buechel, R.; Krumeich, F.; Pratsinis, S.E.; Baiker, A. Oxidative dehydrogenation of ethane with CO2 over flame-made Ga-loaded TiO2. ACS Catal. 2015, 5, 690–702. [Google Scholar] [CrossRef]
- Cheng, Y.; Zhang, F.; Zhang, Y.; Miao, C.; Hua, W.; Yue, Y.; Gao, Z. Oxidative dehydrogenation of ethane with CO2 over Cr supported on submicron ZSM-5 zeolite. Chinese J. Catal. 2015, 36, 1242–1248. [Google Scholar] [CrossRef]
- Ramesh, Y.; Thirumala Bai, P.; Hari Babu, B.; Lingaiah, N.; Rama Rao, K.S.; Prasad, P.S.S. Oxidative dehydrogenation of ethane to ethylene on Cr2O3/Al2O3–ZrO2 catalysts: The influence of oxidizing agent on ethylene selectivity. Appl. Petrochem. Res. 2014, 4, 247–252. [Google Scholar] [CrossRef] [Green Version]
- Deng, S.; Li, H.; Li, S.; Zhang, Y. Activity and characterization of modified Cr2O3/ZrO2 nano-composite catalysts for oxidative dehydrogenation of ethane to ethylene with CO2. J. Mol. Catal. A Chem. 2007, 268, 169–175. [Google Scholar] [CrossRef]
- Yashima, T.; Sato, K.; Hayasaka, T.; Hara, N. Alkylation on synthetic zeolites. III. Alkylation of toluene with methanol and formaldehyde on alkali cation exchanged zeolites. J. Catal. 1972, 26, 303–312. [Google Scholar] [CrossRef]
- Rossetti, I.; Bencini, E.; Trentini, L.; Forni, L. Study of the deactivation of a commercial catalyst for ethylbenzene dehydrogenation to styrene. Appl. Catal. A Gen. 2005, 292, 118–123. [Google Scholar] [CrossRef]
- Meima, G.R.; Menon, P.G. Catalyst deactivation phenomena in styrene production. Appl. Catal. A Gen. 2001, 212, 239–245. [Google Scholar] [CrossRef]
- Sindorenko, L.N.; Galich, P.N.; Gutirya, V.S. Condensation of toluene and methanol upon synthetic zeolites containing ion-exchange cations of alkali metals. Dokl. Akad. Nauk. SSSR 1967, 173, 132–133. [Google Scholar]
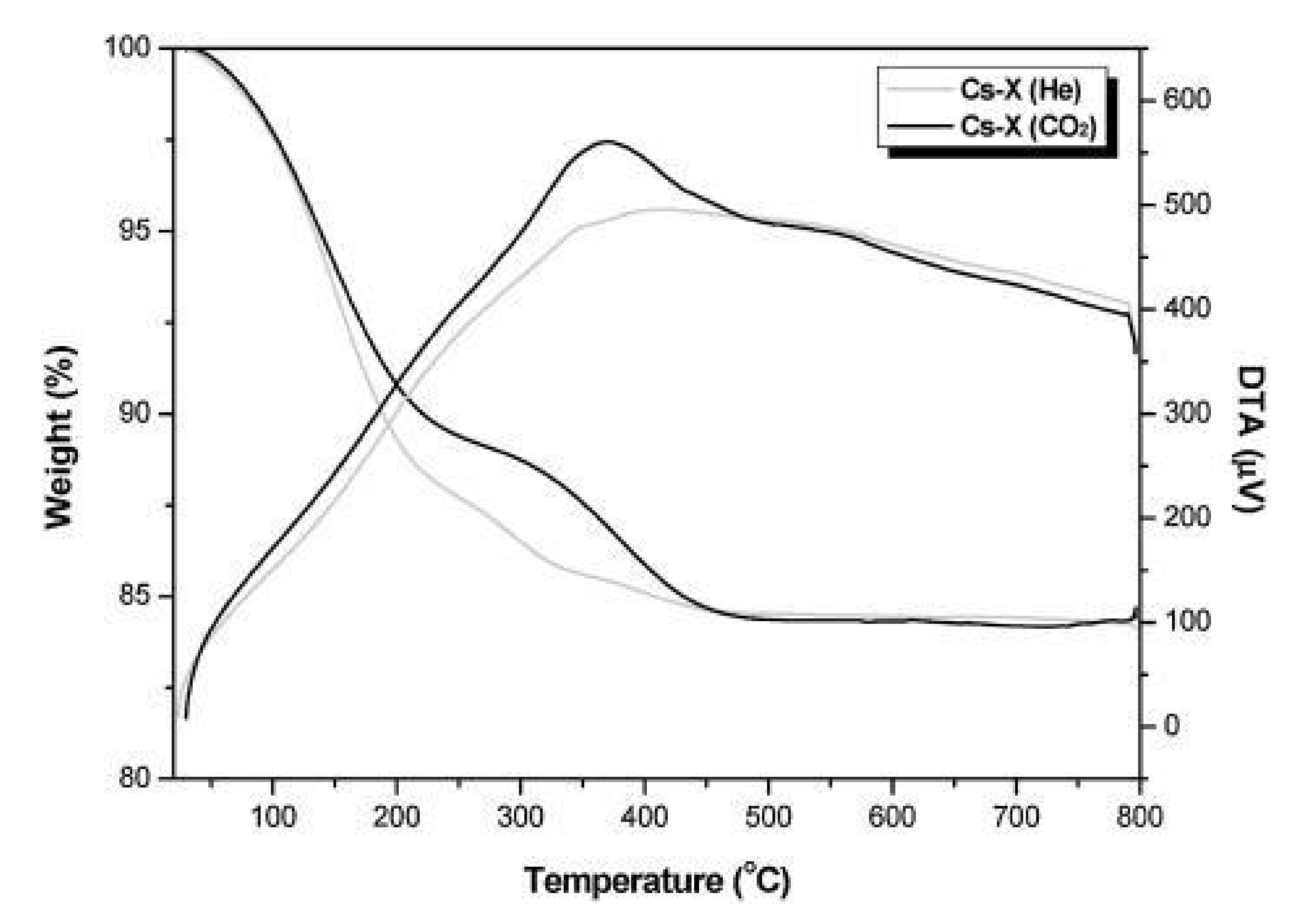
- Seo, D.W.; Rahman, S.T.; Reddy, B.M.; Park, S.-E. Carbon dioxide assisted toluene side-chain alkylation with methanol over Cs-X zeolite catalyst. J. CO2 Util. 2018, 26, 254–261. [Google Scholar] [CrossRef]
- Hattori, H. Solid base catalysts: Fundamentals and their applications in organic reactions. Appl. Catal. A Gen. 2015, 504, 103–109. [Google Scholar] [CrossRef]
- Yoo, K.S.; Smirniotis, P.G. Zeolites-catalyzed alkylation of isobutane with 2-butene: Influence of acidic properties. Catal. Lett. 2005, 103, 249–255. [Google Scholar] [CrossRef]
- Alabi, W.O.; Tope, B.B.; Jermy, R.B.; Aitani, A.M.; Hattori, H.; Al-Khattaf, S.S. Modification of Cs-X for styrene production by side-chain alkylation of toluene with methanol. Catal. Today 2014, 226, 117–123. [Google Scholar] [CrossRef]
- Yoo, K.; Smirniotis, P.G. The deactivation pathway of one-dimensional zeolites, LTL and ZSM-12, for alkylation of isobutane with 2-butene. Appl. Catal. A Gen. 2003, 246, 243–251. [Google Scholar] [CrossRef]
- Yoo, K.; Burckle, E.C.; Smirniotis, P.G. Isobutane/2-butene alkylation using large-pore zeolites: Influence of pore structure on activity and selectivity. J. Catal. 2002, 211, 6–18. [Google Scholar] [CrossRef]
- Han, H.; Liu, M.; Nie, X.; Ding, F.; Wang, Y.; Li, J.; Guo, X.; Song, C. The promoting effects of alkali metal oxide in side-chain alkylation of toluene with methanol over basic zeolite X. Microporous Mesoporous Mater. 2016, 234, 61–72. [Google Scholar] [CrossRef]
- Itoh, H.; Hattori, T.; Suzuki, K.; Murakami, Y. Role of acid and base sites in the side-chain alkylation of alkylbenzenes with methanol on two-ion-exchanged zeolites. J. Catal. 1983, 79, 21–33. [Google Scholar] [CrossRef]
- Tope, B.B.; Alabi, W.O.; Aitani, A.M.; Hattori, H.; Al-Khattaf, S.S. Side-chain alkylation of toluene with methanol to styrene over cesium ion-exchanged zeolite X modified with metal borates. Appl. Catal. A Gen. 2012, 443–444, 214–220. [Google Scholar] [CrossRef]
- Philippou, A.; Anderson, M.W. Solid-State NMR Investigation of the Alkylation of Toluene with Methanol over Basic Zeolite X. J. Am. Chem. Soc. 1994, 116, 5774–5783. [Google Scholar] [CrossRef]
- Jiang, N.; Jin, H.; Jeong, E.-Y.; Park, S.-E. Mgo Encapsulated Mesoporous Zeolite for the Side Chain Alkylation of Toluene with Methanol. J. Nanosci. Nanotechnol. 2010, 10, 227–232. [Google Scholar] [CrossRef]
- Hattori, H.; Alabi, W.O.; Jermy, B.R.; Aitani, A.M.; Al-Khattaf, S.S. Pathway to ethylbenzene formation in side-chain alkylation of toluene with methanol over cesium ion-exchanged zeolite X. Catal. Lett. 2013, 143, 1025–1029. [Google Scholar] [CrossRef]
- Mikkelsen, M.; Jørgensen, M.; Krebs, F.C. The teraton challenge. A review of fixation and transformation of carbon dioxide. Energy Environ. Sci. 2010, 3, 43–81. [Google Scholar] [CrossRef]
- Cavani, F.; Ballarini, N.; Cericola, A. Oxidative dehydrogenation of ethane and propane: How far from commercial implementation? Catal. Today 2007, 127, 113–131. [Google Scholar] [CrossRef]
- Ren, Y.; Zhang, F.; Hua, W.; Yue, Y.; Gao, Z. ZnO supported on high silica HZSM-5 as new catalysts for dehydrogenation of propane to propene in the presence of CO2. Catal. Today 2009, 148, 316–322. [Google Scholar] [CrossRef]
- Chen, M.; Xu, J.; Liu, Y.M.; Cao, Y.; He, H.Y.; Zhuang, J.H. Supported indium oxide as novel efficient catalysts for dehydrogenation of propane with carbon dioxide. Appl. Catal. A Gen. 2010, 377, 35–41. [Google Scholar] [CrossRef]
- Schimmoeller, B.; Jiang, Y.; Pratsinis, S.E.; Baiker, A. Structure of flame-made vanadia/silica and catalytic behavior in the oxidative dehydrogenation of propane. J. Catal. 2010, 274, 64–75. [Google Scholar] [CrossRef]
- Liu, Y.M.; Cao, Y.; Yan, S.R.; Dai, W.L.; Fan, K.N. Highly effective oxidative dehydrogenation of propane over vanadia supported on mesoporous SBA-15 silica. Catal. Lett. 2003, 88, 61–67. [Google Scholar] [CrossRef]
- Santamaría-González, J.; Mérida-Robles, J.; Alcántara-Rodríguez, M.; Maireles-Torres, P.; Rodríguez-Castellón, E.; Jiménez-López, A. Catalytic behaviour of chromium supported mesoporous MCM-41 silica in the oxidative dehydrogenation of propane. Catal. Lett. 2000, 64, 209–214. [Google Scholar] [CrossRef]
- Davies, T.; Taylor, S.H. The oxidative dehydrogenation of propane using gallium-molybdenum oxide-based catalysts. J. Mol. Catal. A Chem. 2004, 220, 77–84. [Google Scholar] [CrossRef]
- Raju, G.; Reddy, B.M.; Abhishek, B.; Mo, Y.H.; Park, S.-E. Synthesis of C4 olefins from n-butane over a novel VOx/SnO2-ZrO2 catalyst using CO2 as soft oxidant. Appl. Catal. A Gen. 2012, 423–424, 168–175. [Google Scholar] [CrossRef]
- Atanga, M.A.; Rezaei, F.; Jawad, A.; Fitch, M.; Rownaghi, A.A. Oxidative dehydrogenation of propane to propylene with carbon dioxide. Appl. Catal. B Environ. 2018, 220, 429–445. [Google Scholar] [CrossRef]
- Reddy, B.M.; Lee, S.C.; Han, D.S.; Park, S.-E. Utilization of carbon dioxide as soft oxidant for oxydehydrogenation of ethylbenzene to styrene over V2O5-CeO2/TiO2-ZrO2 catalyst. Appl. Catal. B Environ. 2009, 87, 230–238. [Google Scholar] [CrossRef]
- Uy, D.; O’Neill, A.E.; Xu, L.; Weber, W.H.; McCabe, R.W. Observation of cerium phosphate in aged automotive catalysts using Raman spectroscopy. Appl. Catal. B Environ. 2003, 41, 269–278. [Google Scholar] [CrossRef]
- Armaroli, T.; Simon, L.J.; Digne, M.; Montanari, T.; Bevilacqua, M.; Valtchev, V.; Patarin, J.; Busca, G. Effects of crystal size and Si/Al ratio on the surface properties of H-ZSM-5 zeolites. Appl. Catal. A Gen. 2006, 306, 78–84. [Google Scholar] [CrossRef]
- Thakkar, H.; Eastman, S.; Hajari, A.; Rownaghi, A.A.; Knox, J.C.; Rezaei, F. 3D-Printed Zeolite Monoliths for CO2 Removal from Enclosed Environments. ACS Appl. Mater. Interfaces 2016, 8, 27753–27761. [Google Scholar] [CrossRef]
- Thakkar, H.; Eastman, S.; Al-Mamoori, A.; Hajari, A.; Rownaghi, A.A.; Rezaei, F. Formulation of Aminosilica Adsorbents into 3D-Printed Monoliths and Evaluation of Their CO2 Capture Performance. ACS Appl. Mater. Interfaces 2017, 9, 7489–7498. [Google Scholar] [CrossRef]
- Ren, Y.; Wang, J.; Hua, W.; Yue, Y.; Gao, Z. Ga2O3/HZSM-48 for dehydrogenation of propane: Effect of acidity and pore geometry of support. J. Ind. Eng. Chem. 2012, 18, 731–736. [Google Scholar] [CrossRef]
- Abello, M.C.; Gomez, M.F.; Ferretti, O. Oxidative conversion of propane over Al2O3-supported molybdenum and chromium oxides. Catal. Lett. 2003, 87, 43–49. [Google Scholar] [CrossRef]
- Cherian, M.; Rao, M.S.; Hirt, A.M.; Wachs, I.E.; Deo, G. Oxidative dehydrogenation of propane over supported chromia catalysts: Influence of oxide supports and chromia loading. J. Catal. 2002, 211, 482–495. [Google Scholar] [CrossRef]
- Rao, T.V.M.; Zahidi, E.M.; Sayari, A. Ethane dehydrogenation over pore-expanded mesoporous silica-supported chromium oxide: 2. Catalytic properties and nature of active sites. J. Mol. Catal. A Chem. 2009, 301, 159–165. [Google Scholar] [CrossRef]
- Hakuli, A.; Kytökivi, A.; Krause, A.O.I. Dehydrogenation of i-butane on CrOx/Al2O3 catalysts prepared by ALE and impregnation techniques. Appl. Catal. A Gen. 2000, 190, 219–232. [Google Scholar] [CrossRef]
- Liu, L.; Li, H.; Zhang, Y. Effect of synthesis parameters on the chromium content and catalytic activities of mesoporous Cr-MSU-x prepared under acidic conditions. J. Phys. Chem. B 2006, 110, 15478–15485. [Google Scholar] [CrossRef] [PubMed]
- Santhosh Kumar, M.; Hammer, N.; Rønning, M.; Holmen, A.; Chen, D.; Walmsley, J.C.; Øye, G. The nature of active chromium species in Cr-catalysts for dehydrogenation of propane: New insights by a comprehensive spectroscopic study. J. Catal. 2009, 261, 116–128. [Google Scholar] [CrossRef]
- Weckhuysen, B.M.; Wachs, I.E.; Schoonheydt, R.A. Surface chemistry and spectroscopy of chromium in inorganic oxides. Chem. Rev. 1996, 96, 3327–3349. [Google Scholar] [CrossRef] [Green Version]
- Burri, A.; Hasib, M.A.; Mo, Y.H.; Reddy, B.M.; Park, S.-E. An Efficient Cr-TUD-1 Catalyst for Oxidative Dehydrogenation of Propane to Propylene with CO2 as Soft Oxidant. Catal. Lett. 2018, 148, 576–585. [Google Scholar] [CrossRef]
- Park, S.-J.; Kim, K.D. Adsorption behaviors of CO2 and NH3 on chemically surface-treated activated carbons. J. Colloid Interface Sci. 1999, 212, 186–189. [Google Scholar] [CrossRef]
- Michorczyk, P.; Pietrzyk, P.; Ogonowski, J. Preparation and characterization of SBA-1-supported chromium oxide catalysts for CO2 assisted dehydrogenation of propane. Microporous Mesoporous Mater. 2012, 161, 56–66. [Google Scholar] [CrossRef]
- COSIA. $20M NRG COSIA Carbon XPRIZE Finalists Announced; Teams Ready to Test Transformative CO2 Technologies at Alberta’s Carbon Conversion Centre. Available online: www.cosia.ca/resources/newsreleases/20m-nrg-cosia-carbon-xprize-finalists-announcedteams-ready-test (accessed on 9 April 2018).
- Zhang, D.; Wang, J.; Lin, Y.; Si, Y.; Huang, C.; Yang, J.; Huang, B.; Li, W. Present situation and future prospect of renewable energy in China. Renew. Sustain. Energy Rev. 2017, 76, 865–871. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Gas Ratio (PSI) a | Conversion (%) O2/CO2 | Conversion (%) O2/N2 | ΔC(%) b |
|---|---|---|---|---|
| 1 | 0.066 | 0 | 0 | 0 |
| 2 | 0.142 | 16 | 9 | 28 |
| 3 | 0.230 | 25 | 18 | 16.3 |
| 4 | 0.333 | 33 | 24 | 15.7 |
| 5 | 0.454 | 34 | 24 | 15.7 |

| Entry | n | Gas | Conversion of 3 (%) | Selectivity (%) | |||
|---|---|---|---|---|---|---|---|
| 4 | 5 | 6 | ΔC (%) | ||||
| 1 | 1 | O C | 40 | 37 | 24 | 29 | 12.6 |
| O | 31 | 30 | 22 | 40 | - | ||
| 2 | 2 | O C | 33 | 30 | 21 | 49 | 15.8 |
| O | 24 | 25 | 16 | 53 | - | ||
| 3 | 4 | O C | 21 | > 99 | - | - | 27.0 |
| - | - | O | 12 | > 99 | - | - | - |
| 4 | 8 | O C | 17 | > 99 | - | - | 30.7 |
| - | - | O | 9 | > 99 | - | - | - |

| Gas | Conversion of 1 (%) | Yield mol (%) | ||||
|---|---|---|---|---|---|---|
| 2 | 3 | 4 | 5 | 6 | ||
| O2 | 57.2 | 17.7 | 47.9 | 2.8 | 1.7 | 29.2 |
| O2/CO2 | 66.8 | 34.8 | 36.9 | 1.7 | 2.4 | 24.2 |
| Temperature (°C) Feed a | 300 | 350 | 375 | |||
|---|---|---|---|---|---|---|
| 1 | 2 | 1 | 2 | 1 | 2 | |
| p-Xylene (Con.%) | 17.6 | 33.3 | 41.2 | 65.5 | 60.7 | 84.1 |
| Product selectivity (mol%) | - | - | - | - | - | - |
| p-Tolu-aldehyde | 57.9 | 57.2 | 50.2 | 40.9 | 40.6 | 40.6 |
| Terephthaldehyde | 16.4 | 27.5 | 23.5 | 33.6 | 32.6 | 30.2 |
| Benzaldehyde | 1.3 | 1.5 | 2.4 | 2.7 | 2.7 | 3.1 |
| Maleic anhydride | 0.0 | 0.0 | 2.4 | 6.0 | 5.8 | 13.7 |
| Toluene | 6.2 | 6.9 | 3.5 | 4.8 | 3.1 | 4.8 |
| Trimethyl biphenyl methane | 7.5 | 6.8 | 1.7 | 0.6 | 0.4 | 0.0 |
| CO | 0.0 | 0.0 | 0.6 | 3.4 | 4.7 | 7.5 |
| CO2 | 10.7 | 0.0 | 15.6 | 0.0 | 20.2 | 0.0 |

| Gas | Conversion of 1 (1%) | Yield mol (%) | |
|---|---|---|---|
| 2 | 3 | ||
| O2 | 60.9 | 58.2 | 3.7 |
| O2/CO2 | 72.7 | 64.9 | 10.6 |

| Gas | Conversion of 1 (%) | Yield mol (%) | ||
|---|---|---|---|---|
| 2 | 3 | 4 | ||
| O2 | 94.9 | 2.05 | 0.83 | 92 |
| O2/CO2 | 98 | 7.7 | 0.38 | 90 |
| Catalyst | Reaction Temperature (°C) | EB Conversion (%) | ST Selectivity (%) | ST Yield (%) | Ref. |
|---|---|---|---|---|---|
| Co3O4/COK-12 | 600 | 57.5 | 95.5 | 54.9 | [59] |
| CeZrO4-δ | 550 | 7 | 97 | 6.8 | [61] |
| Na X zeolite | 545 | 9.4 | 89.6 | 8.4 | [60] |
| K X zeolite | 545 | 10.5 | 92.1 | 9.6 | [60] |
| VOx/Al MCM-41 | 550 | 52.3 | 96.7 | 50.6 | [51] |
| TiO2-ZrO2 | 600 | 69.3 | 96.2 | 66.6 | [67] |
| V2O5/SiO2 | 550 | 50.5 | 96.8 | 48.8 | [64] |
| SnO2-ZrO2 | 600 | 61.1 | 97.6 | 59.6 | [68] |
| MnO2-ZrO2 | 600 | 51.1 | 99.1 | 50.9 | [69] |
| Catalyst | In the Presence of CO2 | In the Presence of Ar | |||||
|---|---|---|---|---|---|---|---|
| Conv. (%) | Selectivity (%) | Conv. (%) | Selectivity (%) | ||||
| C2H6 | CO2 | C2H4 | CH4 | C2H6 | C2H4 | CH4 | |
| SBA-15 | 2.7 | 0.04 | 93.5 | 6.5 | 2.4 | 93.0 | 7.0 |
| 2.5Cr/SBA-15 | 39.6 | 15.9 | 95.5 | 4.5 | 30.2 | 89.7 | 10.3 |
| 5.0Cr/SBA-15 | 46.3 | 16.6 | 94.7 | 5.3 | 34.1 | 91.4 | 8.6 |
| 7.5Cr/SBA-15 | 45.3 | 18.8 | 92.2 | 7.8 | 33.9 | 92.8 | 7.2 |
| 10Cr/SBA-15 | 44.2 | 18.9 | 92.0 | 8.0 | 31.2 | 90.9 | 9.1 |
| 5Cr-5Ce/SBA-15 | 48.4 | 17.9 | 96.4 | 4.6 | 35.8 | 87.6 | 12.4 |
| 5Cr-7.5Ce/SBA-15 | 50.0 | 20.9 | 96.0 | 4.0 | 37.9 | 88.2 | 11.8 |
| 5Cr-10Ce/SBA-15 | 55.0 | 21.9 | 96.0 | 4.0 | 40.8 | 83.1 | 16.9 |
| 5Cr-15Ce/SBA-15 | 52.2 | 21.2 | 95.5 | 4.5 | 40.1 | 82.4 | 17.6 |
| Catalyst | Ethane Conversion (%) | Ethylene Selectivity (%) | Ethylene Yield (%) | Ref. |
|---|---|---|---|---|
| Cr2O3 (5 wt.%) CLT-IA | 39.7 | 98.8 | 39.2 | [70] |
| 3Cr/NaZSM-5-160 | 65.5 | 75.4 | 49.3 | [84] |
| Cr2O3/Al2O3-ZrO2 | 36.0 | 56.2 | 20.2 | [85] |
| Cr/MSU-1 | 68.1 | 81.6 | 55.6 | [72] |
| Cr2O3/ZrO2 | 77.4 | 46.3 | 35.8 | [86] |
| 2.5 Cr/SBA-15 | 46.3 | 94.7 | 43.8 | [75] |
| 5 Cr-10Ce/SBA-15 | 55.0 | 96.0 | 52.8 | [73] |
| 5% Cr2O3/Al2O3 | 19.2 | 56.5 | 10.8 | [77] |
| Catalyst | Carrier Gas | MeOH Conv. (%) | Toluene Conv. (%) | Selectivity (%) | ||
|---|---|---|---|---|---|---|
| SM | EB | Others | ||||
| Ce-X | He | 12.54 | 1.42 | 78.61 | 15.32 | 6.07 |
| CO2 | 35.35 | 3.48 | 45.83 | 33.36 | 20.81 | |
| Cs2O/ | He | 46.48 | 3.59 | 28.76 | 68.02 | 3.22 |
| Cs-X | CO2 | 39.16 | 2.52 | 36.02 | 43.02 | 20.78 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, S.T.; Choi, J.-R.; Lee, J.-H.; Park, S.-J. The Role of CO2 as a Mild Oxidant in Oxidation and Dehydrogenation over Catalysts: A Review. Catalysts 2020, 10, 1075. https://doi.org/10.3390/catal10091075
Rahman ST, Choi J-R, Lee J-H, Park S-J. The Role of CO2 as a Mild Oxidant in Oxidation and Dehydrogenation over Catalysts: A Review. Catalysts. 2020; 10(9):1075. https://doi.org/10.3390/catal10091075
Chicago/Turabian StyleRahman, Sheikh Tareq, Jang-Rak Choi, Jong-Hoon Lee, and Soo-Jin Park. 2020. "The Role of CO2 as a Mild Oxidant in Oxidation and Dehydrogenation over Catalysts: A Review" Catalysts 10, no. 9: 1075. https://doi.org/10.3390/catal10091075
APA StyleRahman, S. T., Choi, J. -R., Lee, J. -H., & Park, S. -J. (2020). The Role of CO2 as a Mild Oxidant in Oxidation and Dehydrogenation over Catalysts: A Review. Catalysts, 10(9), 1075. https://doi.org/10.3390/catal10091075