Asymmetric Cyanation of Activated Olefins with Ethyl Cyanoformate Catalyzed by Ti(IV)-Catalyst: A Theoretical Study



Abstract
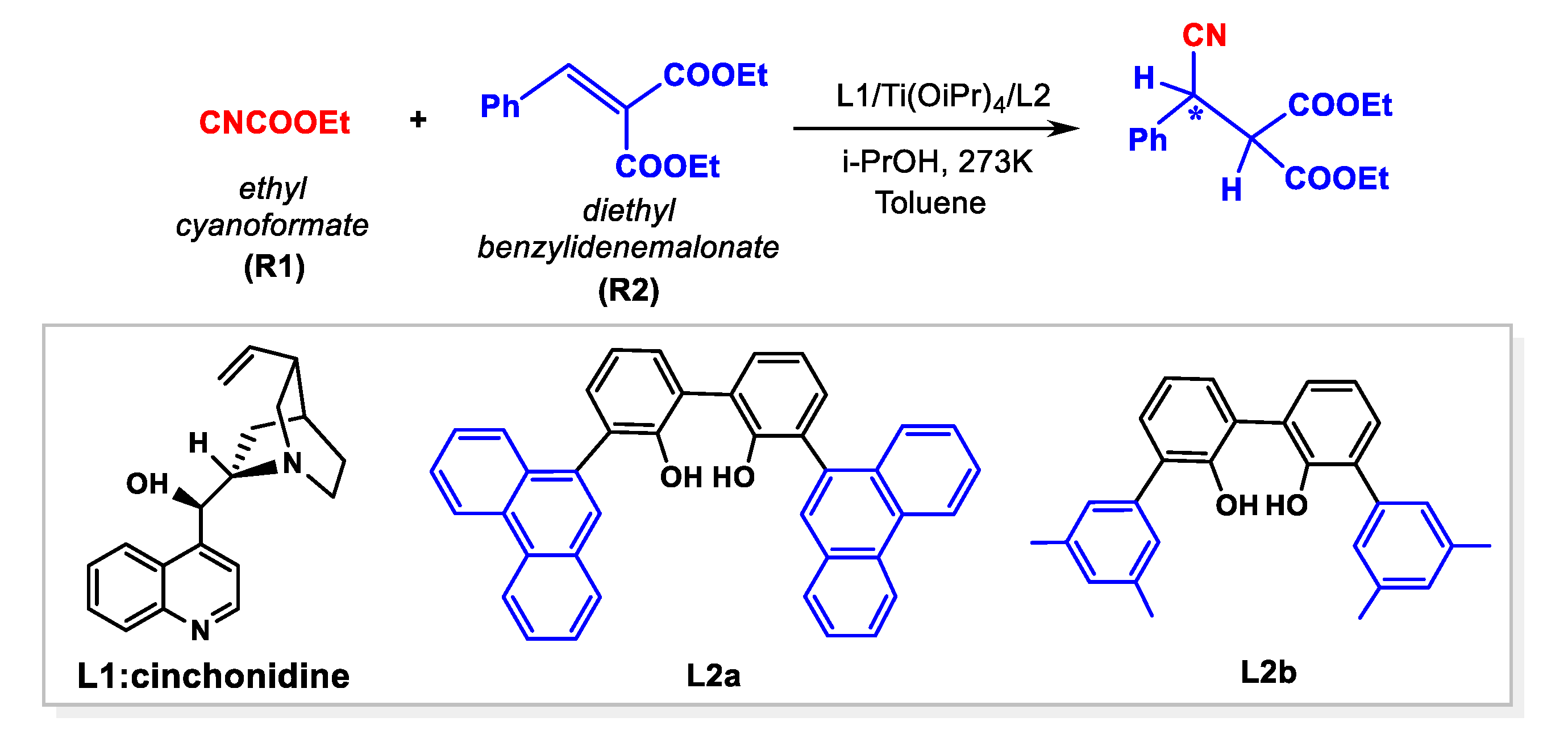
:1. Introduction
2. Computational Details
3. Results and Discussion
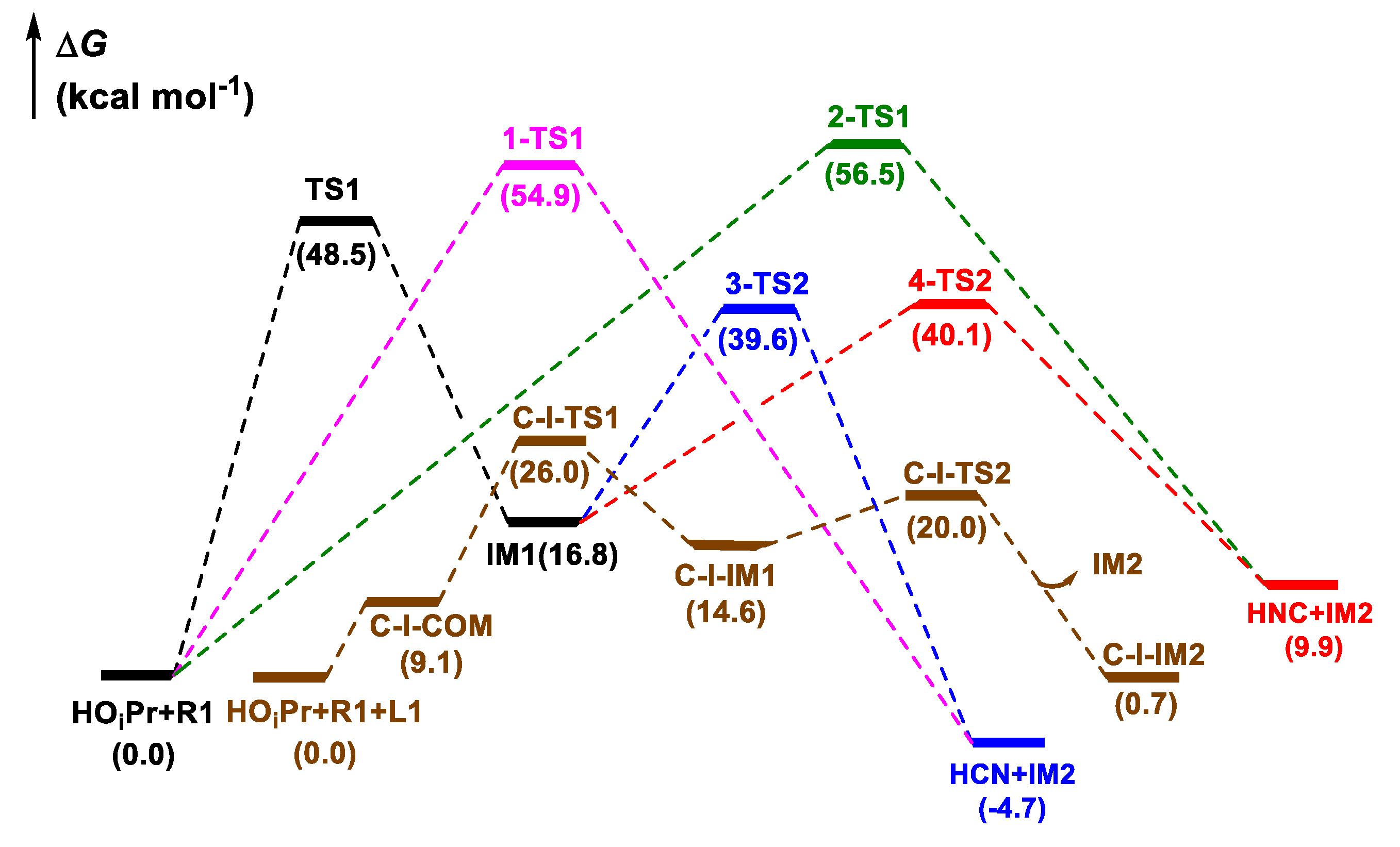
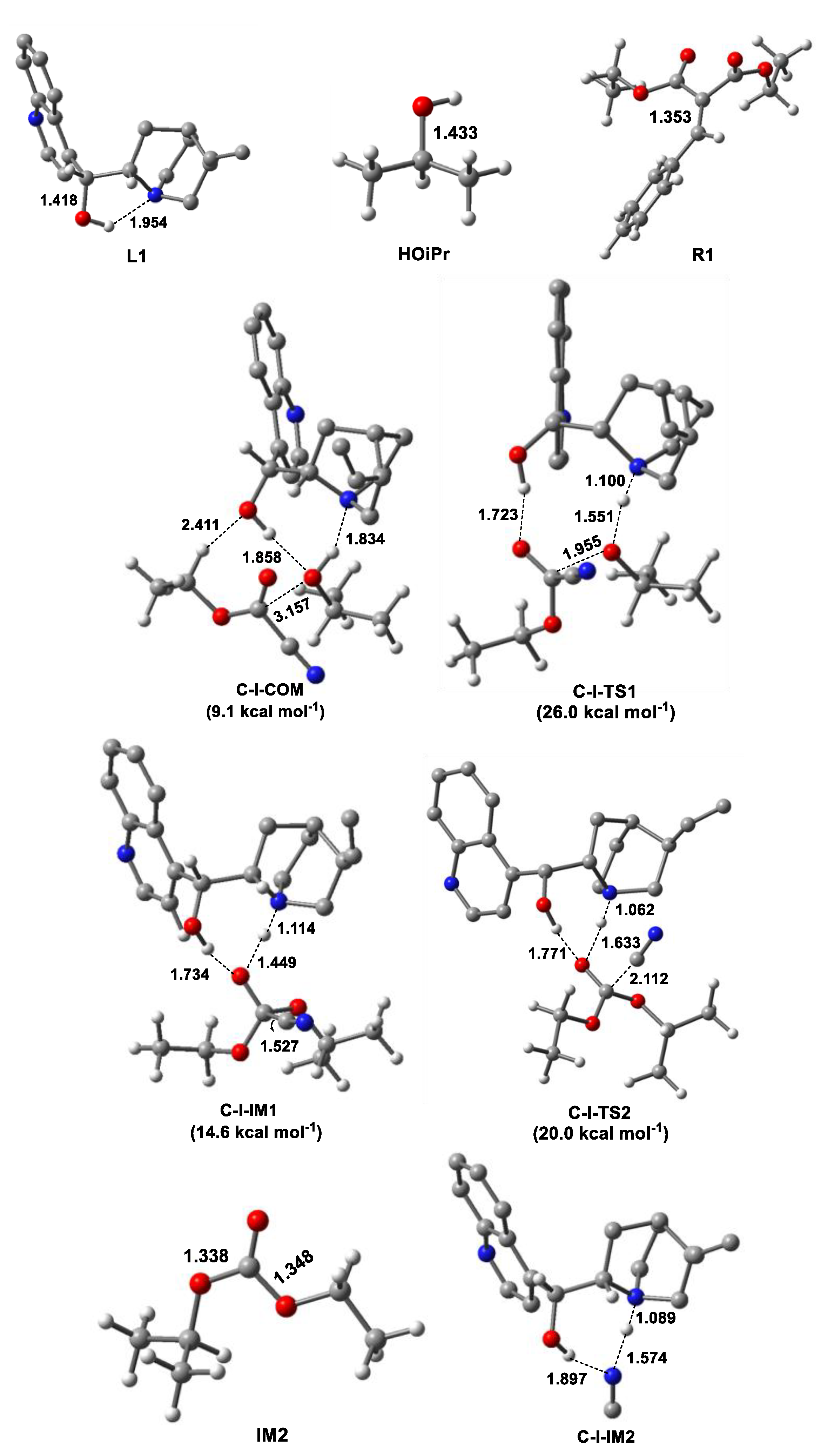
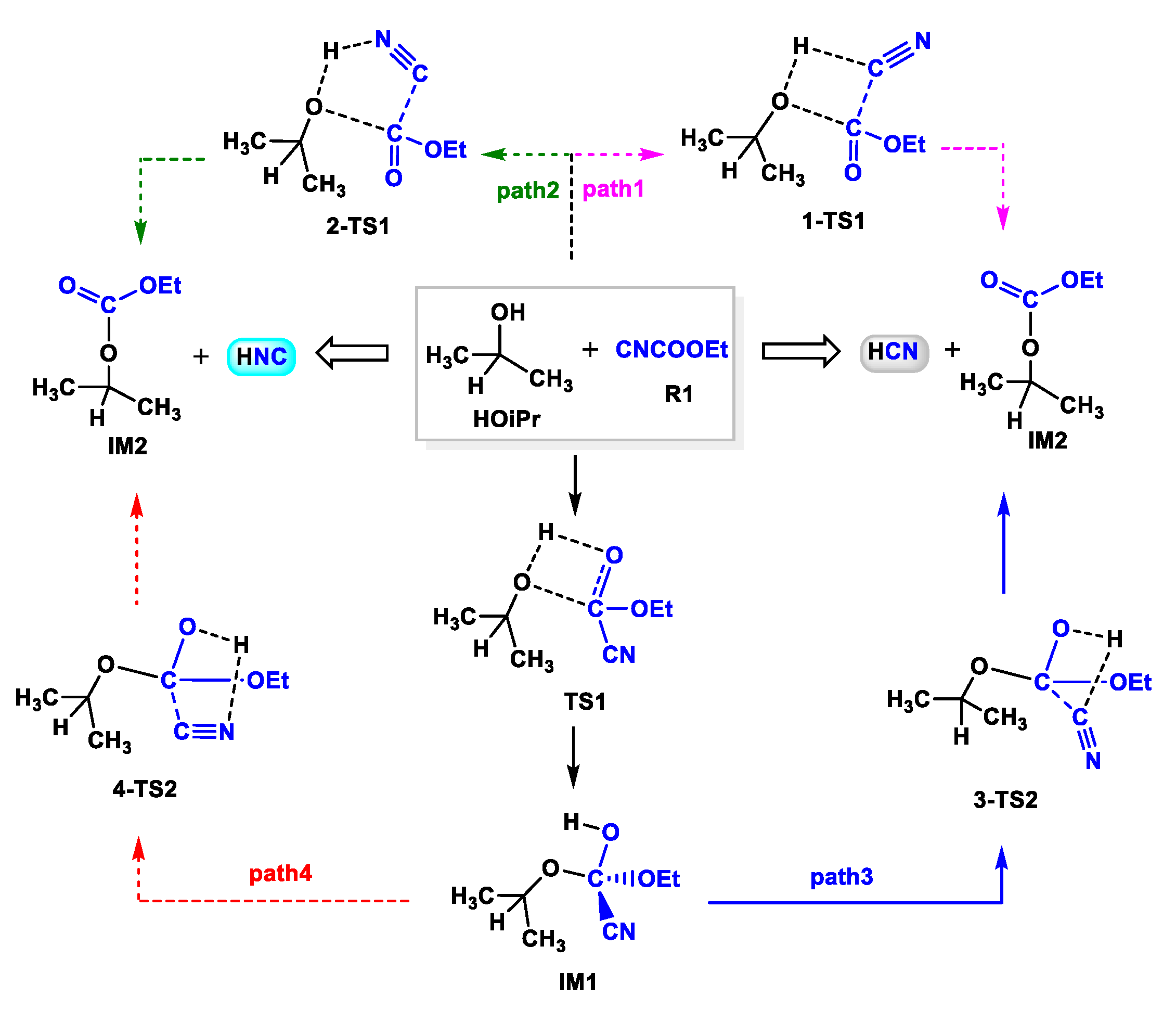
3.1. Release of HCN or HNC Species from CNCOOEt
3.2. Reaction Mechanism
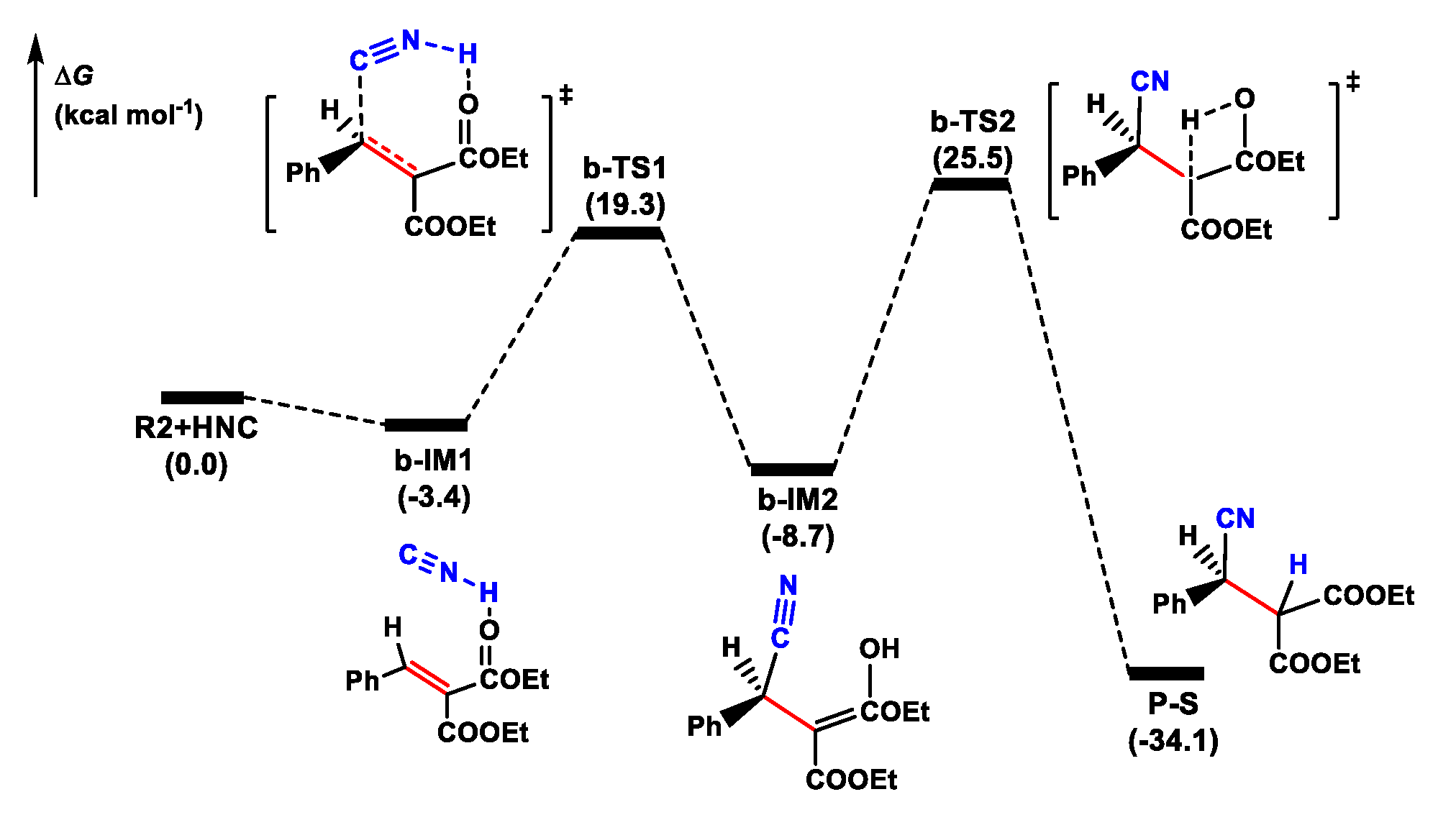
3.2.1. Noncatalytic Reaction
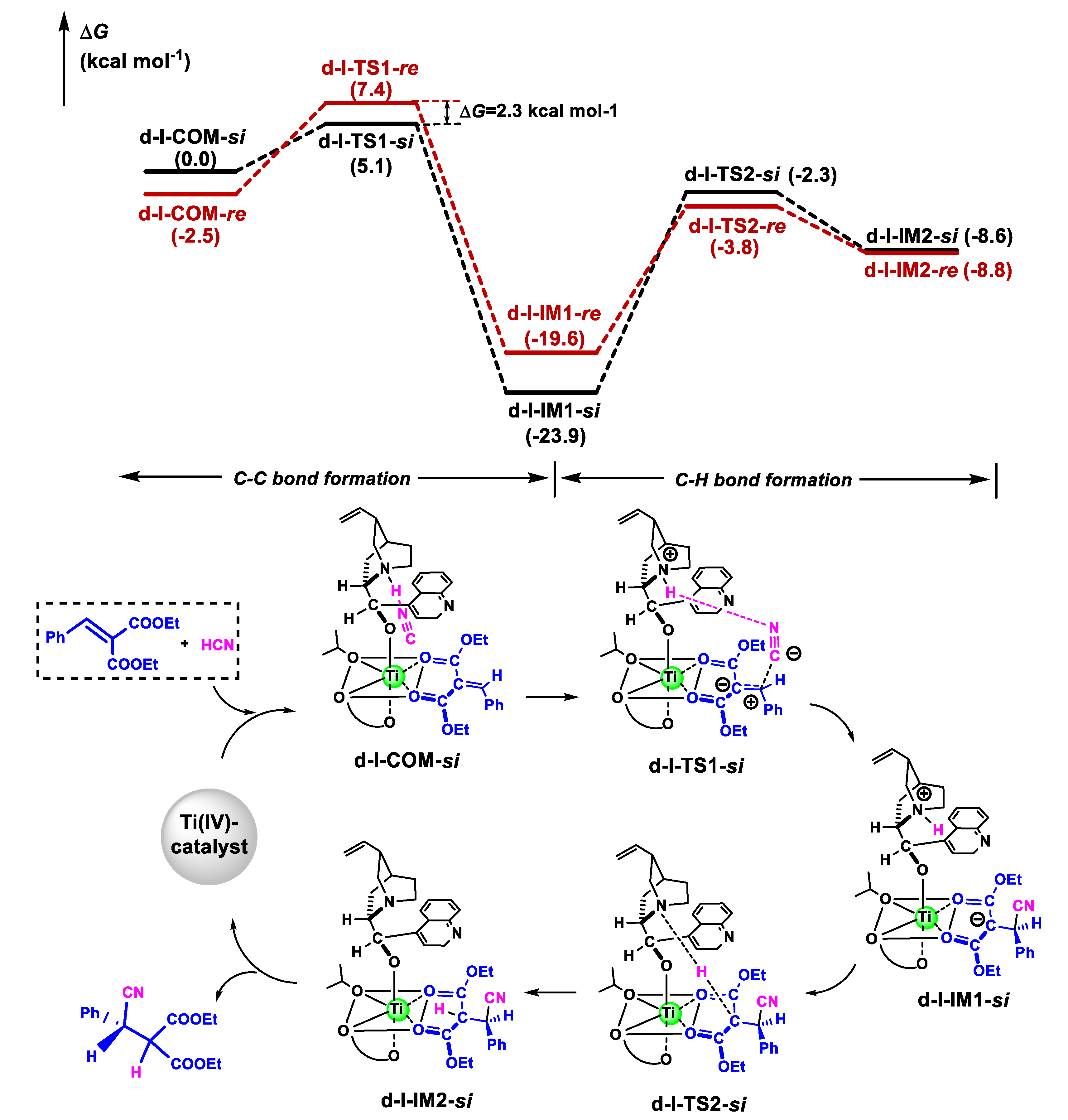
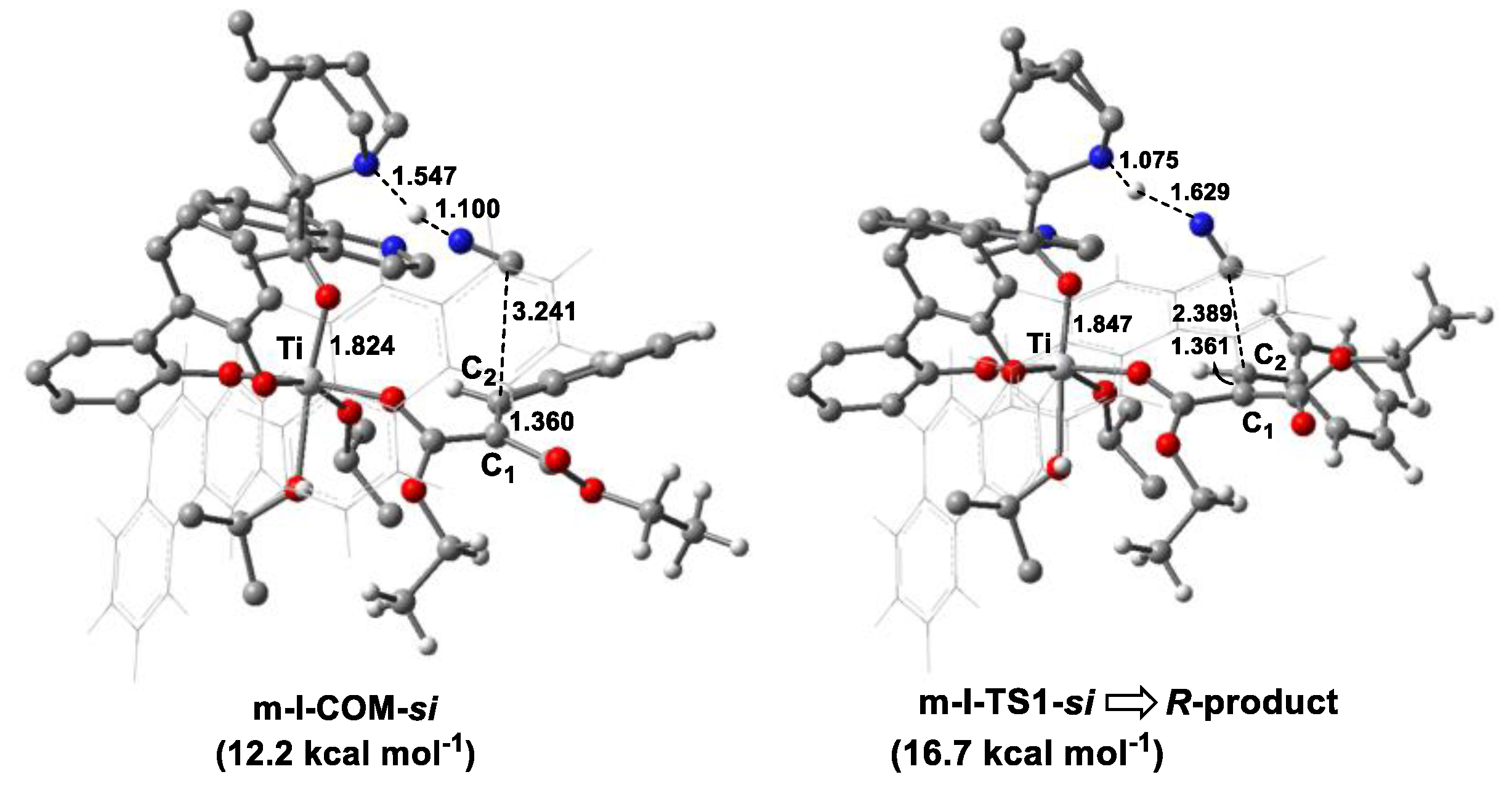
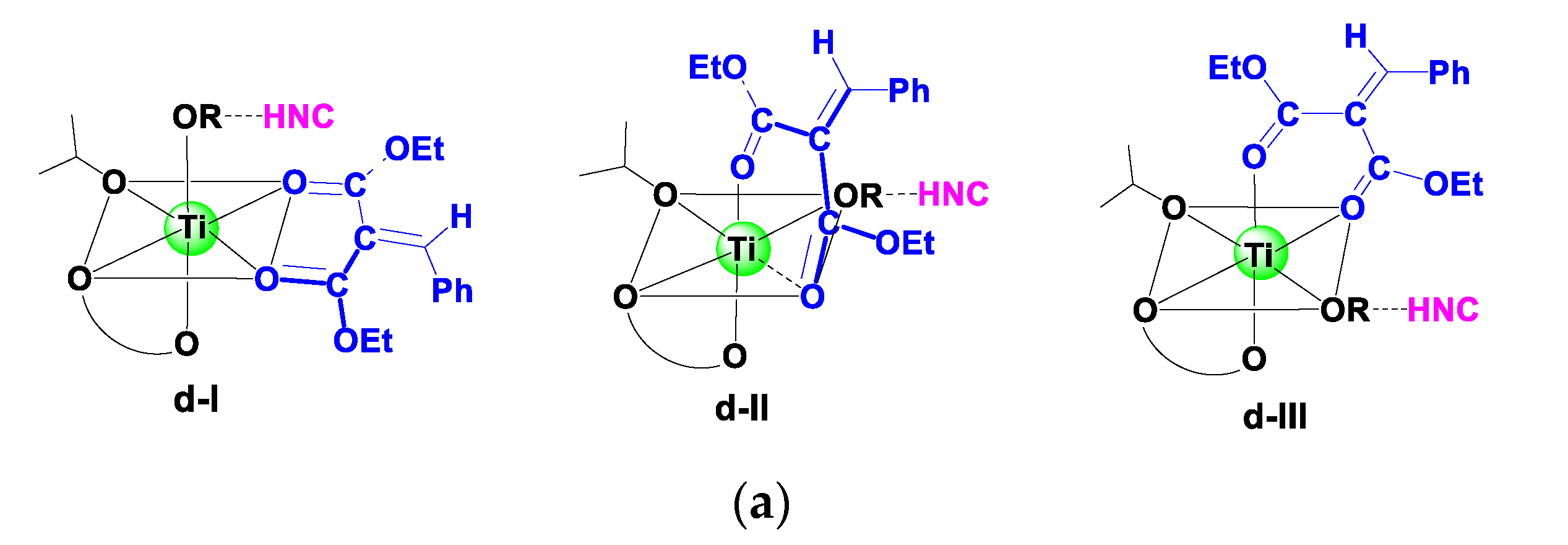
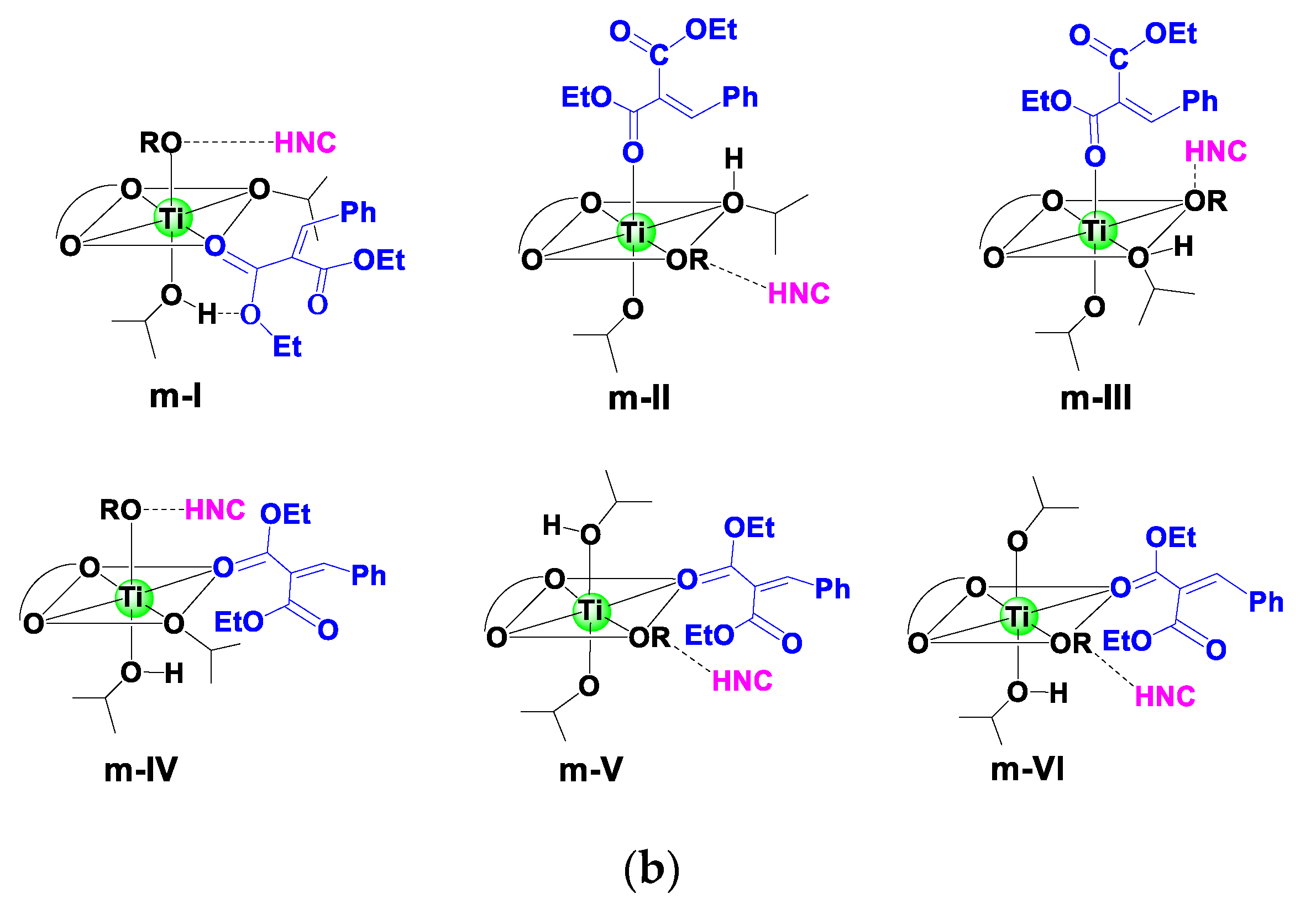
3.2.2. Catalytic Reaction
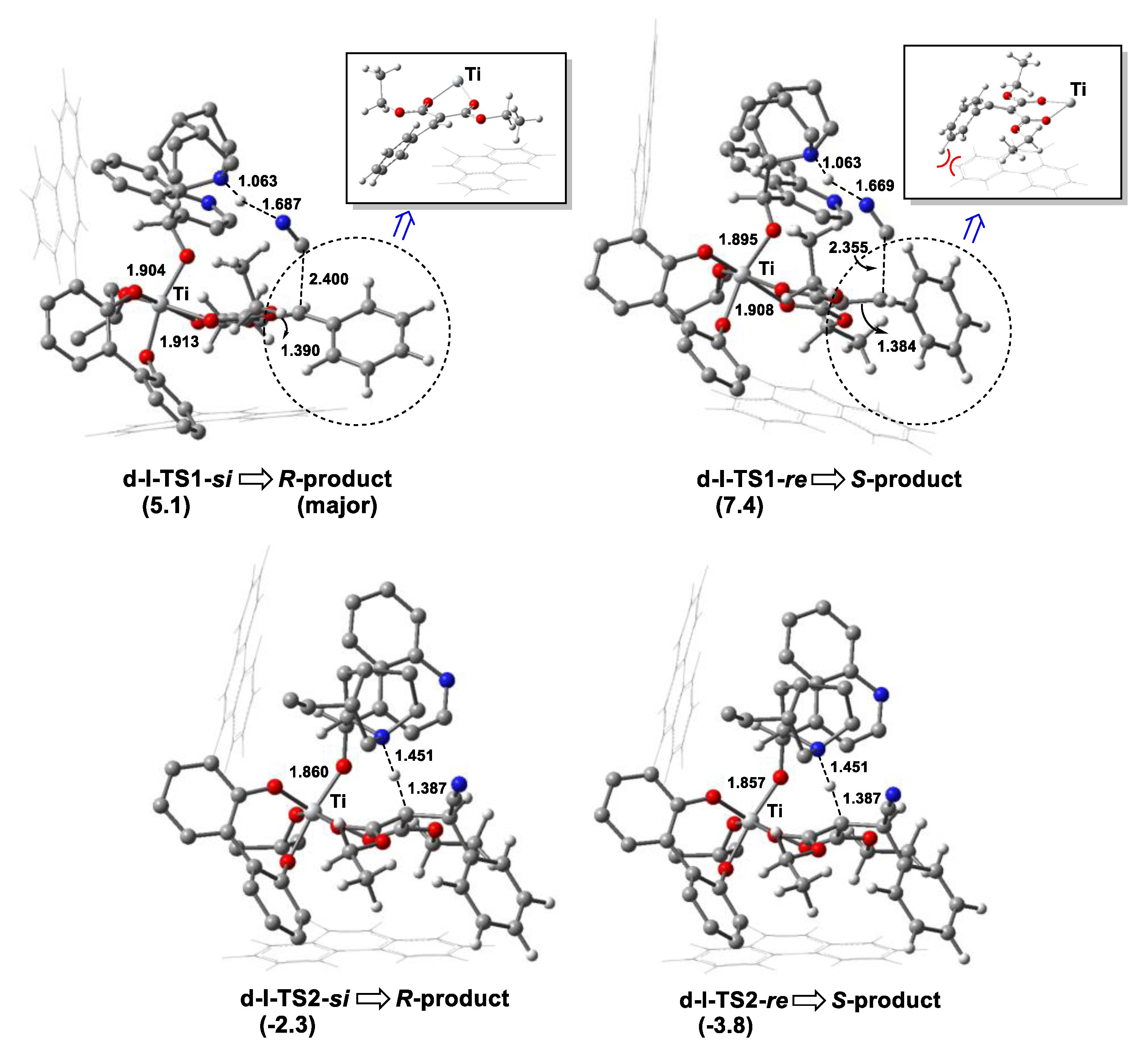
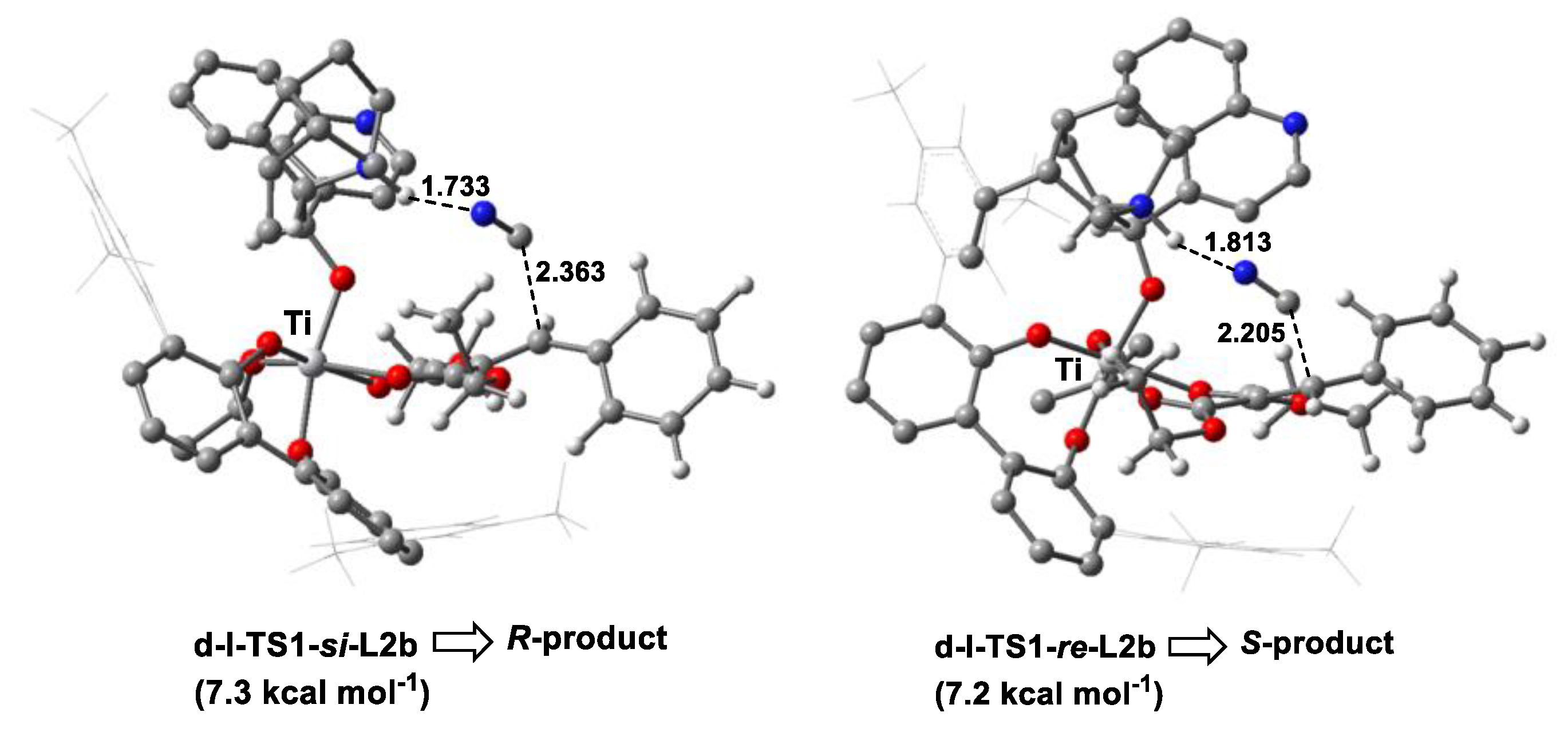
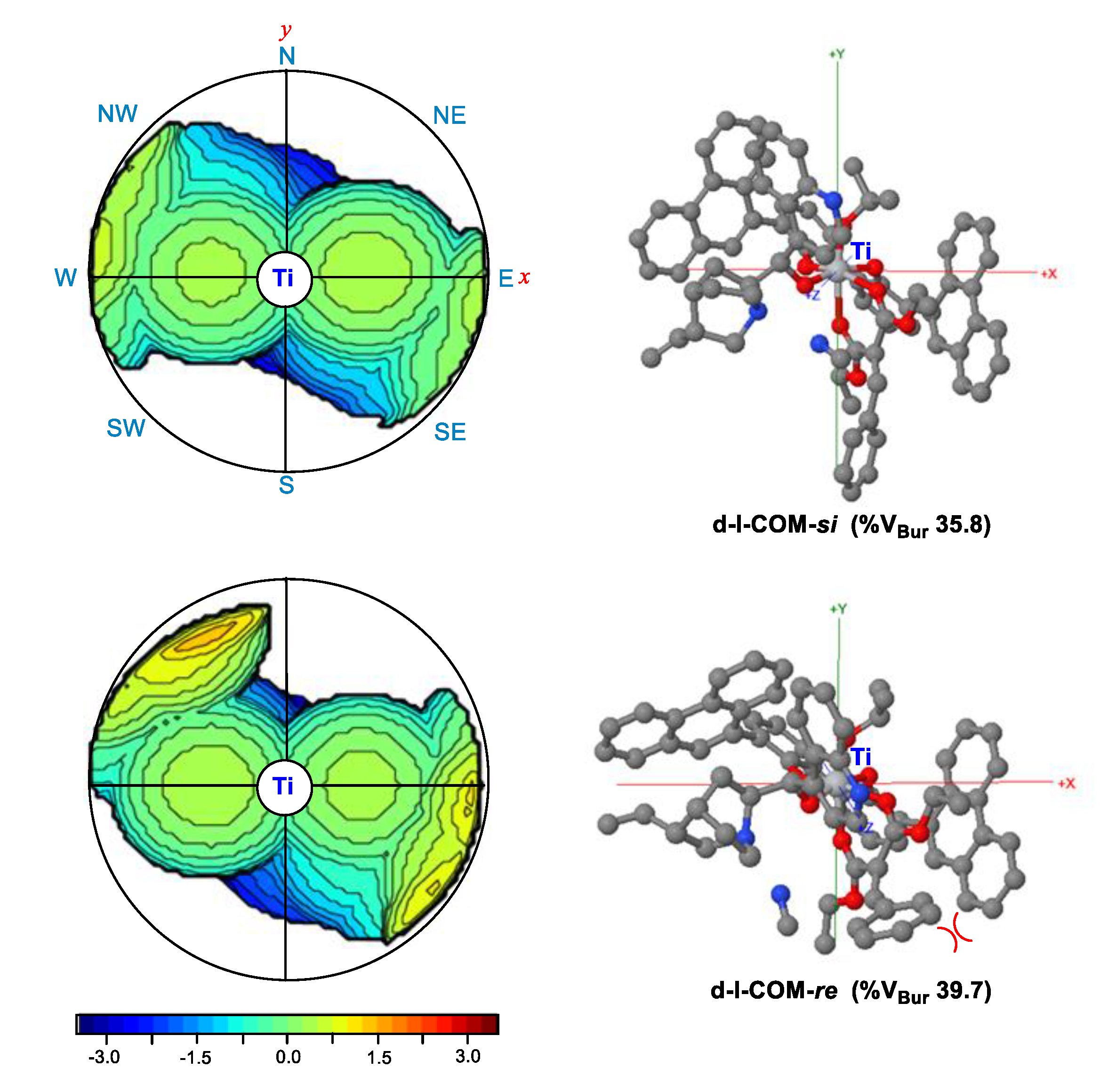
3.3. Origin of Stereoselectivity
4. Conclusions
- (i).
- Cinchona alkaloid facilitated the reaction between HOiPr and ethyl cyanoformate (CNCOOEt) to release the reacting species HCN (or HNC) by organocatalysis with free energy barrier of 26.0 kcal mol−1.
- (ii).
- The cyanation reaction of olefin proceeded via a two-step mechanism, in which the C-C bond construction was followed by H-transfer to generate a cyanide adduct. For noncatalytic reaction, the ∆G≠ for the rate-determining C-H bond construction step was up to 34.2 kcal mol−1, through a four-membered TS. In the catalytic reaction, the olefin coordinated to the self-assembly cinchonidine/Ti(IV)/(R)-3,3′-disubstituted biphenol catalyst in the bidentate model, forming a highly reactive hexacoordinated Ti(IV)-complex. The HNC activated by the quinuclidine tertiary amine moiety of cinchonidine ligand performed a nucleophilic attack towards the activated C=C bond of olefin, generating a cyanide adduct. The catalytic reaction required about 19.9 kcal mol−1 lower energy barrier compared to the noncatalytic reaction.
- (iii).
- EDA showed that the steric repulsion between the bulky group (e.g., 9-phenanthryl substituent) at the 3-position in the biphenol ligand and the phenyl group in olefin raised the Pauli energy (∆E≠Pauli) of the reacting fragments at the re-face attack TS, leading to the predominant R-product through the si-face attack, as observed in the experiment.
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Khan, N.H.; Kureshy, R.I.; Abdi, S.H.R.; Agrawal, S.; Jasra, R.V. Metal catalyzed asymmetric cyanation reactions. Coord. Chem. Rev. 2008, 252, 593–623. [Google Scholar] [CrossRef]
- North, M.; Usanov, D.L.; Young, C. Lewis acid catalyzed asymmetric cyanohydrin synthesis. Chem. Rev. 2008, 108, 5146–5226. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, X.H.; Feng, X.M. Asymmetric strecker reactions. Chem. Rev. 2011, 111, 6947–6983. [Google Scholar] [CrossRef] [PubMed]
- Kurono, N.; Ohkuma, T. Catalytic Asymmetric Cyanation Reactions. ACS Catal. 2016, 6, 989–1023. [Google Scholar] [CrossRef]
- Zeng, X.P.; Sun, J.C.; Liu, C.; Ji, C.B.; Peng, Y.Y. Catalytic Asymmetric Cyanation Reactions of Aldehydes and Ketones in Total Synthesis. Adv. Synth. Catal. 2019, 361, 3281–3305. [Google Scholar] [CrossRef]
- Kouznetsov, V.V.; Galvis, C.E.P. Strecker reaction and α-amino nitriles: Recent advances in their chemistry, synthesis, and biological properties. Tetrahedron 2018, 74, 773–810. [Google Scholar] [CrossRef]
- Mazet, C.; Jacobsen, E.N. Dinuclear {(salen)Al} complexes display expanded scope in the conjugate cyanation of α,β-unsaturated imides. Angew. Chem. Int. Ed. 2008, 47, 1762–1765. [Google Scholar] [CrossRef]
- Sammis, G.M.; Danjo, H.; Jacobsen, E.N. Cooperative dual catalysis: Application to the highly enantioselective conjugate cyanation of unsaturated imides. J. Am. Chem. Soc. 2004, 126, 9928–9929. [Google Scholar] [CrossRef]
- Madhavan, N.; Weck, M. Highly Active Polymer-Supported (Salen)Al Catalysts for the Enantioselective Addition of Cyanide to α,β-Unsaturated Imides. Adv. Synth. Catal. 2008, 350, 419–425. [Google Scholar] [CrossRef]
- Mita, T.; Sasaki, K.; Kanai, M.; Shibasaki, M. Catalytic enantioselective conjugate addition of cyanide to α,β-unsaturated N-acylpyrroles. J. Am. Chem. Soc. 2005, 127, 514–515. [Google Scholar] [CrossRef]
- Fujimori, I.; Mita, T.; Maki, K.; Shiro, M.; Sato, A.; Furusho, S.; Kanai, M.; Shibasaki, M. Toward a rational design of the assembly structure of polymetallic asymmetric catalysts: Design, synthesis, and evaluation of new chiral ligands for catalytic asymmetric cyanation reactions. Tetrahedron 2007, 63, 5820–5831. [Google Scholar] [CrossRef]
- Tanaka, Y.; Kanai, M.; Shibasaki, M. A catalytic enantioselective conjugate addition of cyanide to enones. J. Am. Chem. Soc. 2008, 130, 6072–6073. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Kanai, M.; Shibasaki, M. Catalytic Enantioselective Construction of β-Quaternary Carbons via a Conjugate Addition of Cyanide to β,β-Disubstituted α,β-Unsaturated Carbonyl Compounds. J. Am. Chem. Soc. 2010, 132, 8862–8863. [Google Scholar] [CrossRef] [PubMed]
- Kurono, N.; Nii, N.; Sakaguchi, Y.; Uemura, M.; Ohkuma, T. Asymmetric hydrocyanation of alpha, beta-unsaturated ketones into beta-cyano ketones with the [Ru(phgly)2(binap)]/C6H5OLi catalyst system. Angew. Chem. Int. Ed. 2011, 50, 5541–5544. [Google Scholar] [CrossRef]
- Sakaguchi, Y.; Kurono, N.; Yamauchi, K.; Ohkuma, T. Asymmetric conjugate hydrocyanation of alpha, beta-unsaturated N-acylpyrroles with the Ru(phgly)2(binap)-CH3OLi catalyst system. Org. Lett. 2014, 16, 808–811. [Google Scholar] [CrossRef]
- Zhang, J.L.; Liu, X.H.; Wang, R. Magnesium complexes as highly effective catalysts for conjugate cyanation of α,β-unsaturated amides and ketones. Chem. Eur. J. 2014, 20, 4911–4915. [Google Scholar] [CrossRef]
- Dong, C.; Song, T.; Bai, X.F.; Cui, Y.M.; Xu, Z.; Xu, L.W. Enantioselective conjugate addition of cyanide to chalcones catalyzed by a magnesium-Py-BINMOL complex. Catal. Sci. Technol. 2015, 5, 4755–4759. [Google Scholar] [CrossRef]
- Hatano, M.; Yamakawa, K.; Ishihara, K. Enantioselective Conjugate Hydrocyanation of α,β-Unsaturated N-Acylpyrroles Catalyzed by Chiral Lithium(I) Phosphoryl Phenoxide. ACS Catal. 2017, 7, 6686–6690. [Google Scholar] [CrossRef]
- Kawai, H.; Okusu, S.; Tokunaga, E.; Sato, H.; Shiro, M.; Shibata, N. Organocatalytic asymmetric synthesis of trifluoromethyl-substituted diarylpyrrolines: Enantioselective conjugate cyanation of beta-aryl-beta-trifluoromethyl-disubstituted enones. Angew. Chem. Int. Ed. 2012, 51, 4959–4962. [Google Scholar] [CrossRef]
- Wang, Y.F.; Zeng, W.; Sohail, M.; Guo, J.; Wu, S.; Chen, F.X. Highly Efficient Asymmetric Conjugate Hydrocyanation of Aromatic Enones by an Anionic Chiral Phosphate Catalyst. Eur. J. Org. Chem. 2013, 2013, 4624–4633. [Google Scholar] [CrossRef]
- Liu, Y.; Shirakawa, S.; Maruoka, K. Phase-Transfer-Catalyzed Asymmetric Conjugate Cyanation of Alkylidenemalonates with KCN in the Presence of a Brønsted Acid Additive. Org. Lett. 2013, 15, 1230–1233. [Google Scholar] [CrossRef] [PubMed]
- Provencher, B.A.; Bartelson, K.J.; Liu, Y.; Foxman, B.M.; Deng, L. Structural study-guided development of versatile phase-transfer catalysts for asymmetric conjugate additions of cyanide. Angew. Chem. Int. Ed. 2011, 50, 10565–10569. [Google Scholar] [CrossRef] [PubMed]
- Jakhar, A.; Sadhukhan, A.; Khan, N.H.; Saravanan, S.; Kureshy, R.I.; Abdi, S.H.R.; Bajaj, H.C. Asymmetric Hydrocyanation of Nitroolefins Catalyzed by an Aluminum(III) Salen Complex. ChemCatChem 2014, 6, 2656–2661. [Google Scholar] [CrossRef]
- Nagata, T.; Tamaki, A.; Kiyokawa, K.; Tsutsumi, R.; Yamanaka, M.; Minakata, S. Enantioselective Electrophilic Cyanation of Boron Enolates: Scope and Mechanistic Studies. Chem. Eur. J. 2018, 24, 17027–17032. [Google Scholar] [CrossRef]
- Jakhar, A.; Ansari, A.; Nandi, S.; Gupta, N.; Khan, N.H.; Kureshy, R.I. Bronsted Basic Ionic Liquid as Catalytic and Reusable Media for Conjugate Cyanation of CF3-Substituted Alkylidenemalonates Using Acetone Cyanohydrin. ChemistrySelect 2017, 2, 11346–11351. [Google Scholar] [CrossRef]
- Wang, J.; Hu, X.L.; Jiang, J.; Gou, S.H.; Huang, X.; Liu, X.H.; Feng, X.M. Asymmetric Activation of Tropos 2,2′-Biphenol with Cinchonine Generates an Effective Catalyst for the Asymmetric Strecker Reaction of N-Tosyl-Protected Aldimines and Ketoimines. Angew. Chem. Int. Ed. 2007, 46, 8468–8470. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.T.; Li, W.; Hu, X.L.; Shen, K.; Tan, C.; Liu, X.H.; Feng, X.M. Asymmetric cyanation of aldehydes, ketones, aldimines, and ketimines catalyzed by a versatile catalyst generated from cinchona alkaloid, achiral substituted 2,2′-biphenol and tetraisopropyl titanate. Chem. Eur. J. 2009, 15, 11642–11659. [Google Scholar] [CrossRef]
- Wang, J.; Li, W.; Liu, Y.L.; Chu, Y.Y.; Lin, L.L.; Liu, X.H.; Feng, X.M. Asymmetric Cyanation of Activated Olefifins with Ethyl Cyanoformate Catalyzed by a Modular Titanium Catalyst. Org. Lett. 2010, 12, 1280–1283. [Google Scholar] [CrossRef]
- Su, Z.S.; Li, W.Y.; Wang, J.; Hu, C.W.; Feng, X.M. A theoretical investigation on the Strecker reaction catalyzed by a Ti(IV)-complex catalyst generated from a cinchona alkaloid, achiral substituted 2,2′-biphenol, and tetraisopropyl titanate. Chem. Eur. J. 2013, 19, 1637–1646. [Google Scholar] [CrossRef]
- Frisch, J.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; Millam, J.M.; et al. Pople, Gaussian 09 (Revision D.01); Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. An improved algorithm for reaction path following. J. Chem. Phys. 1989, 90, 2154. [Google Scholar] [CrossRef]
- Fernandez, I. Combined activation strain model and energy decomposition analysis methods: A new way to understand pericyclic reactions. Phys. Chem. Chem. Phys. 2014, 16, 7662–7671. [Google Scholar] [CrossRef]
- Zeist, W.J.; Bickelhaupt, F.M. The activation strain model of chemical reactivity. Org. Biomol. Chem. 2010, 8, 3118–3127. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, I.; Bickelhaupt, F.M. The activation strain model and molecular orbital theory: Understanding and designing chemical reactions. Chem. Soc. Rev. 2014, 43, 4953–4967. [Google Scholar] [CrossRef] [PubMed]
- Ess, D.H.; Houk, K.N. Distortion/Interaction Energy Control of 1,3-Dipolar Cycloaddition Reactivity. J. Am. Chem. Soc. 2007, 129, 10646–10647. [Google Scholar] [CrossRef]
- Thomas, B.E.; Loncharich, R.J.; Houk, K.N. Force Field Modeling of Transition Structures of Intramolecular Ene Reactions and ab Initio Transition Structures for an Activated Enophile. J. Org. Chem. 1992, 57, 1354–1362. [Google Scholar] [CrossRef]
- Hong, X.; Liang, Y.; Griffith, A.K.; Lambert, T.H.; Houk, K.N. Distortion-accelerated cycloadditions and strain-release-promoted cycloreversions in the organocatalytic carbonyl-olefin metathesis. Chem. Sci. 2014, 5, 471–475. [Google Scholar] [CrossRef]
- Houk, K.N.; Beno, B.R.; Nendel, M.; Black, K.; Yoo, H.Y.; Wilsey, S.; Lee, J.K. Exploration of pericyclic reaction transition structures by quantum mechanical methods: Competing concerted and stepwise mechanisms. J. Mol. Struct. Theochem. 1997, 398, 169–179. [Google Scholar] [CrossRef]
- Hopffgarten, M.; Frenking, G. Energy decomposition analysis. WIREs Comput. Mol. Sci. 2012, 2, 43–62. [Google Scholar] [CrossRef]
- Baerends, J.; Autschbach, J.; Bashford, D.; Bérces, A.; Bickelhaupt, F.M.; Bo, C.; Boerrigter, P.M.; Cavallo, L.; Chong, D.P.; Deng, L.; et al. ADF2016, Theoretical Chemistry; Vrije Universiteit: Amsterdam, The Netherlands, 2016; Available online: http://www.scm.com (accessed on 10 July 2020).
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Sammis, G.M.; Jacobsen, E.N. Highly Enantioselective, Catalytic Conjugate Addition of Cyanide to α,β-Unsaturated Imides. J. Am. Chem. Soc. 2003, 125, 4442–4443. [Google Scholar] [CrossRef] [PubMed]
- Falivene, L.; Cao, Z.; Petta, A.; Serra, L.; Poater, A.; Oliva, R.; Scarano, V.; Cavallo, L. Towards the online computer-aided design of catalytic pockets. Nat. Chem. 2019, 11, 872–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poater, A.; Ragone, F.; Giudice, S.; Costabile, C.; Dorta, R.; Nolan, S.P.; Cavallo, L. Thermodynamics of N-Heterocyclic Carbene Dimerization: The Balance of Sterics and Electronics. J. Am. Chem. Soc. 2008, 27, 2679–2681. [Google Scholar] [CrossRef]
- Poater, A.; Ragone, F.; Mariz, R.; Dorta, R.; Cavallo, L. Comparing the enantioselective power of steric and electrostatic effects in transition-metal-catalyzed asymmetric synthesis. Chem. Eur. J. 2010, 16, 14348–14353. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F.W. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Species | ΔG (kcal mol−1) 1 | |
|---|---|---|---|
| Monodentate | m-I | m-I-COM-si | 12.2 |
| m-I-COM-re | 7.5 | ||
| m-II | m-II-COM-si | 12.6 | |
| m-II-COM-re | 6.9 | ||
| m-III | m-III-COM-si | 12.6 | |
| m-III-COM-re | 16.0 | ||
| m-IV | m-IV-COM-si | 14.4 | |
| m-IV-COM-re | 13.5 | ||
| m-V | m-V-COM-si | 16.1 | |
| m-V-COM-re | 20.0 | ||
| m-VI | m-VI-COM-si | 16.2 | |
| m-VI-COM-re | 11.0 | ||
| Bidentate | d-I | d-I-COM-si | 0.0 |
| d-I-COM-re | −2.5 | ||
| d-II | d-II-COM-si | 5.5 | |
| d-II-COM-re | −0.6 | ||
| d-III | d-III-COM-si | 2.7 | |
| d-III-COM-re | 7.4 |
| TS | ΔE≠strain | ΔE≠int | ΔE≠Pauli | ΔE≠oi | ΔV≠elstat | ΔE≠disp |
|---|---|---|---|---|---|---|
| d-I-TS1-si | 8.8 | −5.6 | 130.4 | −77.9 | −99.9 | −28.7 |
| d-I-TS1-re | 8.6 | −4.9 | 139.0 | −82.6 | −104.0 | −29.4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, Z.; Hu, C.; Shahzad, N.; Kim, C.K. Asymmetric Cyanation of Activated Olefins with Ethyl Cyanoformate Catalyzed by Ti(IV)-Catalyst: A Theoretical Study. Catalysts 2020, 10, 1079. https://doi.org/10.3390/catal10091079
Su Z, Hu C, Shahzad N, Kim CK. Asymmetric Cyanation of Activated Olefins with Ethyl Cyanoformate Catalyzed by Ti(IV)-Catalyst: A Theoretical Study. Catalysts. 2020; 10(9):1079. https://doi.org/10.3390/catal10091079
Chicago/Turabian StyleSu, Zhishan, Changwei Hu, Nasir Shahzad, and Chan Kyung Kim. 2020. "Asymmetric Cyanation of Activated Olefins with Ethyl Cyanoformate Catalyzed by Ti(IV)-Catalyst: A Theoretical Study" Catalysts 10, no. 9: 1079. https://doi.org/10.3390/catal10091079
APA StyleSu, Z., Hu, C., Shahzad, N., & Kim, C. K. (2020). Asymmetric Cyanation of Activated Olefins with Ethyl Cyanoformate Catalyzed by Ti(IV)-Catalyst: A Theoretical Study. Catalysts, 10(9), 1079. https://doi.org/10.3390/catal10091079