Palladium PEPPSI-IPr Complex Supported on a Calix[8]arene: A New Catalyst for Efficient Suzuki–Miyaura Coupling of Aryl Chlorides

 ,
,  , and
, and
Abstract
:1. Introduction
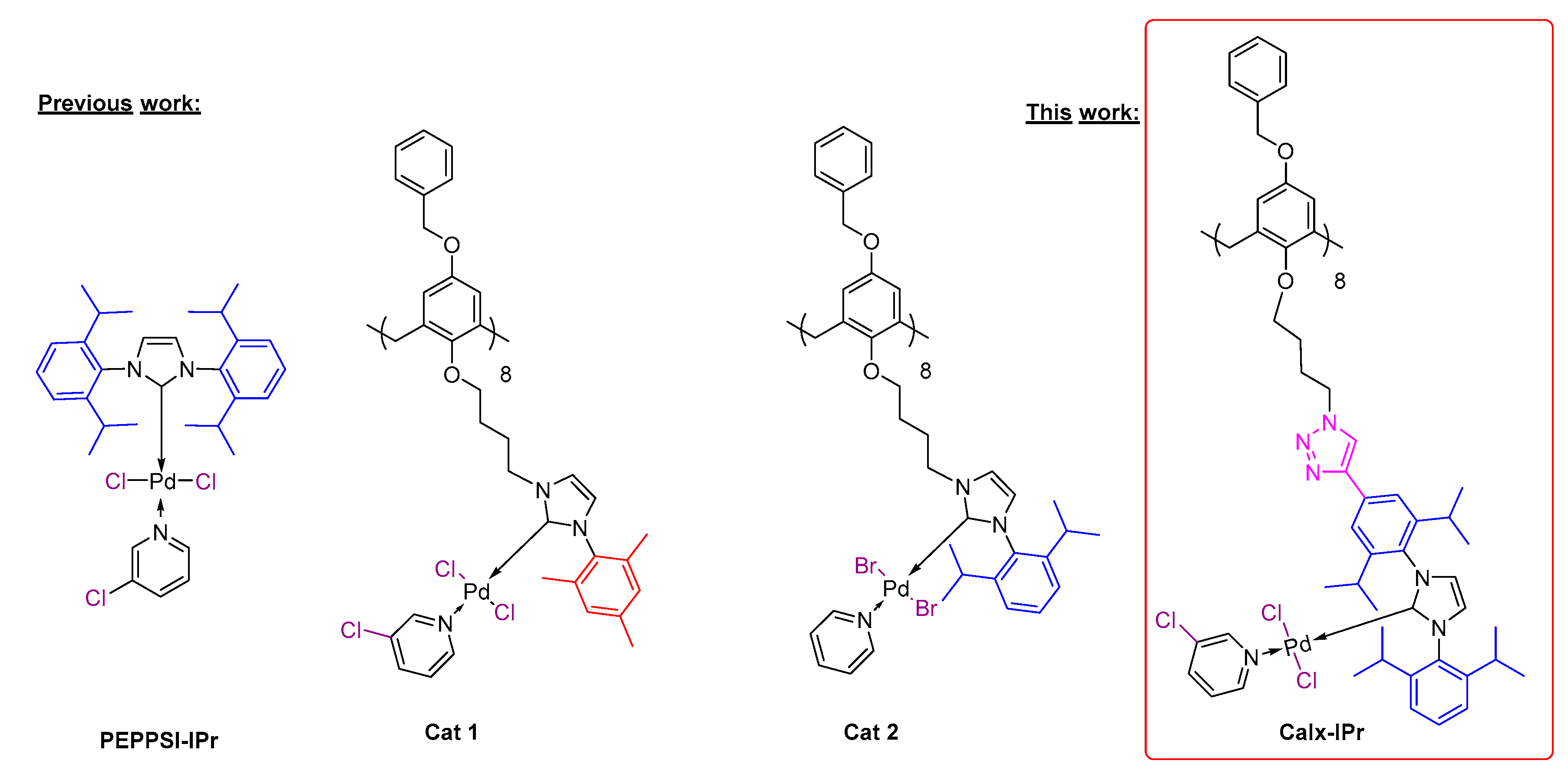
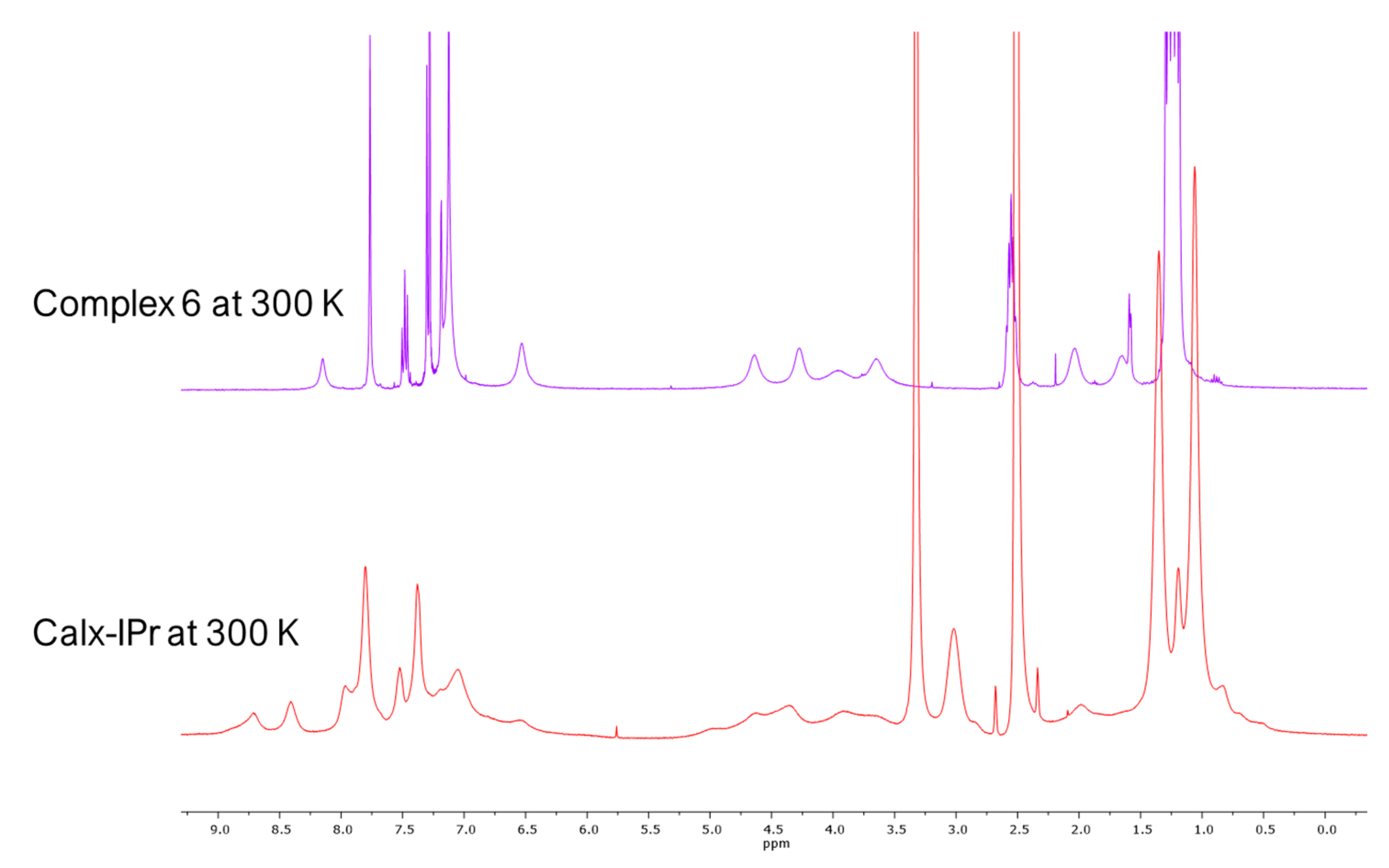
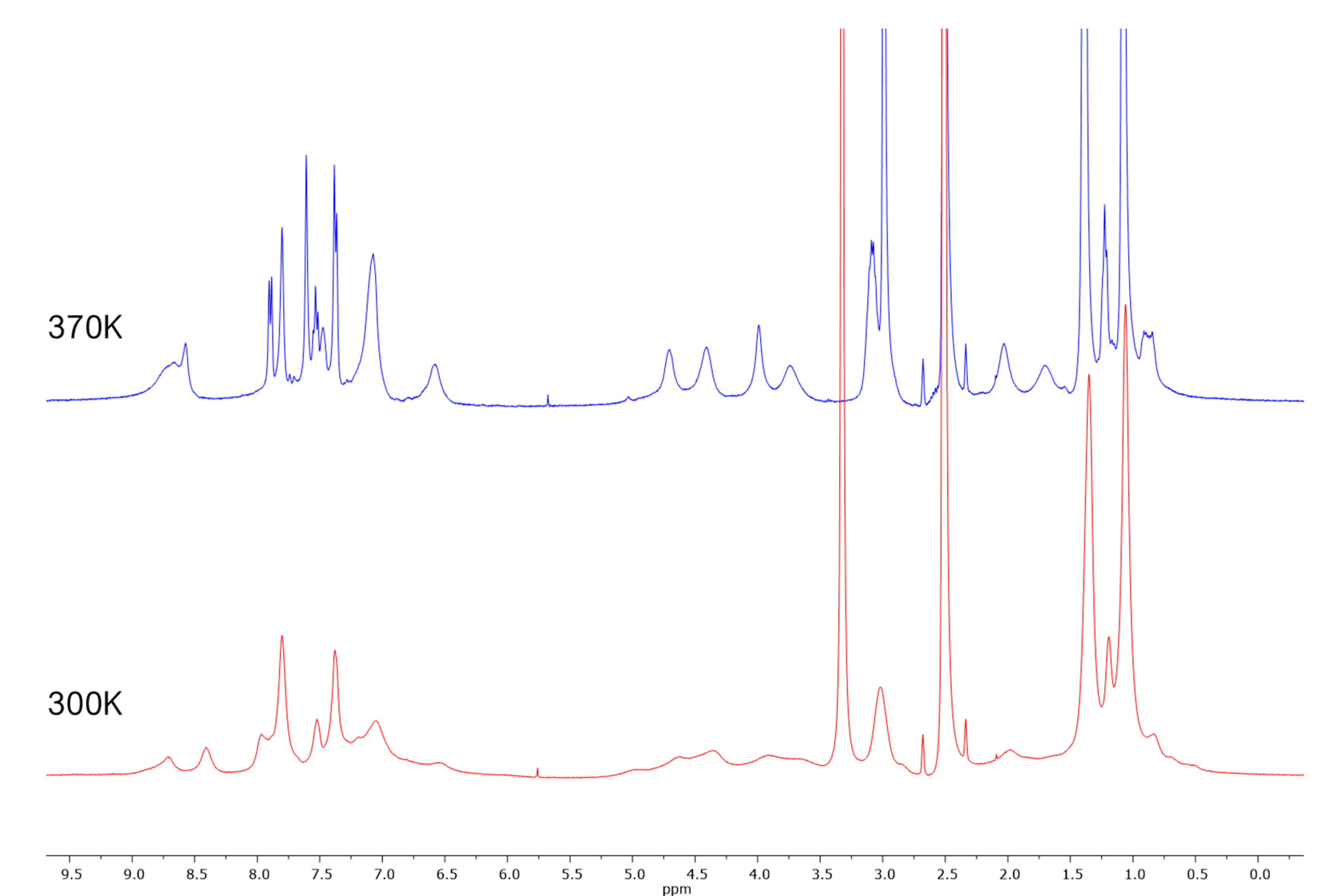
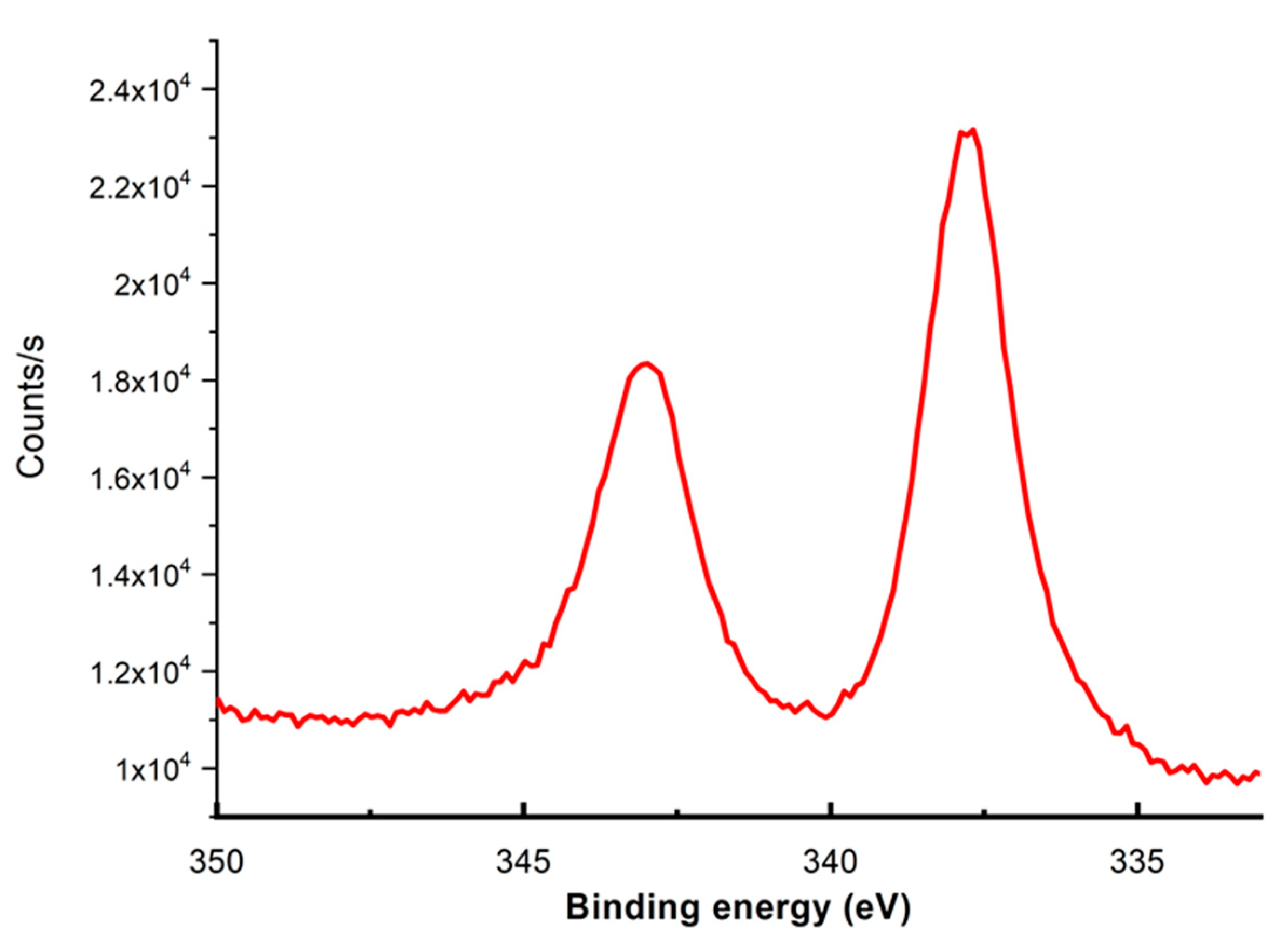
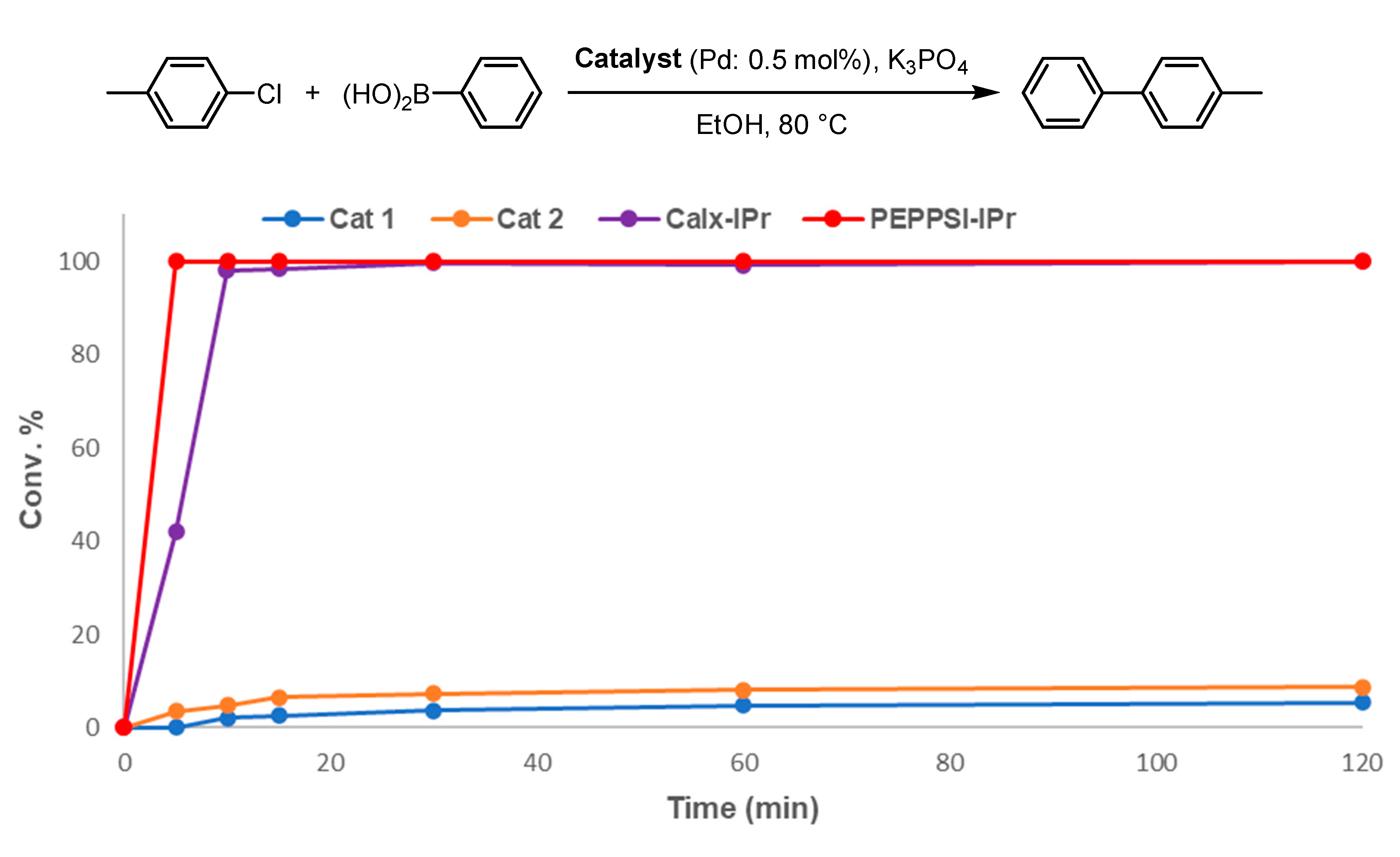
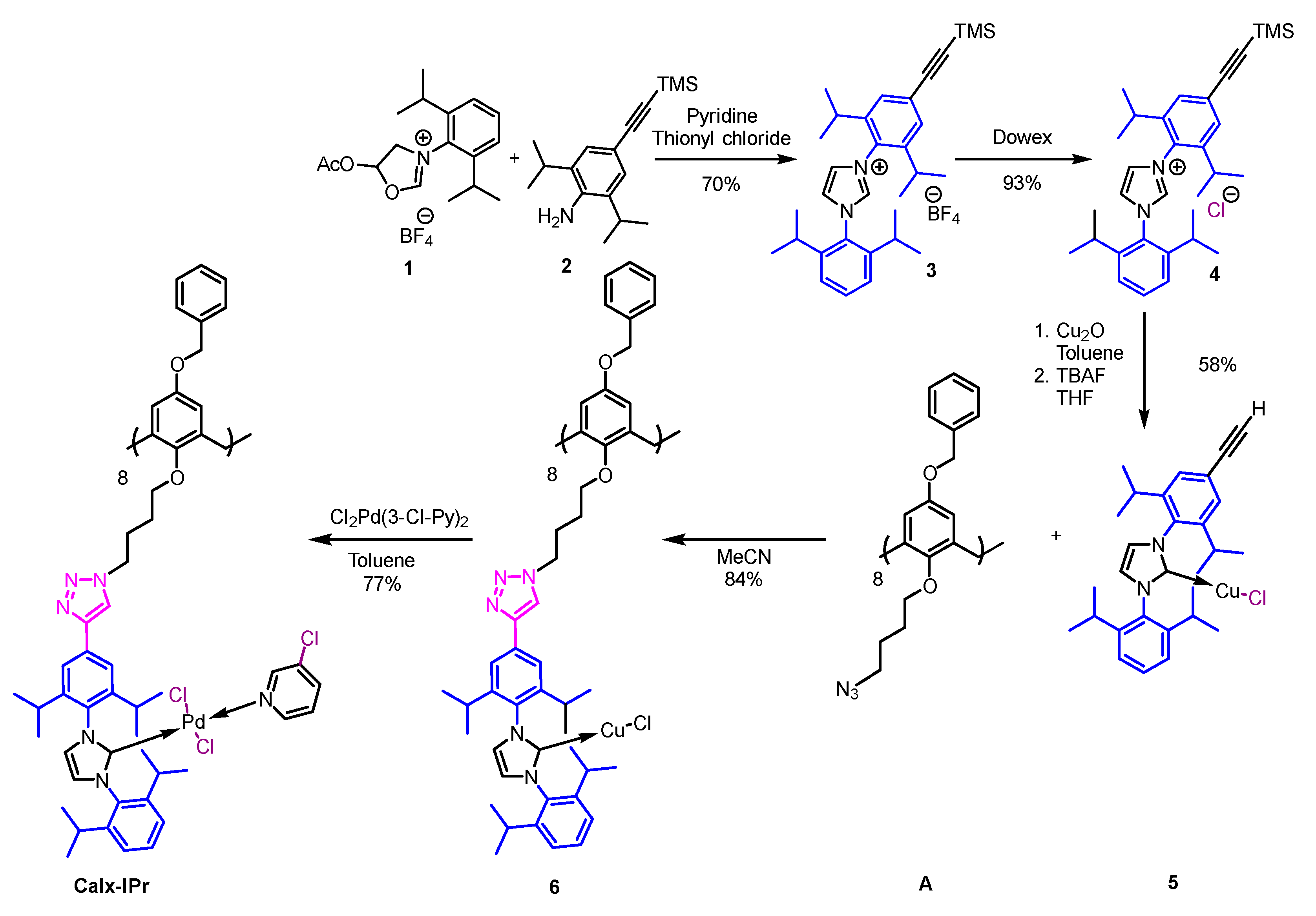
2. Results
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Beller, M.; Bolm, C. Transition Metals for Organic Synthesis, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2004; Volume 1, pp. I–XIX. [Google Scholar]
- Hafner, W.; Jira, R.; Sedlmeier, J.; Smidt, J. Über die Reaktionen von Olefinen mit wassrigen Lösungen von Palladiumsalzen. Chem. Ber. 1962, 95, 1575–1581. [Google Scholar] [CrossRef]
- Smidt, J.; Hafner, W.; Jira, R.; Sieber, R.; Sedlmeier, J.; Sabel, A. The Oxidation of Olefins with Palladium Chloride Catalysts. Angew. Chem. Int. Ed. 1962, 1, 80–88. [Google Scholar] [CrossRef]
- Jira, R. Acetaldehyde from Ethylene. Angew. Chem. Int. Ed. 2009, 48, 9034–9037. [Google Scholar] [CrossRef] [PubMed]
- Mann, S.E.; Benhamou, L.; Sheppard, T.D. Palladium(II)-Catalysed Oxidation of Alkenes. Synthesis 2015, 47, 3079–3117. [Google Scholar]
- Johansson Seechurn, C.C.C.; Kitching, M.O.; Colacot, T.J.; Snieckus, V. Palladium-Catalyzed Cross-Coupling: A Historical Contextual Perspective to the 2010 Nobel Prize. Angew. Chem. Int. Ed. 2012, 51, 5062–5085. [Google Scholar] [CrossRef]
- Gildner, P.G.; Colacot, T.J. Reactions of the 21st Century: Two Decades of Innovative Catalyst Design for Palladium-Catalyzed Cross-Couplings. Organometallics 2015, 34, 5497–5508. [Google Scholar] [CrossRef]
- Biffis, A.; Centomo, P.; Del Zotto, A.; Zecca, M. Pd Metal Catalysts for Cross-Couplings and Related Reactions in the 21st Century: A Critical Review. Chem. Rev. 2018, 118, 2249–2295. [Google Scholar] [CrossRef]
- Torborg, C.; Beller, M. Recent Applications of Palladium-Catalyzed Coupling Reactions in the Pharmaceutical, Agrochemical, and Fine Chemical Industries. Adv. Synth. Catal. 2009, 351, 3027–3043. [Google Scholar] [CrossRef]
- Magano, J.; Dunetz, J.R. Large-Scale Applications of Transition Metal-Catalyzed Couplings for the Synthesis of Pharmaceuticals. Chem. Rev. 2011, 111, 2177–2250. [Google Scholar] [CrossRef]
- Busacca, A.C.; Fan-drick, R.D.; Song, J.J.; Senanayake, C.H. Applications of Transition Metal Catalysis in Drug Discovery and Development; John Wiley & Sons: New York, NY, USA, 2012; pp. 1–24. [Google Scholar]
- Hayler, D.; Leahy, K.D.; Simmons, E.M. A Pharmaceutical Industry Perspective on Sustainable Metal Catalysis. Organometallics 2019, 38, 36–46. [Google Scholar] [CrossRef]
- Diner, C.; Organ, M.G. What Industrial Chemists Want—Are Academics Giving It to Them? Organometallics 2019, 38, 66–75. [Google Scholar] [CrossRef]
- Borths, C.J.; Walker, S.D. Accelerating Pharmaceutical Development via Metal-Mediated Bond Formation. Isr. J. Chem. 2020, 60, 1–12. [Google Scholar] [CrossRef]
- Cheng, F.; Adronov, A. Suzuki Coupling Reactions for the Surface Functionalization of Single-Walled Carbon Nanotubes. Chem. Mater. 2006, 18, 5389–5391. [Google Scholar] [CrossRef]
- Fei, Z.; Soo Kim, J.; Smith, J.; Buchaca Domingo, E.; Anthopoulos, T.D.; Stingelin, N.; Watkins, S.E.; Kim, J.S.; Heeney, M. A low band gap co-polymer of dithienogermole and 2,1,3-benzothiadiazole by Suzuki polycondensation and its application in transistor and photovoltaic cells. J. Mater. Chem. 2011, 21, 16257–16263. [Google Scholar] [CrossRef] [Green Version]
- Jabara, M.; Kumar Maity, S.; Brik, A. Palladium in the Chemical Synthesis and Modification of Proteins. Angew. Chem. Int. Ed. 2017, 56, 10644–10655. [Google Scholar] [CrossRef]
- Isengger, P.G.; Davis, G.B. Concepts of Catalysis in Site-Selective Protein Modifications. J. Am. Chem. Soc. 2019, 141, 8005–8013. [Google Scholar] [CrossRef] [Green Version]
- Ananikov, V.P.; Beletskaya, I.P. Toward the Ideal Catalyst: From Atomic Centers to a “Cocktail” of Catalysts. Organometallics 2012, 31, 1595–1604. [Google Scholar] [CrossRef]
- ICH Guideline Q3D (R1) on Elemental Impurities. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/international-conference-harmonisation-technical-requirements-registration-pharmaceuticals-human-use_en-32.pdf (accessed on 28 March 2019).
- Bourouina, A.; Meille, V.; de Bellefon, C. About Solid Phase vs. Liquid Phase in Suzuki-Miyaura Reaction. Catalysts 2019, 9, 60. [Google Scholar] [CrossRef] [Green Version]
- Phan, N.T.S.; Van Der Sluys, M.; Jones, C.W. On the Nature of the Active Species in Palladium Catalyzed Mizoroki–Heck and Suzuki–Miyaura Couplings -Homogeneous or Heterogeneous Catalysis, A Critical Review. Adv. Synth. Catal. 2006, 348, 609–679. [Google Scholar] [CrossRef]
- Das, P.; Linert, W. Schiff base-derived homogeneous and heterogeneous palladium catalysts for the Suzuki-Miyaura reaction. Coord. Chem. Rev. 2016, 311, 1–23. [Google Scholar] [CrossRef]
- Abdellah, I.; Kasongo, P.; Labattut, A.; Guillot, R.; Schulz, E.; Martini, C.; Huc, V. Benzyloxycalix[8]arene: A new valuable support for NHC palladium complexes in C–C Suzuki–Miyaura couplings. Dalton Trans. 2018, 47, 13843–13848. [Google Scholar] [CrossRef] [PubMed]
- Labattut, A.; Abi Fayssal, S.; Buendia, J.; Abdellah, I.; Huc, V.; Martini, C.; Schulz, E. Calixarene-supported Pd-NHC complexes as efficient catalysts for scalable Suzuki-Miyaura cross-couplings. React. Chem. Eng. 2020, 5, 1509–1514. [Google Scholar] [CrossRef]
- Peramo, A.; Abdellah, I.; Pecnard, S.; Mougin, J.; Martini, C.; Couvreur, P.; Huc, V.; Desmaële, D. A Self-Assembling NHC-Pd-Loaded Calixarene as a Potent Catalyst for the Suzuki-Miyaura Cross-Coupling Reaction in Water. Molecules 2020, 25, 1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzmán-Percástegui, E.; Hernándeza, D.J.; Castillo, I. Calix[8]arene nanoreactor for Cu(I)-catalysed C–S coupling. Chem. Commun. 2016, 52, 3111–3114. [Google Scholar] [CrossRef] [PubMed]
- Abdellah, I.; Martini, C.; Dos Santos, A.; Dragoe, D.; Guérineau, V.; Huc, V.; Schulz, E. Calix[8]arene as New Platform for Cobalt-Salen Complexes Immobilization and Use in Hydrolytic Kinetic Resolution of Epoxides. ChemCatChem 2018, 10, 4761–4767. [Google Scholar] [CrossRef]
- Reyes-Mata, C.A.; Castillo, I. Calix[8]arene-based Ni(II) complexes for electrocatalytic CO2 reduction. Inorg. Chim. Acta 2020, 507, 119607. [Google Scholar] [CrossRef]
- Frank, M.; Maas, G.; Schatz, J. Calix[4]arene-Supported N-Heterocyclic Carbene Ligands as Catalysts for Suzuki Cross-Coupling Reactions of Chlorotoluene. Eur. J. Org. Chem. 2004, 2004, 607–613. [Google Scholar] [CrossRef]
- Brenner, E.; Matt, D.; Henrion, M.; Teci, M.; Toupet, L. Calix[4]arenes with one and two N-linked imidazolium units as precursors of N-heterocyclic carbene complexes. Coordination chemistry and use in Suzuki-Miyaura cross-coupling. Dalton Trans. 2011, 40, 9889–9898. [Google Scholar] [CrossRef]
- Szilvási, T.; Veszprémi, T. Internal Catalytic Effect of Bulky NHC Ligands in Suzuki—Miyaura Cross-Coupling Reaction. ACS Catal. 2013, 3, 1984–1991. [Google Scholar] [CrossRef]
- O’Brien, C.J.; Kantchev, E.A.B.; Valente, C.; Hadei, N.; Chass, G.A.; Lough, A.; Hopkinson, A.C.; Organ, M.G. Easily Prepared Air- and Moisture-Stable Pd–NHC (NHC=N-Heterocyclic Carbene) Complexes: A Reliable, User-Friendly, Highly Active Palladium Precatalyst for the Suzuki–Miyaura Reaction. Chem. Eur. J. 2006, 12, 4743–4748. [Google Scholar] [CrossRef]
- Nasielski, J.; Hadei, N.; Achonduh, G.; Kantchev, E.A.B.; O’Brien, C.J.; Lough, A.; Organ, M.G. Structure–Activity Relationship Analysis of Pd–PEPPSI Complexes in Cross-Couplings: A Close Inspection of the Catalytic Cycle and the Precatalyst Activation Model. Chem. Eur. J. 2010, 16, 10844–10853. [Google Scholar] [CrossRef] [PubMed]
- Price, G.A.; Bogdan, A.R.; Aguirre, A.L.; Iwai, T.; Djuric, S.W.; Organ, M.G. Continuous flow Negishi cross-couplings employing silica-supported Pd-PEPPSI–IPr precatalyst. Catal. Sci. Technol. 2016, 6, 4733–4742. [Google Scholar] [CrossRef]
- Fürstner, A.; Alcarazo, M.; César, V.; Lehmann, C.W. Convenient, scalable and flexible method for the preparation of imidazolium salts with previously inaccessible substitution patterns. Chem. Commun. 2006, 20, 2176–2178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, G.A.; Hassan, A.; Chandrasoma, N.; Bogdan, A.R.; Djuric, S.W.; Organ, M.G. Pd-PEPPSI-IPent-SiO2: A Supported Catalyst for Challenging Negishi Coupling Reactions in Flow. Angew. Chem. Int. Ed. 2017, 56, 1–5. [Google Scholar] [CrossRef]
- Beutler, U.; Boehm, M.; Fuenfschilling, P.C.; Heinz, T.; Mutz, J.P.; Onken, U.; Mueller, M.; Zaugg, W. A High-Throughput Process for Valsartan. Org. Process Res. Dev. 2007, 11, 892–898. [Google Scholar] [CrossRef]
- Fan, X.; Song, Y.-L.; Long, Y.-Q. An Efficient and Practical Synthesis of the HIV Protease Inhibitor Atazanavir via a Highly Diastereoselective Reduction Approach. Org. Process Res. Dev. 2008, 12, 69–75. [Google Scholar] [CrossRef]
- Krogul, A.; Skupinska, J.; Litwinienko, G. Catalytic activity of PdCl2 complexes with pyridines in nitrobenzene carbonylation. J. Mol. Catal. A Chem. 2011, 337, 9–16. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element | Theoretical (%) | Experimental (%) |
|---|---|---|
| C1S | 81 | 85 |
| Cl2p | 5 | 5 |
| N1s | 9 | 8 |
| Pd3d | 2 | 2 |

| Solvent | Base | Conv (%) [a,b] |
|---|---|---|
| EtOH | K3PO4 | 95 |
| THF | K3PO4 | <5 |
| DMF | K3PO4 | <5 |
| Toluene | K3PO4 | <5 |
| EtOH | K2CO3 | 88 |
| EtOH | KOH | 88 (90/4 h) |
| EtOH | Cs2CO3 | 73 |

| Pd (mol%) | T (°C) | Conv (%) [a,b] |
|---|---|---|
| 1 | 80 | 100 |
| 0.5 | 80 | 99 |
| 0.5 | 50 | 70 |
| 0.4 | 25 | 15 (98/20 h) |
| 0.2 | 80 | 95 |
| 0.1 | 80 | 80 |

| Entry | Aromatic Halide | Boronic Acid | T (°C) | [Pd] (mol%) | Conv (%) [a,b] | Yield (%) |
|---|---|---|---|---|---|---|
| 1 |  |  | 80 | 0.5 | 95 | 92 |
| 2 |  |  | 80 | 0.2 | 100 | 93 |
| 3 |  |  | 80 | 1 | 100 | 90 |
| 4 |  |  | 80 | 1 | 74 | 67 |
| 5 |  |  | 80 | 1 | 73 | 56 |
| 6 |  |  | 80 | 0.5 | 100 | 98 |
| 7 |  |  | 80 | 0.5 | 100 | 89 |
| 8 |  |  | 80 | 1 | 83 | 68 |
| 9 |  |  | 80 | 0.5 | 100 | 87 |
| 10 |  |  | 80 | 0.5 | 96 | 93 |

| Aromatic Halide | Boronic Acid | Pd (mol%) | Conversion (%) | Selectivity (%) | Pd (ppm) |
|---|---|---|---|---|---|
 |  | 1 | 74 | 100 | 39.2 |
 |  | 0.1 | 80 | 100 | 5.6 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Labattut, A.; Abdellah, I.; Buendia, J.; Abi Fayssal, S.; Adhel, E.; Dragoe, D.; Martini, C.; Schulz, E.; Huc, V. Palladium PEPPSI-IPr Complex Supported on a Calix[8]arene: A New Catalyst for Efficient Suzuki–Miyaura Coupling of Aryl Chlorides. Catalysts 2020, 10, 1081. https://doi.org/10.3390/catal10091081
Labattut A, Abdellah I, Buendia J, Abi Fayssal S, Adhel E, Dragoe D, Martini C, Schulz E, Huc V. Palladium PEPPSI-IPr Complex Supported on a Calix[8]arene: A New Catalyst for Efficient Suzuki–Miyaura Coupling of Aryl Chlorides. Catalysts. 2020; 10(9):1081. https://doi.org/10.3390/catal10091081
Chicago/Turabian StyleLabattut, Axel, Ibrahim Abdellah, Julien Buendia, Sandra Abi Fayssal, Erika Adhel, Diana Dragoe, Cyril Martini, Emmanuelle Schulz, and Vincent Huc. 2020. "Palladium PEPPSI-IPr Complex Supported on a Calix[8]arene: A New Catalyst for Efficient Suzuki–Miyaura Coupling of Aryl Chlorides" Catalysts 10, no. 9: 1081. https://doi.org/10.3390/catal10091081
APA StyleLabattut, A., Abdellah, I., Buendia, J., Abi Fayssal, S., Adhel, E., Dragoe, D., Martini, C., Schulz, E., & Huc, V. (2020). Palladium PEPPSI-IPr Complex Supported on a Calix[8]arene: A New Catalyst for Efficient Suzuki–Miyaura Coupling of Aryl Chlorides. Catalysts, 10(9), 1081. https://doi.org/10.3390/catal10091081