Effects of Framework Disruption of Ga and Ba Containing Zeolitic Materials by Thermal Treatment


Abstract
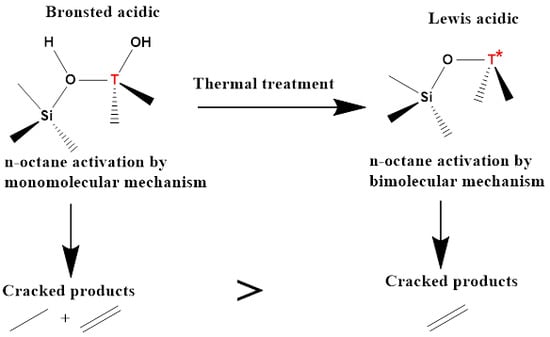
:
1. Introduction
2. Results and Discussion
2.1. Catalysts Characterization
2.1.1. Powder XRD
2.1.2. FT-IR
2.1.3. Scanning Electron Microscopy
2.1.4. Pyridine FT-IR
2.1.5. Temperature-Programmed Desorption with NH3
2.1.6. Thermogravimetric Analysis
2.1.7. Brunauer, Emmett, and Teller (BET) Studies
2.2. Catalytic Performance
3. Materials and Experimental Methods
3.1. Catalysts Preparation
3.2. Catalysts Characterization
3.3. Catalytic Testing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kosinov, N.; Liu, C.; Hensen, E.J.M.; Pidko, E.A. Engineering of Transition Metal Catalysts Confined in Zeolites. Chem. Mater. 2018, 30, 3177–3198. [Google Scholar] [CrossRef] [PubMed]
- Moshoeshoe, M.; Nadiye-Tabbiruka, M.S.; Obuseng, V. A Review of the Chemistry, Structure, Properties and Applications of Zeolites. Am. J. Mater. Sci. 2017, 7, 196–221. [Google Scholar]
- Aliev, A.M.; Shabanova, Z.A.; Nadzhaf-Kuliev, U.M.; Medzhidova, S.M. Oxidative Dehydrogenation of Cyclohexane over Modified Zeolite Catalysts. Petrol. Chem. 2016, 56, 639–645. [Google Scholar] [CrossRef]
- Alamdari, A.; Karimzadeh, R. Oxidative Dehydrogenation of Liquefied Petroleum Gas on Copper, Zinc and Iron Oxide Impregnated on MFI Zeolite Assisted by Electric Power. Catalysts 2018, 8, 270. [Google Scholar] [CrossRef] [Green Version]
- Bakare, A.; Muraza, O.; Yoshioka, M.; Yamani, Z.H.; Yokoi, T. Conversion of Methanol to Olefins over Al-rich ZSM-5 Modified with Alkaline Earth Metal oxides. Catal. Sci. Technol. 2016, 6, 7852. [Google Scholar] [CrossRef]
- Shamzhy, M.; Opanasenko, M.; Concepción, P.; Martínez, A. New Trends in Tailoring Active Sites in Zeolite-based Catalysts. Chem. Soc. Rev. 2019, 48, 1095. [Google Scholar] [CrossRef]
- Li, G.; Liu, C.; Rohling, R.; Hensen, E.J.M.; Pidko, E.A. Lewis Acid Catalysis by Zeolites. In Modelling and Simulation in the Science of Micro- and Meso-Porous Materials; Richard, C., Catlow, A., Van Speybroeck, V., van Santen, R.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 229–263. [Google Scholar]
- Almutairi, S.M.T. The Role of Lewis and Brønsted Acidity for Alkane Activation OVER Zeolites. Ph.D. Thesis, Eindhoven University of Technology, Eindhoven, The Netherlands, 2013. [Google Scholar]
- Areán, C.O.; Palomino, G.T.; Geobaldo, F.; Zecchina, A. Characterization of Gallosilicate MFI-Type Zeolites by IR Spectroscopy of Adsorbed Probe Molecules. J. Phys. Chem. 1996, 100, 6678–6690. [Google Scholar] [CrossRef]
- Krannila, H.; Haag, W.O.; Gates, B.C. Monomolecular and Bimolecular Mechanisms of Paraffin Cracking: N-butane Cracking Catalyzed by HZSM-5. J. Catal. 1992, 135, 115–124. [Google Scholar] [CrossRef]
- Corma, A.; Orchillés, A.V. Current Views on the Mechanism of Catalytic Cracking. Micropor. Mesopor. Mater. 2000, 35, 21–30. [Google Scholar] [CrossRef]
- Ndlela, S.S.; Friedrich, H.B.; Cele, M.N. Effects of modifying acidity and reducibility on the activity of NaY zeolite in the oxidative dehydrogenation of n-octane. Catalysts 2020, 10, 363. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Y.; Walker, H.; Zhu, Q. Reduction of Nitrate by NaY Zeolite Supported Fe, Cu/Fe and Mn/Fe Nanoparticles. J. Hazard. Mater. 2016, 324, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, J.; Dong, M.; Fan, S.; Zhao, T.; Wang, J.; Fan, W. Strategies to control Zeolite Particle Morphology. Chem. Soc. Rev. 2019, 48, 885–907. [Google Scholar] [CrossRef] [PubMed]
- Suresh, S.; Teja, K.R.; Chand, S. Catalytic Wet Peroxide Oxidation of Azo Dye (Acid Orange 7) Using NaY Zeolite from Coal Fly Ash. Int. J. Environ. Waste Manag. 2014, 14, 338–357. [Google Scholar] [CrossRef]
- Zhao, J.; Yin, Y.; Li, Y.; Chen, W.; Liu, B. Synthesis and Characterization of Mesoporous Zeolite Y by Using Block Copolymers as Templates. Chem. Eng. J. 2016, 284, 405–411. [Google Scholar] [CrossRef]
- Pal, N.; Pramanik, M.; Bhaumik, A.; Ali, M. Highly Selective and Direct Oxidation of Cyclohexane to Cyclohexanone over Vanadium Exchanged NaY at Room Temperature under Solvent-free Conditions. J. Mol. Catal. A Chem. 2014, 392, 299–307. [Google Scholar] [CrossRef]
- Lercher, J.A.; Gründling, C.; Eder-Mirth, G. Infra-red Studies of The Surface Acidity of Oxides and Zeolites using Adsorbed Probe Molecular. Catal. Today 1996, 27, 353–376. [Google Scholar] [CrossRef] [Green Version]
- Cele, M.N.; Friedrich, H.B.; Bala, M.D. A Study of Fe(III)TPPCl Encapsulated in Zeolite NaY and Fe(III)NaY in The Oxidation of n-octane, Cyclohexane, 1-octene and 4-octene. Reac. Kinet. Mech. Catal. 2014, 111, 737–750. [Google Scholar] [CrossRef]
- Mu, L.; Feng, W.; Zhang, H.; Hu, X.; Cui, Q. Synthesis and Catalytic Performance of a Small Crystal NaY Zeolite with High SiO2/Al2O3 Ratio. RSC Adv. 2019, 9, 20528. [Google Scholar] [CrossRef] [Green Version]
- Shao, C.; Lang, W.; Yan, X.; Guo, Y. Catalytic performance of gallium oxide based-catalysts for the propane dehydrogenation reaction: Effects of support and loading amount. RSC Adv. 2017, 7, 4710–4723. [Google Scholar] [CrossRef] [Green Version]
- El-Malki, M.; van Santen, R.A.; Sachtler, W.M.H. Introduction of Zn, Ga, and Fe into HZSM-5 Cavities by Sublimation: Identification of Acid Sites. J. Phys. Chem. B 1999, 103, 4611–4622. [Google Scholar] [CrossRef]
- Niwa, M.; Katada, N. New Method for the Temperature- Programmed Desorption (TPD) of Ammonia Experiment for Characterization of Zeolite Acidity: A Review. Chem. Rec. 2013, 13, 432–455. [Google Scholar] [CrossRef] [PubMed]
- Loiland, J.; Zhao, Z.; Patel, A.; Hazin, P. Boron-Containing Catalysts for the Oxidative Dehydrogenation of Ethane/Propane Mixtures. Ind. Eng. Chem. Res. 2019, 58, 19818–19824. [Google Scholar] [CrossRef]
- Bish, D.L.; Carey, J.W. Thermal Behavior of Natural Zeolites. Rev. Miner. Geochem. 2001, 45, 403–452. [Google Scholar] [CrossRef]
- Sing, K.S.W.; Everett, D.H.; Haul, R.A.W.; Moscou, L.; Pierotti, R.A.; Rouquérol, J.; Siemieniewska, T. Reporting Physisorption Data for Gas/Solid Systems with Special Reference to The Determination of Surface Area and Porosity (Recommendations 1984). Pure Appl. Chem. 1985, 57, 603–619. [Google Scholar] [CrossRef]
- Rutkowska, M.; Chmielarz, L.; Jabłońska, M.; Van Oers, C.J.; Cool, P. Iron Exchanged ZSM-5 and Y Zeolites Calcined at Different Temperatures: Activity in N2O Decomposition. J. Porous Mater. 2014, 21, 91–98. [Google Scholar] [CrossRef]
- Lu, J.; Zhao, Z.; Xu, C.; Duan, A.; Zhang, P. Effects of Calcination Temperature on the Acidity and Catalytic Performances of HZSM-5 Zeolite Catalysts for the Catalytic Cracking of n-Butane. J. Nat. Gas Chem. 2005, 14, 213–220. [Google Scholar]
- Huang, Y.; Wan, K.; Dong, D.; Li, D.; Hill, M.R.; Hill, A.J.; Wang, H. Synthesis of Hierarchical Porous Zeolite NaY Particles with Controllable Particle Sizes. Micropor. Mesopor. Mater. 2010, 127, 167–175. [Google Scholar] [CrossRef]
- Elkhalifa, E.A.; Friedrich, H.B. Effects of Boron and Barium Dopants on VMgO Catalysts Employed in the Oxidative Dehydrogenation of n-octane. Kinet. Catal. 2015, 56, 212–221. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Acidity Amount (µmol g−1) | ||||
|---|---|---|---|---|
| Catalyst | W (100–200 °C) | M (200–400 °C) | S (400–600 °C) | Total Acidity |
| Ga-BaY(IS)-550 | 2.5 | 0.6 | 2.4 | 5.5 |
| Ga-BaY(IS)-750 | 0.7 | 1.5 | 3.8 | 6.0 |
| Ga-BaY(Sil)-550 | 1.0 | 0 | 1.1 | 2.1 |
| Ga-BaY(Sil)-750 | 0.7 | 1.1 | 1.6 | 3.4 |
| Catalysts | BET (m2/g) | Pore Volume (cm3/g) | Pore Size (nm) |
|---|---|---|---|
| Ga-BaY(IS)-250 | 734 | 0.33 | 17 |
| Ga-BaY(IS)-750 | 709 | 0.32 | 18 |
| Ga-BaY(Sil)-250 | 492 | 0.22 | 18 |
| Ga-BaY(Sil)-750 | 472 | 0.21 | 19 |
| Catalysts | %Alkanes | %Alkenes |
|---|---|---|
| Ga-BaY(IS)-250 | 11 | 89 |
| Ga-BaY(IS)-550 | 3.0 | 97 |
| Ga-BaY(IS)-750 | 100 | |
| Ga-BaY(Sil)-250 | 6 | 94 |
| Ga-BaY(Sil)-550 | 1.0 | 99 |
| Ga-BaY(Sil)-750 | 100 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ndlela, S.S.; Friedrich, H.B.; Cele, M.N. Effects of Framework Disruption of Ga and Ba Containing Zeolitic Materials by Thermal Treatment. Catalysts 2020, 10, 975. https://doi.org/10.3390/catal10090975
Ndlela SS, Friedrich HB, Cele MN. Effects of Framework Disruption of Ga and Ba Containing Zeolitic Materials by Thermal Treatment. Catalysts. 2020; 10(9):975. https://doi.org/10.3390/catal10090975
Chicago/Turabian StyleNdlela, Siyabonga S., Holger B. Friedrich, and Mduduzi N. Cele. 2020. "Effects of Framework Disruption of Ga and Ba Containing Zeolitic Materials by Thermal Treatment" Catalysts 10, no. 9: 975. https://doi.org/10.3390/catal10090975
APA StyleNdlela, S. S., Friedrich, H. B., & Cele, M. N. (2020). Effects of Framework Disruption of Ga and Ba Containing Zeolitic Materials by Thermal Treatment. Catalysts, 10(9), 975. https://doi.org/10.3390/catal10090975