
Theoretical Study on Epoxide Ring-opening in CO2/Epoxide Copolymerization Catalyzed by Bifunctional Salen-Type Cobalt(III) Complexes: Influence of Stereoelectronic Factors

 , ,
, ,  and
and
Abstract
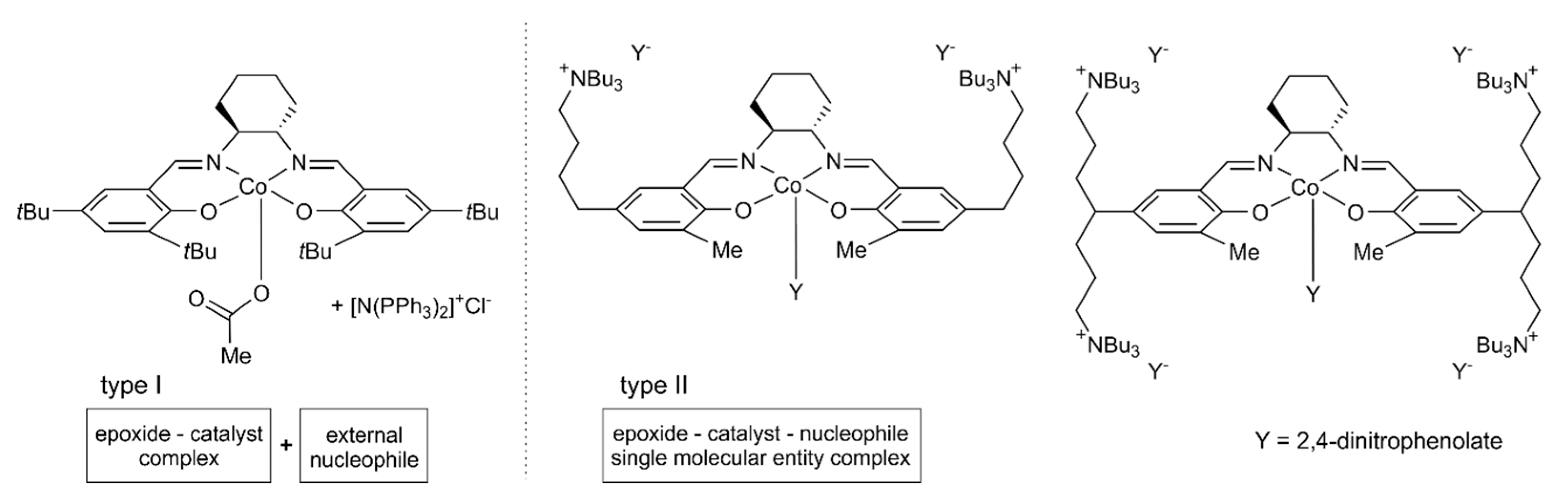
:1. Introduction
2. Results and Discussion
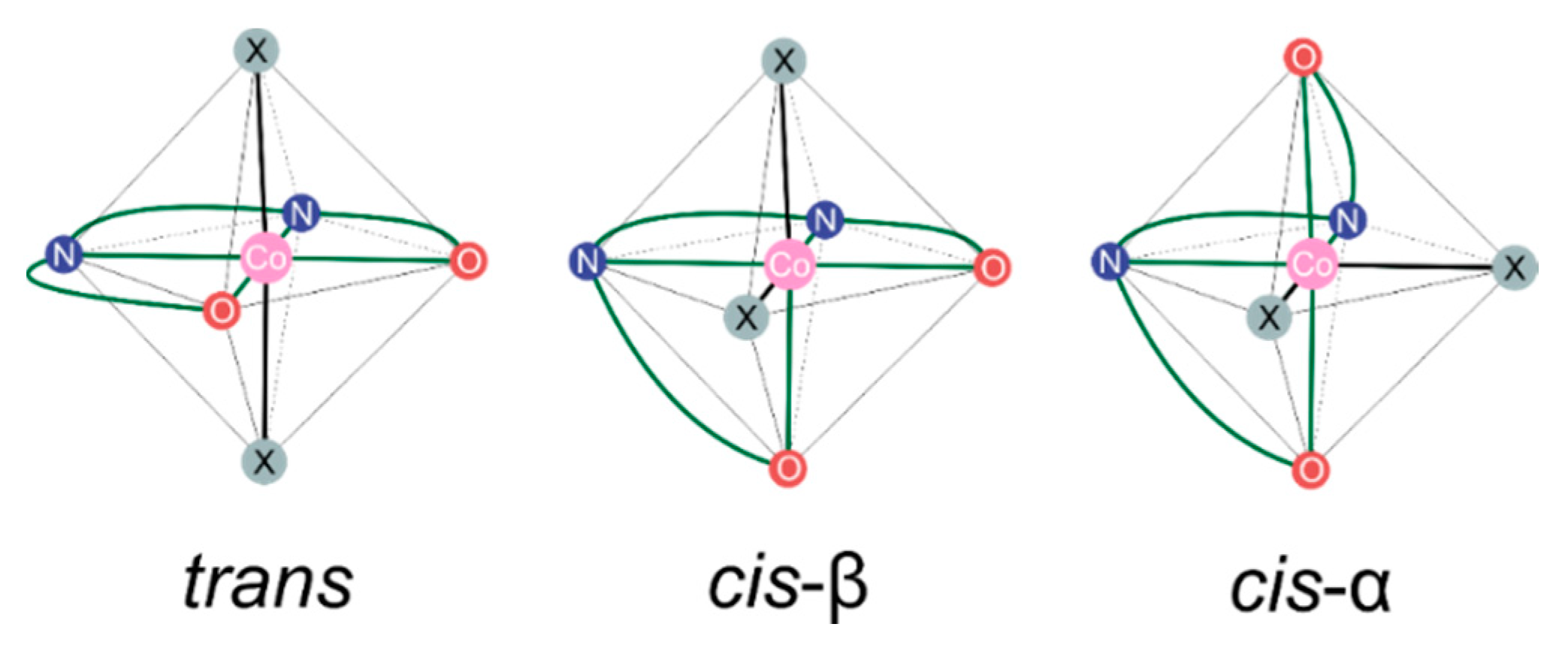
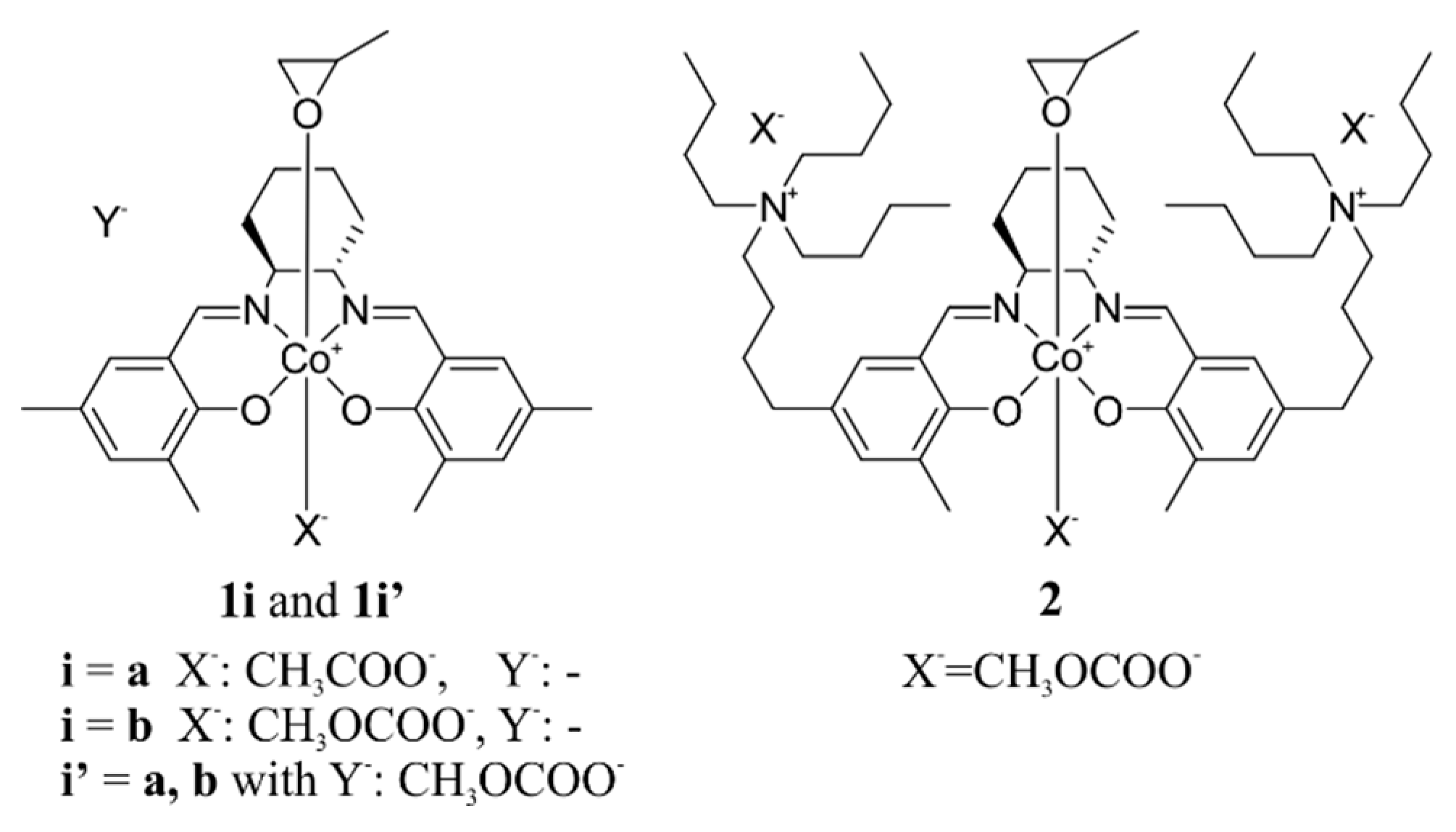
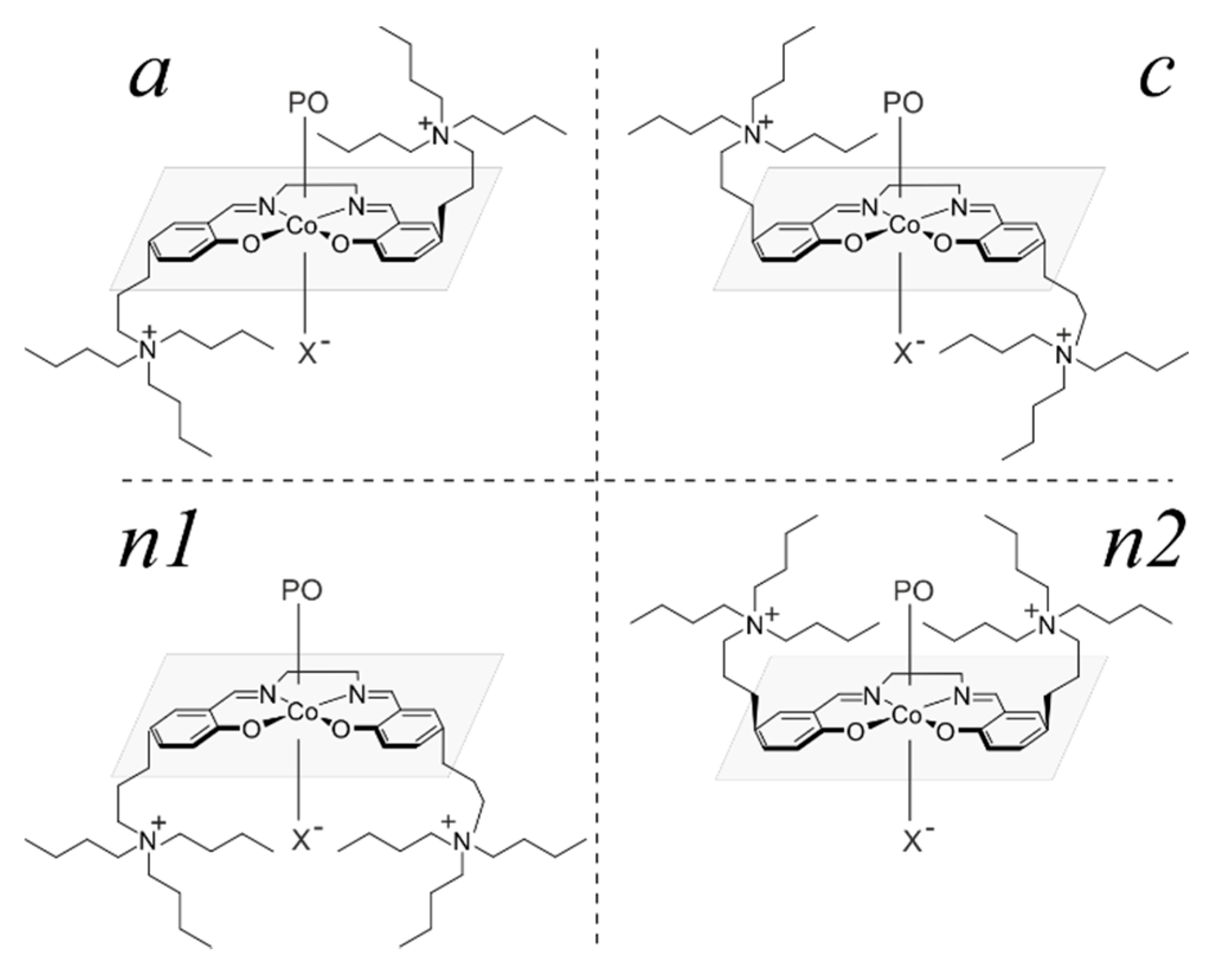
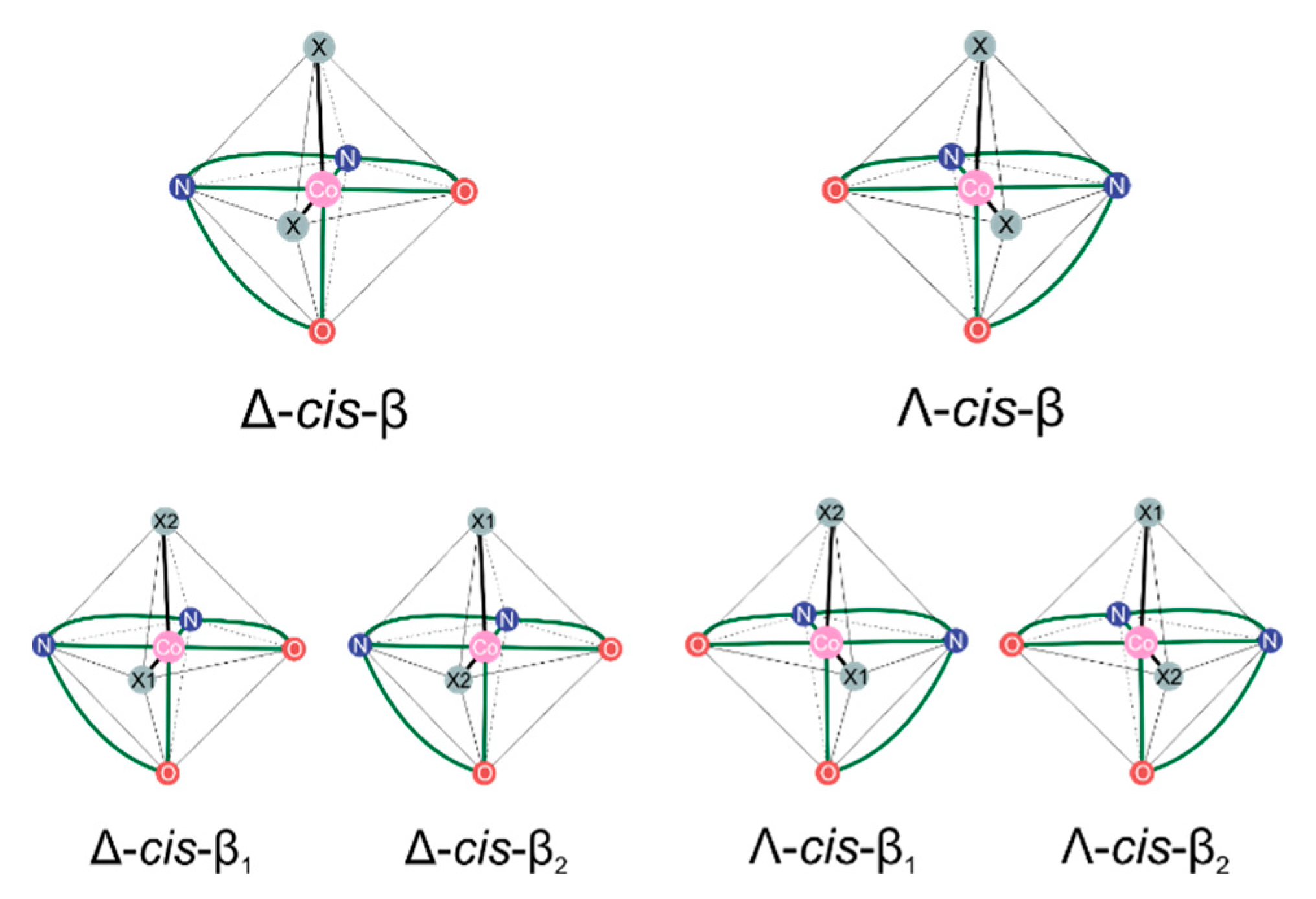
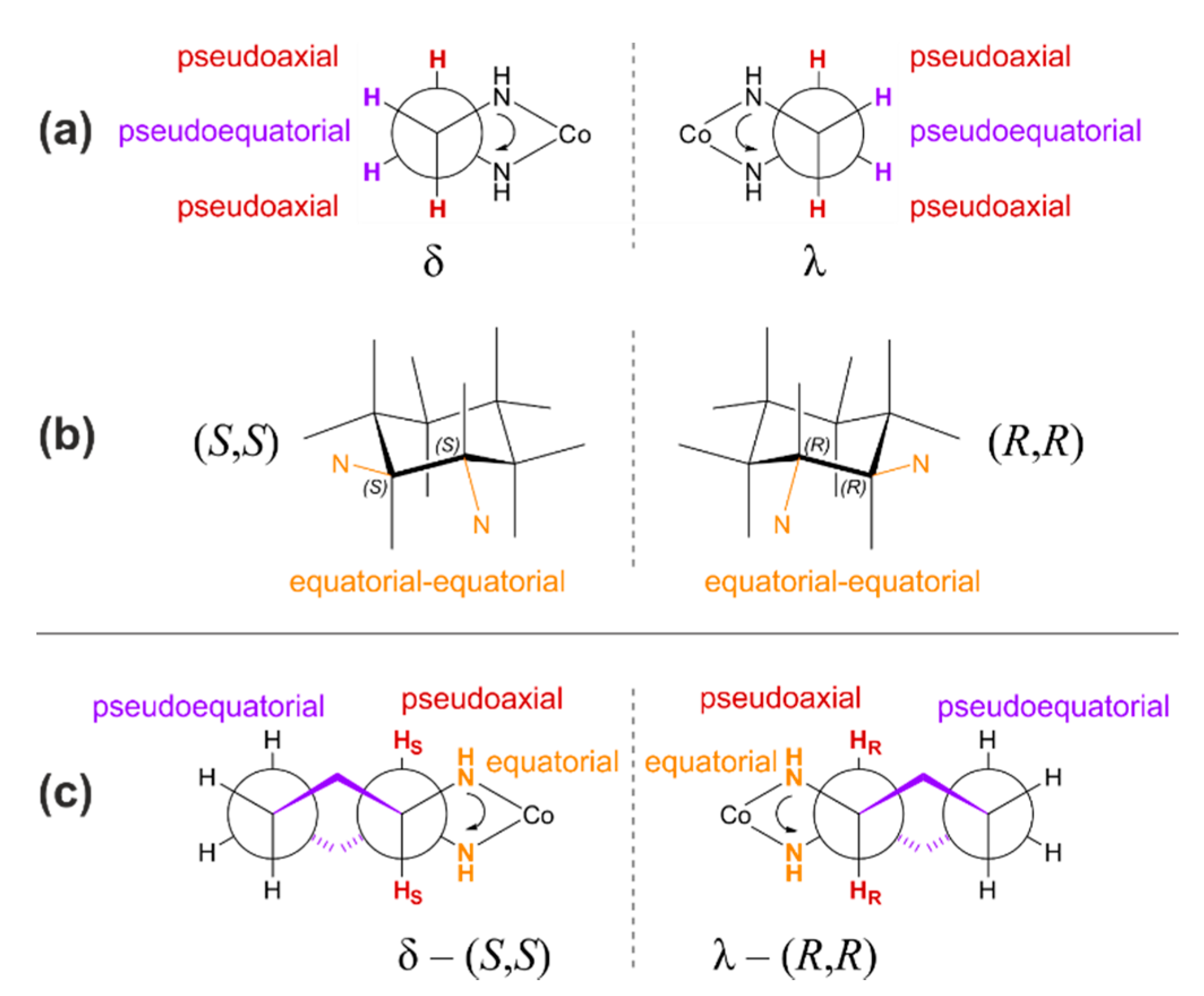
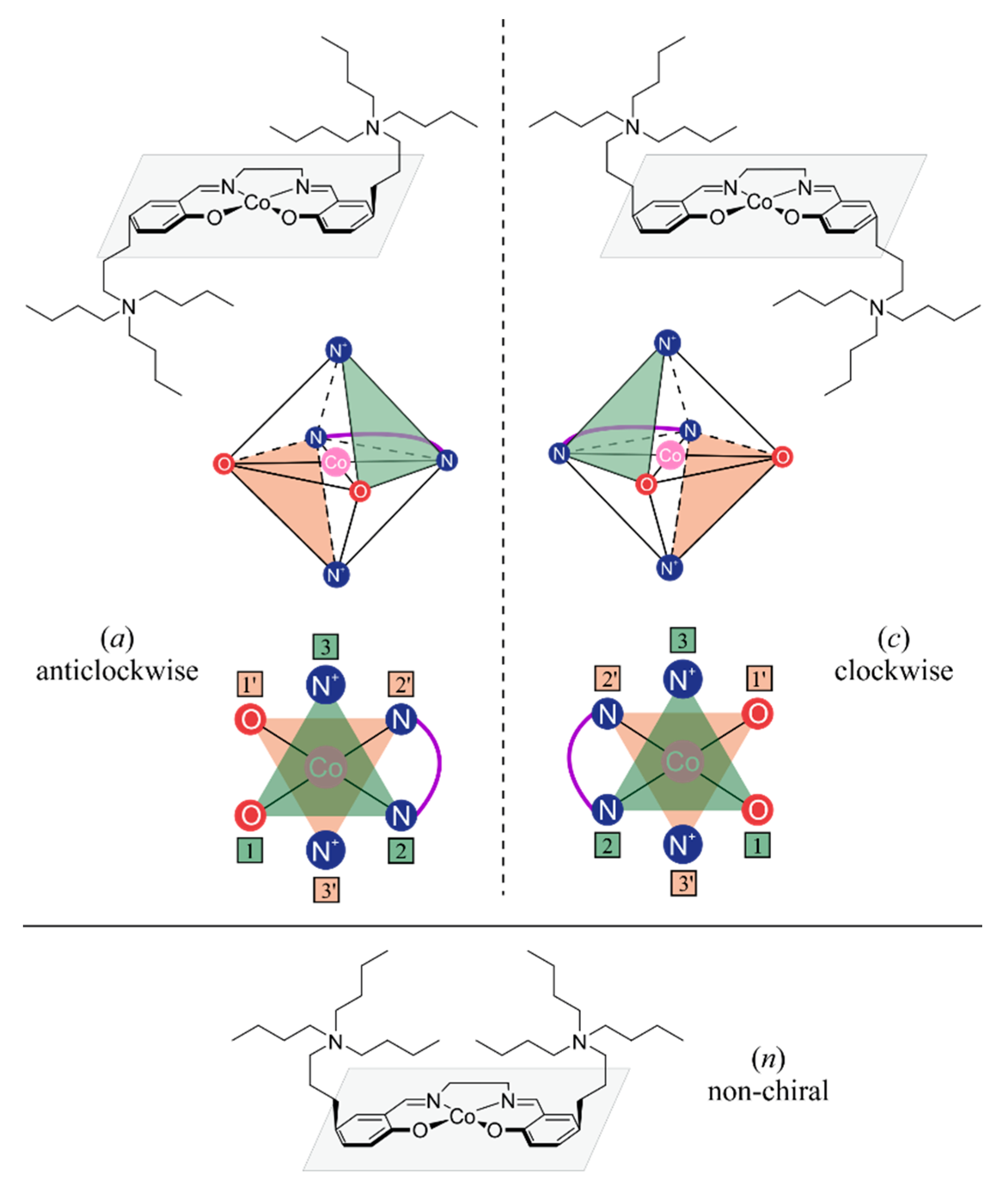
2.1. Considered Models and Stereoisomers
2.2. Epoxide Binding in Model Sytems 1: Stereochemical Complementarity of Epoxide and Catalyst
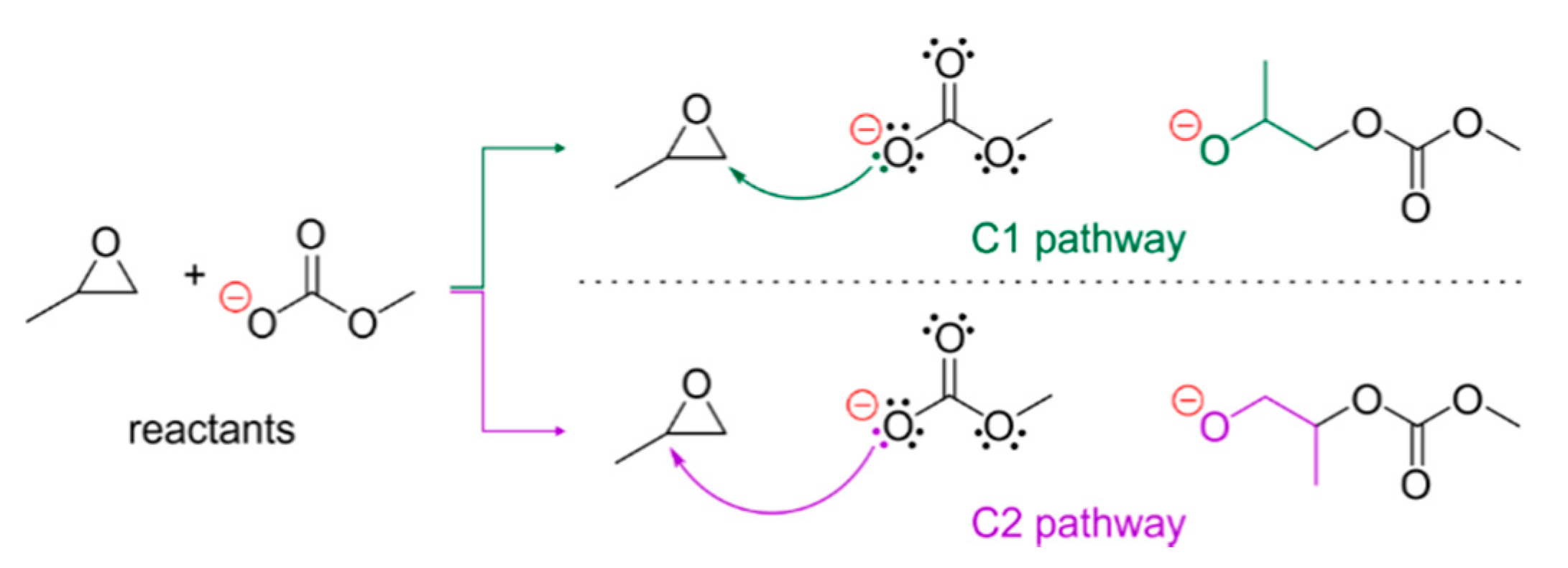
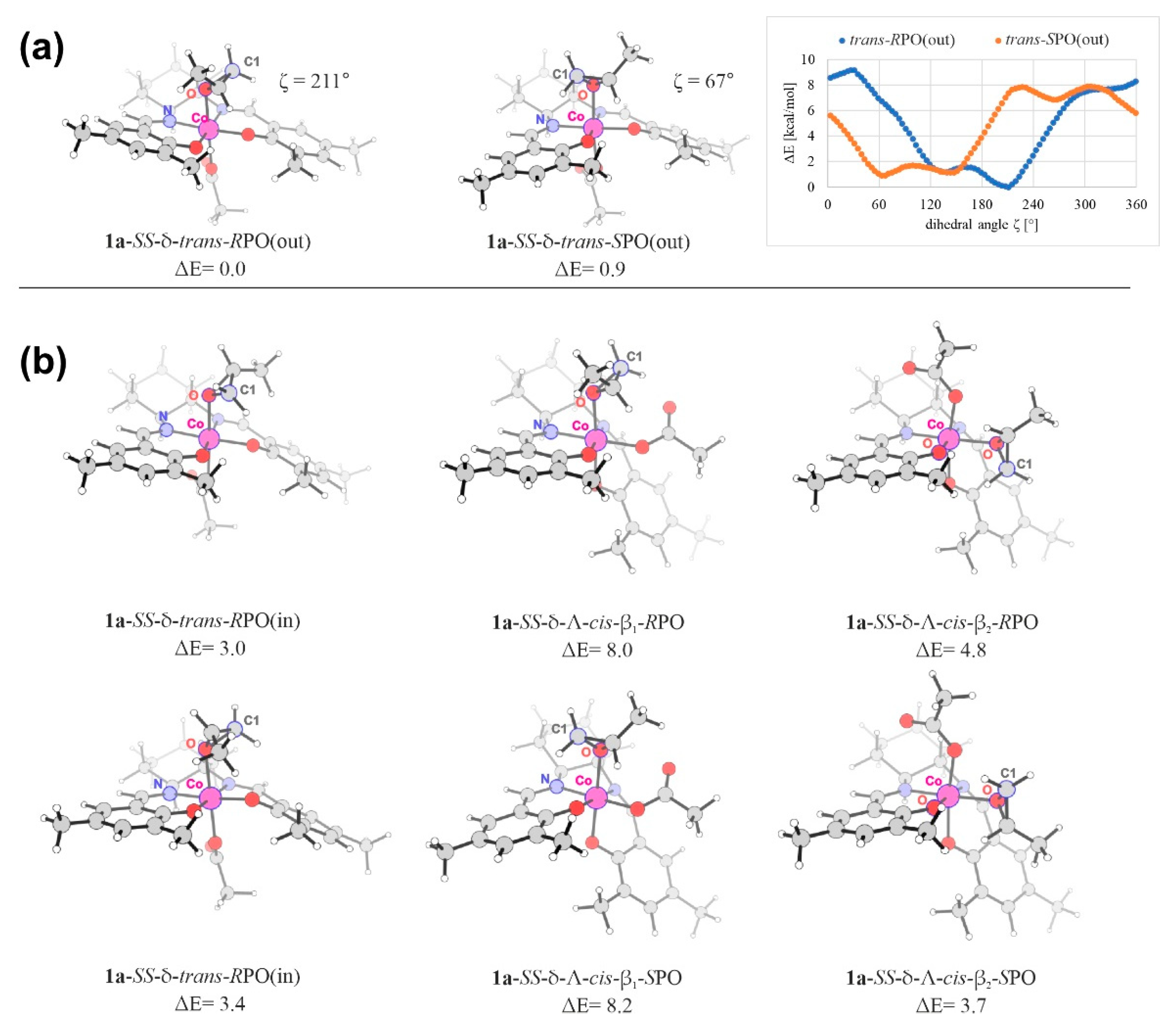
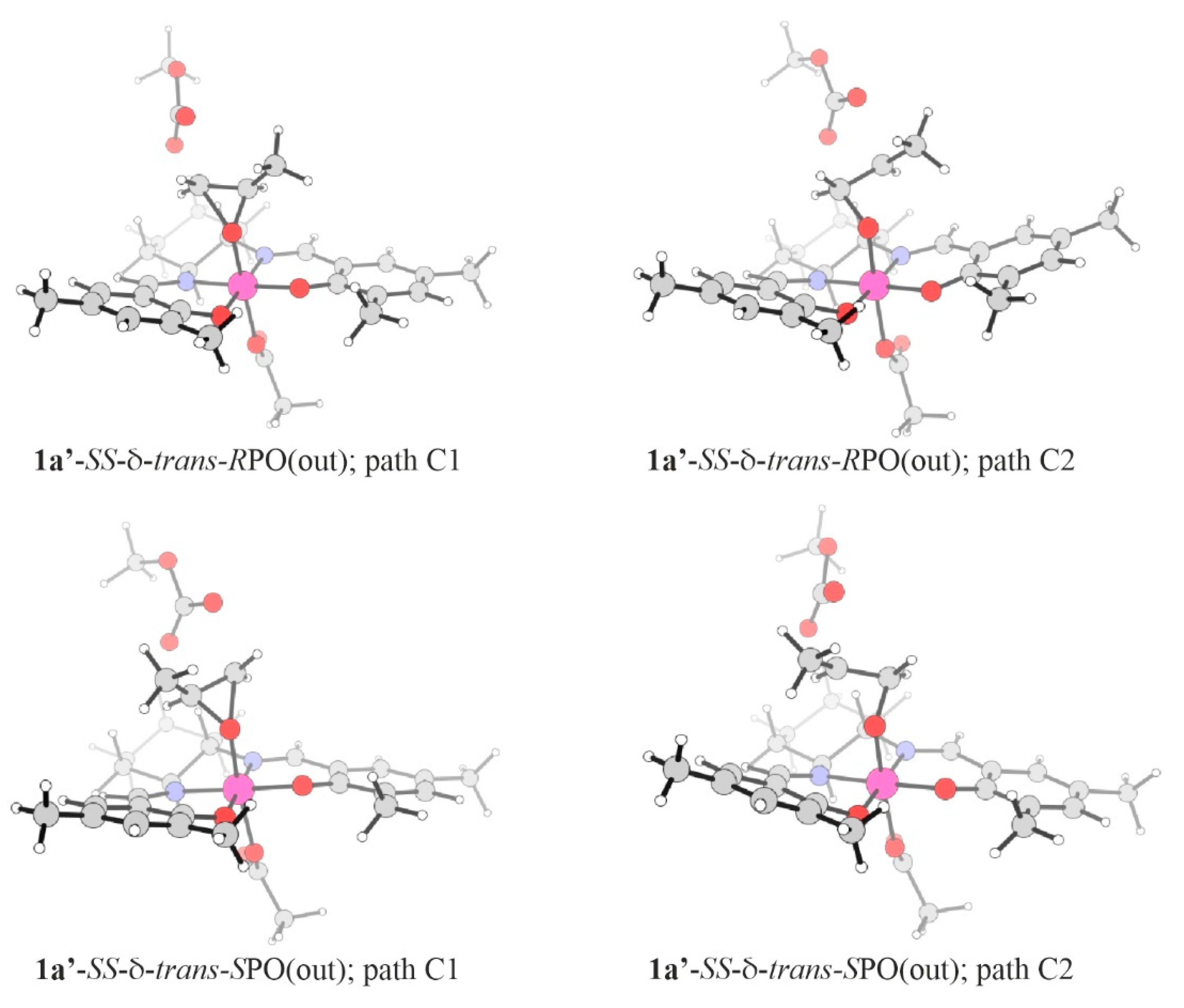
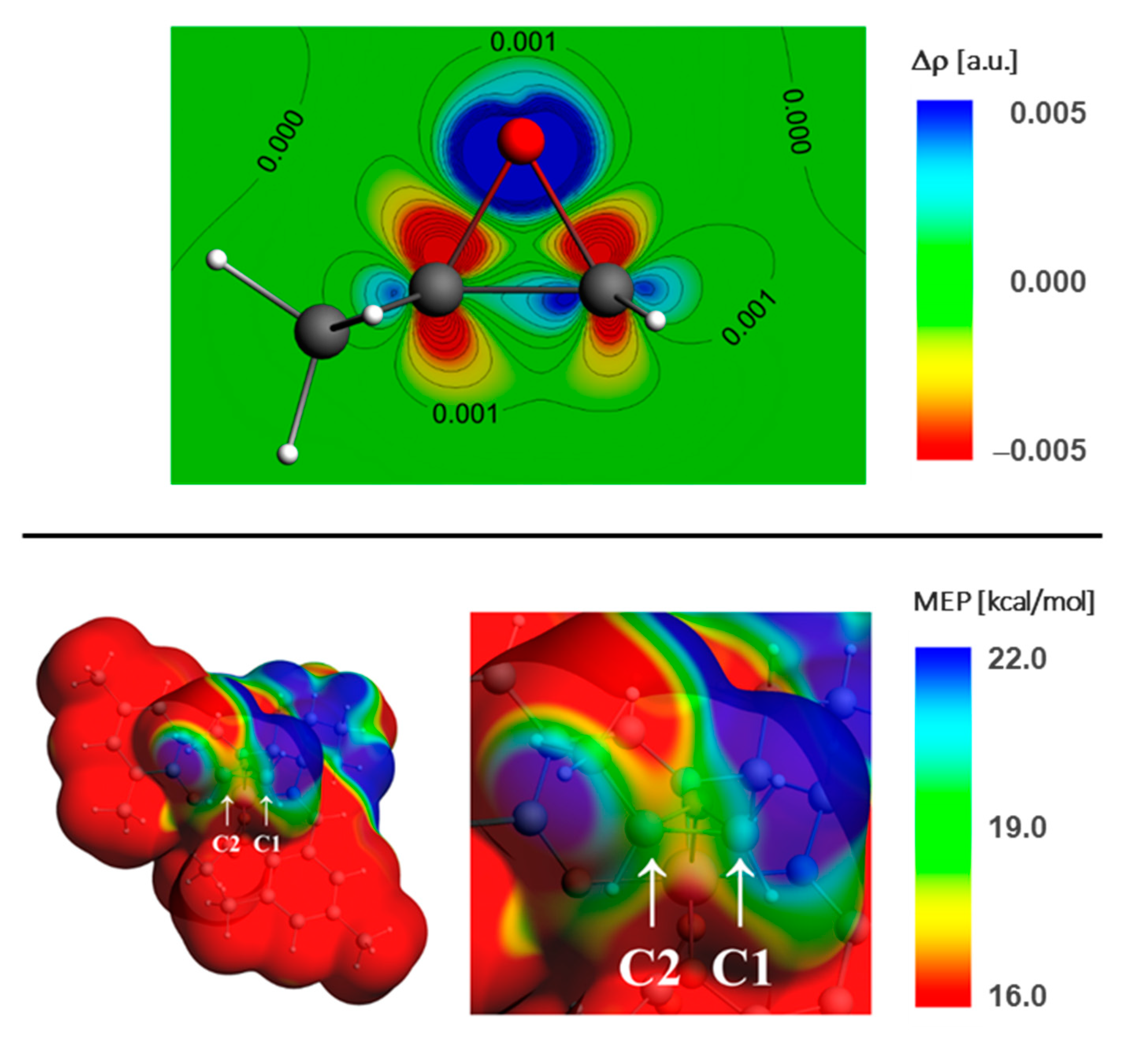
2.3. Epoxide Ring-Opening in the Model Systems 1
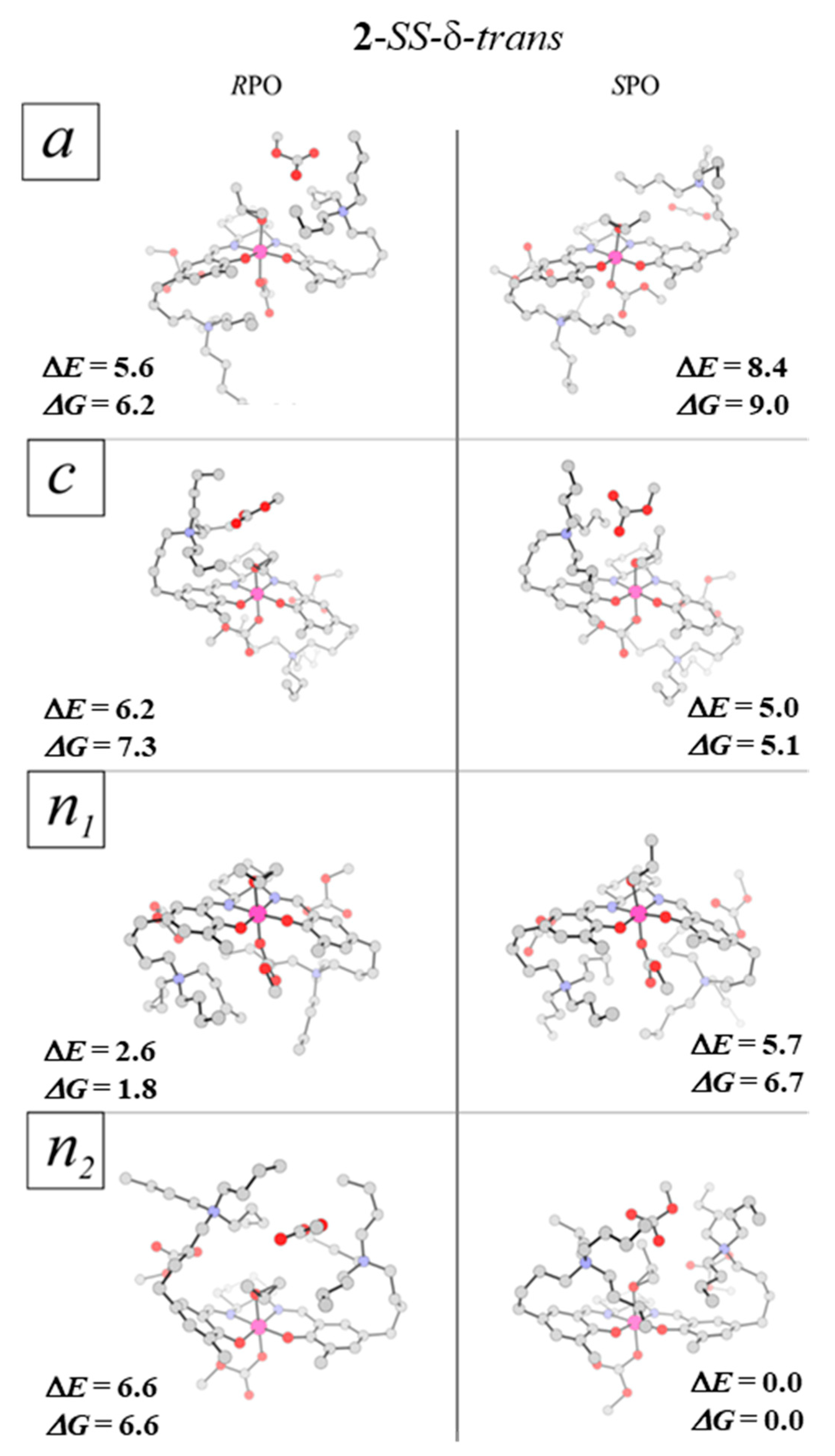
2.4. Epoxide Binding in the Real System 2
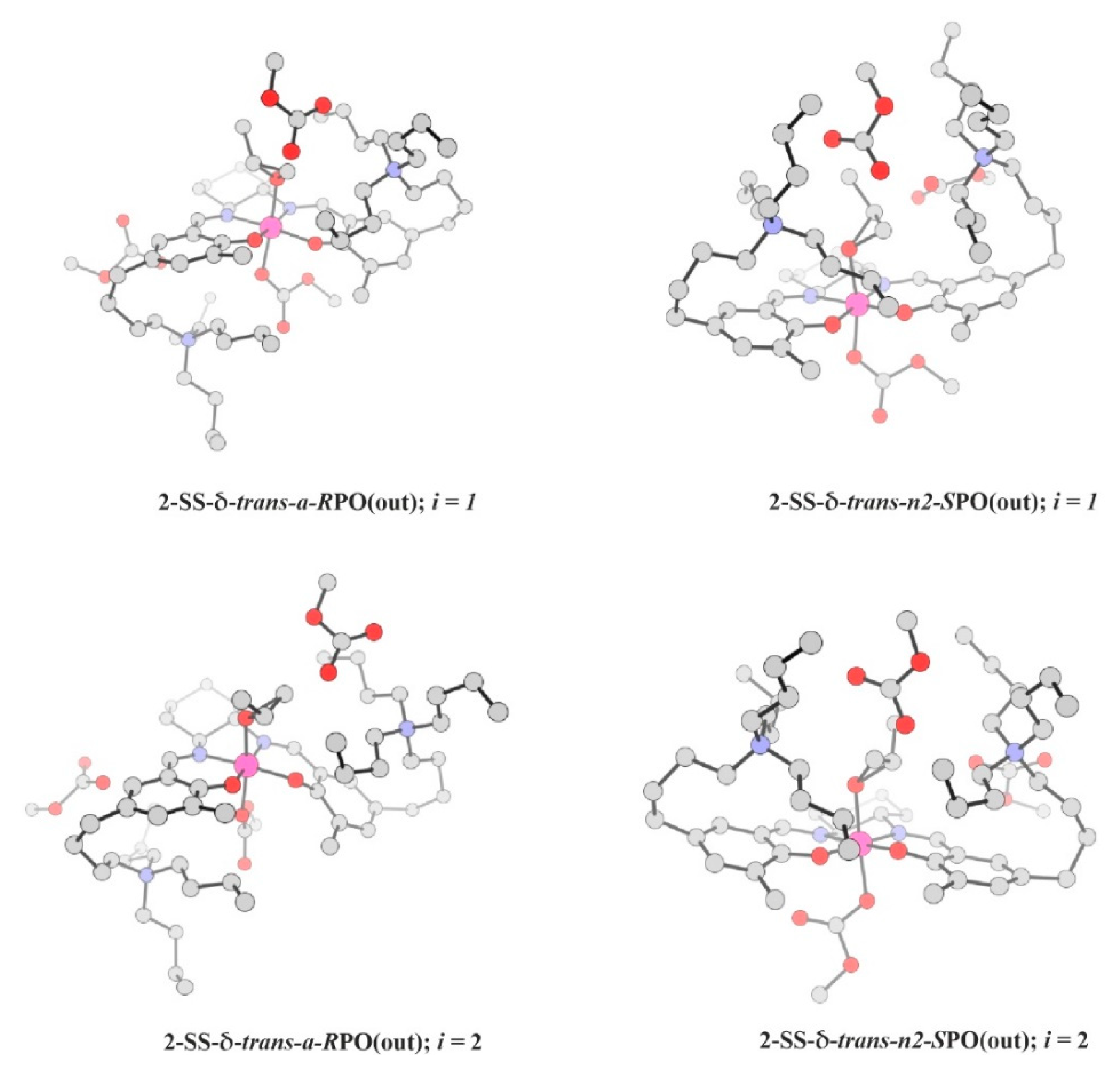
2.5. Epoxide Ring-Opening in the Real Catalytic System 2
3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Stereochemistry of Metal-Salen-Type CO2/epoxide Copolymerization Catalytic Systems



Appendix B
Estimation of the Epoxide Ring-Opening Effective Rate Constant
References
- Inoue, S.; Koinuma, H.; Tsuruta, T. Copolymerization of carbon dioxide and epoxide. J. Polym. Sci. Part B Polym. Lett. 1969, 7, 287–292. [Google Scholar] [CrossRef]
- Sugimoto, H.; Inoue, S. Copolymerization of carbon dioxide and epoxide. J. Polym. Sci. Part A Polym. Chem. 2004, 42, 5561–5573. [Google Scholar] [CrossRef]
- Klaus, S.; Lehenmeier, M.W.; Anderson, C.E.; Rieger, B. Recent advances in CO2/epoxide copolymerization—New strategies and cooperative mechanisms. Coord. Chem. Rev. 2011, 255, 1460–1479. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Wilson, S.J. What’s new with CO2? Recent advances in its copolymerization with oxiranes. Green Chem. 2012, 14, 2665–2671. [Google Scholar] [CrossRef]
- Taherimehr, M.; Pescarmona, P.P. Green Polycarbonates Prepared by the Copolymerization of CO2 with Epoxides. J. Appl. Polym. Sci. 2014, 131, 41141. [Google Scholar] [CrossRef]
- Ang, R.-R.; Sin, L.T.; Bee, S.-T.; Tee, T.-T.; Kadhum, A.A.H.; Rahmat, A.R.; Wasmi, B.A. A review of copolymerization of green house gas carbon dioxide and oxiranes to produce polycarbonate. J. Clean. Prod. 2015, 102, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Darensbourg, D.J. Carbon dioxide-based functional polycarbonates: Metal catalysed copolymerization of CO2 and epoxides. Coord. Chem. Rev. 2018, 372, 85–100. [Google Scholar] [CrossRef]
- Kamphuis, A.J.; Picchioni, F.; Pescarmona, P.P. CO2-fixation into cyclic and polymeric carbonates: Principles and applications. Green Chem. 2019, 21, 406–448. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-Y.; Wu, G.-P.; Darensbourg, D.J. CO2-Based Block Copolymers: Present and Future Designs. Trends Chem. 2020, 2, 750–763. [Google Scholar] [CrossRef]
- Huang, J.; Worch, J.C.; Dove, A.P.; Coulembier, O. Update and Challenges in Carbon Dioxide-Based Polycarbonate Synthesis. ChemSusChem 2020, 13, 469–487. [Google Scholar] [CrossRef]
- Sakakura, T.; Choi, J.-C.; Yasuda, H. Transformation of Carbon Dioxide. Chem. Rev. 2007, 107, 2365–2387. [Google Scholar] [CrossRef]
- Mikkelsen, M.; Jørgensen, M.M.; Krebs, F.C. The teraton challenge. A review of fixation and transformation of carbon dioxide. Energy Environ. Sci. 2010, 3, 43–81. [Google Scholar] [CrossRef]
- Cokoja, M.; Bruckmeier, C.; Rieger, B.; Herrmann, W.A.; Kühn, F.E. Transformation of Carbon Dioxide with Homogeneous Transition-Metal Catalysts: A Molecular Solution to a Global Challenge? Angew. Chem. Int. Ed. 2011, 50, 8510–8537. [Google Scholar] [CrossRef]
- Liu, Q.; Wu, L.; Jackstell, R.; Beller, M. Using carbon dioxide as a building block in organic synthesis. Nat. Commun. 2015, 6, 5933. [Google Scholar] [CrossRef] [PubMed]
- Burkart, M.D.; Hazari, N.; Tway, C.L.; Zeitler, E.L. Opportunities and Challenges for Catalysis in Carbon Dioxide Utilization. ACS Catal. 2019, 9, 7937–7956. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Holtcamp, M.W. Catalysts for the reactions of epoxides and carbon dioxide. Coord. Chem. Rev. 1996, 153, 155–174. [Google Scholar] [CrossRef]
- Coates, G.W.; Moore, D.R. Discrete Metal-Based Catalysts for the Copolymerization of CO2 and Epoxides: Discovery, Reactivity, Optimization, and Mechanism. Angew. Chem. Int. Ed. 2004, 43, 6618–6639. [Google Scholar] [CrossRef] [PubMed]
- Darensbourg, D.J.; Mackiewicz, R.M.; Phelps, A.L.; Billodeaux, D.R. Copolymerization of CO2 and Epoxides Catalyzed by Metal Salen Complexes. Acc. Chem. Res. 2004, 37, 836–844. [Google Scholar] [CrossRef] [PubMed]
- Darensbourg, D.J. Making Plastics from Carbon Dioxide: Salen Metal Complexes as Catalysts for the Production of Polycarbonates from Epoxides and CO2. Chem. Rev. 2007, 107, 2388–2410. [Google Scholar] [CrossRef] [PubMed]
- Kember, M.R.; Buchard, A.; Williams, C.K. Catalysts for CO2/epoxide copolymerization. Chem. Commun. 2011, 47, 141–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.-B.; Darensbourg, D.J. Cobalt catalysts for the coupling of CO2 and epoxides to provide polycarbonates and cyclic carbonates. Chem. Soc. Rev. 2012, 41, 1462–1484. [Google Scholar] [CrossRef]
- Childers, M.I.; Longo, J.M.; Van Zee, N.J.; LaPointe, A.M.; Coates, G.W. Stereoselective Epoxide Polymerization and Copolymerization. Chem. Rev. 2014, 114, 8129–8152. [Google Scholar] [CrossRef] [Green Version]
- Trott, G.; Saini, P.K.; Williams, C.K. Catalysts for CO2/epoxide ring-opening copolymerization. Phil. Trans. R. Soc. A 2016, 374, 20150085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozak, C.M.; Ambrose, K.; Anderson, T.S. Copolymerization of carbon dioxide and epoxides by metal coordination complexes. Coord. Chem. Rev. 2018, 376, 565–587. [Google Scholar] [CrossRef]
- Mandal, M. Group 4 complexes as catalysts for the transformation of CO2 into polycarbonates and cyclic carbonates. J. Organomet. Chem. 2020, 907, 121067. [Google Scholar] [CrossRef]
- Zhang, W.; Loebach, J.L.; Wilson, S.R.; Jacobsen, E.N. Enantioselective epoxidation of unfunctionalized olefins catalyzed by salen manganese complexes. J. Am. Chem. Soc. 1990, 112, 2801–2803. [Google Scholar] [CrossRef]
- Jacobsen, E.R.; Zhang, W.; Muci, A.R.; Ecker, J.R.; Deng, L. Highly enantioselective epoxidation catalysts derived from 1,2-diaminocyclohexane. J. Am. Chem. Soc. 1991, 113, 7063–7064. [Google Scholar] [CrossRef]
- McGarrigle, E.M.; Gilheany, D.G. Chromium- and Manganese-salen Promoted Epoxidation of Alkenes. Chem. Rev. 2005, 105, 1563–1602. [Google Scholar] [CrossRef]
- Jacobsen, E.R. Asymmetric Catalysis of Epoxide Ring-Opening Reactions. Acc. Chem. Res. 2000, 33, 421–431. [Google Scholar] [CrossRef]
- Cohen, C.T.; Chu, T.; Coates, G.W. Cobalt Catalysts for the Alternating Copolymerization of Propylene Oxide and Carbon Dioxide: Combining High Activity and Selectivity. J. Am. Chem. Soc. 2005, 127, 10869–10878. [Google Scholar] [CrossRef]
- Sujith, S.; Min, J.K.; Seong, J.E.; Na, S.J.; Lee, B.Y. A Highly Active and Recyclable Catalytic System for CO2/Propylene Oxide Copolymerization. Angew. Chem. Int. Ed. 2008, 47, 7306–7309. [Google Scholar]
- Nakano, K.; Kamada, T.; Nozaki, K. Selective Formation of Polycarbonate over Cyclic Carbonate: Copolymerization of Epoxides with Carbon Dioxide Catalyzed by a Cobalt(III) Complex with a Piperidinium End-Capping Arm. Angew. Chem. Int. Ed. 2006, 45, 7274–7277. [Google Scholar] [CrossRef] [PubMed]
- Noh, E.K.; Na, S.J.; Sujith, S.; Kim, S.-W.; Lee, B.Y. Two Components in a Molecule: Highly Efficient and Thermally Robust Catalytic System for CO2/Epoxide Copolymerization. J. Am. Chem. Soc. 2007, 129, 8082–8083. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.-M.; Liu, Z.-W.; Wen, Y.-Q.; Zhang, R.; Lu, X.-B. Mechanistic Aspects of the Copolymerization of CO2 with Epoxides Using a Thermally Stable Single-Site Cobalt(III) Catalyst. J. Am. Chem. Soc. 2009, 131, 11509–11518. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.-M.; Zhang, X.; Liu, Y.; Li, J.-F.; Wang, H.; Lu, X.-B. Highly Active, Bifunctional Co(III)-Salen Catalyst for Alternating Copolymerization of CO2 with Cyclohexene Oxide and Terpolymerization with Aliphatic Epoxides. Macromolecules 2010, 43, 1396–1402. [Google Scholar] [CrossRef]
- Jeon, J.Y.; Lee, J.J.; Varghese, J.K.; Na, S.J.; Sujith, S.; Go, M.J.; Lee, J.; Ok, M.-A.; Lee, B.J. CO2/ethylene oxide copolymerization and ligand variation for a highly active salen-cobalt(III) complex tethering 4 quaternary ammonium salts. Dalton Trans. 2013, 42, 9245–9254. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.-B.; Wang, Y. Highly Active, Binary Catalyst Systems for the Alternating Copolymerization of CO2 and Epoxides under Mild Conditions. Angew. Chem. Int. Ed. 2004, 43, 3574–3577. [Google Scholar] [CrossRef]
- Lu, X.-B.; Shi, L.; Wang, Y.-M.; Zhang, R.; Zhang, Y.-J.; Peng, X.-J.; Zhang, Z.-C.; Li, B. Design of Highly Active Binary Catalyst Systems for CO2/Epoxide Copolymerization: Polymer Selectivity, Enantioselectivity, and Stereochemistry Control. J. Am. Chem. Soc. 2006, 128, 1664–1674. [Google Scholar] [CrossRef]
- Shi, L.; Lu, X.-B.; Zhang, R.; Peng, X.-J.; Zhang, C.-Q.; Li, J.-F.; Peng, X.-M. Asymmetric Alternating Copolymerization and Terpolymerization of Epoxides with Carbon Dioxide at Mild Conditions. Macromolecules 2006, 39, 5679–5685. [Google Scholar] [CrossRef]
- Na, S.J.; Sujith, S.; Cyriac, A.; Kim, B.E.; Yoo, J.; Kang, Y.K.; Han, S.J.; Lee, C.; Lee, B.Y. Elucidation of the Structure of a Highly Active Catalytic System for CO2/Epoxide Copolymerization: A salen-Cobaltate Complex of an Unusual Binding Mode. Inorg. Chem. 2009, 48, 10455–10465. [Google Scholar] [CrossRef]
- Lu, X.-B.; Ren, W.-M.; Wu, G.-P. CO2 Copolymers from Epoxides: Catalyst Activity, Product Selectivity, and Stereochemistry Control. Acc. Chem. Res. 2012, 45, 1721–1735. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ren, W.M.; Liu, Y.; Lu, X.-B. Kinetic Study on the Coupling of CO2 and Epoxides Catalyzed by Co(III) Complex with an Inter- or Intramolecular Nucleophilic Cocatalyst. Macromolecules 2013, 46, 1343–1349. [Google Scholar] [CrossRef]
- Fu, X.; Jing, H. Quaternary onium modified SalenCoXY catalysts for alternating copolymerization of CO2 and propylene oxide: A kinetic study. J. Catal. 2015, 329, 317–324. [Google Scholar] [CrossRef]
- Qin, X.; Du, L.; Wang, C.; Yang, Z.; Zhang, M. Copolymerization of Carbon Dioxide and Propylene Oxide by Binary Cobalt Salen Complexes with Various Anion Groups. J. Chin. Chem. Soc. 2017, 64, 547–556. [Google Scholar] [CrossRef]
- Zhu, W.; Du, L.; Qian, S.; Yang, Q.; Song, W. Copolymerization of carbon dioxide and propylene oxide by several metallosalen-based bifunctional catalysts. J. Chin. Chem. Soc. 2018, 65, 841–849. [Google Scholar] [CrossRef]
- Du, L.; Wang, C.; Zhu, W.; Zhang, J. Copolymerization of carbon dioxide and propylene oxide catalyzed by two kinds of bifunctional salen-cobalt(III) complexes bearing four quaternary ammonium salts. J. Chin. Chem. Soc. 2020, 67, 72–79. [Google Scholar] [CrossRef]
- Hu, Y.; Du, L.; Li, C.; Shen, J.; Zhu, W.; Shao, C. Study of electronic effect in bifunctional catalysts for the copolymerization of CO2 and PO/CHO. J. Chin. Chem. Soc. 2020, 67, 1818–1826. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Yeung, A.D. A concise review of computational studies of the carbon dioxide–epoxide copolymerization reactions. Polym. Chem. 2014, 5, 3949–3962. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Yeung, A.D. Kinetics of the (salen)Cr(III)- and (salen)Co(III)-catalyzed copolymerization of epoxides with CO2, and of the accompanying degradation reactions. Polym. Chem. 2015, 6, 1103–1117. [Google Scholar] [CrossRef]
- Liu, C.; Luo, Y.; Lu, X.B. DFT Studies on the Origin of Regioselective Ring-opening of Terminal Epoxides during Copolymerization with CO2. Chin. J. Polym. Sci. 2016, 34, 439–445. [Google Scholar] [CrossRef]
- Dyduch, K.; Srebro-Hooper, M.; Lee, B.Y.; Michalak, A. Exploring the Conformational Space of Cobalt(III)–Salen Catalyst for CO2/Epoxide Copolymerization: Effect of Quaternary Ammonium Salts on Preference of Alternative Isomers. J. Comput. Chem. 2018, 39, 1854–1867. [Google Scholar] [CrossRef]
- Roznowska, A.; Dyduch, K.; Lee, B.Y.; Michalak, A. Theoretical study on preference of open polymer vs. cyclic products in CO2/epoxide copolymerization with cobalt(III)-salen bifunctional catalysts. J. Mol. Model. 2020, 26, 113. [Google Scholar] [CrossRef] [PubMed]
- Calligaris, M.; Manzini, G.; Nardin, G.; Randaccio, L. Ligand properties of quadridentate Schiff’s bases. The crystal and molecular structure of the mixed-ligand complex [NN’-ethylenebis(salicylideneiminato)](acetylacetonato)cobalt(III)–0·7 water. J. Chem. Soc., Dalton Trans. 1972, 4, 543–547. [Google Scholar] [CrossRef]
- Hirota, S.; Kosugi, E.; Marzilli, L.G.; Yamauchi, O. The Co–CH3 bond in Shiff base B12 models: Influence of the trans and equatorial ligands as assessed by Fourier transform Raman spectroscopy. Inorg. Chim. Acta 1998, 275–276, 90–97. [Google Scholar] [CrossRef]
- Blaauw, R.; van der Baan, J.L.; Balt, S.; de Bolster, M.W.G.; Klumpp, G.W.; Kooijman, H.; Spek, A.L. Bridged (alkoxo)CoIII(salen) complexes: Synthesis and structure. Inorg. Chim. Acta 2002, 336, 29–38. [Google Scholar] [CrossRef]
- Dreos, R.; Nardin, G.; Randaccio, L.; Siega, P.; Tauzher, G.; Vrdoljak, V. New β Cis Folded Organocobalt Derivatives with a Salen-Type Ligand. Inorg. Chem. 2003, 42, 6805–6811. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-L.; Ruan, W.-J.; Zhao, X.-J.; Wang, H.-G.; Zhu, Z.-A. Synthesis and characterization of axial coordination cobalt(III) complexes containing chiral Salen ligands. Polyhedron 2003, 22, 1535–1545. [Google Scholar] [CrossRef]
- Khandar, A.A.; Shaabani, B.; Belaj, F.; Bakhtiari, A. Synthesis, characterization and spectroscopic and electrochemical studies of new axially coordinated cobalt(III) salen (salen = N,N′-bis(salicylidene)-1,2-ethylenediamine) complexes. The crystal structure of [CoIII(salen)(aniline)2]ClO4. Polyhedron 2006, 25, 1893–1900. [Google Scholar] [CrossRef]
- Ajiro, H.; Peretti, K.L.; Lobkovsky, E.B.; Coates, G.W. On the mechanism of isospecific epoxidepolymerization by salen cobalt(III) complexes: Evidence for solid-state catalysis. Dalton Trans. 2009, 8828–8830. [Google Scholar] [CrossRef]
- Niu, Y.; Li, H.; Chen, X.; Zhang, W.; Zhuang, X.; Jing, X. Alternating Copolymerization of Carbon Dioxide and Propylene Oxide Catalyzed by Cobalt Schiff Base Complex. Macromol. Chem. Phys. 2009, 210, 1224–1229. [Google Scholar] [CrossRef]
- Cyriac, A.; Jeon, J.Y.; Varghese, J.K.; Park, J.H.; Choi, S.Y.; Chung, Y.K.; Lee, B.Y. Unusual coordination mode of tetradentate Schiff base cobalt(III) complexes. Dalton Trans. 2012, 41, 1444–1447. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Thomas, C.M.; Lee, S.; Coates, G.W. Cobalt-Based Complexes for the Copolymerization of Propylene Oxide and CO2: Active and Selective Catalysts for Polycarbonate Synthesis. Angew. Chem. Int. Ed. 2003, 42, 5484–5487. [Google Scholar] [CrossRef]
- Muetterties, E.L. Topological Representation of Stereoisomerism. I. Polytopal Rearrangements. J. Am. Chem. Soc. 1969, 91, 1636–1643. [Google Scholar] [CrossRef]
- McKee, M.L. Fluctional molecules. WIREs Comput. Mol. Sci. 2011, 1, 943–951. [Google Scholar] [CrossRef]
- Alvarez, S.; Alemany, P.; Casanova, D.; Cirera, J.; Llunell, M.; Avnir, D. Shape maps and polyhedral interconversion paths in transition metal chemistry. Coord. Chem. Rev. 2005, 249, 1693–1708. [Google Scholar] [CrossRef]
- Rzepa, H.S.; Cass, M.E. A Computational Study of the Nondissociative Mechanisms that Interchange Apical and Equatorial Atoms in Square Pyramidal Molecules. Inorg. Chem. 2006, 45, 3958–3963. [Google Scholar] [CrossRef]
- Zou, W.; Tao, Y.; Kraka, E. Describing Polytopal Rearrangements of Fluxional Molecules with Curvilinear Coordinates Derived from Normal Vibrational Modes: A Conceptual Extension of Cremer-Pople Puckering Coordinates. J. Chem. Theory Comput. 2020, 16, 3162–3193. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, D.; Nguyen, S.T.; Baik, M.-H. A computational study of the mechanism of the [(salen)Cr + DMAP]-catalyzed formation of cyclic carbonates from CO2 and epoxide. Chem. Commun. 2014, 50, 2676–2678. [Google Scholar] [CrossRef] [PubMed]
- Ford, D.D.; Nielsen, L.P.C.; Zuend, S.J.; Musgrave, C.B.; Jacobsen, E.N. Mechanistic Basis for High Stereoselectivity and Broad Substrate Scope in the (salen)Co(III)-Catalyzed Hydrolytic Kinetic Resolution. J. Am. Chem. Soc. 2013, 135, 15595–15608. [Google Scholar] [CrossRef] [Green Version]
- Darensbourg, D.J.; Yeung, A.D. Thermodynamics of the Carbon Dioxide–Epoxide Copolymerization and Kinetics of the Metal-Free Degradation: A Computational Study. Macromolecules 2013, 46, 83–95. [Google Scholar] [CrossRef]
- Te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Fonseca Guerra, C.; Snijders, J.G.; Velde, G.; Baerends, E.J. Regular article Towards an order- N DFT method. Theor. Chem. Acc. 1998, 99, 391–403. [Google Scholar]
- ADF. SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands. Available online: http://www.scm.com (accessed on 3 March 2021).
- Becke, A.D. Density-fnnctional exchange-energy approximation with correct asymptotic behaviour. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic Regular Two-component Hamiltonians. J. Chem. Phys. 1993, 99, 4597–4610. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic Total Energy Using Regular Approximations. J. Chem. Phys. 1994, 101, 9783–9792. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Ehlers, A.; Baerends, E.J. Geometry Optimizations in the Zero Order Regular Approximation for Relativistic Effects. J. Chem. Phys. 1999, 110, 8943–8953. [Google Scholar] [CrossRef] [Green Version]
- Van Lenthe, E.; Snijders, J.G.; Baerends, E.J. The Zero-Order Regular Approximation for Relativistic Effects: The Effect of Spin-Orbit Coupling in Closed Shell Molecules. J. Chem. Phys. 1996, 105, 6505–6516. [Google Scholar] [CrossRef] [Green Version]
- Van Lenthe, E.; van Leeuwen, R.; Baerends, E.J.; Snijders, J.G. Relativistic Regular Two-component Hamiltonians. Int. J. Quant. Chem. 1996, 57, 281–293. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Baerends, E.J. Optimized Slater-Type Basis Sets for the Elements 1-118. J. Comput. Chem. 2003, 24, 1142–1156. [Google Scholar] [CrossRef]
- Bérces, A.; Dickson, R.M.; Fan, L.; Jacobsen, H.; Swerhone, D.; Ziegler, T. An Implementation of the Coupled Perturbed Kohn-Sham Equations: Perturbation Due to Nuclear Displacements. Comput. Phys. Commun. 1997, 100, 247–262. [Google Scholar] [CrossRef]
- Jacobsen, H.; Bérces, A.; Swerhone, D.P.; Ziegler, T. Analytic Second Derivatives of Molecular Energies: A Density Functional Implementation. Comput. Phys. Commun. 1997, 100, 263–276. [Google Scholar] [CrossRef]
- Wolff, S.K. Analytical Second Derivatives in the Amsterdam Density Functional Package. Int. J. Quantum Chem. 2005, 104, 645–659. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of Parameters for Semiempirical Methods VI: More Modifications to the NDDO Approximations and Re-Optimization of Parameters. J. Mol. Model. 2013, 19, 1–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MOPAC2016; Stewart, J.J.P. Stewart Computational Chemistry, Colorado Springs, CO, USA. Available online: http://OpenMOPAC.net (accessed on 3 March 2021).
- Verlet, L. Computer “Experiments” on Classical Fluids. I. Thermodynamical Properties of Lennard-Jones Molecules. Phys. Rev. 1967, 159, 98–103. [Google Scholar] [CrossRef]
- Swope, W.C.; Andersen, H.C.; Berens, P.H.; Wilson, K.R. A Computer Simulation Method for the Calculation of Equilibrium Constants for the Formation of Physical Clusters of Molecules: Application to Small Water Clusters. J. Chem. Phys. 1982, 76, 637–649. [Google Scholar] [CrossRef]
- Morgan, G.T.; Smith, J.D.M. CCLXXVI.—Researches on residual affinity and co-ordination. Part XXV. A quadridentate group contributing four associating units to metallic complexes. J. Chem. Soc. Trans. 1925, 127, 2030–2037. [Google Scholar] [CrossRef]
- Knof, U.; von Zelewsky, A. Predetermined Chirality at Metal Centers. Angew. Chem. Int. Ed. 1999, 38, 302–322. [Google Scholar] [CrossRef]
- Brunner, H. Optically Active Organometallic Compounds of Transition Elements with Chiral Metal Atoms. Angew. Chem. Int. Ed. 1999, 38, 1194–1208. [Google Scholar] [CrossRef]
- Brorson, M.; Damhus, T.; Schaffer, C.E. Exhaustive examination of chiral configurations of edges on a regular octahedron: Analysis of the possibilities of assigning chirality descriptors within a generalized.DELTA./.LAMBDA. system. Inorg. Chem. 1983, 22, 1569–1573. [Google Scholar] [CrossRef]
- Chemistry, I.U. Nomenclature of Inorganic Chemistry—IUPAC Recommendations 2005. Chem. Int. Newsmag. IUPAC 2005, 27, 366. [Google Scholar]
- Marzilli, L.G.; Buckingham, D.A. The stereochemistry of some cobalt(III) triethylenetetramine complexes of glycine and sarcosine. Inorg. Chem. 1967, 6, 1042–1052. [Google Scholar] [CrossRef]
- Cahn, R.S.; Ingold, C.; Prelog, V. Specification of Molecular Chirality. Angew. Chem. Int. Ed. 1966, 5, 385–415. [Google Scholar] [CrossRef]
- Prelog, V.; Helmchen, G. Basic Principles of the CIP-System and Proposals for a Revision. Angew. Chem. Int. Ed. 1982, 21, 567–583. [Google Scholar] [CrossRef]
- IUPAC. Compendium of Chemical Terminology, 2nd ed.; the “Gold Book”; McNaught, A.D., Wilkinson, A., Eds.; Blackwell Scientific Publications: Oxford, UK, 1997; ISBN 0-9678550-9-8. XML on-line Corrected Version; Available online: http://goldbook.iupac.org (accessed on 3 March 2021). [CrossRef]
- Weil, M.; Khalaji, A.D. Crystal Structure of the Salen Cobalt(III) Azido Complex Na[Co(salen)(N3)2]. Anal. Sci. X-Ray Struct. Anal. Online 2008, 24, x19–x20. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.-J.; Kim, S.-K.; Kim, B.-J.; Hahn, J.S.; Ok, M.-A.; Song, J.H.; Shin, D.-H.; Ko, J.; Cheong, M.; Kim, J.; et al. Half-Metallocene Titanium(IV) Phenyl Phenoxide for High Temperature Olefin Polymerization: Ortho-Substituent Effect At Ancillary o-Phenoxy Ligand for Enhanced Catalytic Performance. Macromolecules 2009, 42, 6932–6943. [Google Scholar] [CrossRef]
- Srebro-Hooper, M.; Michalak, A. ‘Computational Modeling of Polymerization Catalysts’ in ‘Handbook of Transition Metal Polymerization Catalysts’, 2nd ed.; Hoff, R., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2018; pp. 67–130. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stereoisomer | 1a | 1b | ||||
|---|---|---|---|---|---|---|
| ζ | ΔE | ΔG | ζ | ΔE | ΔG | |
| SS-δ-trans-RPO(out) | 211 | 0.00 | 0.00 (0.47) | 206 | 0.00 | 0.00 |
| SS-δ-trans-RPO(in) | 146 | 3.04 | 2.69 (3.16) | 146 | 3.08 | 1.92 |
| SS-δ-Λ-cis-β1-RPO | 213 | 7.99 | 7.21 (7.68) | 212 | 7.25 | 6.96 |
| SS-δ-Λ-cis-β2-RPO | 34 | 4.75 | 3.67 (4.14) | 31 | 2.44 | 2.58 |
| SS-δ-trans-SPO(out) | 67 | 0.00 (0.88) | 0.00 | 66 | 0.00 (0.92) | 0.00 (0.20) |
| SS-δ-trans-SPO(in) | 209 | 2.55 (3.44) | 3.34 | 207 | 3.44 (4.35) | 5.00 (5.20) |
| SS-δ-Λ-cis-β1-SPO | 71 | 7.32 (8.21) | 7.54 | 72 | 5.75 (6.67) | 6.42 (6.62) |
| SS-δ-Λ-cis-β2-SPO | 322 | 2.79 (3.68) | 3.82 | 318 | 0.92 (1.84) | 0.15 (0.35) |
| System/Stereoisomer | Path | Relative Electronic-Energies | Relative Free-Energies | ||||
|---|---|---|---|---|---|---|---|
| 1a’-RPO: | |||||||
| 1a’-SS-δ-trans-RPO(out) | C1 | 5.26 (7.95) | 0.00 (2.69) | 5.26 | 5.00 (8.73) | 0.27 (4.00) | 4.73 |
| 1a’-SS-δ-trans-RPO(out) | C2 | 7.66 (10.35) | 0.00 (2.69) | 7.66 | 6.38 (10.11) | 0.27 (4.00) | 6.11 |
| 1a’-SS-δ-Λ-cis-β1-RPO | C1 | 20.33 (23.02) | 9.31 (12.00) | 11.02 | 17.72 (21.45) | 8.38 (12.11) | 9.34 |
| 1a’-SS-δ-Λ-cis-β1-RPO | C2 | 19.09 (21.78) | 6.16 (8.85) | 12.93 | 16.92 (20.65) | 11.61 (15.34) | 5.31 |
| 1a’-SS-δ-Λ-cis-β2-RPO | C1 | 16.90 (19.59) | 3.58 (6.27) | 13.32 | 13.91 (17.64) | 1.21 (4.94) | 12.70 |
| 1a’-SS-δ-Λ-cis-β2-RPO | C2 | 17.05 (19.74) | 10.99 (13.68) | 6.06 | 16.04 (19.77) | 5.21 (8.94) | 10.83 |
| 1a’-SPO: | |||||||
| 1a’-SS-δ-trans-SPO(out) 1 | C1 | 8.19 | 2.26 | 5.93 | 8.89 | 1.57 | 7.32 |
| 1a’-SS-δ-trans-SPO(out) | C2 | 8.61 | 2.26 | 6.35 | 8.89 | 1.57 | 7.54 |
| 1a’-SS-δ-Λ-cis-β1-SPO | C1 | 19.59 | 10.86 | 8.73 | 20.84 | 10.68 | 10.16 |
| 1a’-SS-δ-Λ-cis-β1-SPO | C2 | 19.27 | 10.86 | 8.41 | 20.84 | 10.68 | 7.59 |
| 1a’-SS-δ-Λ-cis-β2-SPO | C1 | 16.83 | 14.45 | 2.38 | 17.44 | 14.27 | 3.17 |
| 1a’-SS-δ-Λ-cis-β2-SPO | C2 | 16.14 | 14.45 | 1.69 | 18.70 | 14.27 | 4.43 |
| 1b’-RPO: | |||||||
| 1b’-SS-δ-trans-RPO(out) | C1 | 4.10 (4.82) | 0.00 (0.72) | 4.10 | 5.84 | 0.00 | 5.84 |
| 1b’-SS-δ-trans-RPO(out) | C2 | 6.69 (7.41) | 0.00 (0.72) | 6.69 | 6.87 | 0.00 | 6.87 |
| 1b’-SS-δ-Λ-cis-β1-RPO | C1 | 15.03 (15.75) | 9.00 (9.72) | 6.03 | 17.00 | 12.53 | 4.47 |
| 1b’-SS-δ-Λ-cis-β1-RPO | C2 | 17.09 (17.81) | 6.70 (7.42) | 10.39 | 18.99 | 9.00 | 9.99 |
| 1b’-SS-δ-Λ-cis-β2-RPO | C1 | 12.45 (13.17) | 10.33 (11.05) | 2.12 | 14.41 | 12.55 | 1.86 |
| 1b’-SS-δ-Λ-cis-β2-RPO | C2 | 15.02 (15.74) | 10.33 (11.05) | 4.69 | 14.92 | 12.55 | 2.37 |
| 1b’-SPO: | |||||||
| 1b’-SS-δ-trans-SPO(out) | C1 | 5.10 | 0.00 | 5.10 | 5.50 (6.06) | 0.00 (0.56) | 5.50 |
| 1b’-SS-δ-trans-SPO(out) | C2 | 5.37 | 0.00 | 5.37 | 5.63 (6.19) | 0.00 (0.56) | 5.63 |
| 1b’-SS-δ-Λ-cis-β1-SPO | C1 | 16.31 | 7.03 | 9.28 | 16.82 (17.38) | 8.04 (8.60) | 8.78 |
| 1b’-SS-δ-Λ-cis-β1-SPO | C2 | 17.80 | 7.03 | 10.77 | 15.54 (16.10) | 8.04 (8.60) | 7.50 |
| 1b’-SS-δ-Λ-cis-β2-SPO | C1 | 16.56 | 9.64 | 6.92 | 17.46 (18.02) | 10.34 (10.90) | 7.12 |
| 1b’-SS-δ-Λ-cis-β2-SPO | C2 | 15.70 | 9.64 | 6.06 | 15.76 (16.32) | 10.34 (10.90) | 5.42 |
| System/Stereoisomer | ΔE | ΔG | ||
|---|---|---|---|---|
| 2-SS-δ-trans-a-RPO | 2.93 (5.57) | 4.39 (6.16) | 0.98 | 1.51 |
| 2-SS-δ-trans-c-RPO | 3.58 (6.22) | 5.49 (7.26) | 0.23 | 0.97 |
| 2-SS-δ-trans-n1-RPO | 0.00 (2.64) | 0.00 (1.77) | 64.25 | 97.24 |
| 2-SS-δ-trans-n2-RPO | 3.95 (6.59) | 4.86 (6.63) | 0.39 | 0.28 |
| 2-SS-δ-trans-a-SPO | 8.40 | 8.99 | 0.00 | 0.00 |
| 2-SS-δ-trans-c-SPO | 5.00 | 5.07 | 0.03 | 0.06 |
| 2-SS-δ-trans-n1-SPO | 5.68 | 6.74 | 0.01 | 0.02 |
| 2-SS-δ-trans-n2-SPO | 0.00 | 0.00 | 31.23 | 99.92 |
| System / i / Path | Rate-Constant Contribution | Relative Energies | Relative Free-Energies | ||||
|---|---|---|---|---|---|---|---|
| × 10−3 | |||||||
| 2-SS-δ-trans-a-RPO: | |||||||
| 1 / C1 | 15.92 | 13.68 | 3.89 | 9.79 | 16.73 | 5.11 | 11.62 |
| 2 / C1 | 6.30 | 14.33 | 8.83 | 5.50 | 17.48 | 11.13 | 6.35 |
| 3 / C1 | 2.07 | 15.11 | 6.62 | 8.49 | 18.79 | 10.25 | 8.54 |
| 4 / C1 | 1.80 | 15.21 | 10.34 | 4.87 | 20.32 | 13.19 | 7.13 |
| 5 / C1 | 1.70 | 15.25 | 13.19 | 2.06 | 17.65 | 14.81 | 2.84 |
| Total RPO: | 29.83 | ||||||
| 2-SS-δ-trans-n2-SPO: | |||||||
| 1 / C2 | 15.79 | 13.18 | 6.26 | 6.92 | 14.69 | 5.81 | 8.88 |
| 2 / C1 | 4.70 | 14.03 | 0.97 | 13.06 | 14.34 | 1.21 | 13.13 |
| 3 / C1 | 1.48 | 14.84 | 2.45 | 12.39 | 15.80 | 3.28 | 12.52 |
| 4 / C1 | 0.72 | 15.35 | 0.00 | 15.35 | 15.81 | 0.00 | 15.81 |
| 5 / C2 | 0.62 | 15.45 | 5.59 | 9.86 | 16.42 | 6.33 | 10.09 |
| Total SPO: | 25.80 | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dyduch, K.; Roznowska, A.; Srebro-Hooper, M.; Lee, B.Y.; Michalak, A. Theoretical Study on Epoxide Ring-opening in CO2/Epoxide Copolymerization Catalyzed by Bifunctional Salen-Type Cobalt(III) Complexes: Influence of Stereoelectronic Factors. Catalysts 2021, 11, 328. https://doi.org/10.3390/catal11030328
Dyduch K, Roznowska A, Srebro-Hooper M, Lee BY, Michalak A. Theoretical Study on Epoxide Ring-opening in CO2/Epoxide Copolymerization Catalyzed by Bifunctional Salen-Type Cobalt(III) Complexes: Influence of Stereoelectronic Factors. Catalysts. 2021; 11(3):328. https://doi.org/10.3390/catal11030328
Chicago/Turabian StyleDyduch, Karol, Aleksandra Roznowska, Monika Srebro-Hooper, Bun Yeoul Lee, and Artur Michalak. 2021. "Theoretical Study on Epoxide Ring-opening in CO2/Epoxide Copolymerization Catalyzed by Bifunctional Salen-Type Cobalt(III) Complexes: Influence of Stereoelectronic Factors" Catalysts 11, no. 3: 328. https://doi.org/10.3390/catal11030328
APA StyleDyduch, K., Roznowska, A., Srebro-Hooper, M., Lee, B. Y., & Michalak, A. (2021). Theoretical Study on Epoxide Ring-opening in CO2/Epoxide Copolymerization Catalyzed by Bifunctional Salen-Type Cobalt(III) Complexes: Influence of Stereoelectronic Factors. Catalysts, 11(3), 328. https://doi.org/10.3390/catal11030328