
Functionalization of Ruthenium Olefin-Metathesis Catalysts for Interdisciplinary Studies in Chemistry and Biology


Abstract
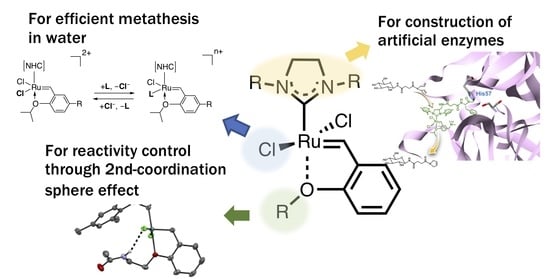
:
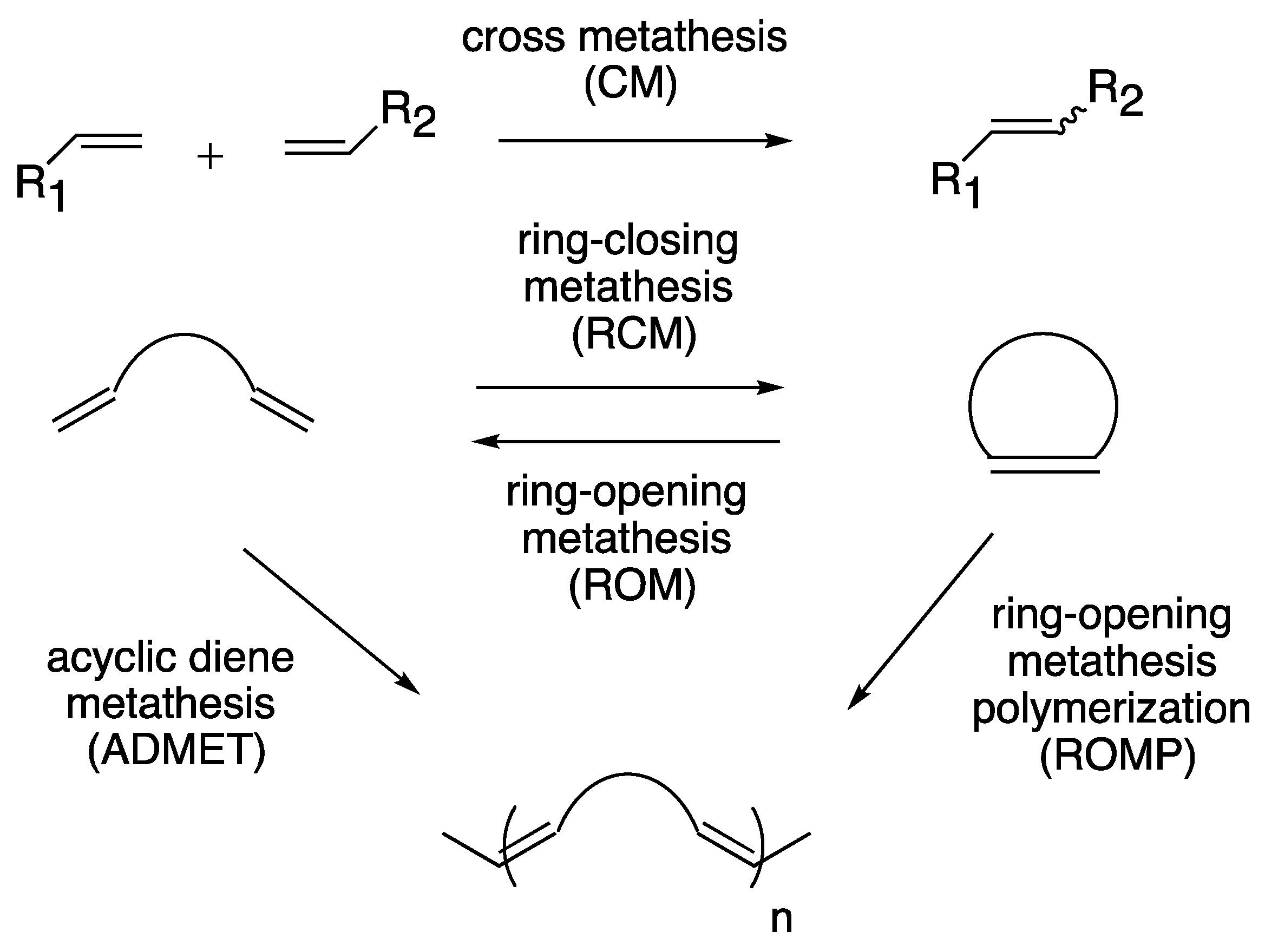
1. Introduction
2. Protein-Based Artificial Metalloenzymes with Olefin Metathesis Activity
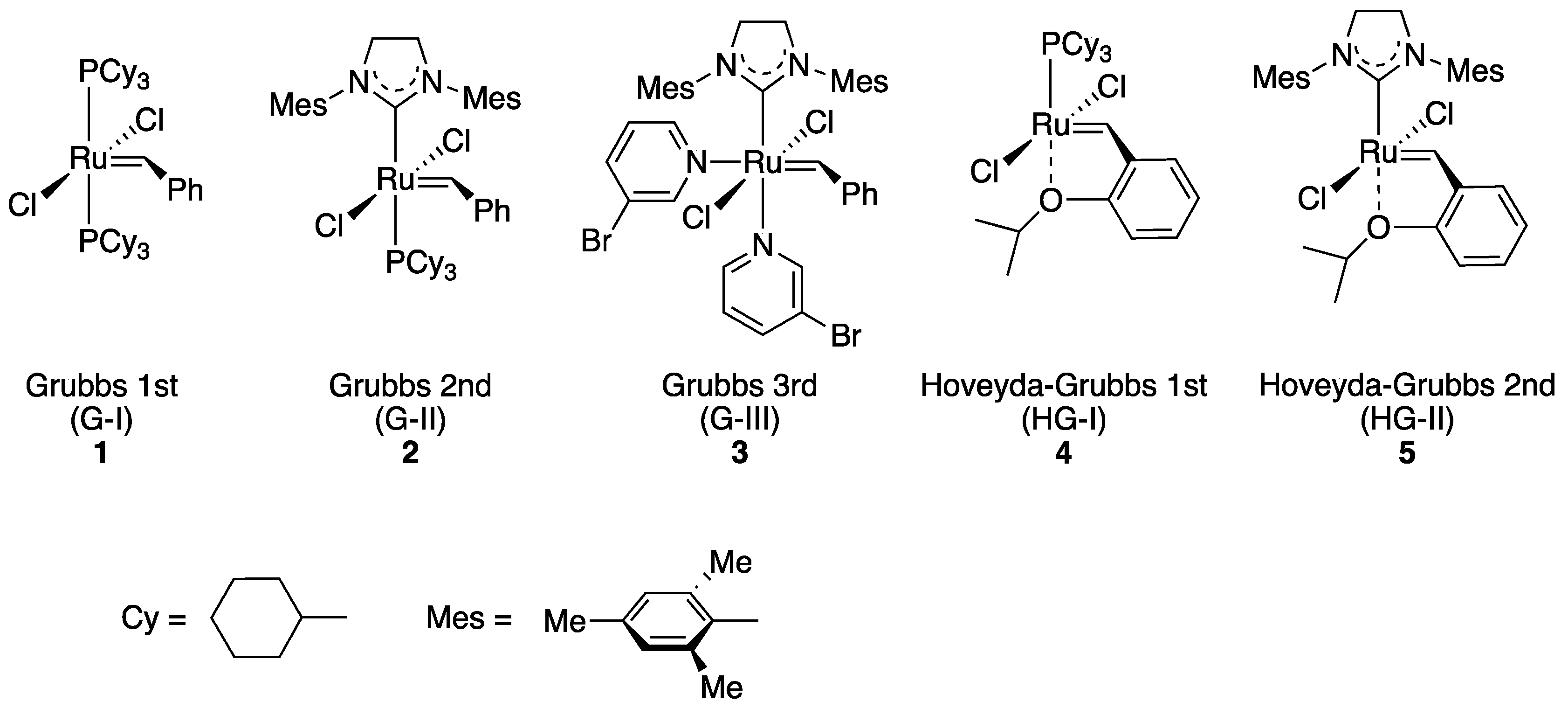
2.1. Choice of Host Proteins and Design of Hoveyda–Grubbs-Type Complexes
- Sufficient space to accommodate a HG-II-type complex inside the protein core;
- Possession of the structural mechanism for regioselective binding of a HG-II-type complex to a protein part;
- Proper thermostability.
2.2. Modulation of Catalytic Activities
3. In-Cell Olefin Metathesis Reactions
4. Application of Ruthenium Complexes for Sensing Small Molecules In Vitro and In Vivo
5. Optimization of Catalytic Activities for Olefin Metathesis in Aqueous Media
6. Reactivity Modulation for Hoveyda–Grubbs-Type Complexes through Second-Coordination Sphere Effect: Utility of Structural Modification of Phenolic Moiety
7. Immobilization of Hoveyda–Grubbs-Type Complexes onto Biomolecules through Ruthenium–Olefin Specific Interaction
8. Conclusion and Perspective
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hughes, D.; Wheeler, P.; Ene, D. Olefin Metathesis in Drug Discovery and Development-Examples from Recent Patent Literature. Org. Process Res. Dev. 2017, 21, 1938–1962. [Google Scholar] [CrossRef]
- Ravindar, L.; Lekkala, R.; Rakesh, K.P.; Asiri, A.M.; Marwani, H.M.; Qin, H.L. Carbonyl-Olefin Metathesis: A Key Review. Org. Chem. Front. 2018, 5, 1381–1391. [Google Scholar] [CrossRef]
- Lecourt, C.; Dhambri, S.; Allievi, L.; Sanogo, Y.; Zeghbib, N.; Ben Othman, R.; Lannou, M.I.; Sorin, G.; Ardisson, J. Natural Products And Ring-Closing Metathesis: Synthesis of Sterically Congested Olefins. Nat. Prod. Rep. 2018, 35, 105–124. [Google Scholar] [CrossRef]
- Grubbs, R.H. Olefin Metathesis. Tetrahedron 2004, 60, 7117–7140. [Google Scholar] [CrossRef]
- Garber, S.B.; Kingsbury, J.S.; Gray, B.L.; Hoveyda, A.H. Efficient and Recyclable Monomeric and Dendritic Ru-Based Metathesis Catalysts. J. Am. Chem. Soc. 2001, 123, 3186. [Google Scholar] [CrossRef] [Green Version]
- Grela, K.; Harutyunyan, S.; Michrowska, A. A Highly Efficient Ruthenium Catalyst for Metathesis Reaction. Angew. Chem. Int. Ed. 2002, 41, 4038–4040. [Google Scholar] [CrossRef] [Green Version]
- Bieniek, M.; Samojłowicz, C.; Sashuk, V.; Bujok, R.; Śledź, P.; Lugan, N.; Lavigne, G.; Arlt, D.; Grela, K. Rational Design and Evaluation of Upgraded Grubbs/Hoveyda Olefin Metathesis Catalysts: Polyfunctional Benzylidene Ethers on the Test Bench. Organometallics 2011, 30, 4144–4158. [Google Scholar] [CrossRef]
- Keitz, B.K.; Endo, K.; Patel, P.R.; Herbert, M.B.; Grubbs, R.H. Improved Ruthenium Catalysts for Z-Selective Olefin Metathesis. J. Am. Chem. Soc. 2012, 134, 693–699. [Google Scholar] [CrossRef] [Green Version]
- Szczepaniak, G.; Kosiński, K.; Grela, K. Towards “Cleaner” Olefin Metathesis: Tailoring the NHC Ligand of Second Generation Ruthenium Catalysts to Afford Auxiliary Traits. Green Chem. 2014, 16, 4474–4492. [Google Scholar] [CrossRef]
- Wang, Z.J.; Jackson, W.R.; Robinson, A.J. A Simple and Practical Preparation of an Efficient Water Soluble Olefin Metathesis Catalyst. Green Chem. 2015, 17, 3407–3414. [Google Scholar] [CrossRef]
- Paradiso, V.; Bertolasi, V.; Costabile, C.; Caruso, T.; Dąbrowski, M.; Grela, K.; Grisi, F. Expanding the Family of Hoveyda-Grubbs Catalysts Containing Unsymmetrical NHC Ligands. Organometallics 2017, 36, 3692–3708. [Google Scholar] [CrossRef]
- Trnka, T.M.; Grubbs, R.H. The Development Of L2X2Ru = CHR Olefin Metathesis Catalysts: An Organometallic Success Story. Acc. Chem. Res. 2001, 34, 18–29. [Google Scholar] [CrossRef]
- Fish, R.H.; Jaouen, G. Bioorganometallic Chemistry: Structural Diversity of Organometallic Complexes with Bioligands and Molecular Recognition Studies of Several Supramolecular Hosts with Biomolecules, Alkali-Metal Ions, and Organometallic Pharmaceuticals. Organometallics 2003, 22, 2166–2177. [Google Scholar] [CrossRef]
- Hartwig, J.F.; Ward, T.R. New “Cats” in the House: Chemistry Meets Biology in Artificial Metalloenzymes and Repurposed Metalloenzymes. Acc. Chem. Res. 2019, 52, 1145. [Google Scholar]
- Binder, J.B.; Raines, R.T. Olefin Metathesis for Chemical Biology. Curr. Opin. Chem. Biol. 2008, 12, 767–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosati, F.; Roelfes, G. Artificial Metalloenzymes. ChemCatChem 2010, 2, 916–927. [Google Scholar] [CrossRef] [Green Version]
- Lewis, J.C. Artificial Metalloenzymes and Metallopeptide Catalysts for Organic Synthesis. ACS Catal. 2013, 3, 2954–2975. [Google Scholar] [CrossRef]
- Matsuo, T.; Hirota, S. Artificial Enzymes with Protein Scaffolds: Structural Design and Modification. Bioorg. Med. Chem. 2014, 22, 5638–5656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauer, D.F.; Gotzen, S.; Okuda, J. Metatheases: Artificial Metalloproteins for Olefin Metathesis. Org. Biomol. Chem. 2016, 14, 9174–9183. [Google Scholar] [CrossRef] [PubMed]
- Schwizer, F.; Okamoto, Y.; Heinisch, T.; Gu, Y.; Pellizzoni, M.M.; Lebrun, V.; Reuter, R.; Köhler, V.; Lewis, J.C.; Ward, T.R. Artificial Metalloenzymes: Reaction Scope and Optimization Strategies. Chem. Rev. 2018, 118, 142–231. [Google Scholar] [CrossRef] [Green Version]
- Sauer, D.F.; Matsuo, T.; Onoda, A.; Okuda, J.; Hayashi, T. Artificially Created Metathease Consisting of an Organometallic Complex Immobilized to a Protein Matrix. In Advances in Bioorganometallic Chemistry; Hirao, T., Moriuchi, T., Eds.; Elsevier Science Publishing Co Inc.: Chennai, India, 2018; pp. 302–326. [Google Scholar]
- Sauer, D.F.; Schiffels, J.; Hayashi, T.; Schwaneberg, U.; Okuda, J. Olefin Metathesis Catalysts Embedded In Beta-Barrel Proteins: Creating Artificial Metalloproteins for Olefin Metathesis. Beilstein J. Org. Chem. 2018, 14, 2861–2871. [Google Scholar] [CrossRef] [Green Version]
- Sabatino, V.; Ward, T.R. Aqueous Olefin Metathesis: Recent Developments and Applications. Beilstein J. Org. Chem. 2019, 15, 445–468. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, T.; Miyake, T.; Hirota, S. Recent Developments on Creation of Artificial Metalloenzymes. Tetrahedron Lett. 2019, 60, 151226. [Google Scholar] [CrossRef]
- Matsuo, T.; Imai, C.; Yoshida, T.; Saito, T.; Hayashi, T.; Hirota, S. Creation of an Artificial Metalloprotein With a Hoveyda-Grubbs Catalyst Moiety through the Intrinsic Inhibition Mechanism of α-Chymotrypsin. Chem. Commun. 2012, 48, 1662–1664. [Google Scholar] [CrossRef] [PubMed]
- Mayer, C.; Gillingham, D.G.; Ward, T.R.; Hilvert, D. An Artificial Metalloenzyme for Olefin Metathesis. Chem. Commun. 2011, 47, 12068–12070. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.; Ringenberg, M.R.; Gnandt, D.; Wilson, Y.; Ward, T.R. Artificial Metalloenzymes for Olefin Metathesis Based on the Biotin-(strept)Avidin Technology. Chem. Commun. 2011, 47, 12065–12067. [Google Scholar] [CrossRef] [PubMed]
- Philippart, F.; Arlt, M.; Gotzen, S.; Tenne, S.J.; Bocola, M.; Chen, H.H.; Zhu, L.L.; Schwaneberg, U.; Okuda, J. A Hybrid Ring-Opening Metathesis Polymerization Catalyst Based on an Engineered Variant of the β-Barrel Protein FhuA. Chem. Eur. J. 2013, 19, 13865–13871. [Google Scholar] [CrossRef] [PubMed]
- Sauer, D.F.; Himiyama, T.; Tachikawa, K.; Fukumoto, K.; Onoda, A.; Mizohata, E.; Inoue, T.; Bocola, M.; Schwaneberg, U.; Hayashi, T. A Highly Active Biohybrid Catalyst for Olefin Metathesis in Water: Impact of a Hydrophobic Cavity in a β-Barrel Protein. ACS Catal. 2015, 5, 7519–7522. [Google Scholar] [CrossRef]
- Kraut, J. Chymotrypsin-Chemical Properties and Catalysis. In The Enzymes; Paul, D.B., Krebs, E.G., Eds.; Academic Press: Cambdige, MA, USA, 1971; Volume 3, pp. 213–248. [Google Scholar]
- Basauri-Molina, M.; Verhoeven, D.G.A.; van Schaik, A.J.; Kleijn, H.; Klein Gebbink, R.J.M. Ring-Closing and Cross-Metathesis with Artificial Metalloenzymes Created by Covalent Active Site-Directed Hybridization of a Lipase. Chem. Eur. J. 2015, 21, 15676–15685. [Google Scholar] [CrossRef]
- Zhao, J.; Kajetanowicz, A.; Ward, T.R. Carbonic Anhydrase II as Host Protein for the Creation of a Biocompatible Artificial Metathesase. Org. Biomol. Chem. 2015, 13, 5652–5655. [Google Scholar] [CrossRef] [Green Version]
- Mingotaud, A.F.; Mingotaud, C.; Moussa, W. Characterization of the Micellar Ring Opening Metathesis Polymerization in Water of a Norbornene Derivative Initiated by Hoveyda-Grubbs’ Catalyst. J. Polym. Sci. Pol. Chem. 2008, 46, 2833–2844. [Google Scholar] [CrossRef]
- Thiel, V.; Hendann, M.; Wannowius, K.J.; Plenio, H. On the Mechanism of the Initiation Reaction in Grubbs-Hoveyda Complexes. J. Am. Chem. Soc. 2012, 134, 1104–1114. [Google Scholar] [CrossRef]
- Grimm, A.R.; Sauer, D.F.; Davari, M.D.; Zhu, L.L.; Bocola, M.; Kato, S.; Onoda, A.; Hayashi, T.; Okuda, J.; Schwaneberg, U. Cavity Size Engineering of a β-Barrel Protein Generates Efficient Biohybrid Catalysts for Olefin Metathesis. ACS Catal. 2018, 8, 3358–3364. [Google Scholar] [CrossRef]
- Lutz, S. Beyond Directed Evolution—Semi-Rational Protein Engineering and Design. Curr. Opin. Biotechnol. 2010, 21, 734–743. [Google Scholar] [CrossRef] [Green Version]
- Reetz, M.T. Directed Evolution of Selective Enzymes: Catalysts for Organic Chemistry and Biotechnology; Wiley-VCH: Weinheim, Germany, 2016. [Google Scholar]
- Jeschek, M.; Reuter, R.; Heinisch, T.; Trindler, C.; Klehr, J.; Panke, S.; Ward, T.R. Directed Evolution Of Artificial Metalloenzymes For In Vivo Metathesis. Nature 2016, 537, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Samanta, A.; Sabatino, V.; Ward, T.R.; Walther, A. Functional and Morphological Adaptation in DNA Protocells via Signal Processing Prompted by Artificial Metalloenzymes. Nat. Nanotechnol. 2020, 15, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Sabatino, V.; Rebelein, J.G.; Ward, T.R. “Close-to-Release”: Spontaneous Bioorthogonal Uncaging Resulting from Ring-Closing Metathesis. J. Am. Chem. Soc. 2019, 141, 17048–17052. [Google Scholar] [CrossRef] [Green Version]
- Eda, S.; Nasibullin, I.; Vong, K.; Kudo, N.; Yoshida, M.; Kurbangalieva, A.; Tanaka, K. Biocompatibility and Therapeutic Potential of Glycosylated Albumin Artificial Metalloenzymes. Nat. Catal. 2019, 2, 780–792. [Google Scholar] [CrossRef]
- Vong, K.; Nasibullin, I.; Tanaka, K. Exploring and Adapting the Molecular Selectivity of Artificial Metalloenzymes. Bull. Chem. Soc. Jpn. 2021, 94, 382–396. [Google Scholar] [CrossRef]
- Sohn, J.H.; Kim, K.H.; Lee, H.Y.; No, Z.S.; Ihee, H. Initial Catalyst-Substrate Association Step in Enyne Metathesis Catalyzed by Grubbs Ruthenium Complex Probed by Time-Dependent Fluorescence Quenching. J. Am. Chem. Soc. 2008, 130, 16506–16507. [Google Scholar] [CrossRef]
- Toussaint, S.N.W.; Calkins, R.T.; Lee, S.; Michel, B.W. Olefin Metathesis-based Fluorescent Probes for the Selective Detection of Ethylene in Live Cells. J. Am. Chem. Soc. 2018, 140, 13151–13155. [Google Scholar] [CrossRef]
- Vong, K.; Eda, S.; Kadota, Y.; Nasibullin, I.; Wakatake, T.; Yokoshima, S.; Shirasu, K.; Tanaka, K. An Artificial Metalloenzyme Biosensor can Detect Ethylene Gas in Fruits and Arabidopsis Leaves. Nat. Commun. 2019, 10, 5746. [Google Scholar] [CrossRef] [Green Version]
- Kirkland, T.A.; Lynn, D.M.; Grubbs, R.H. Ring-closing Metathesis in Methanol And Water. J. Org. Chem. 1998, 63, 9904–9909. [Google Scholar] [CrossRef]
- Lynn, D.M.; Grubbs, R.H. Novel Reactivity Of Ruthenium Alkylidenes in Protic Solvents: Degenerate Alkylidene Proton Exchange. J. Am. Chem. Soc. 2001, 123, 3187–3193. [Google Scholar] [CrossRef]
- Hong, S.H.; Wenzel, A.G.; Salguero, T.T.; Day, M.W.; Grubbs, R.H. Decomposition of Ruthenium Olefin Metathesis Catalysts. J. Am. Chem. Soc. 2007, 129, 7961–7968. [Google Scholar] [CrossRef] [Green Version]
- Jawiczuk, M.; Marczyk, A.; Trzaskowski, B. Decomposition of Ruthenium Olefin Metathesis Catalyst. Catalysts 2020, 10, 887. [Google Scholar] [CrossRef]
- Dinger, M.B.; Mol, J.C. Degradation of The First-Generation Grubbs Metathesis Catalyst with Primary Alcohols, Water, and Oxygen. Formation and Catalytic Activity of Ruthenium(II) Monocarbonyl Species. Organometallics 2003, 22, 1089–1095. [Google Scholar] [CrossRef]
- Goudreault, A.Y.; Walden, D.M.; Nascimento, D.L.; Botti, A.G.; Steinmann, S.N.; Michel, C.; Fogg, D.E. Hydroxide-induced Degradation of Olefin Metathesis Catalysts: A Challenge for Metathesis in Alkaline Media. ACS Catal. 2020, 10, 3838–3843. [Google Scholar] [CrossRef]
- Dinger, M.B.; Mol, J.C. Degradation of the Second-generation Grubbs Metathesis Catalyst with Primary Alcohols and Oxygen—Isomerization and Hydrogenation Activities of Monocarbonyl Complexes. Eur. J. Inorg. Chem. 2003, 2003, 2827–2833. [Google Scholar] [CrossRef]
- Banti, D.; Mol, J.C. Degradation of the Ruthenium-based Metathesis Catalyst [RuCl2(CHPh)(H2IPr)(PCy3)] with Primary Alcohols. J. Organomet. Chem. 2004, 689, 3113–3116. [Google Scholar] [CrossRef]
- Zirngast, M.; Pump, E.; Leitgeb, A.; Albering, J.H.; Slugovc, C. Pyridine as Trigger for Chloride Isomerisation in Chelated Ruthenium Benzylidene Complexes: Implications for Olefin Metathesis. Chem. Commun. 2011, 47, 2261–2263. [Google Scholar] [CrossRef] [PubMed]
- Blanco, C.O.; Sims, J.; Nascimento, D.L.; Goudreault, A.Y.; Steinmann, S.N.; Michel, C.; Fogg, D.E. The Impact of Water on Ru-catalyzed Olefin Metathesis: Potent Deactivating Effects Even at Low Water Concentrations. ACS Catal. 2021, 11, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Wappel, J.; Urbina-Blanco, C.A.; Abbas, M.; Albering, J.H.; Saf, R.; Nolan, S.P.; Slugovc, C. Halide Exchanged Hoveyda-type Complexes in Olefin Metathesis. Beilstein J. Org. Chem. 2010, 6, 1091–1098. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, T.; Yoshida, T.; Fujii, A.; Kawahara, K.; Hirota, S. Effect of Added Salt on Ring-closing Metathesis Catalyzed by a Water-Soluble Hoveyda-Grubbs Type Complex To Form N-Containing Heterocycles in Aqueous Media. Organometallics 2013, 32, 5313–5319. [Google Scholar] [CrossRef]
- Foster, J.C.; Grocott, M.C.; Arkinstall, L.A.; Varlas, S.; Redding, M.J.; Grayson, S.M.; O’Reilly, R.K. It is Better with Salt: Aqueous Ring-Opening Metathesis Polymerization at Neutral pH. J. Am. Chem. Soc. 2020, 142, 13878–13885. [Google Scholar] [CrossRef]
- Michrowska, A.; Bujok, R.; Harutyunyan, S.; Sashuk, V.; Dolgonos, G.; Grela, K. Nitro-substituted Hoveyda−Grubbs Ruthenium Carbenes: Enhancement of Catalyst Activity through Electronic Activation. J. Am. Chem. Soc. 2004, 126, 9318–9325. [Google Scholar] [CrossRef] [Green Version]
- Michrowska, A.; Gułajski, Ł.; Kaczmarska, Z.; Mennecke, K.; Kirschning, A.; Grela, K. A Green Catalyst for Green Chemistry: Synthesis and Application of an Olefin Metathesis Catalyst Bearing a Quaternary Ammonium Group. Green Chem. 2006, 8, 685–688. [Google Scholar] [CrossRef]
- Gawin, R.; Makal, A.; Wozniak, K.; Mauduit, M.; Grela, K. A Dormant Ruthenium Catalyst Bearing A Chelating Carboxylate Ligand: In Situ Activation and Application in Metathesis Reactions. Angew. Chem. Int. Ed. 2007, 46, 7206–7209. [Google Scholar] [CrossRef]
- Engle, K.M.; Lu, G.; Luo, S.X.; Henling, L.M.; Takase, M.K.; Liu, P.; Houk, K.N.; Grubbs, R.H. Origins of Initiation Rate Differences in Ruthenium Olefin Metathesis Catalysts Containing Chelating Benzylidenes. J. Am. Chem. Soc. 2015, 137, 5782–5792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zieliński, A.; Szczepaniak, G.; Gajda, R.K.W.; Trzaskowski, B.; Vidović, D.; Kajetanowicz, A.; Grela, K. Ruthenium Olefin Metathesis Catalysts Systematically Modified in Chelating Benzylidene Ether Fragment: Experiment and Computations. Eur. J. Inorg. Chem. 2018, 3675–3685. [Google Scholar] [CrossRef]
- Skowerski, K.; Kasprzycki, P.; Bieniek, M.; Olszewski, T.K. Efficient, Durable and Reusable Olefin Metathesis Catalysts with High Affinity to Silica Gel. Tetrahedron 2013, 69, 7408–7415. [Google Scholar] [CrossRef]
- Jatmika, C.; Goshima, K.; Wakabayashi, K.; Akiyama, N.; Hirota, S.; Matsuo, T. Second-coordination Sphere Effects on The Reactivities of Hoveyda-Grubbs-Type Catalysts: A Ligand Exchange Study Using Phenolic Moiety-Functionalized Ligands. Dalton Trans. 2020, 49, 11618–11627. [Google Scholar] [CrossRef]
- Jatmika, C.; Wakabayashi, K.; Tamaki, R.; Akiyama, N.; Nakamura, I.; Hirota, S.; Yamaguchi, H.; Matsuo, T. Ligand Exchange Strategy for Delivery of Ruthenium Complex Unit to Biomolecules Based on Ruthenium-Olefin Specific Interactions. Chem. Lett. 2020, 49, 1490–1493. [Google Scholar] [CrossRef]
- Fujii, A.; Hirota, S.; Matsuo, T. Reversible Switching of Fluorophore Property Based on Intrinsic Conformational Transition of Adenylate Kinase During Its Catalytic Cycle. Bioconjugate Chem. 2013, 24, 1218–1225. [Google Scholar] [CrossRef] [PubMed]
- Fujii, A.; Sekiguchi, Y.; Matsumura, H.; Inoue, T.; Chung, W.S.; Hirota, S.; Matsuo, T. Excimer Emission Properties on Pyrene-Labeled Protein Surface: Correlation Between Emission Spectra, Ring Stacking Modes, and Flexibilities of Pyrene Probes. Bioconjugate Chem. 2015, 26, 537–548. [Google Scholar] [CrossRef]
- Miyake, T.; Tamaki, R.; Asanuma, M.; Fukada, Y.; Hirota, S.; Matsuo, T. Regioselective Chemical Modification of Cysteine Residues on Protein Surfaces Focusing on Local Environment around the Conjugation Site. Bioconjugate Chem. 2020, 31, 794–802. [Google Scholar] [CrossRef]
- Lin, Y.A.; Chalker, J.M.; Floyd, N.; Bernardes, G.J.; Davis, B.G. Allyl Sulfides Are Privileged Substrates in Aqueous Cross-Metathesis: Application to Site-Selective Protein Modification. J. Am. Chem. Soc. 2008, 130, 9642–9643. [Google Scholar] [CrossRef] [Green Version]
- Chalker, J.M.; Lin, Y.A.; Boutureira, O.; Davis, B.G. Enabling Olefin Metathesis on Proteins: Chemical Methods for Installation of S-Allyl Cysteine. Chem. Commun. 2009, 3714–3716. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.A.; Chalker, J.M.; Davis, B.G. Olefin Metathesis for Site-Selective Protein Modification. ChemBioChem 2009, 10, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Bhushan, B.; Lin, Y.A.; Bak, M.; Phanumartwiwath, A.; Yang, N.; Bilyard, M.K.; Tanaka, T.; Hudson, K.L.; Lercher, L.; Stegmann, M. Genetic Incorporation of Olefin Cross-Metathesis Reaction Tags for Protein Modification. J. Am. Chem. Soc. 2018, 140, 14599–14603. [Google Scholar] [CrossRef]
























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Host Protein (Native or Mutant) | HG-II-Type Complex (See Figure 4) | Fixation of Metal Center into Protein Core | Representative OM Reactivities | Ref |
|---|---|---|---|---|
| MjHSP (G41C) 1 | 6 | Cys conjugation |  | [26] |
| FhuAΔCVFTEV 2 | 6 or 7 | Cys conjugation |  | [28] |
| nitrobindin (H76L/Q98C/ H158L; NB4) | 7 | Cys conjugation |  | [29] |
| α-chymotrypsin (α-CT) | 8 | His conjugation |  | [25] |
| cutinase | 9 | Ser conjugation |  | [31] |
| (strept)avidin | 10 | biotin-avidin interaction |  | [27] |
| carbonic anhydrase-II | 11 | binding to Zn2+ |  | [32] |

| Ru Catalyst | R | X | TON (No H2O) | TON (1% H2O) | H2O-Tolerance (%) |
|---|---|---|---|---|---|
| 5 | H | Cl | 17,800 | 5600 | 31 |
| 5-NO2 | NO2 | Cl | 18,200 | 1800 | 10 |
| 5′-NO2 | NO2 | I | 17,400 | 9800 | 56 |

| Entry | Additive | Yield (%) |
|---|---|---|
| 1 | none | 50 |
| 2 | 100 mM KCl | 80 |
| 3 | 10 mM MES (pD = 6.4) 1 | 9 |
| 4 | 10 mM MES + 100 mM KCl (pD = 6.4) | 40 |
| 5 | 100 mM KNO3 | 47 |
| Entry | Ligand Precursor | Functional Group at the End of Ligand | Product Complex | Linker Length 2 | Conversion (%) (24 h) 3 |
|---|---|---|---|---|---|
| 1 | 34-L |  | 34 | n = 2 | 82 4 |
| 2 | 35-L |  | 35 | n = 2 | 77 5 |
| 3 | 36-L |  | 36 | n = 3 | 61 4 |
| 4 | 37-L |  | 37 | n = 2 | 31 4 |
| 5 | 38-L |  | 38 | n = 2 | 19 5 |
| 6 | 39-L |  | 39 | n = 3 | 41 5 |
| 7 | 40-L |  | 40 | n = 4 | 51 5 |
| Entry | Reaction | Cat. | Cat. Load | Yield (%) |
|---|---|---|---|---|
| 1 2 |  | 34 | 5 mol% | 15 (2 h); 94 (54 h) |
| 2 2 | 38 | 5 mol% | 53 (2 h); 99 (24 h) | |
| 3 2 | 5 | 5 mol% | 23 (2 h); 95 (24 h) | |
| 4 2 |  | 34 | 5 mol% | 30 (0.5 h); >99 (7 h) |
| 5 2 | 38 | 5 mol% | >99 (0.5 h) | |
| 6 2 | 5 | 5 mol% | >99 (0.5 h) | |
| 7 2 | 34 | 0.1 mol% | 11 (0.5 h); 91 (24 h) | |
| 8 2 | 38 | 0.1 mol% | 85 (0.5 h); >99 (24 h) | |
| 9 2 | 5 | 0.1 mol% | 63 (0.5 h); >99 (24 h) | |
| 10 3 |  | 34 | 1 mol% | 47: 19 (2 h); 37 (24 h) 48: <5 (2 h); 5 (24 h) |
| 11 3 | 38 | 1 mol% | 47: 54 (2 h); 61 (24 h) 48: 7 (2 h); 8 (24 h) | |
| 12 3 | 5 | 1 mol% | 47: 48 (2 h); 74 (24 h) 48: 5 (2 h); 6 (24 h) |

| Entry | Peptide | Ru Complex Donor | Solvent | Additive | Conversion (%) |
|---|---|---|---|---|---|
| 1 2 | 49 | 53 | CDCl3 | - | 91 (24 h) |
| 2 2 | 49 | 54 | CDCl3 | - | 89 (24 h) |
| 3 2 | 49 | 5 | CDCl3 | - | 53 (24 h); 70 (120 h) |
| 4 2 | 49 | 54 | CD3OD | - | 75 (10 h); 86 (48 h) |
| 5 2 | 49 | 5 | CD3OD | - | 46 (10 h); 72 (48 h) |
| 6 2 | 49′ | 54 | D2O 1 | MgCl2 2 | 75 (72 h) |
| 7 2 | 49′ | 54 | D2O 1 | CaCl2 2 | 72 (72 h) |
| 8 2 | 49′ | 54 | D2O 1 | KCl 2 | 64 (72 h) |
| 9 2 | 49′ | 54 | D2O 1 | NaCl 2 | 63 (72 h) |
| 10 | 49′ | 54 | D2O 1 | KNO3 2 | n.r. (72 h) 3 |
| 11 | 49′ | 54 | D2O 1 | none | <5 (72 h) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsuo, T. Functionalization of Ruthenium Olefin-Metathesis Catalysts for Interdisciplinary Studies in Chemistry and Biology. Catalysts 2021, 11, 359. https://doi.org/10.3390/catal11030359
Matsuo T. Functionalization of Ruthenium Olefin-Metathesis Catalysts for Interdisciplinary Studies in Chemistry and Biology. Catalysts. 2021; 11(3):359. https://doi.org/10.3390/catal11030359
Chicago/Turabian StyleMatsuo, Takashi. 2021. "Functionalization of Ruthenium Olefin-Metathesis Catalysts for Interdisciplinary Studies in Chemistry and Biology" Catalysts 11, no. 3: 359. https://doi.org/10.3390/catal11030359
APA StyleMatsuo, T. (2021). Functionalization of Ruthenium Olefin-Metathesis Catalysts for Interdisciplinary Studies in Chemistry and Biology. Catalysts, 11(3), 359. https://doi.org/10.3390/catal11030359