
Organizing Multi-Enzyme Systems into Programmable Materials for Biocatalysis



Abstract
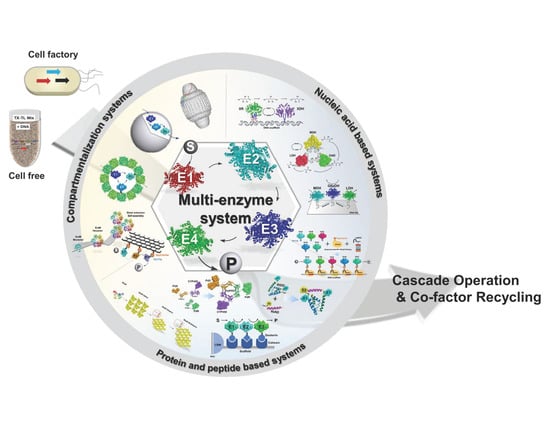
:
1. Introduction
2. Systems Platforms for Multi-Enzyme Self-Organization
3. Nucleic Acid Based Scaffolds
4. Protein and Peptide-Based Systems for Enzyme Co-Localization
4.1. Co-Localization Using Non-Covalent Interaction Domains and Tags
4.2. Co-Localization Using Covalent Conjugation Mechanisms
4.3. Self-Assembling Protein Arrays and Nanostructures as Scaffolds
5. Compartmentalization Systems
5.1. Encapsulation in Proteinaceous Compartments, Cages and Virus-Like Particles
5.2. Encapsulation in Lipid Vesicles and Polymersomes
6. Conclusions and Future Directions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Bornscheuer, U.T.; Huisman, G.W.; Kazlauskas, R.J.; Lutz, S.; Moore, J.C.; Robins, K. Engineering the third wave of biocatalysis. Nature 2012, 485, 185–194. [Google Scholar] [CrossRef]
- Wu, S.; Snajdrova, R.; Moore, J.C.; Baldenius, K.; Bornscheuer, U.T. Biocatalysis: Enzymatic Synthesis for Industrial Applications. Angew. Chem. Int. Ed. Engl. 2021, 60, 88–119. [Google Scholar] [CrossRef]
- Bornscheuer, U.T. The fourth wave of biocatalysis is approaching. Philos. Trans. A Math. Phys. Eng. Sci. 2018, 376. [Google Scholar] [CrossRef] [PubMed]
- Bornscheuer, U.T. Biocatalysis: Successfully Crossing Boundaries. Angew. Chem. Int. Ed. Engl. 2016, 55, 4372–4373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldon, R.A.; Brady, D. Broadening the Scope of Biocatalysis in Sustainable Organic Synthesis. Chemsuschem 2019, 12, 2859–2881. [Google Scholar] [CrossRef] [PubMed]
- Ricca, E.; Brucher, B.; Schrittwieser, J.H. Multi-Enzymatic Cascade Reactions: Overview and Perspectives. Adv. Synth. Catal. 2011, 353, 2239–2262. [Google Scholar] [CrossRef]
- France, S.P.; Hepworth, L.J.; Turner, N.J.; Flitsch, S.L. Constructing Biocatalytic Cascades: In Vitro and in Vivo Approaches to de Novo Multi-Enzyme Pathways. ACS Catal. 2017, 7, 710–724. [Google Scholar] [CrossRef]
- Schrittwieser, J.H.; Velikogne, S.; Hall, M.; Kroutil, W. Artificial Biocatalytic Linear Cascades for Preparation of Organic Molecules. Chem. Rev. 2018, 118, 270–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sperl, J.M.; Sieber, V. Multienzyme Cascade Reactions-Status and Recent Advances. ACS Catal. 2018, 8, 2385–2396. [Google Scholar] [CrossRef]
- Gandomkar, S.; Zadlo-Dobrowolska, A.; Kroutil, W. Extending Designed Linear Biocatalytic Cascades for Organic Synthesis. ChemCatChem 2019, 11, 225–243. [Google Scholar] [CrossRef] [Green Version]
- Kuska, J.; O’Reilly, E. Engineered biosynthetic pathways and biocatalytic cascades for sustainable synthesis. Curr. Opin. Chem. Biol. 2020, 58, 146–154. [Google Scholar] [CrossRef]
- Losada-Garcia, N.; Cabrera, Z.; Urrutia, P.; Garcia-Sanz, C.; Andreu, A.; Palomo, J.M. Recent Advances in Enzymatic and Chemoenzymatic Cascade Processes. Catalysts 2020, 10, 1258. [Google Scholar] [CrossRef]
- Lee, Y.S.; Lim, K.; Minteer, S.D. Cascaded Biocatalysis and Bioelectrocatalysis: Overview and Recent Advances. Annu. Rev. Phys. Chem. 2021. [Google Scholar] [CrossRef]
- Rudroff, F.; Mihovilovic, M.D.; Groger, H.; Snajdrova, R.; Iding, H.; Bornscheuer, U.T. Opportunities and challenges for combining chemo- and biocatalysis. Nat. Catal. 2018, 1, 12–22. [Google Scholar] [CrossRef]
- Dias Gomes, M.; Woodley, J.M. Considerations when Measuring Biocatalyst Performance. Molecules 2019, 24, 3573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Zhang, X.; Yuan, H.; Huang, D.; Wang, R.; Liu, H.; Wang, T. Research progress and the biotechnological applications of multienzyme complex. Appl. Microbiol. Biotechnol. 2021, 105, 1759–1777. [Google Scholar] [CrossRef]
- Mohamad, N.R.; Marzuki, N.H.; Buang, N.A.; Huyop, F.; Wahab, R.A. An overview of technologies for immobilization of enzymes and surface analysis techniques for immobilized enzymes. Biotechnol. Biotechnol. Equip. 2015, 29, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Chen, X.; Zheng, R.; Zheng, Y. Immobilization of Multi-Enzymes on Support Materials for Efficient Biocatalysis. Front. Bioeng. Biotechnol. 2020, 8, 660. [Google Scholar] [CrossRef]
- Wahab, R.A.; Elias, N.; Abdullah, F.; Ghoshal, S.K. On the taught new tricks of enzymes immobilization: An all-inclusive overview. React. Funct. Polym. 2020, 152. [Google Scholar] [CrossRef]
- Romero-Fernandez, M.; Paradisi, F. Protein immobilization technology for flow biocatalysis. Curr. Opin. Chem. Biol. 2020, 55, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.Z.; Li, C.H.; Jiao, X.B.; Jia, S.R.; Jiang, Y.J.; Bilal, M.; Cui, J.D. Recent progress in multienzymes co-immobilization and multienzyme system applications. Chem. Eng. J. 2019, 373, 1254–1278. [Google Scholar] [CrossRef]
- Hwang, E.T.; Lee, S. Multienzymatic Cascade Reactions via Enzyme Complex by Immobilization. ACS Catal. 2019, 9, 4402–4425. [Google Scholar] [CrossRef]
- Petroll, K.; Kopp, D.; Care, A.; Bergquist, P.L.; Sunna, A. Tools and strategies for constructing cell-free enzyme pathways. Biotechnol. Adv. 2019, 37, 91–108. [Google Scholar] [CrossRef]
- Berckman, E.A.; Hartzell, E.J.; Mitkas, A.A.; Sun, Q.; Chen, W. Biological Assembly of Modular Protein Building Blocks as Sensing, Delivery, and Therapeutic Agents. Annu. Rev. Chem. Biomol. Eng. 2020, 11, 35–62. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, P.; de Carvalho, C.C.C.R. Multi-Enzyme Systems in Flow Chemistry. Processes 2021, 9, 225. [Google Scholar] [CrossRef]
- Bugada, L.F.; Smith, M.R.; Wen, F. Engineering Spatially Organized Multienzyme Assemblies for Complex Chemical Transformation. ACS Catal. 2018, 8, 7898–7906. [Google Scholar] [CrossRef] [Green Version]
- Schmid-Dannert, C.; Lopez-Gallego, F. Advances and opportunities for the design of self-sufficient and spatially organized cell-free biocatalytic systems. Curr. Opin. Chem. Biol. 2019, 49, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Ellis, G.A.; Klein, W.P.; Lasarte-Aragones, G.; Thakur, M.; Walper, S.A.; Medintz, I.L. Artificial Multienzyme Scaffolds: Pursuing in Vitro Substrate Channeling with an Overview of Current Progress. ACS Catal. 2019, 9, 10812–10869. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Wu, Y.; Zhang, S.; Tian, Y.; Yang, D.; Jiang, Z. Bioinspired construction of multi-enzyme catalytic systems. Chem. Soc. Rev. 2018, 47, 4295–4313. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Sheng, J.; Feng, X. Mini-review: In vitro Metabolic Engineering for Biomanufacturing of High-value Products. Comput. Struct. Biotechnol. J. 2017, 15, 161–167. [Google Scholar] [CrossRef]
- Tinafar, A.; Jaenes, K.; Pardee, K. Synthetic Biology Goes Cell-Free. BMC Biol. 2019, 17, 64. [Google Scholar] [CrossRef] [Green Version]
- Khambhati, K.; Bhattacharjee, G.; Gohil, N.; Braddick, D.; Kulkarni, V.; Singh, V. Exploring the Potential of Cell-Free Protein Synthesis for Extending the Abilities of Biological Systems. Front. Bioeng. Biotechnol. 2019, 7, 248. [Google Scholar] [CrossRef] [PubMed]
- Noireaux, V.; Liu, A.P. The New Age of Cell-Free Biology. Annu. Rev. Biomed. Eng. 2020, 22, 51–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowie, J.U.; Sherkhanov, S.; Korman, T.P.; Valliere, M.A.; Opgenorth, P.H.; Liu, H. Synthetic Biochemistry: The Bio-inspired Cell-Free Approach to Commodity Chemical Production. Trends Biotechnol. 2020, 38, 766–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grubbe, W.S.; Rasor, B.J.; Kruger, A.; Jewett, M.C.; Karim, A.S. Cell-free styrene biosynthesis at high titers. Metab. Eng. 2020, 61, 89–95. [Google Scholar] [CrossRef]
- Liu, W.Q.; Wu, C.; Jewett, M.C.; Li, J. Cell-free protein synthesis enables one-pot cascade biotransformation in an aqueous-organic biphasic system. Biotechnol. Bioeng. 2020, 117, 4001–4008. [Google Scholar] [CrossRef] [PubMed]
- Ngo, T.A.; Nakata, E.; Saimura, M.; Morii, T. Spatially Organized Enzymes Drive Cofactor-Coupled Cascade Reactions. J. Am. Chem. Soc. 2016, 138, 3012–3021. [Google Scholar] [CrossRef]
- Liu, M.; Fu, J.; Qi, X.; Wootten, S.; Woodbury, N.W.; Liu, Y.; Yan, H. A Three-Enzyme Pathway with an Optimised Geometric Arrangement to Facilitate Substrate Transfer. Chembiochem 2016, 17, 1097–1101. [Google Scholar] [CrossRef]
- Kang, W.; Ma, T.; Liu, M.; Qu, J.; Liu, Z.; Zhang, H.; Shi, B.; Fu, S.; Ma, J.; Lai, L.T.F.; et al. Modular enzyme assembly for enhanced cascade biocatalysis and metabolic flux. Nat. Commun. 2019, 10, 4248. [Google Scholar] [CrossRef]
- Yang, Z.; Gao, X.; Xie, H.; Wang, F.; Ren, Y.; Wei, D. Enhanced itaconic acid production by self-assembly of two biosynthetic enzymes in Escherichia coli. Biotechnol. Bioeng. 2017, 114, 457–462. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, H.; Wang, Y.; Ren, Y.; Wei, D. Manufacturing Multienzymatic Complex Reactors In Vivo by Self-Assembly To Improve the Biosynthesis of Itaconic Acid in Escherichia coli. ACS Synth. Biol. 2018, 7, 1244–1250. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Yang, S.; Zhao, C.; Ren, Y.; Wei, D. Artificial multienzyme supramolecular device: Highly ordered self-assembly of oligomeric enzymes in vitro and in vivo. Angew. Chem. Int. Ed. Engl. 2014, 53, 14027–14030. [Google Scholar] [CrossRef]
- Dueber, J.E.; Wu, G.C.; Malmirchegini, G.R.; Moon, T.S.; Petzold, C.J.; Ullal, A.V.; Prather, K.L.; Keasling, J.D. Synthetic protein scaffolds provide modular control over metabolic flux. Nat. Biotechnol. 2009, 27, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Kibblewhite, R.E.; Paavola, C.D.; Orts, W.J.; Wagschal, K. Production of D-Xylonic Acid from Hemicellulose Using Artificial Enzyme Complexes. J. Microbiol. Biotechn. 2017, 27, 77–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, X.; Tang, S.; Xu, Q.; Huang, H.; Jiang, L. SpyCatcher/SpyTag-Mediated Self-Assembly of a Supramolecular Complex for Improved Biocatalytic Production of Trehalose. Appl. Biochem. Microbiol. 2019, 55, 596–602. [Google Scholar] [CrossRef]
- Zhang, G.Q.; Quin, M.B.; Schmidt-Dannert, C. Self-Assembling Protein Scaffold System for Easy in Vitro Coimmobilization of Biocatalytic Cascade Enzymes. ACS Catal. 2018, 8, 5611–5620. [Google Scholar] [CrossRef]
- Peschke, T.; Skoupi, M.; Burgahn, T.; Gallus, S.; Ahmed, I.; Rabe, K.S.; Niemeyer, C.M. Self-Immobilizing Fusion Enzymes for Compartmentalized Biocatalysis. ACS Catal. 2017, 7, 7866–7872. [Google Scholar] [CrossRef]
- Jiang, W.; Zeng, W. Construction of a Self-Purification and Self-Assembly Coenzyme Regeneration System for the Synthesis of Chiral Drug Intermediates. ACS Omega 2021, 6, 1911–1916. [Google Scholar] [CrossRef]
- Qu, J.; Cao, S.; Wei, Q.; Zhang, H.; Wang, R.; Kang, W.; Ma, T.; Zhang, L.; Liu, T.; Wing-Ngor Au, S.; et al. Synthetic Multienzyme Complexes, Catalytic Nanomachineries for Cascade Biosynthesis In Vivo. ACS Nano 2019, 13, 9895–9906. [Google Scholar] [CrossRef]
- Lim, S.; Kim, J.; Kim, Y.; Xu, D.; Clark, D.S. CRISPR/Cas-directed programmable assembly of multi-enzyme complexes. Chem. Commun. (Camb.) 2020, 56, 4950–4953. [Google Scholar] [CrossRef]
- Patterson, D.P.; Schwarz, B.; Waters, R.S.; Gedeon, T.; Douglas, T. Encapsulation of an Enzyme Cascade within the Bacteriophage P22 Virus-Like Particle. ACS Chem. Biol. 2014, 9, 359–365. [Google Scholar] [CrossRef]
- Myhrvold, C.; Polka, J.K.; Silver, P.A. Synthetic Lipid-Containing Scaffolds Enhance Production by Colocalizing Enzymes. ACS Synth. Biol. 2016, 5, 1396–1403. [Google Scholar] [CrossRef] [PubMed]
- van Dongen, S.F.; Nallani, M.; Cornelissen, J.J.; Nolte, R.J.; van Hest, J.C. A Three-enzyme cascade reaction through positional assembly of enzymes in a polymersome nanoreactor. Chemistry 2009, 15, 1107–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, R.J.; Marguet, M.; Marais, S.; Fraaije, M.W.; van Hest, J.C.; Lecommandoux, S. Cascade Reactions in Multicompartmentalized Polymersomes. Angew. Chem. Int. Ed. Engl. 2014, 53, 146–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klermund, L.; Poschenrieder, S.T.; Castiglione, K. Biocatalysis in Polymersomes: Improving Multienzyme Cascades with Incompatible Reaction Steps by Compartmentalization. ACS Catal. 2017, 7, 3900–3904. [Google Scholar] [CrossRef]
- Seeman, N.C. DNA in a material world. Nature 2003, 421, 427–431. [Google Scholar] [CrossRef]
- Linko, V.; Dietz, H. The enabled state of DNA nanotechnology. Curr. Opin. Biotechnol. 2013, 24, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Seeman, N.C. Nanomaterials based on DNA. Annu. Rev. Biochem. 2010, 79, 65–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, H.J.; Wang, Y.Q.; Wang, J.; Li, Z.H.; Yuan, Q. Emerging Biomimetic Applications of DNA Nanotechnology. ACS Appl. Mater. Interfaces 2019, 11, 13859–13873. [Google Scholar] [CrossRef]
- Rothemund, P.W. Folding DNA to create nanoscale shapes and patterns. Nature 2006, 440, 297–302. [Google Scholar] [CrossRef] [Green Version]
- Bush, J.; Singh, S.; Vargas, M.; Oktay, E.; Hu, C.H.; Veneziano, R. Synthesis of DNA Origami Scaffolds: Current and Emerging Strategies. Molecules 2020, 25, 3386. [Google Scholar] [CrossRef]
- Hong, F.; Zhang, F.; Liu, Y.; Yan, H. DNA Origami: Scaffolds for Creating Higher Order Structures. Chem. Rev. 2017, 117, 12584–12640. [Google Scholar] [CrossRef]
- Goldberg, M.; Langer, R.; Jia, X. Nanostructured materials for applications in drug delivery and tissue engineering. J. Biomater. Sci. Polym. Ed. 2007, 18, 241–268. [Google Scholar] [CrossRef] [Green Version]
- Liechty, W.B.; Kryscio, D.R.; Slaughter, B.V.; Peppas, N.A. Polymers for drug delivery systems. Annu. Rev. Chem. Biomol. Eng. 2010, 1, 149–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doll, T.A.; Raman, S.; Dey, R.; Burkhard, P. Nanoscale assemblies and their biomedical applications. J. R. Soc. Interface 2013, 10, 20120740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, J.J.; Elisseeff, J.H. Mimicking biological functionality with polymers for biomedical applications. Nature 2016, 540, 386–394. [Google Scholar] [CrossRef]
- Goldberg, M.S. Improving cancer immunotherapy through nanotechnology. Nat. Rev. Cancer 2019, 19, 587–602. [Google Scholar] [CrossRef]
- Fruk, L.; Muller, J.; Weber, G.; Narvaez, A.; Dominguez, E.; Niemeyer, C.M. DNA-directed immobilization of horseradish peroxidase-DNA conjugates on microelectrode arrays: Towards electrochemical screening of enzyme libraries. Chemistry 2007, 13, 5223–5231. [Google Scholar] [CrossRef] [PubMed]
- Cheglakov, Z.; Weizmann, Y.; Braunschweig, A.B.; Wilner, O.I.; Willner, I. Increasing the complexity of periodic protein nanostructures by the rolling-circle-amplified synthesis of aptamers. Angew. Chem. Int. Ed. Engl. 2008, 47, 126–130. [Google Scholar] [CrossRef] [PubMed]
- McCluskey, J.B.; Clark, D.S.; Glover, D.J. Functional Applications of Nucleic Acid-Protein Hybrid Nanostructures. Trends Biotechnol. 2020, 38, 976–989. [Google Scholar] [CrossRef]
- Fu, J.; Wang, Z.; Liang, X.H.; Oh, S.W.; St Iago-McRae, E.; Zhang, T. DNA-Scaffolded Proximity Assembly and Confinement of Multienzyme Reactions. Top. Curr. Chem. 2020, 378, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilner, O.I.; Weizmann, Y.; Gill, R.; Lioubashevski, O.; Freeman, R.; Willner, I. Enzyme cascades activated on topologically programmed DNA scaffolds. Nat. Nanotechnol. 2009, 4, 249–254. [Google Scholar] [CrossRef]
- Timm, C.; Niemeyer, C.M. Assembly and purification of enzyme-functionalized DNA origami structures. Angew. Chem. Int. Ed. Engl. 2015, 54, 6745–6750. [Google Scholar] [CrossRef]
- Fu, J.; Yang, Y.R.; Johnson-Buck, A.; Liu, M.; Liu, Y.; Walter, N.G.; Woodbury, N.W.; Yan, H. Multi-enzyme complexes on DNA scaffolds capable of substrate channelling with an artificial swinging arm. Nat. Nanotechnol. 2014, 9, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Ke, G.; Liu, M.; Jiang, S.; Qi, X.; Yang, Y.R.; Wootten, S.; Zhang, F.; Zhu, Z.; Liu, Y.; Yang, C.J.; et al. Directional Regulation of Enzyme Pathways through the Control of Substrate Channeling on a DNA Origami Scaffold. Angew. Chem. Int. Ed. Engl. 2016, 55, 7483–7486. [Google Scholar] [CrossRef]
- Fu, J.; Liu, M.; Liu, Y.; Woodbury, N.W.; Yan, H. Interenzyme substrate diffusion for an enzyme cascade organized on spatially addressable DNA nanostructures. J. Am. Chem. Soc. 2012, 134, 5516–5519. [Google Scholar] [CrossRef] [Green Version]
- Sweetlove, L.J.; Fernie, A.R. The role of dynamic enzyme assemblies and substrate channelling in metabolic regulation. Nat. Commun. 2018, 9, 2136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Tsitkov, S.; Hess, H. Proximity does not contribute to activity enhancement in the glucose oxidase–horseradish peroxidase cascade. Nat. Commun. 2016, 7, 13982. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.B.; Mo, S.S.; Liu, M.W.; Liu, L.; Yu, L.L.; Wang, C.X. Rationally Designed Protein Building Blocks for Programmable Hierarchical Architectures. Front. Chem. 2020, 8, 23. [Google Scholar] [CrossRef]
- Gerbelli, B.B.; Vassiliades, S.V.; Rojas, J.E.U.; Pelin, J.; Mancini, R.S.N.; Pereira, W.S.G.; Aguilar, A.M.; Venanzi, M.; Cavalieri, F.; Giuntini, F.; et al. Hierarchical Self-Assembly of Peptides and its Applications in Bionanotechnology. Macromol. Chem. Phys. 2019, 220, 22. [Google Scholar] [CrossRef]
- Shen, H.; Fallas, J.A.; Lynch, E.; Sheffler, W.; Parry, B.; Jannetty, N.; Decarreau, J.; Wagenbach, M.; Vicente, J.J.; Chen, J.; et al. De novo design of self-assembling helical protein filaments. Science 2018, 362, 705–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapenta, F.; Aupic, J.; Strmsek, Z.; Jerala, R. Coiled coil protein origami: From modular design principles towards biotechnological applications. Chem. Soc. Rev. 2018, 47, 3530–3542. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, N.; Arai, R. Design and construction of self-assembling supramolecular protein complexes using artificial and fusion proteins as nanoscale building blocks. Curr. Opin. Biotechnol. 2017, 46, 57–65. [Google Scholar] [CrossRef]
- Magnotti, E.; Conticello, V. Two-Dimensional Peptide and Protein Assemblies. Adv. Exp. Med. Biol. 2016, 940, 29–60. [Google Scholar] [CrossRef]
- Drobnak, I.; Ljubetic, A.; Gradisar, H.; Pisanski, T.; Jerala, R. Designed Protein Origami. Adv. Exp. Med. Biol. 2016, 940, 7–27. [Google Scholar] [CrossRef]
- Yeboah, A.; Cohen, R.I.; Rabolli, C.; Yarmush, M.L.; Berthiaume, F. Elastin-like polypeptides: A strategic fusion partner for biologics. Biotechnol. Bioeng. 2016, 113, 1617–1627. [Google Scholar] [CrossRef]
- Miranda-Nieves, D.; Chaikof, E.L. Collagen and Elastin Biomaterials for the Fabrication of Engineered Living Tissues. ACS Biomater. Sci. Eng. 2017, 3, 694–711. [Google Scholar] [CrossRef]
- Wang, Y.; Katyal, P.; Montclare, J.K. Protein-Engineered Functional Materials. Adv. Healthc. Mater. 2019, e1801374. [Google Scholar] [CrossRef]
- Das, S.; Jacob, R.S.; Patel, K.; Singh, N.; Maji, S.K. Amyloid Fibrils: Versatile Biomaterials for Cell Adhesion and Tissue Engineering Applications. Biomacromolecules 2018, 19, 1826–1839. [Google Scholar] [CrossRef] [PubMed]
- Behrendorff, J.B.Y.H.; Borras-Gas, G.; Pribil, M. Synthetic Protein Scaffolding at Biological Membranes. Trends Biotechnol. 2020, 38, 432–446. [Google Scholar] [CrossRef] [PubMed]
- Alves, V.D.; Fontes, C.; Bule, P. Cellulosomes: Highly Efficient Cellulolytic Complexes. Subcell. Biochem. 2021, 96, 323–354. [Google Scholar] [CrossRef] [PubMed]
- Artzi, L.; Bayer, E.A.; Moraïs, S. Cellulosomes: Bacterial nanomachines for dismantling plant polysaccharides. Nat. Rev. Microbiol. 2017, 15, 83–95. [Google Scholar] [CrossRef]
- You, C.; Myung, S.; Zhang, Y.H. Facilitated substrate channeling in a self-assembled trifunctional enzyme complex. Angew. Chem. Int. Ed. Engl. 2012, 51, 8787–8790. [Google Scholar] [CrossRef]
- You, C.; Zhang, Y.H. Self-assembly of synthetic metabolons through synthetic protein scaffolds: One-step purification, co-immobilization, and substrate channeling. ACS Synth. Biol. 2013, 2, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Mitsuzawa, S.; Kagawa, H.; Li, Y.; Chan, S.L.; Paavola, C.D.; Trent, J.D. The rosettazyme: A synthetic cellulosome. J. Biotechnol. 2009, 143, 139–144. [Google Scholar] [CrossRef]
- Krishna, T.S.; Kong, X.P.; Gary, S.; Burgers, P.M.; Kuriyan, J. Crystal structure of the eukaryotic DNA polymerase processivity factor PCNA. Cell 1994, 79, 1233–1243. [Google Scholar] [CrossRef]
- Williams, G.J.; Johnson, K.; Rudolf, J.; McMahon, S.A.; Carter, L.; Oke, M.; Liu, H.; Taylor, G.L.; White, M.F.; Naismith, J.H. Structure of the heterotrimeric PCNA from Sulfolobus solfataricus. Acta. Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2006, 62, 944–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirakawa, H.; Nagamune, T. Molecular assembly of P450 with ferredoxin and ferredoxin reductase by fusion to PCNA. Chembiochem 2010, 11, 1517–1520. [Google Scholar] [CrossRef]
- Tan, C.Y.; Hirakawa, H.; Nagamune, T. Supramolecular protein assembly supports immobilization of a cytochrome P450 monooxygenase system as water-insoluble gel. Sci. Rep. 2015, 5, 8648. [Google Scholar] [CrossRef] [Green Version]
- Reddington, S.C.; Howarth, M. Secrets of a covalent interaction for biomaterials and biotechnology: SpyTag and SpyCatcher. Curr. Opin. Chem. Biol. 2015, 29, 94–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutherland, A.R.; Alam, M.K.; Geyer, C.R. Post-translational Assembly of Protein Parts into Complex Devices by Using SpyTag/SpyCatcher Protein Ligase. Chembiochem 2019, 20, 319–328. [Google Scholar] [CrossRef]
- Hatlem, D.; Trunk, T.; Linke, D.; Leo, J.C. Catching a SPY: Using the SpyCatcher-SpyTag and Related Systems for Labeling and Localizing Bacterial Proteins. Int. J. Mol. Sci. 2019, 20, 2129. [Google Scholar] [CrossRef] [Green Version]
- Keeble, A.H.; Howarth, M. Power to the protein: Enhancing and combining activities using the Spy toolbox. Chem. Sci. 2020, 11, 7281–7291. [Google Scholar] [CrossRef]
- Zakeri, B.; Fierer, J.O.; Celik, E.; Chittock, E.C.; Schwarz-Linek, U.; Moy, V.T.; Howarth, M. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc. Natl. Acad. Sci. USA 2012, 109, E690–E697. [Google Scholar] [CrossRef] [Green Version]
- Keeble, A.H.; Banerjee, A.; Ferla, M.P.; Reddington, S.C.; Anuar, I.; Howarth, M. Evolving Accelerated Amidation by SpyTag/SpyCatcher to Analyze Membrane Dynamics. Angew. Chem. Int. Ed. Engl. 2017, 56, 16521–16525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keeble, A.H.; Howarth, M. Insider information on successful covalent protein coupling with help from SpyBank. Methods Enzymol. 2019, 617, 443–461. [Google Scholar] [CrossRef] [PubMed]
- Veggiani, G.; Nakamura, T.; Brenner, M.D.; Gayet, R.V.; Yan, J.; Robinson, C.V.; Howarth, M. Programmable polyproteams built using twin peptide superglues. Proc. Natl. Acad. Sci. USA 2016, 113, 1202–1207. [Google Scholar] [CrossRef] [Green Version]
- Khairil Anuar, I.N.A.; Banerjee, A.; Keeble, A.H.; Carella, A.; Nikov, G.I.; Howarth, M. Spy&Go purification of SpyTag-proteins using pseudo-SpyCatcher to access an oligomerization toolbox. Nat. Commun. 2019, 10, 1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beesley, J.L.; Woolfson, D.N. The de novo design of α-helical peptides for supramolecular self-assembly. Curr. Opin. Biotechnol. 2019, 58, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zaro, J.L.; Shen, W.C. Fusion protein linkers: Property, design and functionality. Adv. Drug Deliv. Rev. 2013, 65, 1357–1369. [Google Scholar] [CrossRef] [Green Version]
- Steen Redeker, E.; Ta, D.T.; Cortens, D.; Billen, B.; Guedens, W.; Adriaensens, P. Protein engineering for directed immobilization. Bioconjug. Chem. 2013, 24, 1761–1777. [Google Scholar] [CrossRef]
- Meldal, M.; Schoffelen, S. Recent advances in covalent, site-specific protein immobilization. F1000Research 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Dorr, B.M.; Ham, H.O.; An, C.; Chaikof, E.L.; Liu, D.R. Reprogramming the specificity of sortase enzymes. Proc. Natl. Acad. Sci. USA 2014, 111, 13343–13348. [Google Scholar] [CrossRef] [Green Version]
- Schmohl, L.; Schwarzer, D. Sortase-mediated ligations for the site-specific modification of proteins. Curr. Opin. Chem. Biol. 2014, 22, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Ritzefeld, M. Sortagging: A Robust and Efficient Chemoenzymatic Ligation Strategy. Chem. Eur. J. 2014, 20, 8516–8529. [Google Scholar] [CrossRef]
- Broguiere, N.; Formica, F.; Barreto, G.; Zenobi-Wong, M. Sortase A as a cross-linking enzyme in tissue engineering. Acta Biomater. 2018, 77, 182–190. [Google Scholar] [CrossRef]
- Singh, S.; Gupta, K.; Shukla, S.; Sampathkumar, S.G.; Roy, R.P. Sortase-click strategy for defined protein conjugation on a heptavalent cyclodextrin scaffold. PLoS ONE 2019, 14, e0217369. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Tanaka, T.; Kondo, A. Sortase A-Catalyzed Site-Specific Coimmobilization on Microparticles via Streptavidin. Langmuir 2012, 28, 3553–3557. [Google Scholar] [CrossRef]
- McConnell, S.A.; Cannon, K.A.; Morgan, C.; McAllister, R.; Amer, B.R.; Clubb, R.T.; Yeates, T.O. Designed Protein Cages as Scaffolds for Building Multienzyme Materials. ACS Synth. Biol. 2020, 9, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Mateu, M.G. Assembly, Engineering and Applications of Virus-Based Protein Nanoparticles. Adv. Exp. Med. Biol. 2016, 940, 83–120. [Google Scholar] [CrossRef] [PubMed]
- Perlmutter, J.D.; Hagan, M.F. Mechanisms of virus assembly. Annu. Rev. Phys. Chem. 2015, 66, 217–239. [Google Scholar] [CrossRef] [Green Version]
- Raff, J.; Matys, S.; Suhr, M.; Vogel, M.; Gunther, T.; Pollmann, K. S-Layer-Based Nanocomposites for Industrial Applications. Adv. Exp. Med. Biol. 2016, 940, 245–279. [Google Scholar] [CrossRef]
- Sleytr, U.B.; Schuster, B.; Egelseer, E.M.; Pum, D. S-layers: Principles and applications. Fems Microbiol. Rev. 2014, 38, 823–864. [Google Scholar] [CrossRef] [PubMed]
- Demchuk, A.M.; Patel, T.R. The biomedical and bioengineering potential of protein nanocompartments. Biotechnol. Adv. 2020, 41, 107547. [Google Scholar] [CrossRef]
- Glasgow, J.E.; Capehart, S.L.; Francis, M.B.; Tullman-Ercek, D. Osmolyte-Mediated Encapsulation of Proteins inside MS2 Viral Capsids. ACS Nano 2012, 6, 8658–8664. [Google Scholar] [CrossRef]
- Jones, J.A.; Giessen, T.W. Advances in encapsulin nanocompartment biology and engineering. Biotechnol. Bioeng. 2021, 118, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Kerfeld, C.A.; Sutter, M. Engineered bacterial microcompartments: Apps for programming metabolism. Curr. Opin. Biotechnol. 2020, 65, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Palmer, D.J.; Warren, M.J. Biotechnological Advances in Bacterial Microcompartment Technology. Trends Biotechnol. 2019, 37, 325–336. [Google Scholar] [CrossRef]
- Planamente, S.; Frank, S. Bio-engineering of bacterial microcompartments: A mini review. Biochem. Soc. Trans. 2019, 47, 765–777. [Google Scholar] [CrossRef]
- Yeates, T.O. Geometric Principles for Designing Highly Symmetric Self-Assembling Protein Nanomaterials. Annu. Rev. Biophys. 2017, 46, 23–42. [Google Scholar] [CrossRef]
- Okesola, B.O.; Mata, A. Multicomponent self-assembly as a tool to harness new properties from peptides and proteins in material design. Chem. Soc. Rev. 2018, 47, 3721–3736. [Google Scholar] [CrossRef]
- Kuan, S.L.; Bergamini, F.R.G.; Weil, T. Functional protein nanostructures: A chemical toolbox. Chem. Soc. Rev. 2018, 47, 9069–9105. [Google Scholar] [CrossRef] [Green Version]
- Matsuura, K. Synthetic approaches to construct viral capsid-like spherical nanomaterials. Chem. Commun. 2018, 54, 8944–8959. [Google Scholar] [CrossRef]
- Majerle, A.; Schmieden, D.T.; Jerala, R.; Meyer, A.S. Synthetic Biology for Multiscale Designed Biomimetic Assemblies: From Designed Self-Assembling Biopolymers to Bacterial Bioprinting. Biochemistry 2019, 58, 2095–2104. [Google Scholar] [CrossRef] [Green Version]
- Hamley, I.W. Protein Assemblies: Nature-Inspired and Designed Nanostructures. Biomacromolecules 2019, 20, 1829–1848. [Google Scholar] [CrossRef] [Green Version]
- Cannon, K.A.; Ochoa, J.M.; Yeates, T.O. High-symmetry protein assemblies: Patterns and emerging applications. Curr. Opin. Struct. Biol. 2019, 55, 77–84. [Google Scholar] [CrossRef] [Green Version]
- Bale, J.B.; Gonen, S.; Liu, Y.; Sheffler, W.; Ellis, D.; Thomas, C.; Cascio, D.; Yeates, T.O.; Gonen, T.; King, N.P.; et al. Accurate design of megadalton-scale two-component icosahedral protein complexes. Science 2016, 353, 389–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, N.P.; Sheffler, W.; Sawaya, M.R.; Vollmar, B.S.; Sumida, J.P.; Andre, I.; Gonen, T.; Yeates, T.O.; Baker, D. Computational design of self-assembling protein nanomaterials with atomic level accuracy. Science 2012, 336, 1171–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Y.T.; Cascio, D.; Yeates, T.O. Structure of a 16-nm cage designed by using protein oligomers. Science 2012, 336, 1129. [Google Scholar] [CrossRef] [PubMed]
- Padilla, J.E.; Colovos, C.; Yeates, T.O. Nanohedra: Using symmetry to design self assembling protein cages, layers, crystals, and filaments. Proc. Natl. Acad. Sci. USA 2001, 98, 2217–2221. [Google Scholar] [CrossRef] [Green Version]
- Ben-Sasson, A.J.; Watson, J.L.; Sheffler, W.; Johnson, M.C.; Bittleston, A.; Somasundaram, L.; Decarreau, J.; Jiao, F.; Chen, J.; Mela, I.; et al. Design of biologically active binary protein 2D materials. Nature 2021, 589, 468–473. [Google Scholar] [CrossRef]
- Worsdorfer, B.; Woycechowsky, K.J.; Hilvert, D. Directed evolution of a protein container. Science 2011, 331, 589–592. [Google Scholar] [CrossRef]
- Frey, R.; Mantri, S.; Rocca, M.; Hilvert, D. Bottom-up Construction of a Primordial Carboxysome Mimic. J. Am. Chem. Soc. 2016, 138, 10072–10075. [Google Scholar] [CrossRef]
- Sasaki, E.; Bohringer, D.; van de Waterbeemd, M.; Leibundgut, M.; Zschoche, R.; Heck, A.J.; Ban, N.; Hilvert, D. Structure and assembly of scalable porous protein cages. Nat. Commun. 2017, 8, 14663. [Google Scholar] [CrossRef] [Green Version]
- Terasaka, N.; Azuma, Y.; Hilvert, D. Laboratory evolution of virus-like nucleocapsids from nonviral protein cages. Proc. Natl. Acad. Sci. USA 2018, 115, 5432–5437. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, Y.; Cardone, G.; Restrepo, D.; Zavattieri, P.D.; Baker, T.S.; Tezcan, F.A. Self-assembly of coherently dynamic, auxetic, two-dimensional protein crystals. Nature 2016, 533, 369–373. [Google Scholar] [CrossRef]
- Chen, Z.; Johnson, M.C.; Chen, J.; Bick, M.J.; Boyken, S.E.; Lin, B.; De Yoreo, J.J.; Kollman, J.M.; Baker, D.; DiMaio, F. Self-Assembling 2D Arrays with de Novo Protein Building Blocks. J. Am. Chem. Soc. 2019, 141, 8891–8895. [Google Scholar] [CrossRef] [PubMed]
- Brodin, J.D.; Carr, J.R.; Sontz, P.A.; Tezcan, F.A. Exceptionally stable, redox-active supramolecular protein assemblies with emergent properties. Proc. Natl. Acad. Sci. USA 2014, 111, 2897–2902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberstein, R.; Suzuki, Y.; Paesani, F.; Tezcan, F.A. Engineering the entropy-driven free-energy landscape of a dynamic nanoporous protein assembly. Nat. Chem. 2018, 10, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Golub, E.; Subramanian, R.H.; Esselborn, J.; Alberstein, R.G.; Bailey, J.B.; Chiong, J.A.; Yan, X.D.; Booth, T.; Baker, T.S.; Tezcan, F.A. Constructing protein polyhedra via orthogonal chemical interactions. Nature 2020, 578, 172–176. [Google Scholar] [CrossRef]
- Kakkis, A.; Gagnon, D.; Esselborn, J.; Britt, R.D.; Tezcan, F.A. Metal-Templated Design of Chemically Switchable Protein Assemblies with High-Affinity Coordination Sites. Angew. Chem. Int. Ed. Engl. 2020, 59, 21940–21944. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Alberstein, R.G.; De Yoreo, J.J.; Tezcan, F.A. Assembly of a patchy protein into variable 2D lattices via tunable multiscale interactions. Nat. Commun. 2020, 11, 3770. [Google Scholar] [CrossRef]
- Manea, F.; Garda, V.G.; Rad, B.; Ajo-Franklin, C.M. Programmable assembly of 2D crystalline protein arrays into covalently stacked 3D bionanomaterials. Biotechnol. Bioeng. 2020, 117, 912–923. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.; Quin, M.B.; Sanders, M.A.; Johnson, E.T.; Schmidt-Dannert, C. Engineered protein nano-compartments for targeted enzyme localization. PLoS ONE 2012, 7, e33342. [Google Scholar] [CrossRef] [Green Version]
- Held, M.; Quin, M.B.; Schmidt-Dannert, C. Eut bacterial microcompartments: Insights into their function, structure, and bioengineering applications. J. Mol. Microbiol. Biotechnol. 2013, 23, 308–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quin, M.B.; Perdue, S.A.; Hsu, S.Y.; Schmidt-Dannert, C. Encapsulation of multiple cargo proteins within recombinant Eut nanocompartments. Appl. Microbiol. Biotechnol. 2016, 100, 9187–9200. [Google Scholar] [CrossRef]
- Schmidt-Dannert, S.; Zhang, G.; Johnston, T.; Quin, M.B.; Schmidt-Dannert, C. Building a toolbox of protein scaffolds for future immobilization of biocatalysts. Appl. Microbiol. Biotechnol. 2018, 102, 8373–8388. [Google Scholar] [CrossRef]
- Zhang, G.; Schmidt-Dannert, S.; Quin, M.B.; Schmidt-Dannert, C. Protein-based scaffolds for enzyme immobilization. Methods Enzymol. 2019, 617, 323–362. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Johnston, T.; Quin, M.B.; Schmidt-Dannert, C. Developing a Protein Scaffolding System for Rapid Enzyme Immobilization and Optimization of Enzyme Functions for Biocatalysis. ACS Synth. Biol. 2019, 8, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Schoonen, L.; van Hest, J.C. Compartmentalization Approaches in Soft Matter Science: From Nanoreactor Development to Organelle Mimics. Adv. Mater. 2016, 28, 1109–1128. [Google Scholar] [CrossRef]
- Quin, M.B.; Wallin, K.K.; Zhang, G.; Schmidt-Dannert, C. Spatial organization of multi-enzyme biocatalytic cascades. Org. Biomol. Chem. 2017, 15, 4260–4271. [Google Scholar] [CrossRef]
- Schwarz, B.; Uchida, M.; Douglas, T. Biomedical and Catalytic Opportunities of Virus-Like Particles in Nanotechnology. Adv. Virus Res. 2017, 97, 1–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupka, I.; Heddle, J.G. Artificial protein cages—inspiration, construction, and observation. Curr. Opin. Struct. Biol. 2020, 64, 66–73. [Google Scholar] [CrossRef]
- Lv, C.; Zhang, X.; Liu, Y.; Zhang, T.; Chen, H.; Zang, J.; Zheng, B.; Zhao, G. Redesign of protein nanocages: The way from 0D, 1D, 2D to 3D assembly. Chem. Soc. Rev. 2021. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.M.; Stewart, K.L.; Yeates, T.O.; Bobik, T.A. Advances in the World of Bacterial Microcompartments. Trends Biochem. Sci. 2021. [Google Scholar] [CrossRef]
- Sutter, M.; McGuire, S.; Ferlez, B.; Kerfeld, C.A. Structural Characterization of a Synthetic Tandem-Domain Bacterial Microcompartment Shell Protein Capable of Forming Icosahedral Shell Assemblies. ACS Synth. Biol. 2019, 8, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Hagen, A.R.; Plegaria, J.S.; Sloan, N.; Ferlez, B.; Aussignargues, C.; Burton, R.; Kerfeld, C.A. In Vitro Assembly of Diverse Bacterial Microcompartment Shell Architectures. Nano Lett. 2018, 18, 7030–7037. [Google Scholar] [CrossRef]
- Lončar, N.; Rozeboom, H.J.; Franken, L.E.; Stuart, M.C.A.; Fraaije, M.W. Structure of a robust bacterial protein cage and its application as a versatile biocatalytic platform through enzyme encapsulation. Biochem. Biophys. Res. Commun. 2020, 529, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.M.; Jung, S.M.; Coffman, J.L.; Lutz, S. Pore Engineering for Enhanced Mass Transport in Encapsulin Nanocompartments. ACS Synth. Biol. 2018, 7, 2514–2517. [Google Scholar] [CrossRef] [PubMed]
- Azuma, Y.; Edwardson, T.G.W.; Hilvert, D. Tailoring lumazine synthase assemblies for bionanotechnology. Chem. Soc. Rev. 2018, 47, 3543–3557. [Google Scholar] [CrossRef]
- Uchida, M.; McCoy, K.; Fukuto, M.; Yang, L.; Yoshimura, H.; Miettinen, H.M.; LaFrance, B.; Patterson, D.P.; Schwarz, B.; Karty, J.A.; et al. Modular Self-Assembly of Protein Cage Lattices for Multistep Catalysis. ACS Nano 2018, 12, 942–953. [Google Scholar] [CrossRef]
- Giessen, T.W.; Silver, P.A. A Catalytic Nanoreactor Based on in Vivo Encapsulation of Multiple Enzymes in an Engineered Protein Nanocompartment. Chembiochem 2016, 17, 1931–1935. [Google Scholar] [CrossRef]
- Lizatovic, R.; Assent, M.; Barendregt, A.; Dahlin, J.; Bille, A.; Satzinger, K.; Tupina, D.; Heck, A.J.R.; Wennmalm, S.; Andre, I. A Protein-Based Encapsulation System with Calcium-Controlled Cargo Loading and Detachment. Angew. Chem. Int. Ed. Engl. 2018, 57, 11334–11338. [Google Scholar] [CrossRef]
- Fernandez-Trillo, F.; Grover, L.M.; Stephenson-Brown, A.; Harrison, P.; Mendes, P.M. Vesicles in Nature and the Laboratory: Elucidation of Their Biological Properties and Synthesis of Increasingly Complex Synthetic Vesicles. Angew. Chem. Int. Ed. Engl. 2017, 56, 3142–3160. [Google Scholar] [CrossRef] [Green Version]
- Rideau, E.; Dimova, R.; Schwille, P.; Wurm, F.R.; Landfester, K. Liposomes and polymersomes: A comparative review towards cell mimicking. Chem. Soc. Rev. 2018, 47, 8572–8610. [Google Scholar] [CrossRef] [Green Version]
- Blackman, L.D.; Varlas, S.; Arno, M.C.; Fayter, A.; Gibson, M.I.; O’Reilly, R.K. Permeable Protein-Loaded Polymersome Cascade Nanoreactors by Polymerization-Induced Self-Assembly. ACS Macro Lett. 2017, 6, 1263–1267. [Google Scholar] [CrossRef] [PubMed]
- Gaitzsch, J.; Appelhans, D.; Wang, L.; Battaglia, G.; Voit, B. Synthetic Bio-nanoreactor: Mechanical and Chemical Control of Polymersome Membrane Permeability. Angew. Chem. Int. Ed. Engl. 2012, 51, 4448–4451. [Google Scholar] [CrossRef]
- De Hoog, H.-P.M.; Arends, I.W.C.E.; Rowan, A.E.; Cornelissen, J.J.L.M.; Nolte, R.J.M. A hydrogel-based enzyme-loaded polymersome reactor. Nanoscale 2010, 2, 709–716. [Google Scholar] [CrossRef] [Green Version]
- Alves, N.J.; Moore, M.; Johnson, B.J.; Dean, S.N.; Turner, K.B.; Medintz, I.L.; Walper, S.A. Environmental Decontamination of a Chemical Warfare Simulant Utilizing a Membrane Vesicle-Encapsulated Phosphotriesterase. ACS Appl. Mater. Interfaces 2018, 10, 15712–15719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Gallego, F.; Schmidt-Dannert, C. Multi-enzymatic synthesis. Curr. Opin. Chem. Biol. 2010, 14, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Wegst, U.G.K.; Bai, H.; Saiz, E.; Tomsia, A.P.; Ritchie, R.O. Bioinspired structural materials. Nat. Mater. 2015, 14, 23–36. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biocatalysts | Methods | Substrate | Product | Co-Factor | Ref. |
|---|---|---|---|---|---|
| XR, XDH | DNA origami | Xylose | Xylulose | NAD+, NADH | [37] |
| MDH, OAD, LDH | DNA origami | Malic acid | Lactate | NAD+, NADH | [38] |
| MenF, D, H | RIAD, RIDD | Chorismate | MKH2 | — | [39] |
| ACO, CAD | PDZ, SH3 | Glucose | Itaconic acid | — | [40,41] |
| LeuDH, FDH | PDZ | Trimethlypyruvate (TMP) | l-tert-Leucine | NAD+, NADH | [42] |
| ACAT, HMGS, HMGR | PDZ, SH3, GBD | Acetyl-CoA | Mevalonate | — | [43] |
| Xyn, Agu, β-Xyl, XylB | Cohesin–dockerin | Glucuromoxylan | d-Xylonic acid | — | [44] |
| MTS, MTH | Spy toolbox | Trehalose | Latopentaose | — | [45] |
| ADH, AmDH | Spy toolbox, EutM | (S)-2-Hexanol | (R)-2-Aminohexane | NAD+, NADH | [46] |
| ADH, Gre2P, GDH | SBP, Spy, Halo-tags | 5-Nitrononane-2,8-dione (NDK) | Meso anti-NDK | NADPH, NADP | [47] |
| LeuDH, FDH | Spy toolbox | TMP | l-tert-Leucine | NAD+, NADH | [48] |
| ACAT, HMGS, HMGR | Spy, Snoop toolbox | Acetyl-CoA | Mevalonate | — | [49] |
| VioA, B, C, D, E | Spy toolbox, DNA origami | l-Tryptophan | Violacein | — | [50] |
| CelB, GALK, GLUK | P22 VLPs | Lactose | G-1-P, G-6-P | ATP, ADP, AMP | [51] |
| TnaA, FMO | Liposome | l-Tryptophan | Indigo | — | [52] |
| CalB, GOx, HRP | Polymersome | Glucose acetate | Gluconolactone | — | [53] |
| PAMO, CalB, ADH | Polymersome | 7-((4-Oxopentyl)oxy)-3H-phenoxazin-3-one | Resorufin | NAD+, NADPH | [54] |
| AGE, NAL, CSS | Polymersome | GlgNAc | CMP-Neu5Ac | — | [55] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seo, M.-J.; Schmidt-Dannert, C. Organizing Multi-Enzyme Systems into Programmable Materials for Biocatalysis. Catalysts 2021, 11, 409. https://doi.org/10.3390/catal11040409
Seo M-J, Schmidt-Dannert C. Organizing Multi-Enzyme Systems into Programmable Materials for Biocatalysis. Catalysts. 2021; 11(4):409. https://doi.org/10.3390/catal11040409
Chicago/Turabian StyleSeo, Min-Ju, and Claudia Schmidt-Dannert. 2021. "Organizing Multi-Enzyme Systems into Programmable Materials for Biocatalysis" Catalysts 11, no. 4: 409. https://doi.org/10.3390/catal11040409
APA StyleSeo, M. -J., & Schmidt-Dannert, C. (2021). Organizing Multi-Enzyme Systems into Programmable Materials for Biocatalysis. Catalysts, 11(4), 409. https://doi.org/10.3390/catal11040409