
Stable Performance of Supported PdOx Catalyst on Mesoporous Silica-Alumina of Water Tolerance for Methane Combustion under Wet Conditions

 , , , and
, , , and 
Abstract
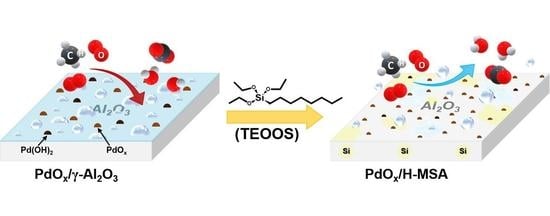
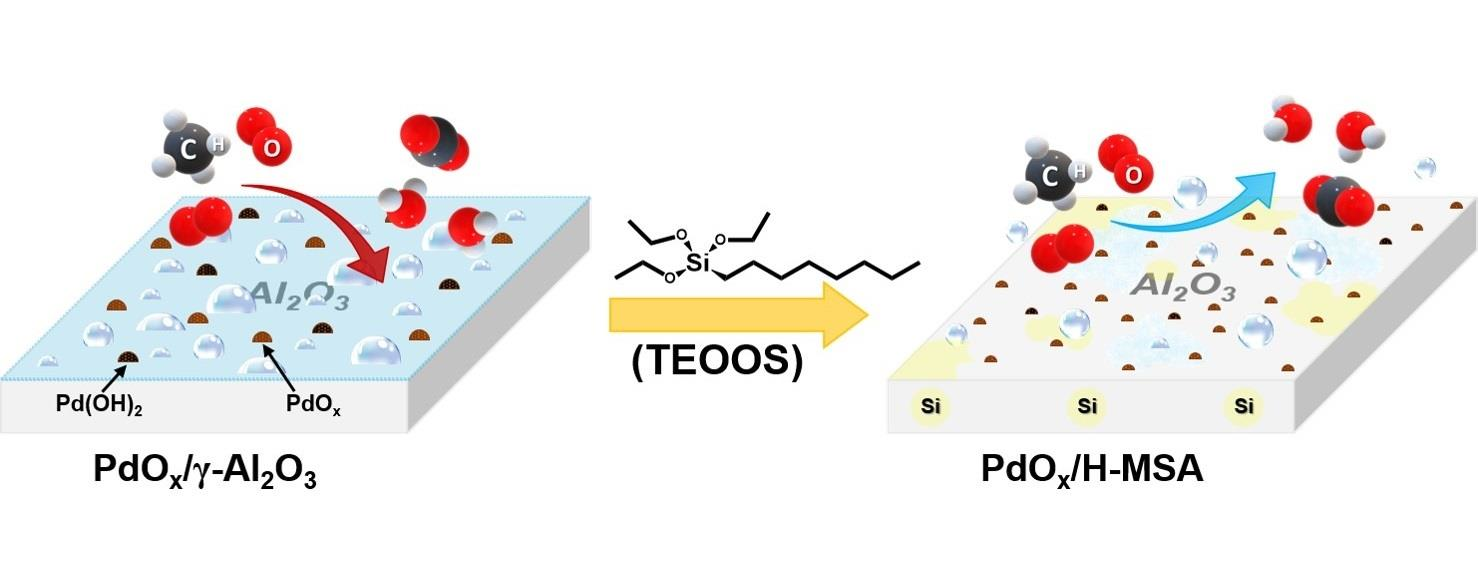
:
1. Introduction
2. Results
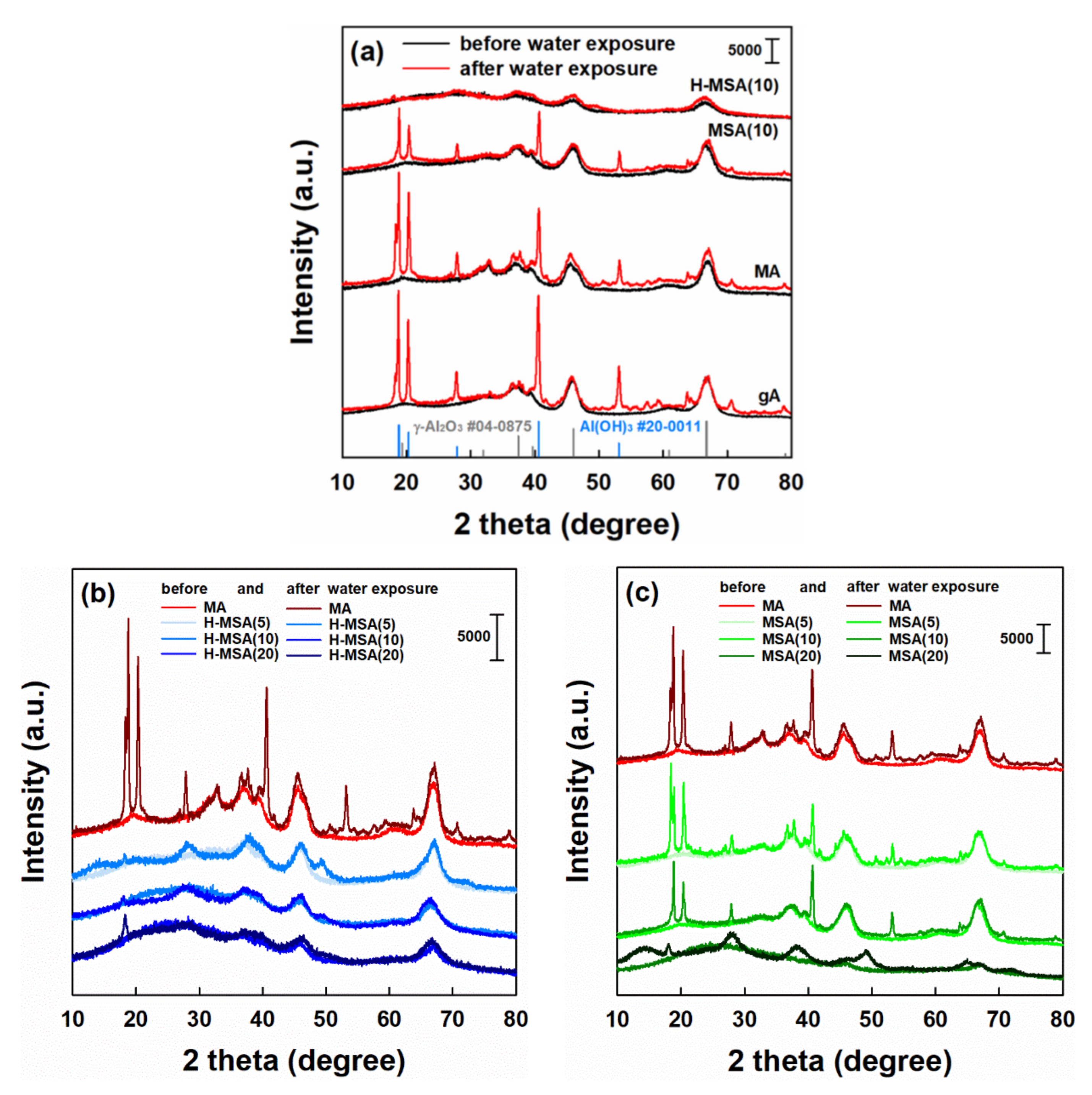
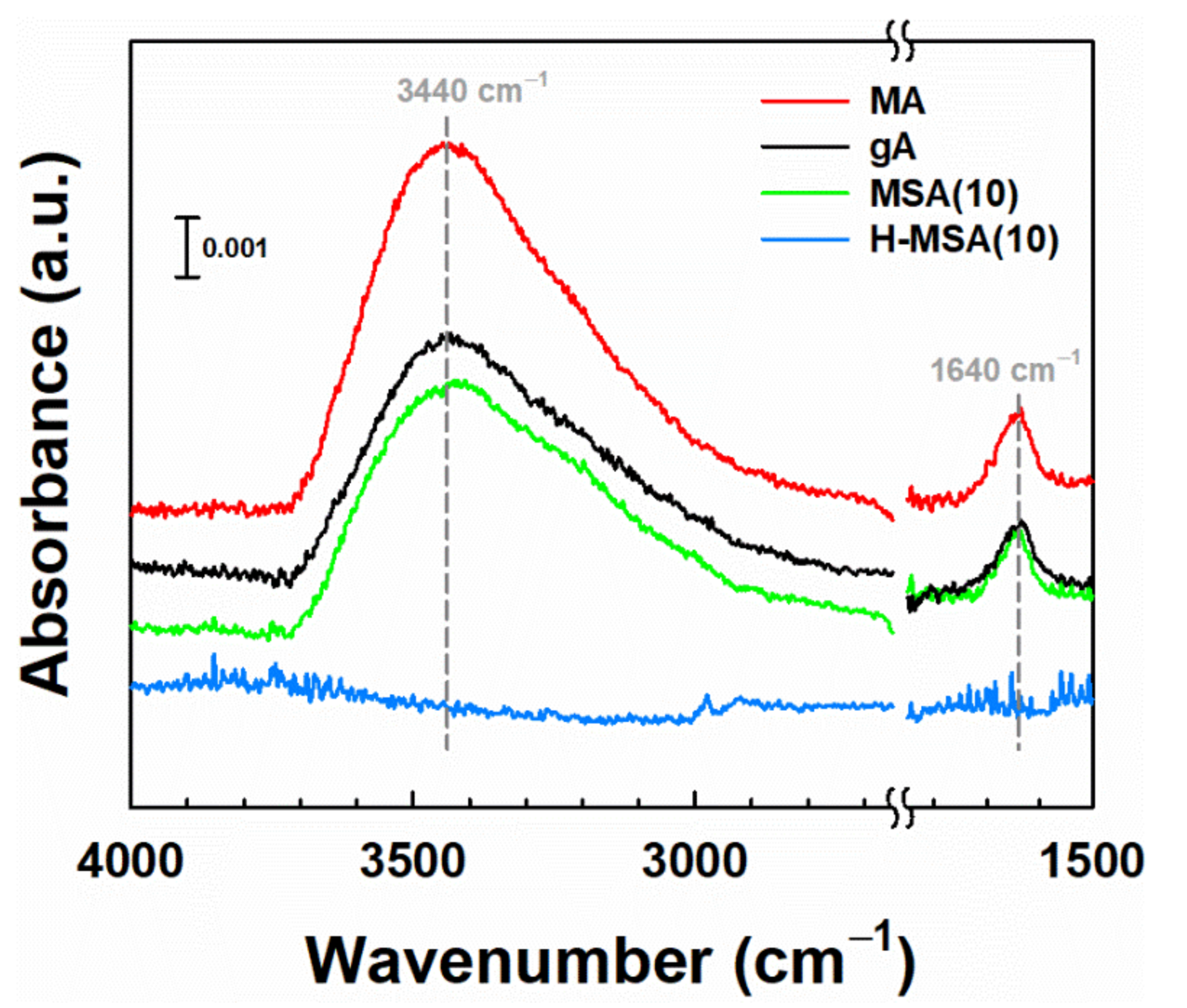
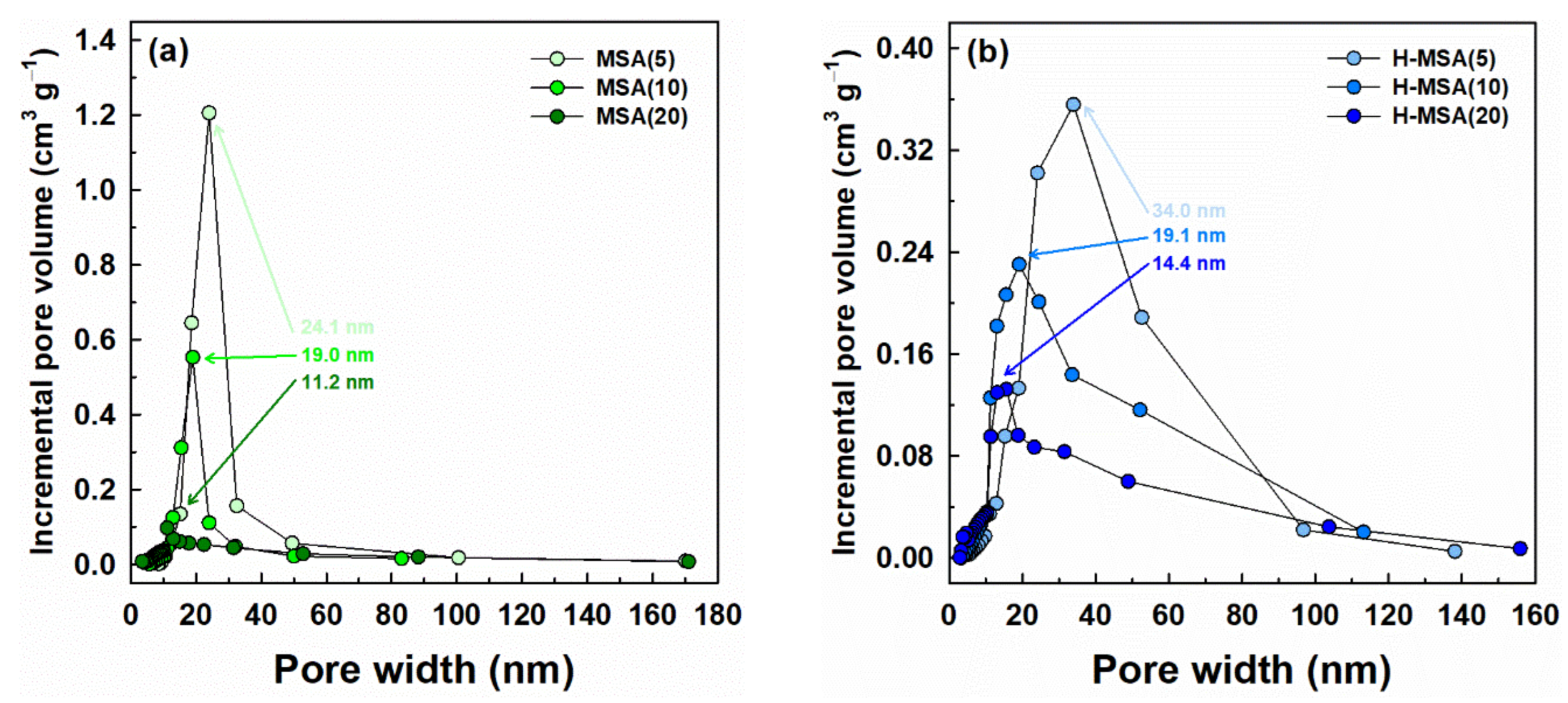
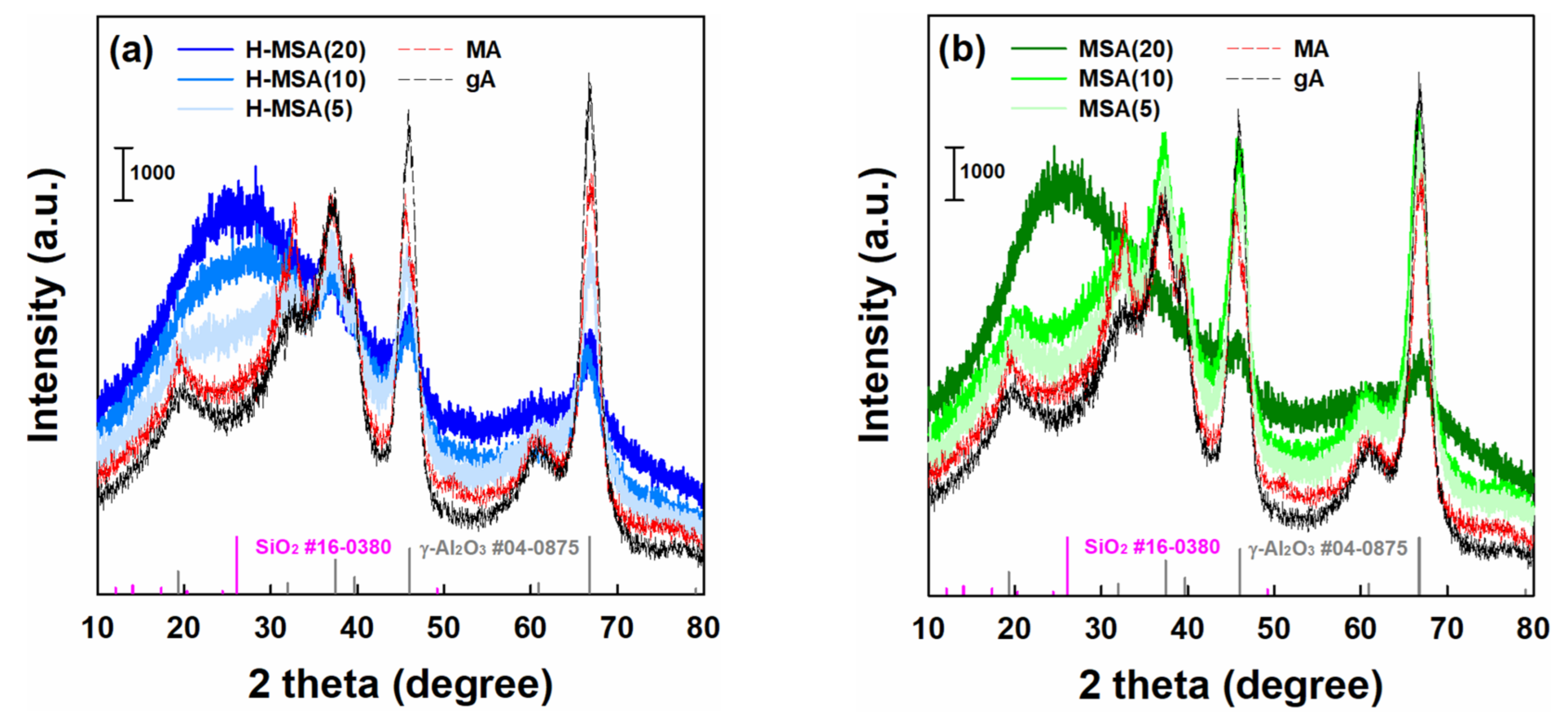
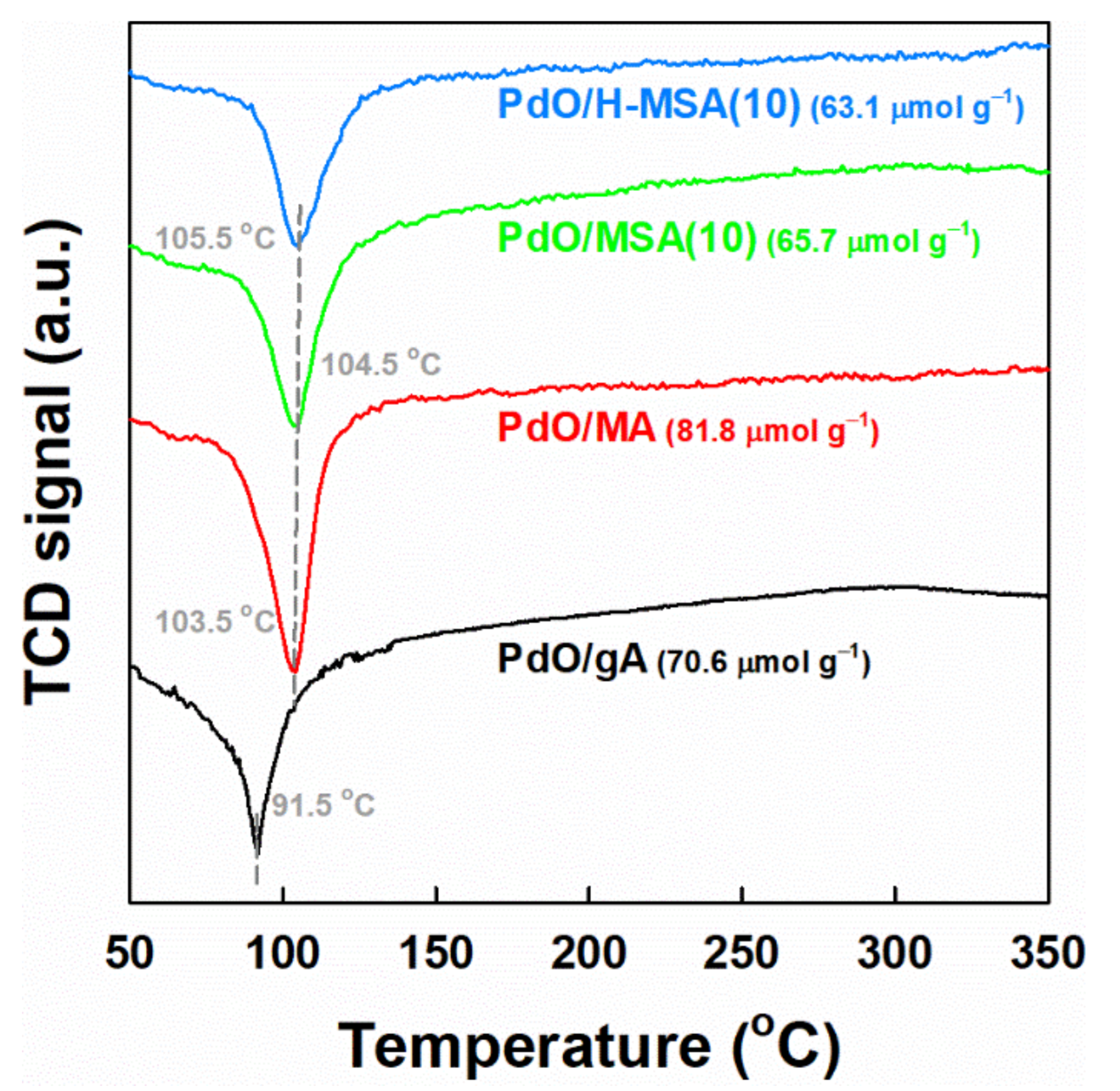
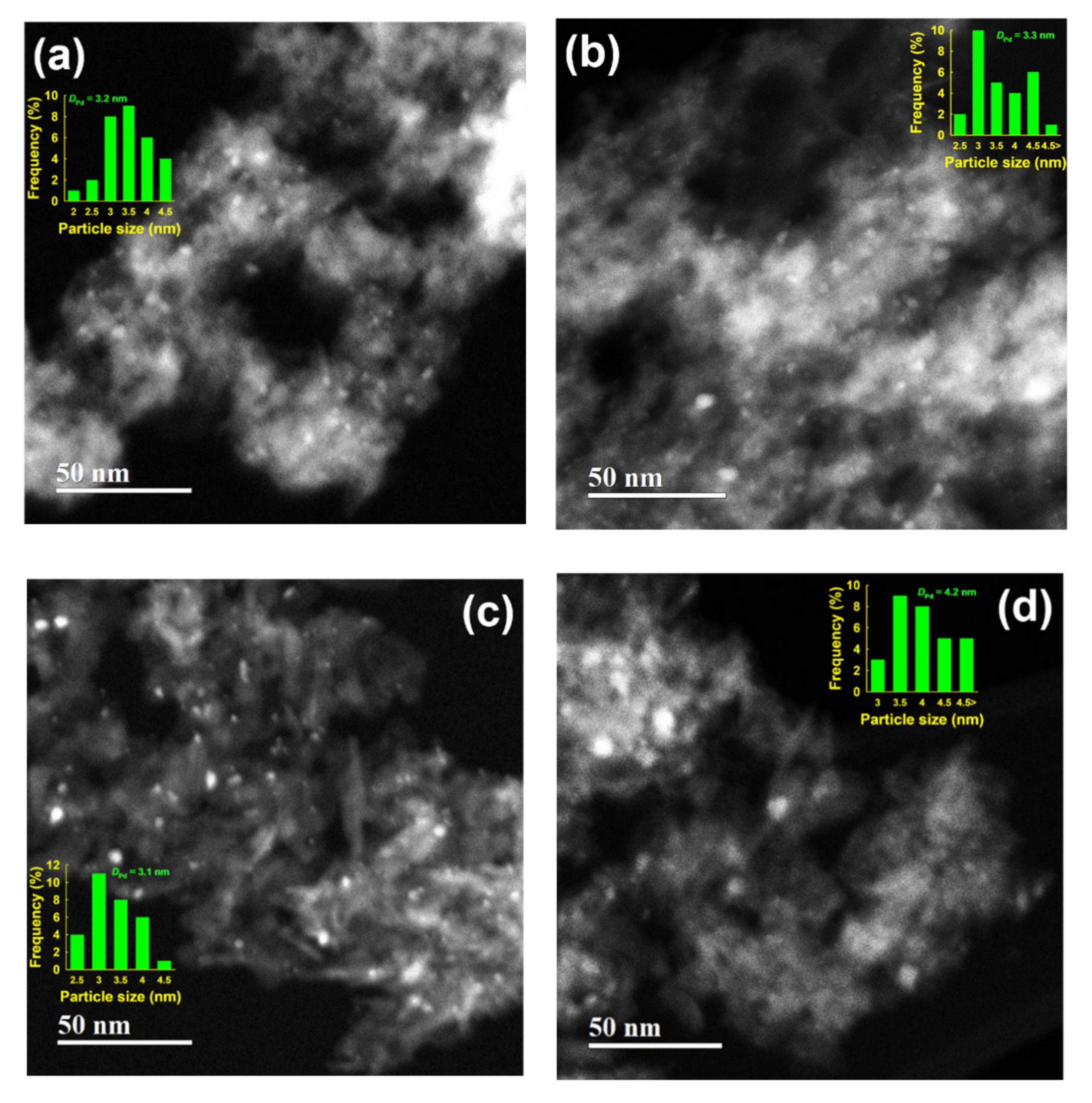
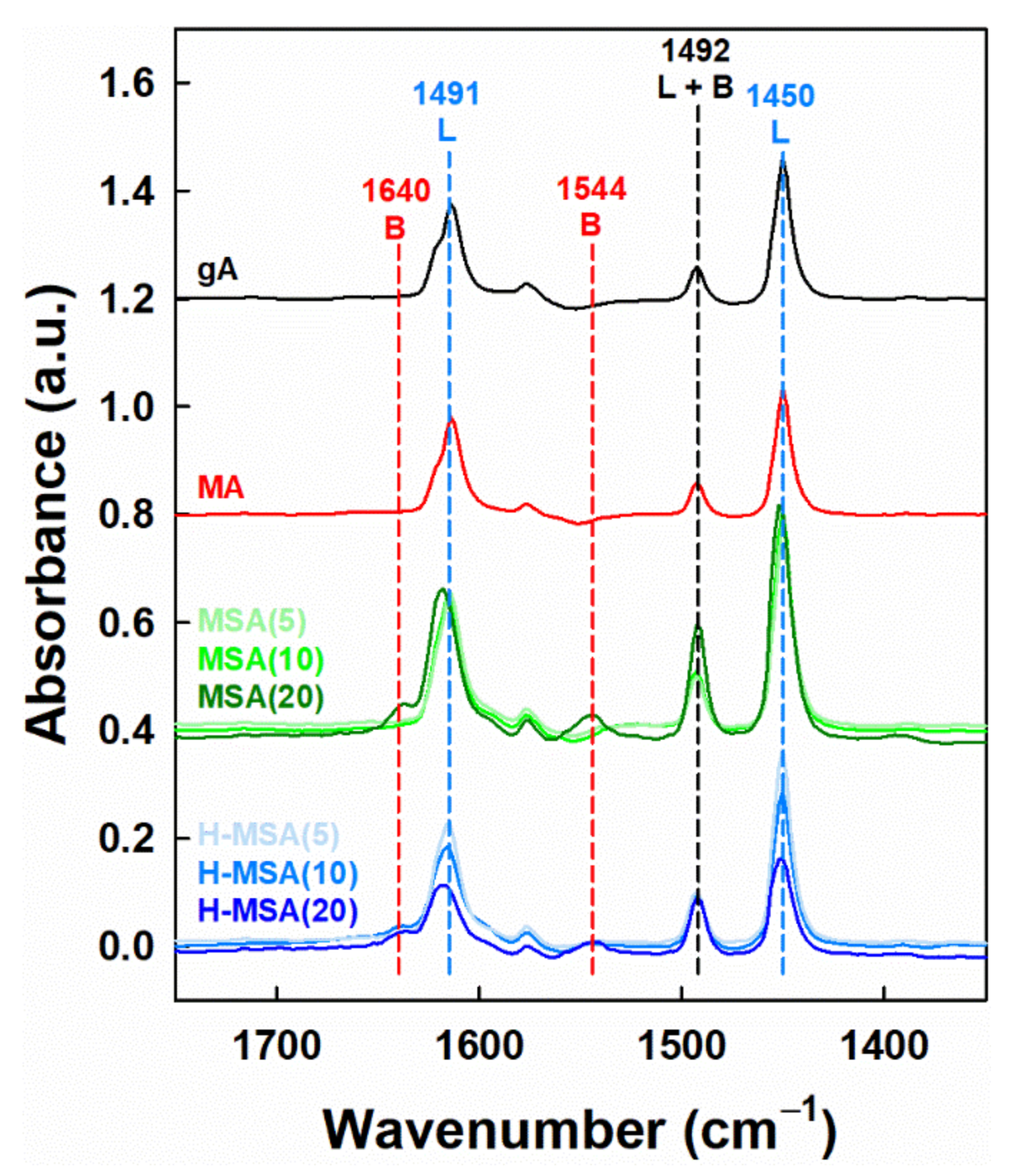
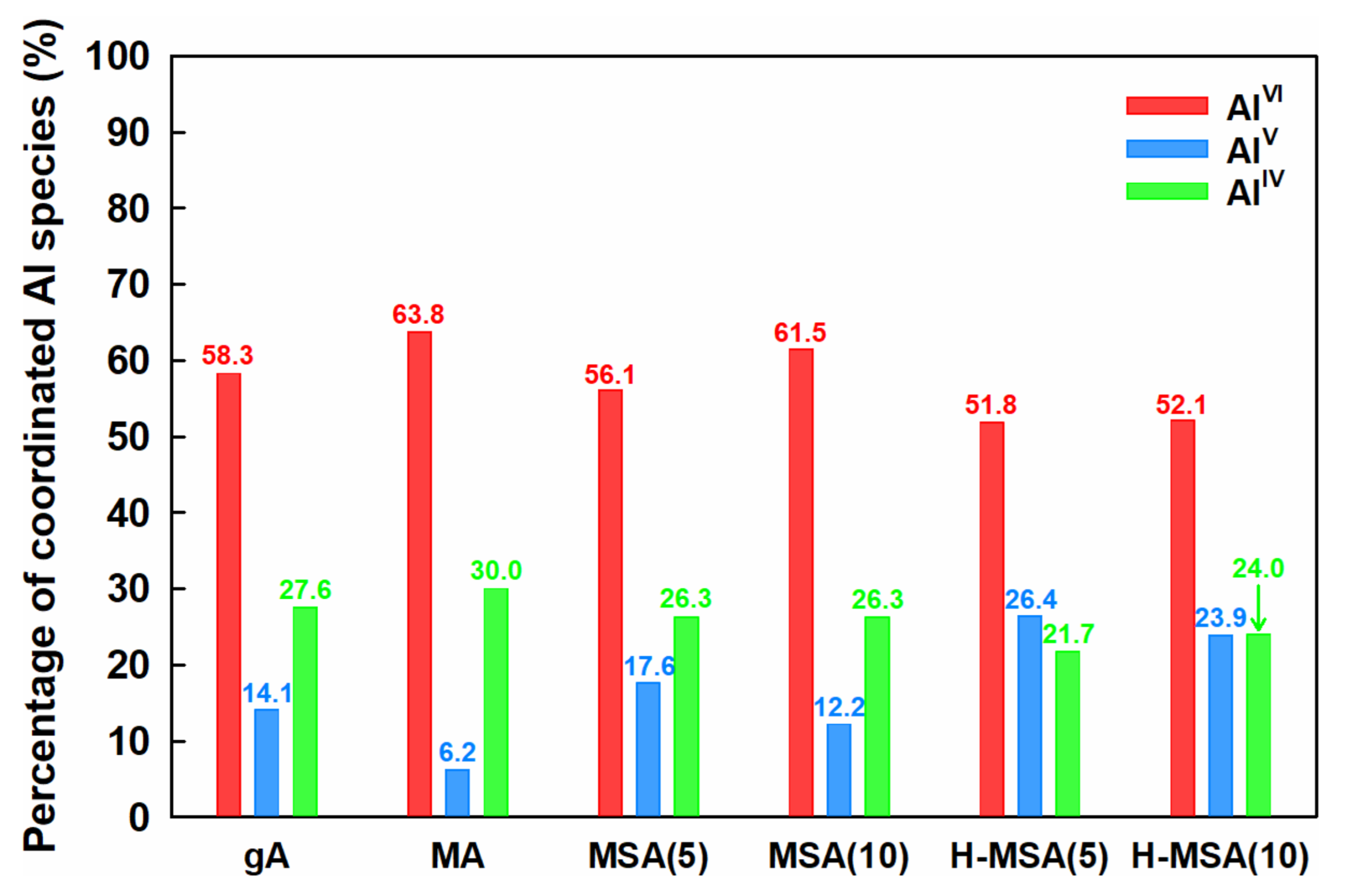
2.1. Characteristics of the Prepared Supports and Supported PdOx Catalysts
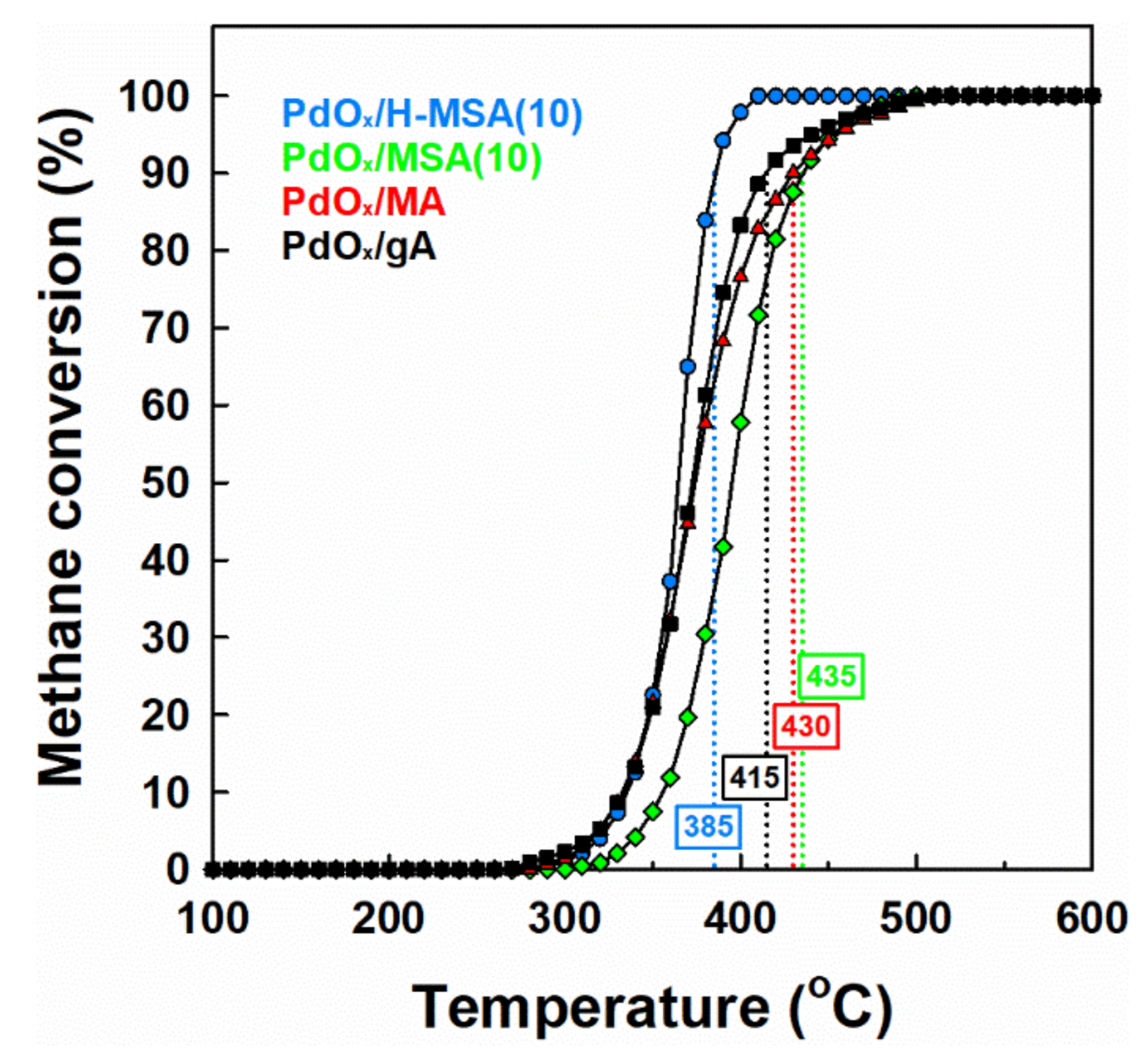
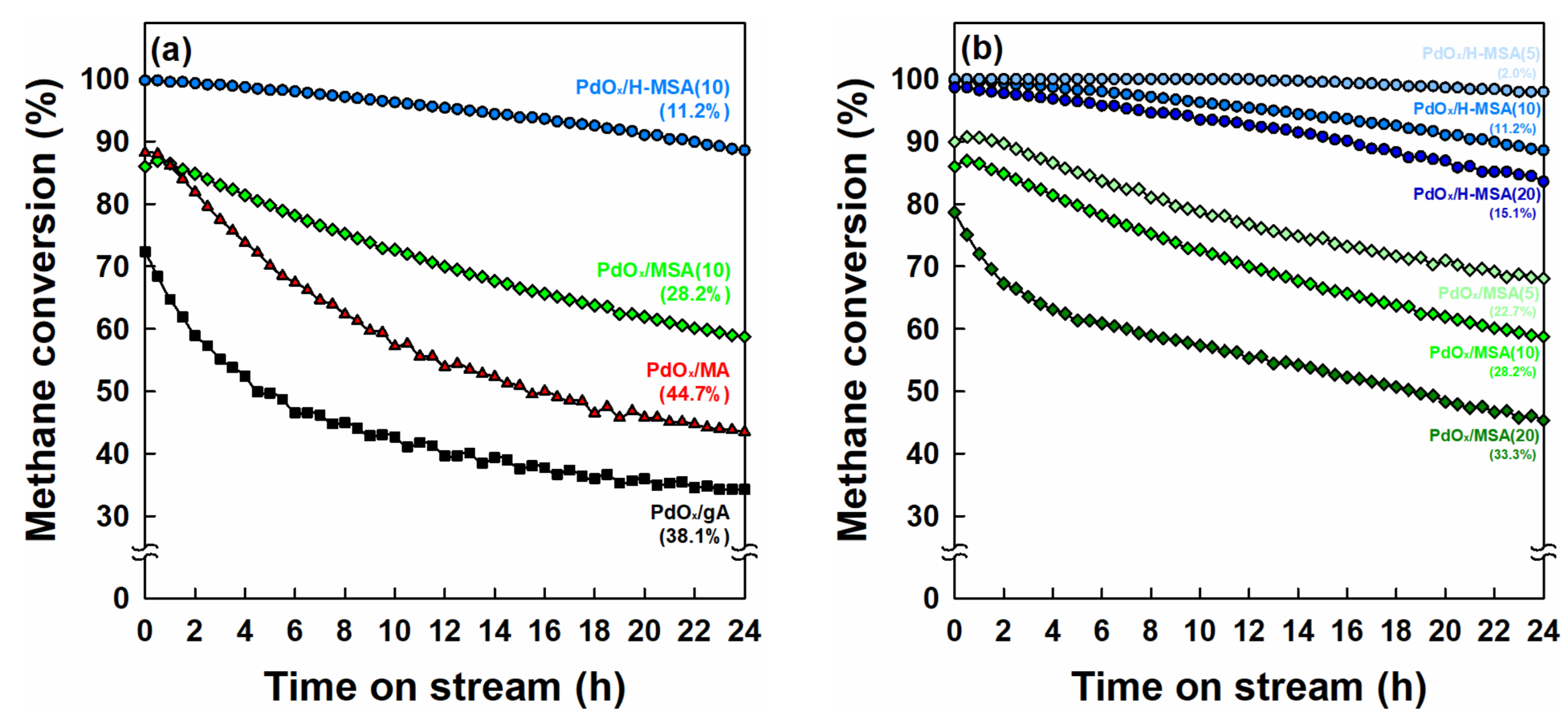
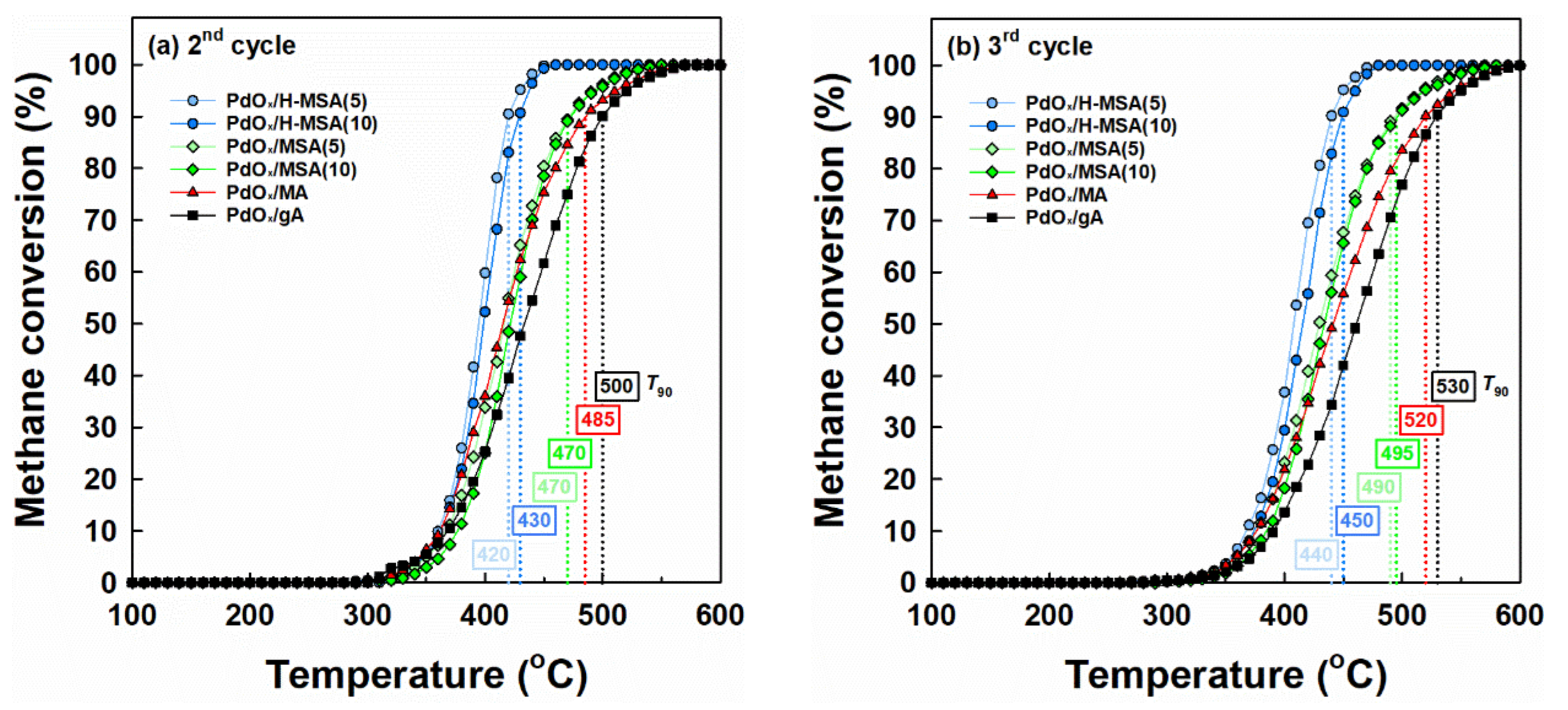
2.2. Catalytic Performance in Methane Combustion under Wet Conditions
2.3. Effect of Alumina Substitution by Silica
3. Materials and Methods
3.1. Catalyst Preparation
3.2. Activity Test for Methane Combustion
3.3. Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mihai, O.; Smedler, G.; Nylén, U.; Olofsson, M.; Olsson, L. The effect of water on methane oxidation over Pd/Al2O3 under lean, stoichiometric and rich conditions. Catal. Sci. Technol. 2017, 7, 3084–3096. [Google Scholar] [CrossRef]
- Nilsson, J.; Carlsson, P.-A.; Martin, N.M.; Adams, E.C.; Agostini, G.; Grönbek, H.; Skoglundh, M. Methane oxidation over Pd/Al2O3 under rich/lean cycling followed by operando XAFS and modulation excitation spectroscopy. J. Catal. 2017, 356, 237–245. [Google Scholar] [CrossRef]
- Baylet, A.; Marecot, P.; Duprez, D.; Castellazzi, P.; Groppi, G.; Forzatti, P. In situ Raman and in situ XRD analysis of PdO reduction and Pd0 oxidation supported on γ-Al2O3 catalyst under different atmospheres. PCCP 2011, 13, 4607–4613. [Google Scholar] [CrossRef] [PubMed]
- Ciuparu, D.; Lyubovsky, M.R.; Altman, E.; Pfefferle, L.D.; Datye, A. Catalytic combustion of methane over palladium-based catalysts. Catal. Rev. 2002, 44, 593–649. [Google Scholar] [CrossRef]
- McCarty, J.G. Kinetics of PdO combustion catalysis. Catal. Today 1995, 26, 283–293. [Google Scholar] [CrossRef]
- Oh, S.H.; Mitchell, P.J.; Siewert, R.M. Methane oxidation over alumina-supported noble metal catalysts with and without cerium additives. J. Catal. 1991, 132, 287–301. [Google Scholar] [CrossRef]
- Baldwin, T.R.; Burch, R. Catalytic combustion of methane over supported palladium catalysts: I. Alumina supported catalysts. Appl. Catal. 1990, 66, 337–358. [Google Scholar]
- Liu, Y.; Wang, S.; Gao, D.; Sun, T.; Zhang, C.; Wang, S. Influence of metal oxides on the performance of Pd/Al2O3 catalysts for methane combustion under lean-fuel conditions. Fuel Process. Technol. 2013, 111, 55–61. [Google Scholar] [CrossRef]
- Lott, P.; Dolcet, P.; Casapu, M.; Grunwaldt, J.-D.; Deutschmann, O. The effect of prereduction on the performance of Pd/Al2O3 and Pd/CeO2 catalysts during methane oxidation. Ind. Eng. Chem. Res. 2019, 58, 12561–12570. [Google Scholar] [CrossRef]
- Losch, P.; Huang, W.; Vozniuk, O.; Goodman, E.D.; Schmidt, W.; Cargnello, M. Modular Pd/zeolite composites demonstrating the key role of support hydrophobic/hydrophilic character in methane catalytic combustion. ACS. Catal. 2019, 9, 4742–4753. [Google Scholar] [CrossRef]
- Burch, R.; Urbano, F.J.; Loader, P.K. Methane combustion over palladium catalysts: The effect of carbon dioxide and water on activity. Appl. Catal. A 1995, 123, 173–184. [Google Scholar] [CrossRef]
- Roth, D.; Gélin, P.; Primet, M.; Tena, E. Catalytic behaviour of Cl-free and Cl-containing Pd/Al2O3 catalysts in the total oxidation of methane at low temperature. Appl. Catal. A 2000, 203, 37–45. [Google Scholar] [CrossRef]
- Schwartz, W.R.; Ciuparu, D.; Pfefferle, L.D. Combustion of methane over palladium-based catalysts: Catalytic deactivation and role of the support. J. Phys. Chem. C 2012, 116, 8587–8593. [Google Scholar] [CrossRef]
- Barrett, W.; Shen, J.; Hu, Y.; Hayes, R.E.; Scott, R.W.J.; Semagina, N. Understanding the role of SnO2 support in water-tolerant methane combustion: In situ observation of Pd(OH)2 and comparison with Pd/Al2O3. ChemCatChem 2019, 11, 1–10. [Google Scholar]
- Cargnello, M.; Jaén, J.J.D.; Garrido, J.C.H.; Bakhmutsky, K.; Montini, T.; Gámez, J.J.C.; Gorte, R.J.; Fornasiero, P. Exceptional activity for methane combustion over modular Pd@CeO2 subunits on functionalized Al2O3. Science 2012, 337, 713–717. [Google Scholar] [CrossRef]
- Smith, S.J.; Huang, B.; Liu, S.; Liu, Q.; Olsen, R.E.; Boerio-Goates, J.; Woodfield, B.F. Synthesis of metal oxide nanoparticles via a robust “solvent-defient” method. Nanoscale 2015, 7, 144–156. [Google Scholar] [CrossRef]
- Huang, B.; Bartholomew, C.H.; Woodfield, B.F. Facile structure-controlled synthesis of mesoporous γ-alumina: Effects of alcohols in precursor formation and calcination. Micropor. Mesopor. Mater. 2013, 177, 37–46. [Google Scholar] [CrossRef]
- Huang, B.; Bartholomew, C.H.; Smith, S.J.; Woodfield, B.F. Facile solvent-deficient synthesis of mesoporous γ-alumina with controlled pore structures. Micropor. Mesopor. Mater. 2013, 165, 70–78. [Google Scholar] [CrossRef]
- Mardkhe, M.K.; Huang, B.; Bartholomew, C.H.; Alam, T.M.; Woodfield, B.F. Synthesis and characterization of silica doped alumina catalyst support with superior thermal stability and unique pore properties. J. Porous. Mater. 2016, 23, 475–487. [Google Scholar] [CrossRef]
- Hong, E.; Kim, C.; Lim, D.-H.; Cho, H.-J.; Shin, C.-H. Catalytic methane combustion over Pd/ZrO2 catalysts: Effect of crystalline structure and textural properties. Appl. Catal. B 2018, 232, 544–552. [Google Scholar] [CrossRef]
- Rytter, E.; Tsakoumis, N.E.; Myrstad, R.; Yang, J.; Lögdberg, S.; Holmen, A.; Rønning, M. Hydrophobic catalyst support surfaces by silylation of γ-alumina for Co/Re Fischer-Tropsch synthesis. Catal. Today 2018, 299, 20–27. [Google Scholar] [CrossRef]
- Al-Abadleh, H.A.; Grassian, V.H. FT-IR study of water adsorption on aluminum oxide surfaces. Langmuir 2003, 19, 341–347. [Google Scholar] [CrossRef]
- Zheng, Q.; Farrauto, R.; Deeba, M. Part II: Oxidative thermal aging of Pd/Al2O3 and Pd/CexOy-ZrO2 in automotive three way catalysts: The effects of fuel shutoff and attempted fuel rich regeneration. Catalysts 2015, 5, 1797–1814. [Google Scholar] [CrossRef] [Green Version]
- Oh, J.; Bathula, H.B.; Park, J.H.; Suh, Y.-W. A sustainable mesoporous palladium-alumina catalyst for efficient hydrogen release from N-heterocyclic liquid organic hydrogen carriers. Commun. Chem. 2019, 2, 68. [Google Scholar] [CrossRef] [Green Version]
- Esteves, L.M.; Brijaldo, M.H.; Passos, F.B. Decomposition of acetic acid for hydrogen production over Pd/Al2O3 and Pd/TiO2: Influence of metal precursor. J. Mol. Catal. A 2016, 422, 275–288. [Google Scholar] [CrossRef]
- Bhogeswararao, S.; Srinivas, D. Catalytic conversion of furfural to industrial chemical over supported Pt and Pd catalysts. J. Catal. 2015, 327, 65–77. [Google Scholar] [CrossRef]
- Murata, K.; Mahara, Y.; Ohyama, J.; Yamamoto, Y.; Arai, S.; Satsuma, A. The metal-support interaction concerning the particle size effect of Pd/Al2O3 on methane combustion. Angew. Chem. Int. Ed. 2017, 56, 15993–15997. [Google Scholar] [CrossRef]
- Chen, J.; Zhong, J.; Wu, Y.; Qu, P.; Xiao, X.; Zhang, G.; Liu, X.; Jiao, Y.; Zhong, L.; Chen, Y. Particle size effects in stoichiometric methane combustion: Structure−activity relationship of Pd catalyst supported on gamma-alumina. ACS Catal. 2020, 10, 10339–10349. [Google Scholar] [CrossRef]
- Toso, A.; Colussi, S.; Llorca, J.; Trovarelli, A. The dynamics of PdO-Pd phase transfromation in the presnece of water over Si-doped Pd/CeO2 methane oxidation catalysts. Appl Catal. A 2019, 574, 79–86. [Google Scholar] [CrossRef]
- Toso, A.; Colussi, S.; Padigapaty, S.; de Leitenburg, C.; Trovarelli, A. High stability and activity of solution combustion synthesized Pd-based catalysts for methane combustion in presence of water. Appl. Catal. A 2018, 230, 237–245. [Google Scholar] [CrossRef]
- Kim, C.; Hong, E.; Shin, C.-H. Improvement of methane combustion activity for Pd/ZrO2 catalyst by simple reduction/reoxidation treatment. Catalysts 2019, 9, 838. [Google Scholar] [CrossRef] [Green Version]
- Emeis, C.A. Determination of integrated molar extinction coefficients for infrared absorption bands of pyridine adsorbed on solid acid catalysts. J. Catal. 1993, 141, 347–354. [Google Scholar] [CrossRef]
- Dong, Y.; Huang, S.; Wang, S.; Zhao, Y.; Gong, J.; Ma, X. Synthesis of dimethyl carbonate through vapor-phase carbonylation catalyzed by Pd-doped zeolites: Interaction of Lewis acidic sites and Pd species. ChemCatChem 2013, 5, 2174–2177. [Google Scholar] [CrossRef]
- Duan, H.; You, R.; Xu, S.; Li, Z.; Qian, K.; Cao, T.; Huang, W.; Bao, X. Pentacoordinated Al3+-stabilized active Pd structure on Al2O3-coated palladium catalysts for methane combustion. Angew. Chem. Int. Ed. 2019, 58, 12043–12048. [Google Scholar] [CrossRef] [PubMed]
- Su, S.C.; Carstens, J.N.; Bell, A.T. A study of the dynamics of Pd oxidation and PdO reduction by H2 and CH4. J. Catal. 1998, 176, 125–135. [Google Scholar] [CrossRef]
- Kim, M.; Lim, S.; Bathula, H.B.; Heo, I.; Kim, J.-R.; Lee, J.H.; Suh, Y.-W. Methane combustion over Pd catalysts supported on mesoporous alumina. KSAE 2020, 28, 353–357. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | BET Surface Area a (m2 g−1) | Pore Volume a (cm3 g−1) | Pore Diameter b (nm) | Lewis Acidity c (μmol g−1) |
|---|---|---|---|---|
| gA | 192 | 0.44 | 7.1 | 59.4 |
| MA | 187 | 1.37 | 24.2 | 51.1 |
| MSA(5) | 276 | 1.87 | 24.1 | 88.1 |
| MSA(10) | 230 | 1.30 | 19.0 | 94.9 |
| MSA(20) | 172 | 0.58 | 11.2 | 109.0 |
| H-MSA(5) | 222 | 1.51 | 34.0 | 82.4 |
| H-MSA(10) | 304 | 1.45 | 19.1 | 69.7 |
| H-MSA(20) | 310 | 1.12 | 14.4 | 48.1 |
| Sample | BET Surface Area a (m2 g−1) | Pore Volume a (cm3 g−1) | Pore Diameter b (nm) | Pd Dispersion c (%) |
|---|---|---|---|---|
| PdOx/gA | 183 | 0.42 | 7.5 | 29.4 (3.8 nm) |
| PdOx/MA | 155 | 0.67 | 15.5 | 18.1 (6.2 nm) |
| PdOx/MSA(5) | 201 | 0.76 | 12.9 | 29.8 (3.7 nm) |
| PdOx/MSA(10) | 187 | 0.78 | 13.9 | 21.8 (5.1 nm) |
| PdOx/MSA(20) | 174 | 0.63 | 10.0 | 17.4 (6.4 nm) |
| PdOx/H-MSA(5) | 217 | 1.36 | 20.4 | 35.2 (3.2 nm) |
| PdOx/H-MSA(10) | 297 | 1.23 | 18.9 | 34.3 (3.3 nm) |
| PdOx/H-MSA(20) | 276 | 0.93 | 12.9 | 37.5 (3.0 nm) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, M.; Lim, S.; Kim, C.; Shin, C.-H.; Baik, J.H.; Suh, Y.-W. Stable Performance of Supported PdOx Catalyst on Mesoporous Silica-Alumina of Water Tolerance for Methane Combustion under Wet Conditions. Catalysts 2021, 11, 670. https://doi.org/10.3390/catal11060670
Kim M, Lim S, Kim C, Shin C-H, Baik JH, Suh Y-W. Stable Performance of Supported PdOx Catalyst on Mesoporous Silica-Alumina of Water Tolerance for Methane Combustion under Wet Conditions. Catalysts. 2021; 11(6):670. https://doi.org/10.3390/catal11060670
Chicago/Turabian StyleKim, Minseok, Suhyun Lim, Chansong Kim, Chae-Ho Shin, Joon Hyun Baik, and Young-Woong Suh. 2021. "Stable Performance of Supported PdOx Catalyst on Mesoporous Silica-Alumina of Water Tolerance for Methane Combustion under Wet Conditions" Catalysts 11, no. 6: 670. https://doi.org/10.3390/catal11060670
APA StyleKim, M., Lim, S., Kim, C., Shin, C. -H., Baik, J. H., & Suh, Y. -W. (2021). Stable Performance of Supported PdOx Catalyst on Mesoporous Silica-Alumina of Water Tolerance for Methane Combustion under Wet Conditions. Catalysts, 11(6), 670. https://doi.org/10.3390/catal11060670