
Epoxidation of Terpenes


Abstract
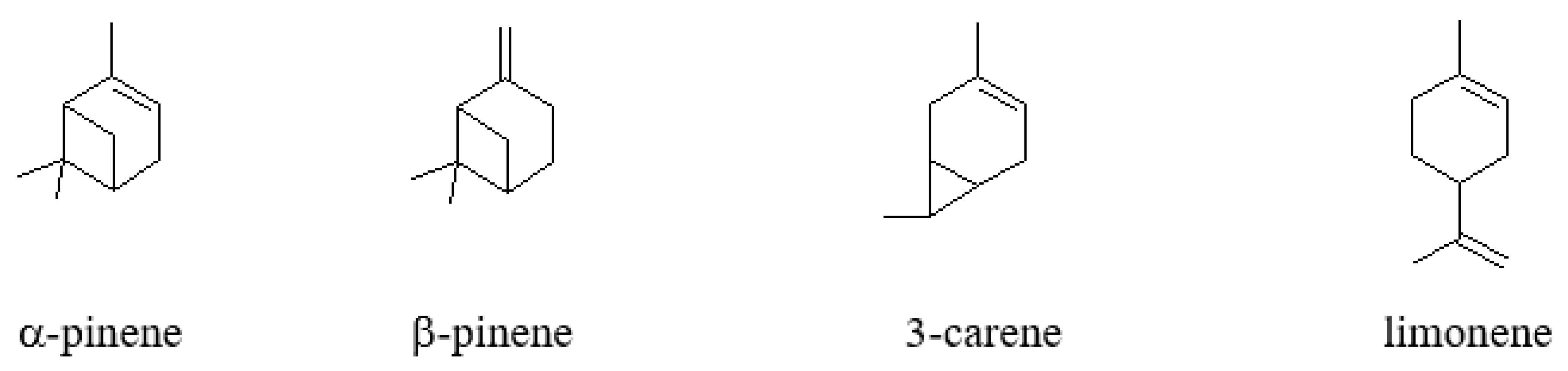
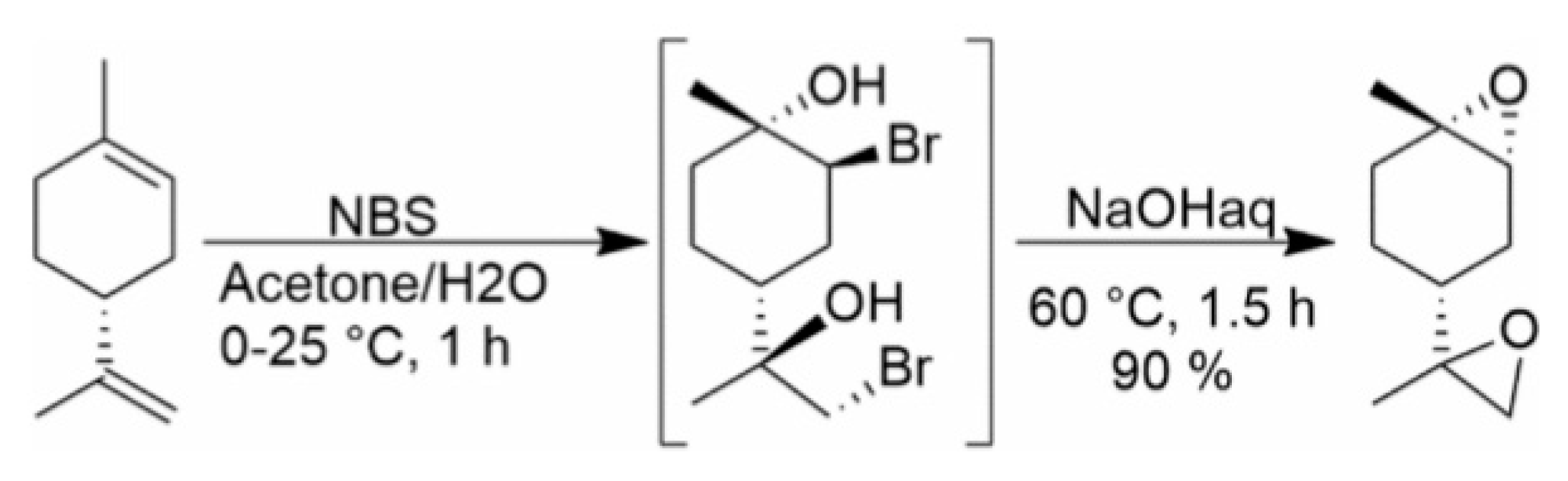
:1. Introduction
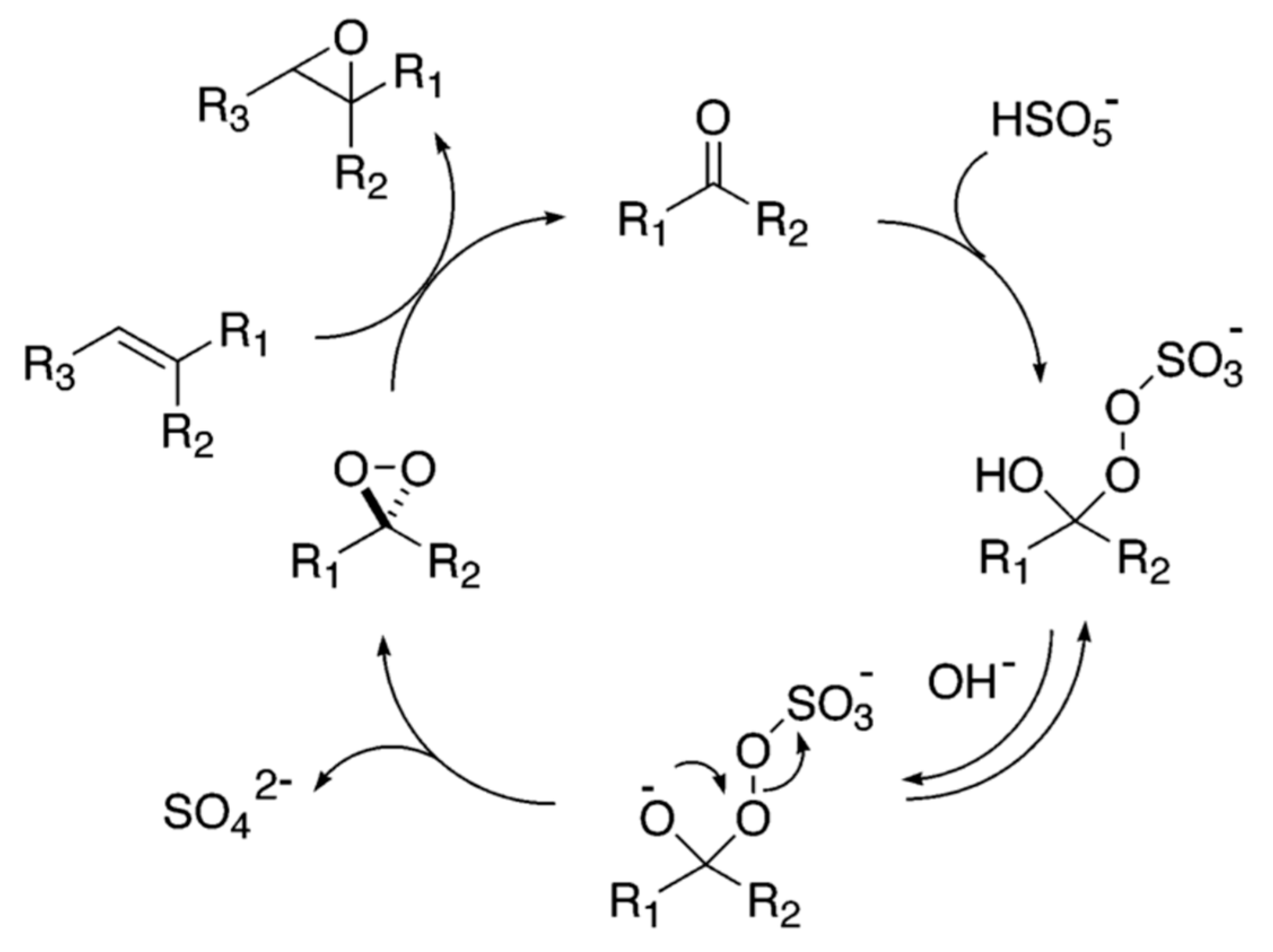
2. Hydroperoxides
3. Dimethyl Dioxirane (DMDO)
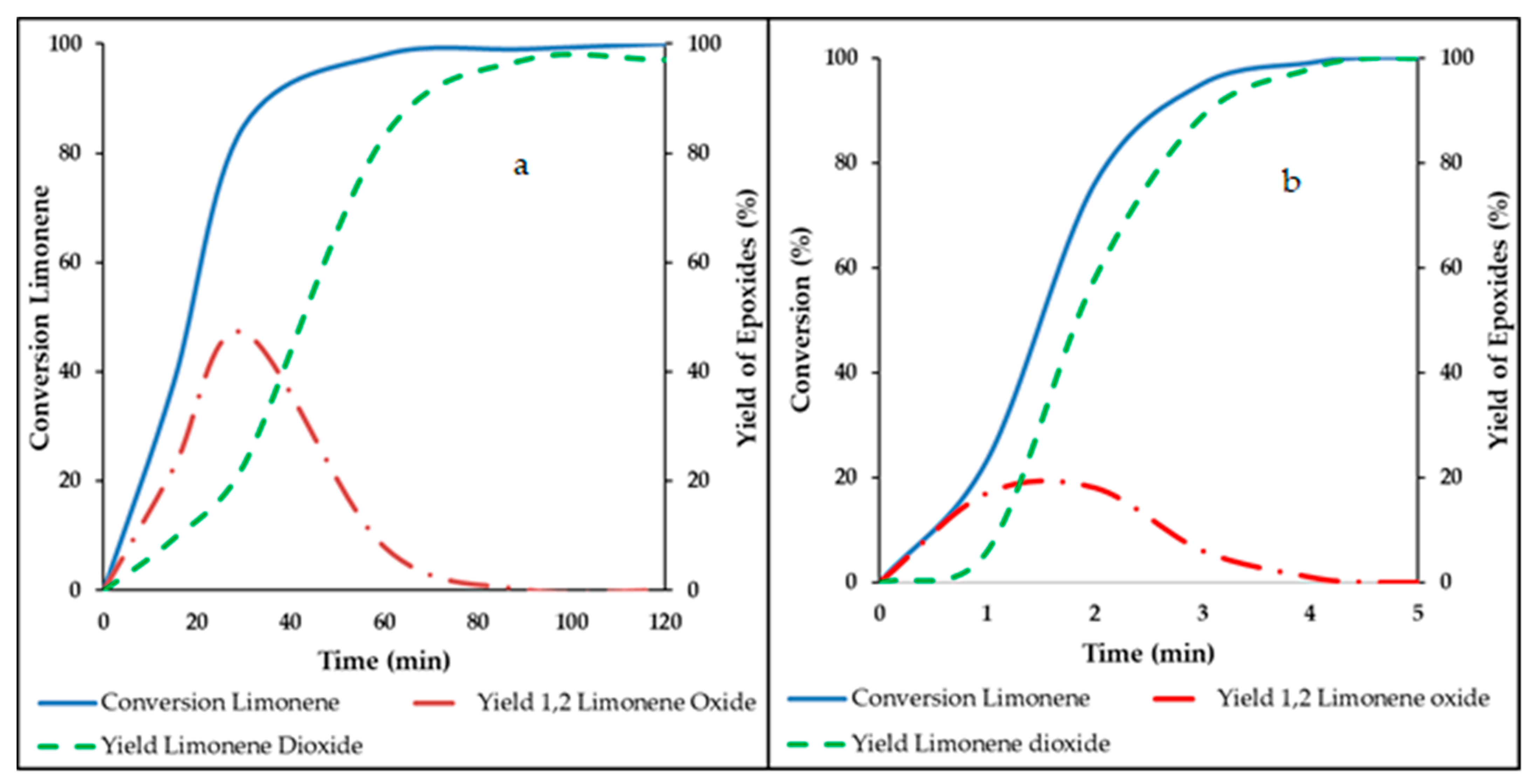
3.1. Epoxidation of Limonene in Semi-Batch Reactor
3.2. Epoxidation under Ultrasound
3.3. Heterogeneous Polydioxiranes
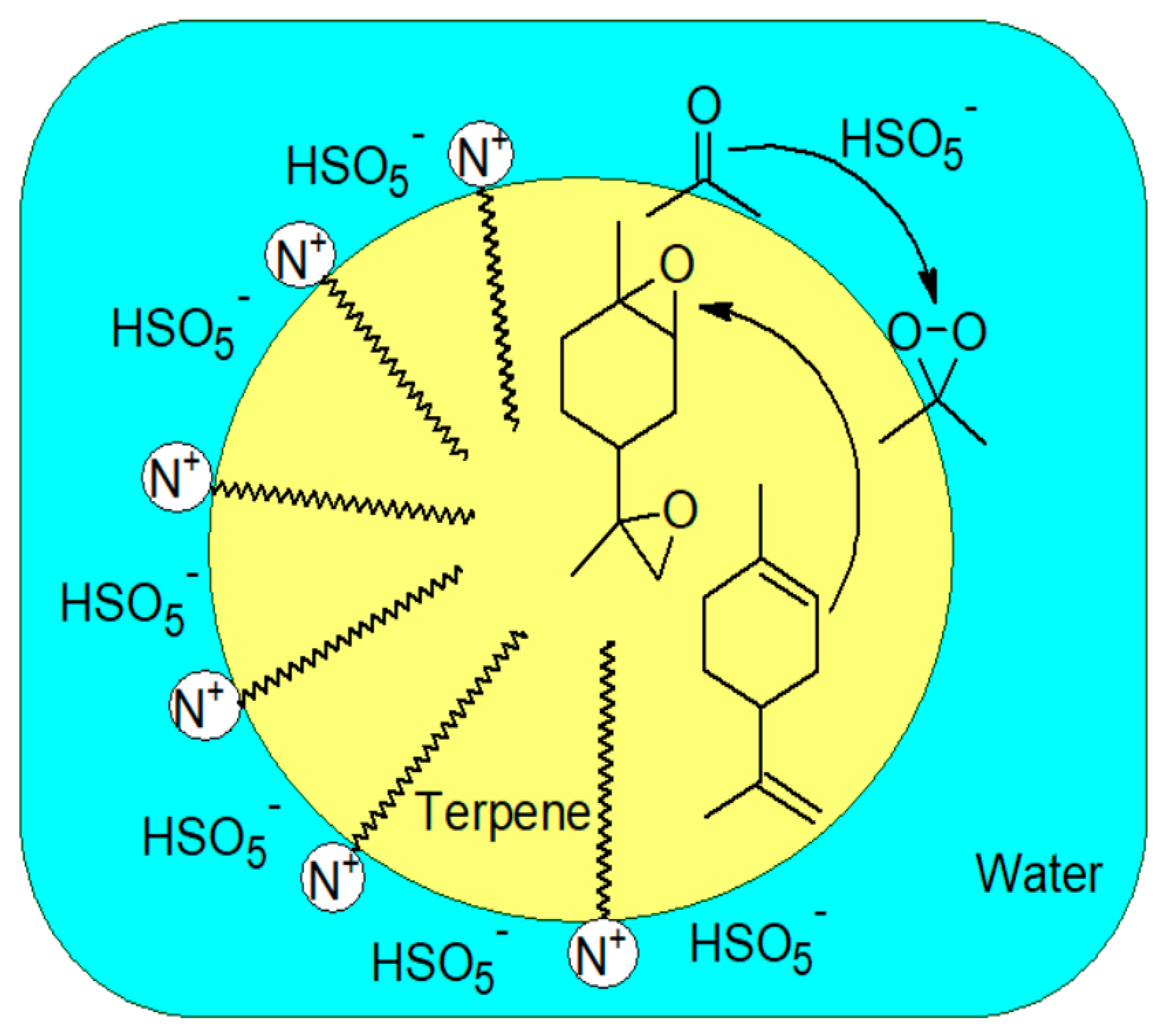
3.4. In Microemulsions
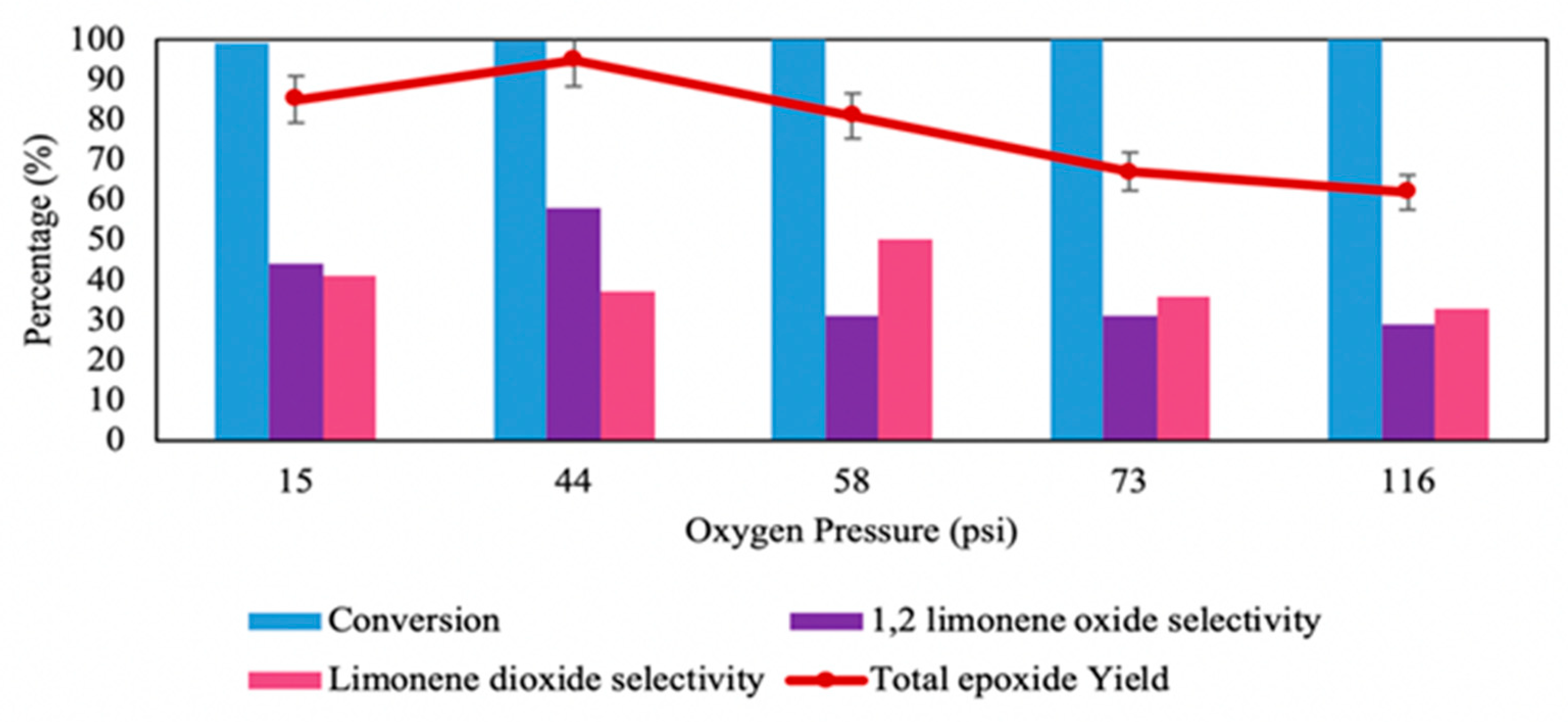
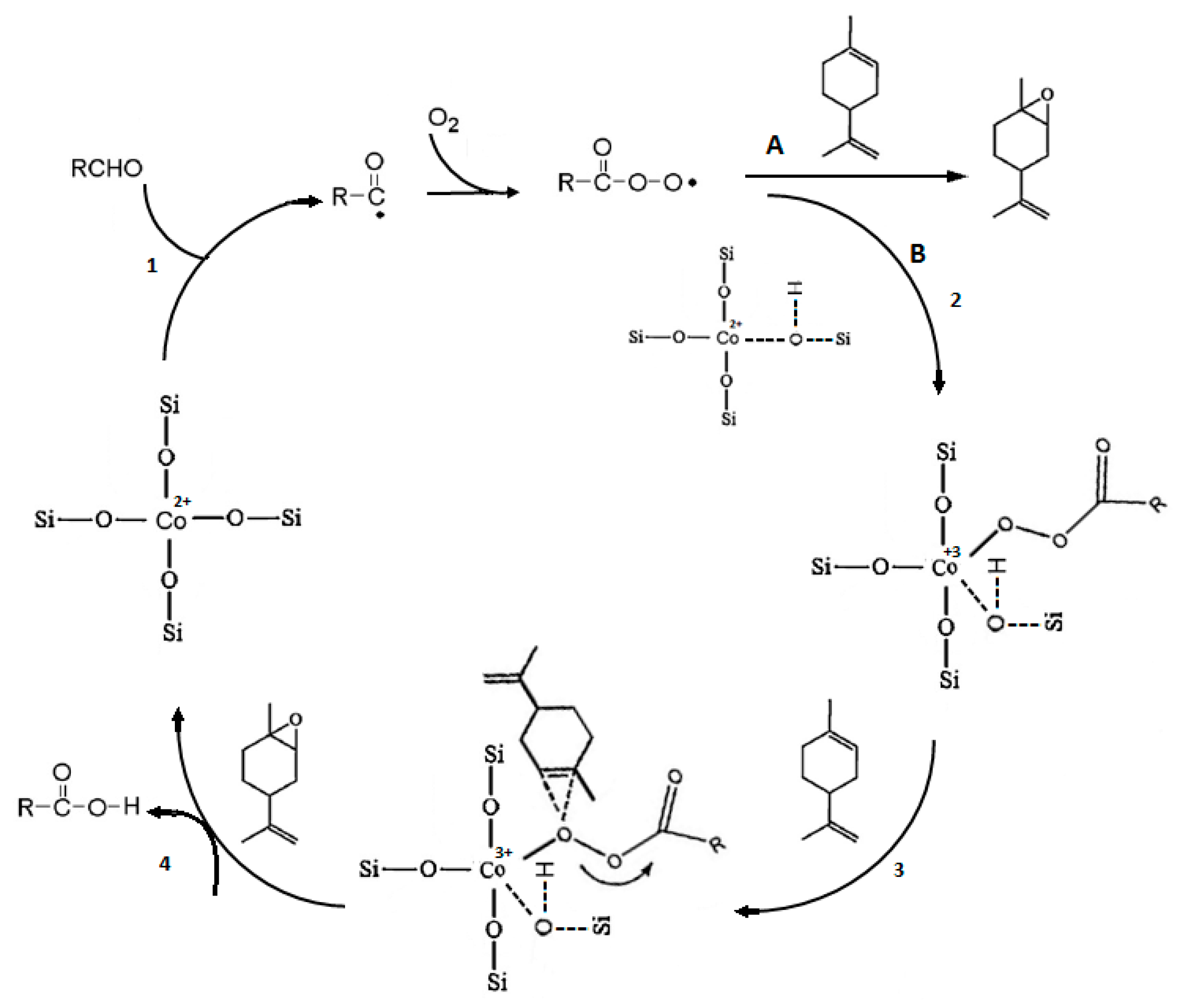
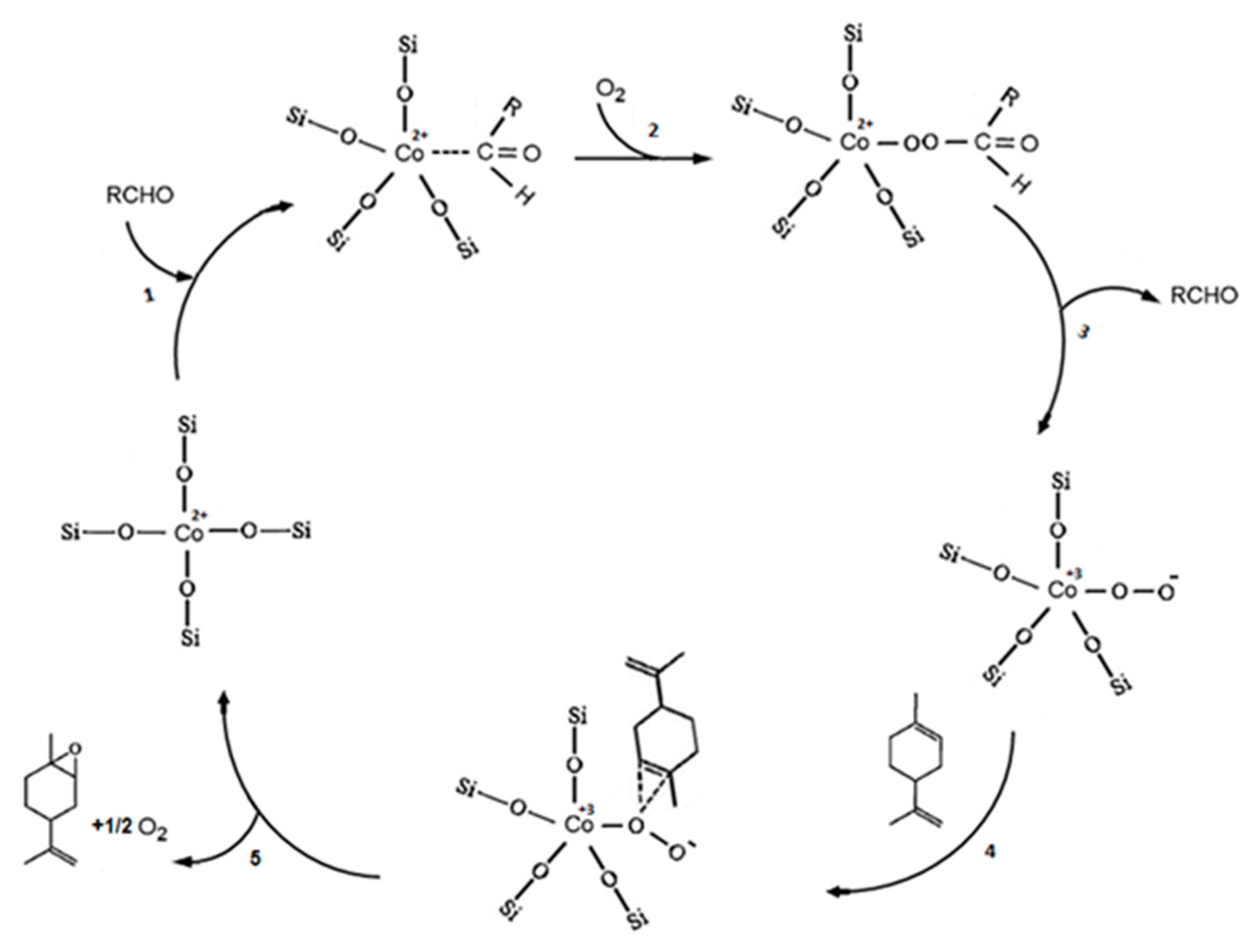
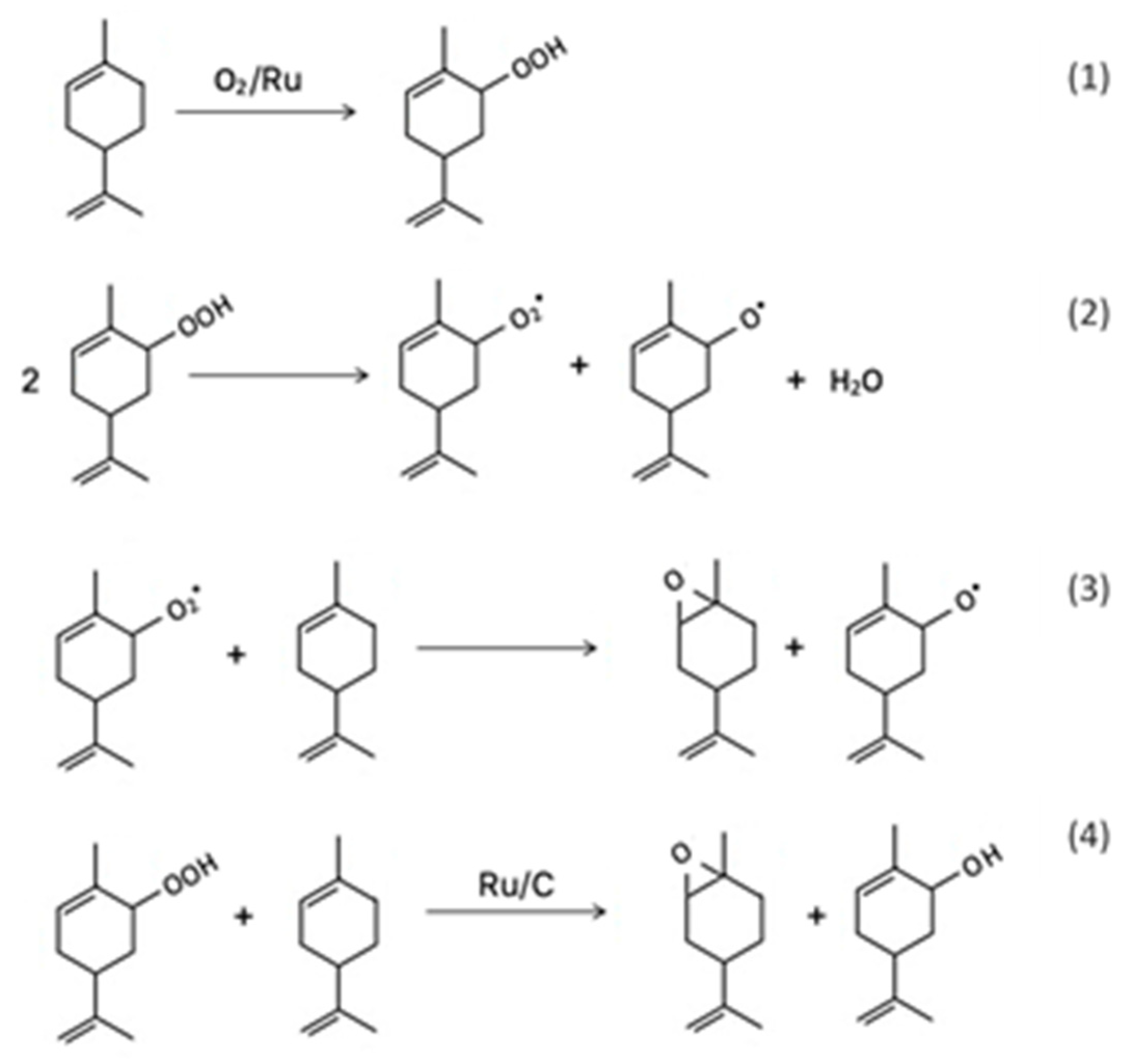
4. Aerobic Epoxidation
4.1. Aerobic Epoxidation Using an Aldehyde as Sacrificial Reductant (Mukaiyama Reaction)
4.2. Solvent-Free and Initiator-Free Epoxidation
5. Importance and Perspective of Stereoselective Epoxidation
Enantioselective Separation
6. Conclusions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nguyen, H.T.H.; Qi, P.; Rostagno, M.; Feteha, A.; Miller, S.A. The quest for high glass transition temperature bioplastics. J. Mater. Chem. A 2018, 6, 9298–9331. [Google Scholar] [CrossRef]
- Kristufek, S.L.; Wacker, K.T.; Tsao, Y.-Y.T.; Su, L.; Wooley, K.L. Monomer design strategies to create natural product-based polymer materials. Nat. Prod. Rep. 2017, 34, 433–459. [Google Scholar] [CrossRef] [PubMed]
- Hernández, N.; Williams, R.C.; Cochran, E.W. The battle for the “green” polymer. Different approaches for biopolymer synthesis: Bioadvantaged vs. bioreplacement. Org. Biomol. Chem. 2014, 12, 2834–2849. [Google Scholar] [CrossRef]
- Harman-Ware, A.E. Conversion of terpenes to chemicals and related products. In Chemical Catalysts for Biomass Upgrading; Crocker, M., Santillan-Jimenez, E., Eds.; Wiley-VCH: Weinheim, Germany, 2020; pp. 529–568. [Google Scholar]
- Ciriminna, R.; Lomeli-Rodriguez, M.; Carà, P.D.; Lopez-Sanchez, J.A.; Pagliaro, M. Limonene: A versatile chemical of the bioe-conomy. Chem. Commun. 2014, 50, 15288–15296. [Google Scholar] [CrossRef]
- Pandarus, V.; Ciriminna, R.; Béland, F.; Pagliaro, M.; Kaliaguine, S. Solvent-Free Chemoselective Hydrogenation of Squalene to Squalane. ACS Omega 2017, 2, 3989–3996. [Google Scholar] [CrossRef] [Green Version]
- Lange, B.M. Biosynthesis and Biotechnology of High-Value p-Menthane Monoterpenes, Including Menthol, Carvone, and Limonene. Biotechnol. Isoprenoids 2015, 148, 319–353. [Google Scholar] [CrossRef]
- Korman, T.P.; Opgenorth, P.H.; Bowie, J.U. A synthetic biochemistry platform for cell free production of monoterpenes from glucose. Nat. Commun. 2017, 8, 15526. [Google Scholar] [CrossRef]
- Korman, T.P.; Sahachartsiri, B.; Li, D.; Vinokur, J.M.; Eisenberg, D.; Bowie, J.U. A synthetic biochemistry system for the in vitro production of isoprene from glycolysis intermediates. Protein Sci. 2014, 23, 576–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Li, R.; Yi, X.; Fang, T.; Yang, J.; Bae, H.-J. Isoprene Production on Enzymatic Hydrolysate of Peanut Hull Using Different Pretreatment Methods. BioMed Res. Int. 2016, 2016, 4342892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, T.; Liu, H.; Zou, H.; Chen, N.; Shi, M.; Xie, C.; Zhao, G.; Xian, M. Enzymatic process optimization for the in vitro production of isoprene from mevalonate. Microb. Cell Factories 2017, 16, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, L.D.; Dauenhauer, P.J. Hybrid routes to biofuels. Nat. Cell Biol. 2007, 447, 914–915. [Google Scholar] [CrossRef]
- Lundberg, D.J.; Lundberg, D.J.; Zhang, K.; Dauenhauer, P.J. Process Design and Economic Analysis of Renewable Isoprene from Biomass via Mesaconic Acid. ACS Sustain. Chem. Eng. 2019, 7, 5576–5586. [Google Scholar] [CrossRef]
- Liang, J.; Zhang, Q.; Wu, H.; Meng, G.; Tang, Q.; Wang, Y. Iron-based heterogeneous catalysts for epoxidation of alkenes using molecular oxygen. Catal. Commun. 2004, 5, 665–669. [Google Scholar] [CrossRef]
- Jiang, J.; Li, R.R.; Wang, H.; Zheng, Y.; Chen, H.; Ma, J. Highly Active/Selective Heterogeneous Catalyst Co/TS-1 for Epoxi-dation of Styrene by Molecular Oxygen: Effects of Catalyst Preparation Conditions and Reaction Conditions on the Reaction. Catal. Lett. 2008, 120, 221–228. [Google Scholar] [CrossRef]
- Clayden, J.; Greeves, N.; Warren, S.G. Organic Chemistry; Oxford University Press: Oxford, UK; New York, NY, USA, 2012. [Google Scholar]
- Cunningham, W. Catalytic Conversion of Terpene Feedstocks into Value-Added Chemicals and Commodity Chemicals. Ph.D. Thesis, University of Bath, Bath, UK, 2018. [Google Scholar]
- Yadav, G.D.; Pujari, A.A. Epoxidation of Styrene to Styrene Oxide: Synergism of Heteropoly Acid and Phase-Transfer Catalyst under Ishii−Venturello Mechanism. Org. Process. Res. Dev. 2000, 4, 88–93. [Google Scholar] [CrossRef]
- Brégeault, J.-M. Transition-metal complexes for liquid-phase catalytic oxidation: Some aspects of industrial reactions and of emerging technologies. Dalton Trans. 2003, 3289–3302. [Google Scholar] [CrossRef]
- Adolfsson, H. Transition Metal Catalyzed Epoxidation of Alkenes. In Modern Oxidation Methods; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- Sheldon, R. Synthetic and mechanistic aspects of metal-catalysed epoxidations with hydroperoxides. J. Mol. Catal. 1980, 7, 107–126. [Google Scholar] [CrossRef]
- Rao, A.S. Addition Reactions with Formation of Carbon–Oxygen Bonds: (i) General Methods of Epoxidation. Oxidation 1991, 357–387. [Google Scholar] [CrossRef]
- Arends, I.; Sheldon, R. Activities and stabilities of heterogeneous catalysts in selective liquid phase oxidations: Recent developments. Appl. Catal. A Gen. 2001, 212, 175–187. [Google Scholar] [CrossRef]
- Sheldon, R. Metal-catalyzed epoxidation of olefins with organic hydroperoxides I. A comparison of various metal catalysts. J. Catal. 1973, 31, 427–437. [Google Scholar] [CrossRef]
- Corma, A.; Esteve, P.; Martinez, A.; Valencia, S. Oxidation of Olefins with Hydrogen Peroxide and tert-Butyl Hydroper-oxide on Ti-Beta Catalyst. J. Catal. 1995, 152, 18–24. [Google Scholar] [CrossRef]
- Chiker, F.; Launay, F.; Nogier, J.P.; Bonardet, J.L. Green and selective epoxidation of alkenes catalysed by new TiO2–SiO2SBA mesoporous solids. Green Chem. 2003, 5, 318–322. [Google Scholar] [CrossRef]
- Kapoor, M.P.; On, D.T.; Gallot, J.E.; Kaliaguine, S. Hydrolytic cleavage of epoxy ring during the epoxidation of al-kenes over boron modified titanium silicalites of MFI topology. Catal. Lett. 1997, 43, 127–131. [Google Scholar] [CrossRef]
- Gallot, J.E.; Kaliaguine, S. Oxidation of hydrocarbons by hydrogen peroxide over Ti catalysts: Kinetics and mechanistic studies. Can. J. Chem. Eng. 1998, 76, 833–852. [Google Scholar] [CrossRef]
- Bérubé, F.; Kleitz, F.; Kaliaguine, S. Surface properties and epoxidation catalytic activity of Ti-SBA15 prepared by direct synthesis. J. Mater. Sci. 2009, 44, 6727–6735. [Google Scholar] [CrossRef]
- Bérubé, F.; Kleitz, F.; Kaliaguine, S. A Comprehensive Study of Titanium-Substituted SBA-15 Mesoporous Materials Prepared by Direct Synthesis. J. Phys. Chem. C 2008, 112, 14403–14411. [Google Scholar] [CrossRef]
- Bérubé, F.; Nohair, B.; Kleitz, F.; Kaliaguine, S. Controlled Postgrafting of Titanium Chelates for Improved Synthesis of Ti-SBA-15 Epoxidation Catalysts. Chem. Mater. 2010, 22, 1988–2000. [Google Scholar] [CrossRef]
- Bérubé, F.; Khadhraoui, A.; Janicke, M.T.; Kleitz, F.; Kaliaguine, S. Optimizing Silica Synthesis for the Preparation of Mesoporous Ti-SBA-15 Epoxidation Catalysts. Ind. Eng. Chem. Res. 2010, 49, 6977–6985. [Google Scholar] [CrossRef]
- Charbonneau, L.; Kaliaguine, S. Epoxidation of limonene over low coordination Ti in Ti-SBA-16. Appl. Catal. A Gen. 2017, 533, 1–8. [Google Scholar] [CrossRef]
- Charbonneau, L.; Foster, X.; Zhao, D.; Kaliaguine, S. Catalyst-Free Epoxidation of Limonene to Limonene Dioxide. ACS Sustain. Chem. Eng. 2018, 6, 5115–5121. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, W.B.; Tibbetts, J.D.; Hutchby, M.; Maltby, K.A.; Davidson, M.G.; Hintermair, U.; Plucinski, P.; Bull, S.D. Sustaina-ble catalytic protocols for the solvent free epoxidation and anti-dihydroxylation of the alkene bonds of biorenewable terpene feedstocks using H2O2 as oxidant. Green Chem. 2020, 22, 513–524. [Google Scholar] [CrossRef] [Green Version]
- Murray, R.W. Chemistry of dioxiranes. 12. Dioxiranes. Chem. Rev. 1989, 89, 1187–1201. [Google Scholar] [CrossRef]
- Shi, Y. Organocatalytic Asymmetric Epoxidation of Olefins by Chiral Ketones. ACC Chem. Res. 2004, 37, 488–496. [Google Scholar] [CrossRef]
- Tu, Y.; Wang, Z.-X.; Shi, Y. An Efficient Asymmetric Epoxidation Method for trans-Olefins Mediated by a Fructose-Derived Ketone. J. Am. Chem. Soc. 1996, 118, 9806–9807. [Google Scholar] [CrossRef]
- Asouti, A.; Hadjiarapoglou, L.P. Substrate-Induced Diastereoselectivity in the Dimethyldioxirane Epoxidation of Simple Al-kenes and Dienes. Synlett 2001, 2001, 1847–1850. [Google Scholar] [CrossRef]
- Wang, Z. Comprehensive Organic Name Reactions and Reagents; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010. [Google Scholar]
- Edwards, J.O.; Pater, R.H.; Curclf, R.; Di Furia, F. On The Formation and Reactivity of Dioxirane Intermediates in the Reaction of Peroxoanions with Organic Substrates. Photochem. Photobiol. 1979, 30, 63–70. [Google Scholar] [CrossRef]
- Charbonneau, L.; Foster, X.; Kaliaguine, S. Ultrasonic and Catalyst-Free Epoxidation of Limonene and Other Terpenes Using Dimethyl Dioxirane in Semibatch Conditions. ACS Sustain. Chem. Eng. 2018, 6, 12224–12231. [Google Scholar] [CrossRef]
- Kazemnejadi, M.; Shakeri, A.; Nikookar, M.; Shademani, R.; Mohammadi, M. Selective and metal-free epoxidation of terminal alkenes by heterogeneous polydioxirane in mild conditions. R. Soc. Open Sci. 2018, 5, 171541. [Google Scholar] [CrossRef] [Green Version]
- Sartori, G.; Armstrong, A.; Maggi, R.; Mazzacani, A.; Sartorio, R.; Bigi, F.; Dominguez-Fernandez, B. α-Fluorotropinone Immobilized on Silica: A New Stereoselective Heterogeneous Catalyst for Epoxidation of Alkenes with Oxone. J. Org. Chem. 2003, 68, 3232–3237. [Google Scholar] [CrossRef]
- D’Accolti, L.; Annese, C.; De Riccardis, A.; De Giglio, E.; Cafagna, D.; Fanelli, F.; Fusco, C. Dioxirane-Mediated Heterogeneous Epoxidations with Potassium Caroate: A Solid Catalyst Bearing Anchored Ketone Moieties. Eur. J. Org. Chem. 2012, 2012, 4616–4621. [Google Scholar] [CrossRef]
- Song, C.E.; Lim, J.S.; Kim, S.C.; Lee, K.-J.; Chi, D.Y. Immobilisation of ketone catalyst: A method to prevent ketone catalyst from decomposing during dioxirane-mediated epoxidation of alkenes. Chem. Commun. 2000, 24, 2415–2416. [Google Scholar] [CrossRef]
- Menger, F.M. Groups of Organic Molecules That Operate Collectively. Angew. Chem. Int. Ed. 1991, 30, 1086–1099. [Google Scholar] [CrossRef]
- Mahamat Ahmat, Y.; Kaliaguine, S. Epoxidation of limonene and pinenes by dimethyldioxirane in microemulsions. Catal. Today. submitted.
- Zhong, W.; Liu, M.; Dai, J.; Yang, J.; Mao, L.; Yin, D. Synergistic hollow CoMo oxide dual catalysis for tandem oxygen transfer: Preferred aerobic epoxidation of cyclohexene to 1,2-epoxycyclohexane. Appl. Catal. B Environ. 2018, 225, 180–196. [Google Scholar] [CrossRef]
- Acharyya, S.S.; Ghosh, S.; Sharma, S.K.; Bal, R. Cetyl alcohol mediated fabrication of forest of Ag/Mn3O4 nanowhiskers catalyst for the selective oxidation of styrene with molecular oxygen. RSC Adv. 2015, 5, 89879–89887. [Google Scholar] [CrossRef]
- Madadi, S.; Charbonneau, L.; Bergeron, J.Y.; Kaliaguine, S. Aerobic epoxidation of limonene using cobalt substituted mesoporous SBA-16 Part 1: Optimization via Response Surface Methodology (RSM). Appl. Catal. B Environ. 2020, 260, 118049. [Google Scholar] [CrossRef]
- Madadi, S.; Bergeron, J.-Y.; Kaliaguine, S. Kinetic investigation of aerobic epoxidation of limonene over cobalt substituted mesoporous SBA-16. Catal. Sci. Technol. 2020, 11, 594–611. [Google Scholar] [CrossRef]
- Madadi, S.; Kaliaguine, S. Activated carbon supported ruthenium as a catalyst for solvent- and initiator-free aerobic epoxidation of limonene. ACS Sustain. Chem. Eng. submitted.
- Schutz, L.; Kazemi, F.; Mackenzie, E.; Bergeron, J.Y.; Gagnon, E.; Claverie, J.P. Trans-limonene dioxide, a promising bio-based epoxy monomer. J. Polym. Sci. 2021, 59, 321–328. [Google Scholar] [CrossRef]
- Byrne, C.M.; Allen, S.D.; Lobkovsky, A.E.B.; Coates, G.W. Alternating Copolymerization of Limonene Oxide and Carbon Dioxide. J. Am. Chem. Soc. 2004, 126, 11404–11405. [Google Scholar] [CrossRef] [Green Version]
- Auriemma, F.; De Rosa, C.; Di Caprio, M.R.; Di Girolamo, R.; Ellis, W.C.; Coates, G.W. Stereocomplexed Poly(Limonene Carbonate): A Unique Example of the Cocrystallization of Amorphous Enantiomeric Polymers. Angew. Chem. 2014, 127, 1231–1234. [Google Scholar] [CrossRef]
- Parrino, F.; Fidalgo, A.; Palmisano, L.; Ilharco, L.M.; Pagliaro, M.; Ciriminna, R. Polymers of Limonene Oxide and Carbon Dioxide: Polycarbonates of the Solar Economy. ACS Omega 2018, 3, 4884–4890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauenstein, O.; Reiter, M.; Agarwal, S.; Rieger, B.; Greiner, A. Bio-based polycarbonate from limonene oxide and CO2 with high molecular weight, excellent thermal resistance, hardness and transparency. Green Chem. 2016, 18, 760–770. [Google Scholar] [CrossRef] [Green Version]
- Rehman, A.; Fernández, A.M.L.; Resul, M.G.; Harvey, A. Highly selective, sustainable synthesis of limonene cyclic carbonate from bio-based limonene oxide and CO2: A kinetic study. J. CO2 Util. 2019, 29, 126–133. [Google Scholar] [CrossRef]
- Schimpf, V.; Ritter, B.S.; Weis, P.; Parison, K.; Mülhaupt, R. High Purity Limonene Dicarbonate as Versatile Building Block for Sustainable Non-Isocyanate Polyhydroxyurethane Thermosets and Thermoplastics. Macromolecules 2017, 50, 944–955. [Google Scholar] [CrossRef]
- Steiner, D.; Ivison, L.; Goralski, C.T.; Appell, R.B.; Gojkovic, J.R.; Singaram, B. A facile and efficient method for the kinetic separation of commercially available cis- and trans-limonene epoxide. Tetrahedron Asymmetry 2002, 13, 2359–2363. [Google Scholar] [CrossRef]
- Morikawa, H.; Minamoto, M.; Gorou, Y.; Yamaguchi, J.-I.; Morinaga, H.; Motokucho, S. Two Diastereomers of d-Limonene-Derived Cyclic Carbonates from d-Limonene Oxide and Carbon Dioxide with a Tetrabutylammonium Chloride Catalyst. Bull. Chem. Soc. Jpn. 2018, 91, 92–94. [Google Scholar] [CrossRef]
- Rehman, A.; Russell, E.; Saleem, F.; Javed, F.; Ahmad, S.; Eze, V.C.; Harvey, A. Synthesis of trans-limonene bis-epoxide by stereoselective epoxidation of (R)-(+)-limonene. J. Environ. Chem. Eng. 2021, 9, 104680. [Google Scholar] [CrossRef]
- Cubillos, J.; Hölderich, W. Jacobsen s catalyst anchored on Al-MCM-41 and NH2 group modified Si-MCM-41 as heterogeneous enantioselective epoxidation catalyst using in situ generated dimethyldioxirane as oxidant. Rev. Fac. Ing. Univ. Antioq. 2007, 41, 31–47. [Google Scholar]
- Tian, H.; She, X.; Shu, L.; Yu, H.; Shi, Y. Highly enantioselective epoxidation of cis-olefins by chiral dioxirane. J. Am. Chem. Soc. 2000, 122, 11551–11552. [Google Scholar] [CrossRef]
- Bérubé, F. Catalyseurs D’époxydation de Type Ti/SBA-15. Ph.D. Thesis, Université Laval, Québec, QC, Canada, 2015. [Google Scholar]
- Charbonneau, L. Époxydation du Limonene. Ph.D. Thesis, Université Laval, Québec, QC, Canada, 2018. [Google Scholar]
- Madadi, S. Aerobic Epoxidation of Terpenes. Ph.D. Thesis, Université Laval, Québec, QC, Canada, 2021. [Google Scholar]
- Jérôme, F. Preface. In Industrial Green Chemistry; Kaliaguine, S., Dubois, J.-L., Eds.; De Gruyter: Berlin, Germany, 2021. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oxone®/Limonene Ratio | pH of Oxone® Solution | Conversion a (%) | 1,2-LO Yield a (%) | Limonene Dioxide Yield a (%) | Oxone Conversion to Epoxide (%) |
|---|---|---|---|---|---|
| 0.65 | 1.78 | 41 | 16 | 22 | 92.6 |
| 1.30 | 1.55 | 65 | 22 | 38 | 75 |
| 2.00 | 1.43 | 98 | 23 | 75 | 86 |
| 2.60 | 1.30 | 100 | 0 | 100 | 77 |
| CTAHS (g) | Vacetone Vwater | noxone (mmol) | Conversion (%) | Yield (%) | Carbon Balance (%) | Oxygen Balance (%) | ||
|---|---|---|---|---|---|---|---|---|
| 1.2 LO | LDO | 1.2 LO + LDO | ||||||
| 0 | 1/11 | 12.4 | 71 | 16 | 56 | 72 | 100 | 33 |
| 0.05 | 100 | 0 | 99 | 99 | 99 | 51 | ||
| 0 | 1/7 | 8.6 | 61 | 25 | 36 | 61 | 100 | 35 |
| 0.05 | 100 | 9 | 89 | 98 | 98 | 67 | ||
| 0 | 1/5 | 6.2 | 55 | 27 | 24 | 51 | 92 | 37 |
| 0.05 | 85 | 26 | 58 | 84 | 98 | 71 | ||
| 0 | 1/3 | 3.7 | 45 | 36 | 10 | 46 | 100 | 46 |
| 0.05 | 52 | 33 | 20 | 53 | 100 | 61 | ||
| Terpene | Reaction Time (min) | CTAHS (g) | Active O/Terpene b | Vacetone/Vwater | Yield (%) c |
|---|---|---|---|---|---|
 | 30 | 0 | 3.2 | 1/9 | 44 |
| 0.05 | 3.2 | 1/9 | 100 | ||
 | 30 | 0 | 1.8 | 1/5 | 54 |
| 0.05 | 1.8 | 1/5 | 98 | ||
 | 45 | 0 | 2 | 1/6 | 47 |
| 0.05 | 2 | 1/6 | 97 |
| Catalyst | Co/Si (%) | Conversion (%) | 1,2-LO Selectivity (%) | LDO Selectivity (%) |
|---|---|---|---|---|
| None | - | 42 | 15 | - |
| Co/SBA16 | 1.1 | 99 | 45 | 25 |
| Co/SBA16 | 2.7 | 99 | 49 | 30 |
| Co/SBA16 | 4.5 | 99 | 50 | 33 |
| Co/SBA16 | 6.9 | 99 | 48 | 31 |
| Co/SBA16 | 9.7 | 99 | 35 | 27 |
| Catalyst | Co/Si (%) | Conversion (%) | 1,2-LO Selectivity (%) | LDO Selectivity (%) | Epoxide Yield (%) |
|---|---|---|---|---|---|
| Co/SBA16 | 1.1 | 99 | 44 | 41 | 85 |
| Co/SBA16 | 3 | 99 | 42 | 31 | 72 |
| Co/SBA16 | 5.2 | 99 | 42 | 28 | 69 |
| Co/SBA16 | 7.5 | 99 | 41 | 25 | 65 |
| Co/SBA16 | 8.8 | 99 | 41 | 25 | 65 |
| Catalysts | Conversion (%) | 1,2-LO (%) | 8,9-LO (%) | LDO (%) | Carvone (%) | Carveol (%) | Others (%) |
|---|---|---|---|---|---|---|---|
| Ru/AC G60 (IWI) | 28 | 21 | 6.5 | 14.6 | 16.7 | 8 | 33.2 |
| Ru/AC MRX (IWI) | 22 | 21 | 6.7 | 13.5 | 17 | 9.1 | 32.7 |
| Ru/AC RB4C (IWI) | 18 | 18 | 6 | 10 | 15 | 7.5 | 43.5 |
| Ru/AC RX3 (IWI) | 21 | 8.7 | 2.6 | 7.9 | 10 | 8 | 62.8 |
| Ru/Graphite (IWI) | 15 | 16 | 4 | 8.5 | 18 | 9.5 | 44 |
| Ru/AC G60 (CE) | 35 | 28 | 10 | 19 | 19 | 6.5 | 17.5 |
| Ru/AC G60 (SI) | 40.4 | 18.5 | 5.5 | 12.8 | 20.7 | 12.9 | 39.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahamat Ahmat, Y.; Madadi, S.; Charbonneau, L.; Kaliaguine, S. Epoxidation of Terpenes. Catalysts 2021, 11, 847. https://doi.org/10.3390/catal11070847
Mahamat Ahmat Y, Madadi S, Charbonneau L, Kaliaguine S. Epoxidation of Terpenes. Catalysts. 2021; 11(7):847. https://doi.org/10.3390/catal11070847
Chicago/Turabian StyleMahamat Ahmat, Yacoub, Sara Madadi, Luc Charbonneau, and Serge Kaliaguine. 2021. "Epoxidation of Terpenes" Catalysts 11, no. 7: 847. https://doi.org/10.3390/catal11070847
APA StyleMahamat Ahmat, Y., Madadi, S., Charbonneau, L., & Kaliaguine, S. (2021). Epoxidation of Terpenes. Catalysts, 11(7), 847. https://doi.org/10.3390/catal11070847