1. Introduction
Considering the necessity to implement efficient methods for the elimination of volatile organic compounds (VOCs) from contaminated air, active and selective catalysts have been extensively developed, which could allow the lowering of the reaction temperature of the total oxidation of VOCs. In general, systems tested so far can be divided into two main types due to the nature of the catalytically active phase, i.e., materials based on noble metals and transition metal oxides. The catalysts containing noble metals (e.g., Pt, Pd, Au or Ag) supported on various oxides (mainly SiO
2 or γ-Al
2O
3) have an obvious advantage due to their extraordinary activity at low temperatures [
1,
2]. However, high production costs and sensitivity to poisoning (by sulfur- and halogen-containing compounds) constitute a serious limitation in their application. A compromise solution is therefore to use transition metal oxides which combine the benefits of low manufacturing costs with relatively high efficiency and enhanced resistance to deactivation. The final performance of these catalysts is influenced by various factors, including properties of active components, dispersion of the active phase, and size and morphology of grains [
1,
2,
3,
4,
5]. Co
3O
4, CuO, MnO
x, Fe
2O
3 and NiO are the most common oxide systems used in the combustion of VOCs. As in the case of noble metals, the involvement of a support also has a positive effect on dispersion and thermal stability of the catalytically active phase. The selection of the appropriate material forming the support depends on many factors (including its porosity, chemical activity and strength of interaction with the active phase), but a typical choice is γ-Al
2O
3, SiO
2, TiO
2, ZrO
2, CeO
2 and zeolites [
1,
5,
6]. Mixed systems containing at least two transition or noble metals in their structure have also been increasingly studied. The effect of improved catalytic activity obtained in such cases is most often related to promoting the mobility of lattice oxygen and facilitating electron transfer [
7].
Particularly promising catalysts for the total oxidation of VOCs could be core–shell composites, which contain a core located inside another domain forming an outer layer [
8,
9]. The structures belonging to the group of core–shell materials also include hollow core–shell and yolk–shell systems [
10]. The advantages of using such composite materials include: (i) ability to protect the core against effects of environmental changes outside the material, (ii) limiting the possibility of increasing the volume and maintaining structural integrity, (iii) protecting the core particles against aggregation into larger clusters, and (iv) selective molecule penetration into the core. The core–shell materials combine properties of both the shell and the core. By covering nanoparticles with one or more layers of other materials, systems with intensified or completely changed physical or chemical properties in relation to the initial structure can be obtained [
11,
12]. Few core–shell structures for VOCs combustion, limited mainly to systems containing noble metal nanoparticles embedded in an oxide shell (e.g., Pd@CeO
2 [
13]), have been described in the literature so far.
Due to availability and the relatively low price of substrates, as well as simplicity and short time of synthesis, polymer templates are often selected as structure-directing agents to obtain core–shell composites by the bottom-up approach [
14,
15]. Oxide layers, e.g., SiO
2, TiO
2, ZrO
2 or CeO
2, are deposited onto the polymer cores, and then the organic interior is easily removed by calcination or chemical etching with acidic, alkaline or organic agents [
16,
17,
18,
19,
20]. In the synthesis of such core–shell systems, among other things, heterophase polymerization (leading to encapsulation of inorganic nanoparticles), sol-gel processes (leading to coating polymer colloids with an inorganic layer) and layer-by-layer deposition are used. Regardless of the choice of the synthesis method, it is necessary to facilitate interaction between a polymer and an inorganic component [
18,
20,
21,
22].
Amines are often used to modify a surface of polystyrene templates [
23]. The amine layer adsorbed on the latex surface induces the formation of the silica layer through electrostatic interactions. Upon dissolution or calcination, the polymer is removed together with the amine, creating a hollow silica sphere. However, the attachment of the amine to the latex surface reduces the integrity of the resulting SiO
2 shell. This effect can be overcome by using silanes or silica nanoparticles as hollow shell precursors, but this drastically increases the cost of synthesis, and the process becomes more complicated.
One of the most frequently used techniques for the preparation of polymer cores is emulsion polymerization. It is a type of radical polymerization which is based on the use of an emulsion containing water, monomer and surfactant. Potassium or ammonium persulfate is widely used as an initiator in the emulsion polymerization [
14]. The process often takes place in an o/w (oil in water) emulsion in which monomer droplets are dispersed in water by surfactants. Other types of polymerization, such as so-called reverse w/o emulsion (water in oil) as well as w/o/w and o/w/o microemulsion, are rarely chosen [
14,
16,
20]. The mechanism of surfactant-free emulsion polymerization has also been described [
24]. The amount of polymer contained in the aqueous phase influences the nucleation mechanism and the change in dispersion of the systems during the polymerization of molecules with different properties [
25,
26]. A common solution used during the polymerization of styrene is an addition of methacrylic or acrylic acid. Ge and co-workers [
15] synthesized poly(styrene-
co-methacrylic acid) spheres by the emulsion polymerization without emulsifiers. The addition of methacrylic acid reduced the size of the spheres formed (to ca. 370 nm) and the thickness of the SiO
2 layer (to ca. 50 nm) compared to the synthesis with the use of a purely polystyrene template. It was unequivocally found that the presence of methacrylic acid in the template has a beneficial effect on the formation of homogeneous silica layers on the surface of latex templates. Thus, in order to increase the degree of uniform bonding of SiO
2 with the surface of polystyrene spheres, they can be functionalized, for example, with acid groups, by copolymerization of styrene with methacrylic acid or sulfonation with sulfuric acid [
14,
15]. The deposition of the SiO
2 shell on the poly(styrene-
co-methacrylic acid) spheres was carried out using various aging conditions—at 150 °C (obtaining materials with a mesoporous shell) or at room temperature (microporous spheres). Moreover, aging at room temperature resulted in a layer of SiO
2 with a thickness of ca. 30 nm, while the higher temperature of the thermal treatment reduced its thickness to ca. 15 nm.
In this paper a pioneering method of synthesis of core–shell catalysts with an active phase in the form of Co3O4 spinel nanoparticles located inside a porous SiO2 shell is described. In the first step of preparation, a spherical copolymer template (namely poly(styrene-co-acrylic acid)) with a defined grain size within the range of 200–300 nm was obtained. The SiO2 coating was deposited on the surface of the latex spheres by performing the hydrolysis and condensation of tetraethyl orthosilicate (TEOS) in a water-isopropanol solution of ammonia. The formed composite particles with a copolymer core retained or removed by calcination were modified with various amounts of cobalt-based active phase. The samples containing Co3O4 nanoparticles were characterized by a wide range of physicochemical techniques, and above all, their activity in the toluene combustion was determined. The role of selected parameters influencing the catalytic performance of the developed composite materials in the studied process is discussed. At the same time, a very significant influence of the proper path selection, resulting in various dispersion of the active component within the Co3O4@SiO2 yolk–shell structure, on the achieved conversion of the organic pollutant is shown.
2. Results and Discussion
The main objective of this study was to obtain composite materials containing Co
3O
4 nanoparticles embedded and protected inside a spherical, porous SiO
2 shell, which would be highly active and selective in the catalytic combustion of VOCs. In the first step of the synthesis, a spherical poly(styrene-
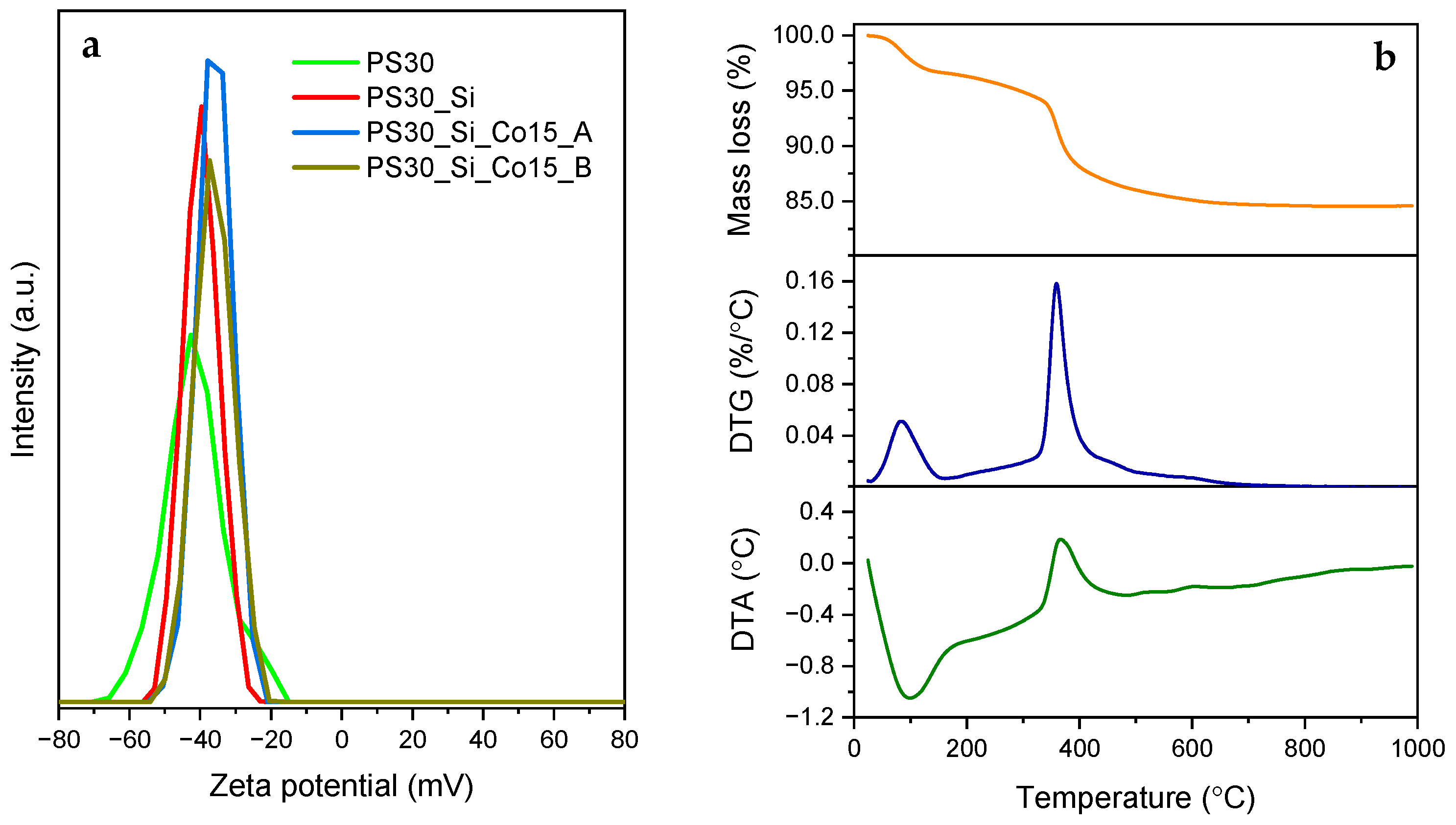
co-acrylic acid) latex containing 30 wt.% acrylic acid in relation to styrene was prepared. The formed PS30 emulsion was examined by dynamic light scattering (DLS), proving the high monodispersity of the particles with diameters within the narrow range of 240–260 nm and the average ζ potential of −42.8 mV measured in deionized water at pH = 7 (
Figure 1a). The latter value is related to the high amount of carboxyl groups on the surface of the copolymer nanoparticles and confirms high colloidal stability of the formed emulsion [
27].
The prepared latex spheres were further modified by the deposition of the silica shell using the Stöber strategy. The coverage of the PS30 surface with the silica coating slightly shifts the average ζ potential to −39.6 mV (
Figure 1a). This change indicates a variation in the chemical composition of the surface. In this case, the negative zeta potential mainly comes from the ionization of silanol groups [
28]. However, since the zeta potential value remains outside the range from −30 mV to +30 mV, it should be considered that the PS30@SiO
2 nanoparticles have sufficient repulsion force to maintain colloidal stability [
29]. In the literature, the correlation between the SiO
2 particle size and the zeta potential value was discussed [
30]. It was postulated that as the size of the spheres decreased, the average ζ potential raised and oscillated in the range from −37.7 mV to −45.5 mV.
The introduction of the inorganic part was additionally confirmed by thermal analysis (
Figure 1b). The mass loss of 3.2% in the temperature range up to approx. 140 °C is associated with removal of residual solvents, unreacted monomers and physically adsorbed water. Desorption of these components is related to the heat consumption, which confirms the endothermic peak observed by differential thermal analysis (DTA). At higher temperatures, the parts of the chain of acrylic acid units slowly decompose and polystyrene depolymerizes. Rapid decomposition begins at ca. 320 °C with a maximum rate at 360 °C. This exothermic step is mainly due to the combustion of the polymer. After the analysis, the copolymer-free SiO
2 remains, the content of which in the PS30_Si material is estimated to be 84.5 wt.%.
The obtained PS30_Si composite was calcined at the oxidizing atmosphere to remove the copolymer core and subsequently studied using scanning electron microscopy (SEM). The SEM image shown in
Figure 2a confirms a well-formed spherical structure of the grains. The particles are homogeneous, and their average size is close to ca. 300 nm. This means that, taking into account the size of the latex spheres determined earlier by DLS (240–260 nm), the thickness of the SiO
2 layer is approx. 20–30 nm.
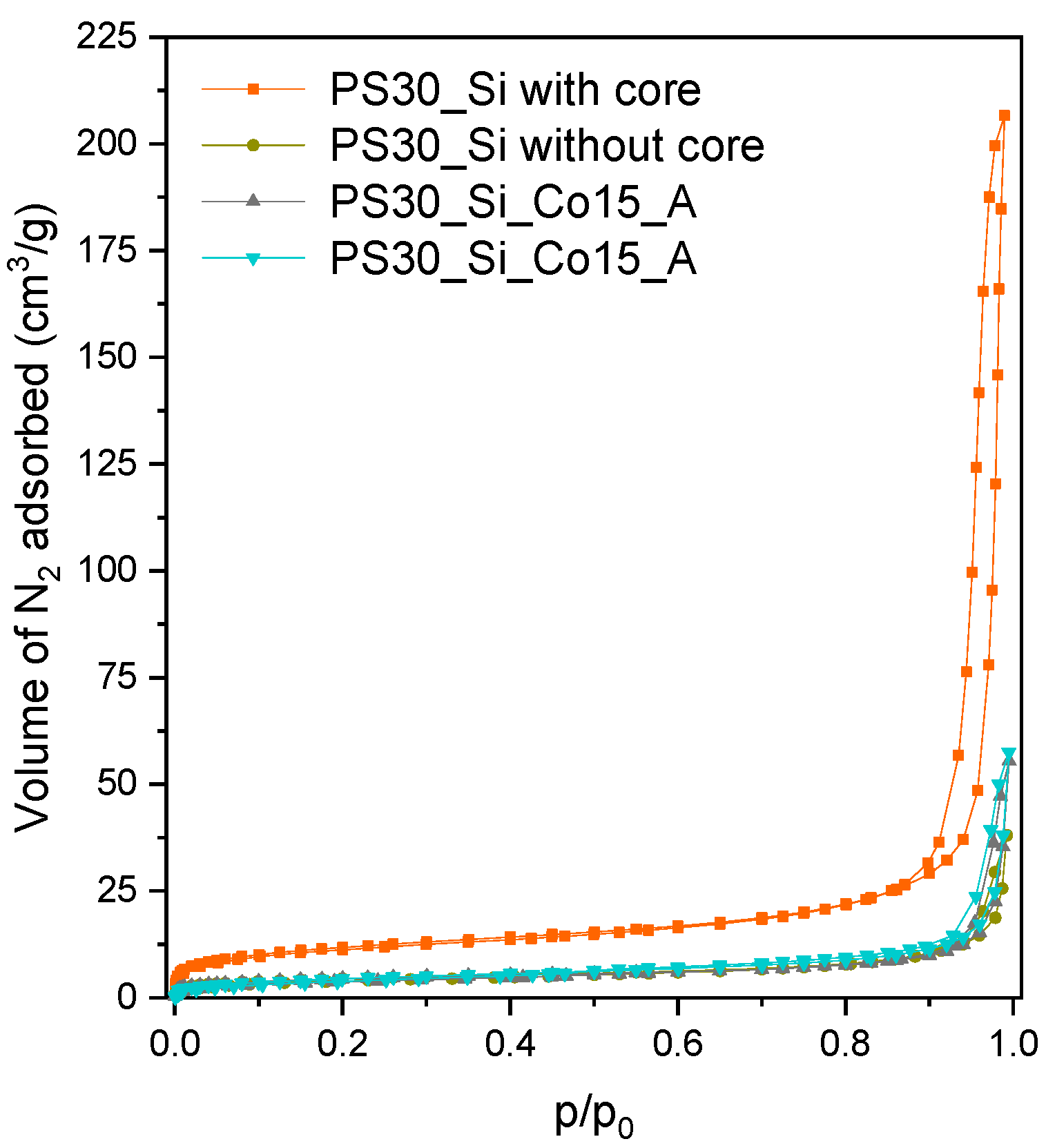
Simultaneously, textural properties of PS30_Si before and after removal of the copolymer core were analyzed. The measured isotherms of N
2 adsorption, shown in
Figure 3, reveal a very interesting conclusion. The shape of the isotherm for the starting PS30@SiO
2 structure can be classified as type II, according to the IUPAC (International Union of Pure and Applied Chemistry, Research Triangle Park, NC, USA) nomenclature, which is characteristic of materials containing pores with diameters within the macroporosity range. It is manifested by a very intense adsorption effect observed at the relative pressure p/p
0 > 0.9. Interestingly, after removing the PS30 template, this effect largely disappears. It should therefore be assumed that the adsorption of N
2 prior to the core elimination corresponds to the condensation of the adsorbate in slits formed between the silica shell and the copolymer core, which during drying shrinks slightly, forming the free spaces. These observations are reflected in the determined textural parameters. For the PS30_Si filled by the copolymer core, the total pore volume (V
total, constituted mainly by wide meso- and macropores) is 0.319 cm
3/g, and consequently the surface area is determined according to the Brunauer–Emmett–Teller theory (S
BET) is 41 m
2/g (
Table 1). The destruction of the copolymer scaffold results in a significant decrease in V
total and S
BET to 0.059 cm
3/g and 15 m
2/g, respectively, since the voids formed inside the hollow SiO
2 spheres are too large for the adsorbate to condense therein.
X-ray fluorescence (XRF) analyses were carried out to confirm the introduction of the Co-containing active phase onto the silica support. The determined Co loadings are presented in
Table 1. It should be stated that the chemical composition of the studied materials is close to the intended values.
The modification of PS30@SiO
2 with the Co
3O
4 phase causes a further change in average ζ potential to a value of ca. −37 mV which is very similar for the calcined PS30_Si_Co15 samples prepared both on path A and B (
Figure 1a). Therefore, it should be stated that a part of the Co modifier was deposited on the outer surface of SiO
2 nanospheres, changing its charge a little.
Figure 2b,c demonstrate the SEM images collected for the calcined PS30_Si_Co15 obtained in the paths A and B, respectively. For PS30_Si_Co15_A no significant differences in morphology are observed compared to the hollow SiO
2 spheres (
Figure 2a). In the case of PS30_Si_Co15_B, a greater aggregation of nanoparticles is noted. They form distinct clusters, and consequently polydispersity increases significantly.
In
Figure 3 two examples of the N
2 adsorption isotherms for the samples with the highest Co loadings are demonstrated, while the textural parameters for all synthesized Co-loaded samples are summarized in
Table 1. It can be concluded that the introduction of cobalt oxide results in a slight increase in the pore volume compared to the hollow SiO
2 spheres. This effect is also correlated with the amount of active phase deposited—the more it is, the greater value of V
total. Therefore, it should be assumed that the cobalt oxide nanoparticles formed as a result of calcination create secondary porosity. These nanograins are arranged partly on the outer surface of the SiO
2 nanospheres, partly inside their pores, but above all on the inner surface of the SiO
2 shells. In the case of the synthesis by the path B, the deposition of the active phase precursor takes place in the completely empty interior of the SiO
2 spheres compared to the path A, in which the Co(II) nitrate solution is introduced into the spaces between the shell and the copolymer core and only to a limited extent inside the PS30 template structure. This leads to different contents of the active phase precursor inside the spheres, and thus various arrangements of the formed oxide nanograins. For the PS30_Si_Co_B materials, they are a bit more loosely aggregated, and therefore in this case the V
total values are slightly higher than in the case of the analogous PS30_Si_Co_A catalysts.
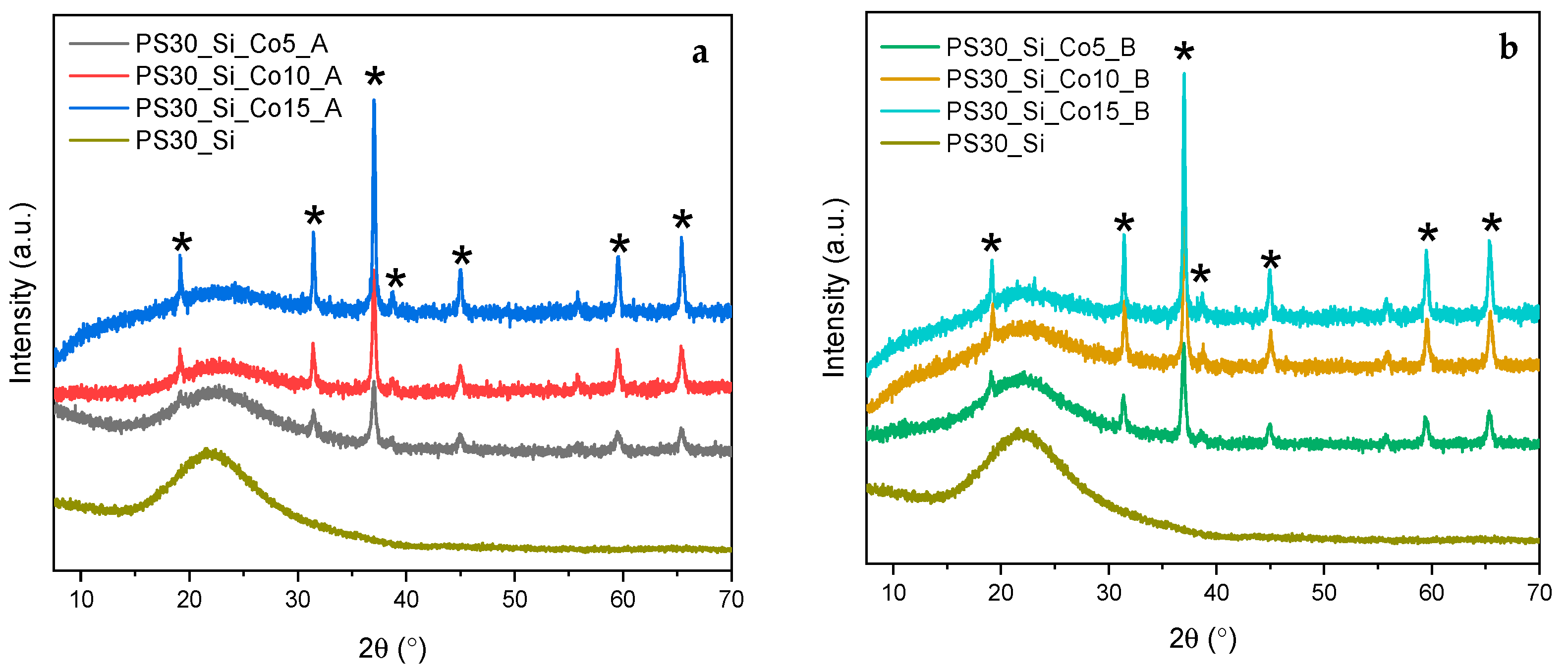
The crystalline structure of the synthesized materials was studied by X-ray diffraction (XRD).
Figure 4a,b present the diffractograms registered for the PS30_Si_Co_A and PS30_Si_Co_B catalysts, respectively.
The diffraction patterns clearly display a broad, low, intense diffraction peak (101) at 2θ~22°, originating from the poorly ordered silica in the composite shells [
31]. The presence of the transition metal oxide in the structure of the developed materials is confirmed by other characteristic reflections observed at 2θ equal to 19.0°, 31.2°, 37.0°, 38.5°, 44.8°, 59.3° and 65.2° with intensities growing with the increasing Co content. They correspond to (111), (220), (311), (222), (400), (511) and (440) lattice planes in the crystal structure of Co
3O
4 spinel phase (space group
Fd3
m), respectively (PDF 00-009-0418). The calculated values of the parameter a of 8.06 Å and the unit cell volume of 523.2 Å are only slightly lower compared to the theoretical data of 8.084 Å and 528.30 Å, respectively [
32]. The mean Co
3O
4 crystallite sizes determined on the basis of the Scherrer equation from the broadening of the most intense (311) diffraction are given in
Table 1. A very clear correlation between the Co
3O
4 mean crystallite size and the Co content for the materials in both series is observed.
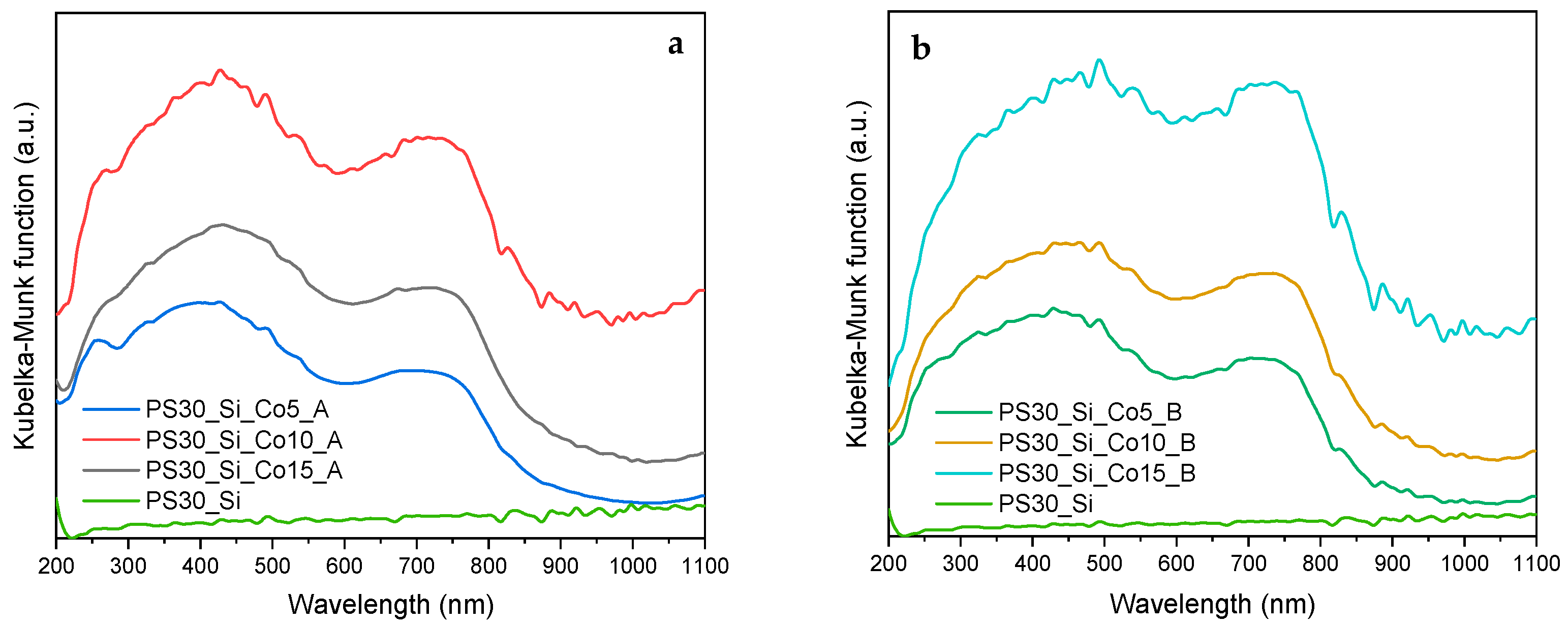
In order to determine the chemical environment of the active phase in the investigated catalysts, UV-Vis-DR measurements were performed. The collected spectra are shown in
Figure 5a,b. The results for both series of the catalysts are very similar, and fully confirm the presence of the Co
3O
4 spinel phase. Around 260 nm, the band corresponding to the O
2− → Co
3+ charge transfer is distinguished. However, the main bands occur at higher wavelengths. Their width is indicative of different ordering of spinel particles, but generally the absorption band in the range of 300–500 nm is attributed to octahedrally coordinated Co
3+ ions, whereas another wide band at 600–800 nm is assigned to the electron transitions characteristic for Co
2+ ions in tetrahedral coordination in the Co
3O
4 spinel [
33,
34,
35].
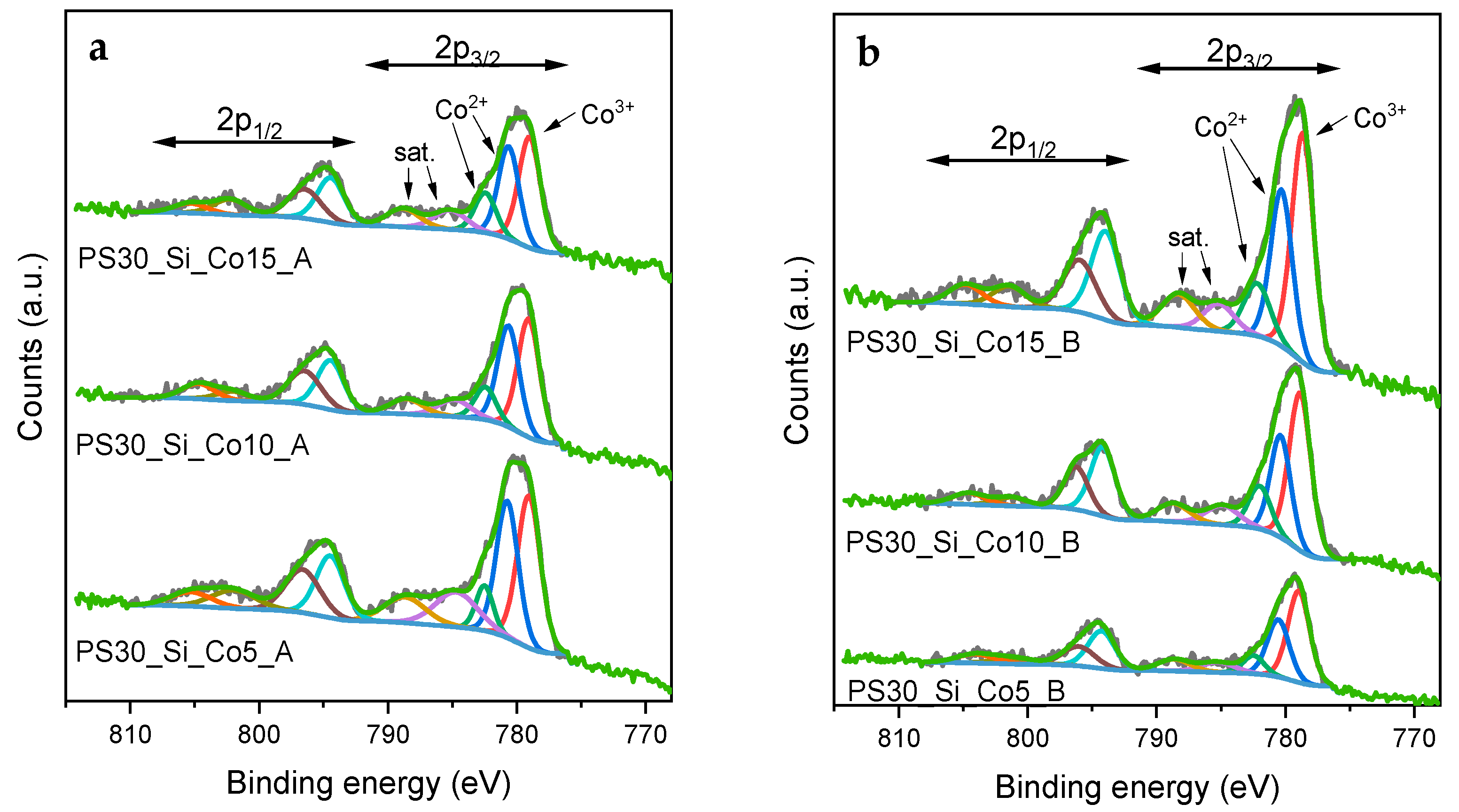
The surface composition of the Co-loaded materials synthesized in the paths A and B was more precisely characterized by X-ray photoelectron spectroscopy (XPS). The high-resolution XPS spectra recorded in the Co 2p region (
Figure 6a,b) display a characteristic doublet of the Co 2p
3/2 and Co 2p
1/2 peaks, which is related to the spin-orbital splitting. The determined distance between the peaks in this doublet for all studied materials is Δ = 15.3–15.4 eV, which is the typical value reported for the Co
3O
4 phase in the literature [
36,
37].
The studied materials are characterized by the presence of Co
3+ ions in octahedral coordination (779.0 ± 0.3 eV) and Co
2+ in tetrahedral coordination (780.6 ± 0.2 eV) [
38]. In the fitting model used, an additional component related to Co
2+ multiplet splitting at 782.4 ± 0.3 eV was added [
38,
39,
40]. In order to describe the surface composition in more detail, the Co
2+/Co
3+ ratio was calculated based on the performed deconvolution. The values for the PS30_Si_Co_A catalysts are close to 1.0, while for PS30_Si_Co_B they are at the level of 0.8, which indicates that in the latter case the Co
3+ ion content is higher. The presence of more Co
3+ ions usually enhances the reducibility and increases the activity of the Co
3O
4 spinel phase in the catalytic combustion of VOCs [
41,
42]. The calculated ratio of the Co concentration on the surface (from the XPS results) to that in the total volume (from the XRF results) provides clear differences in the cobalt deposition on the surface of the SiO
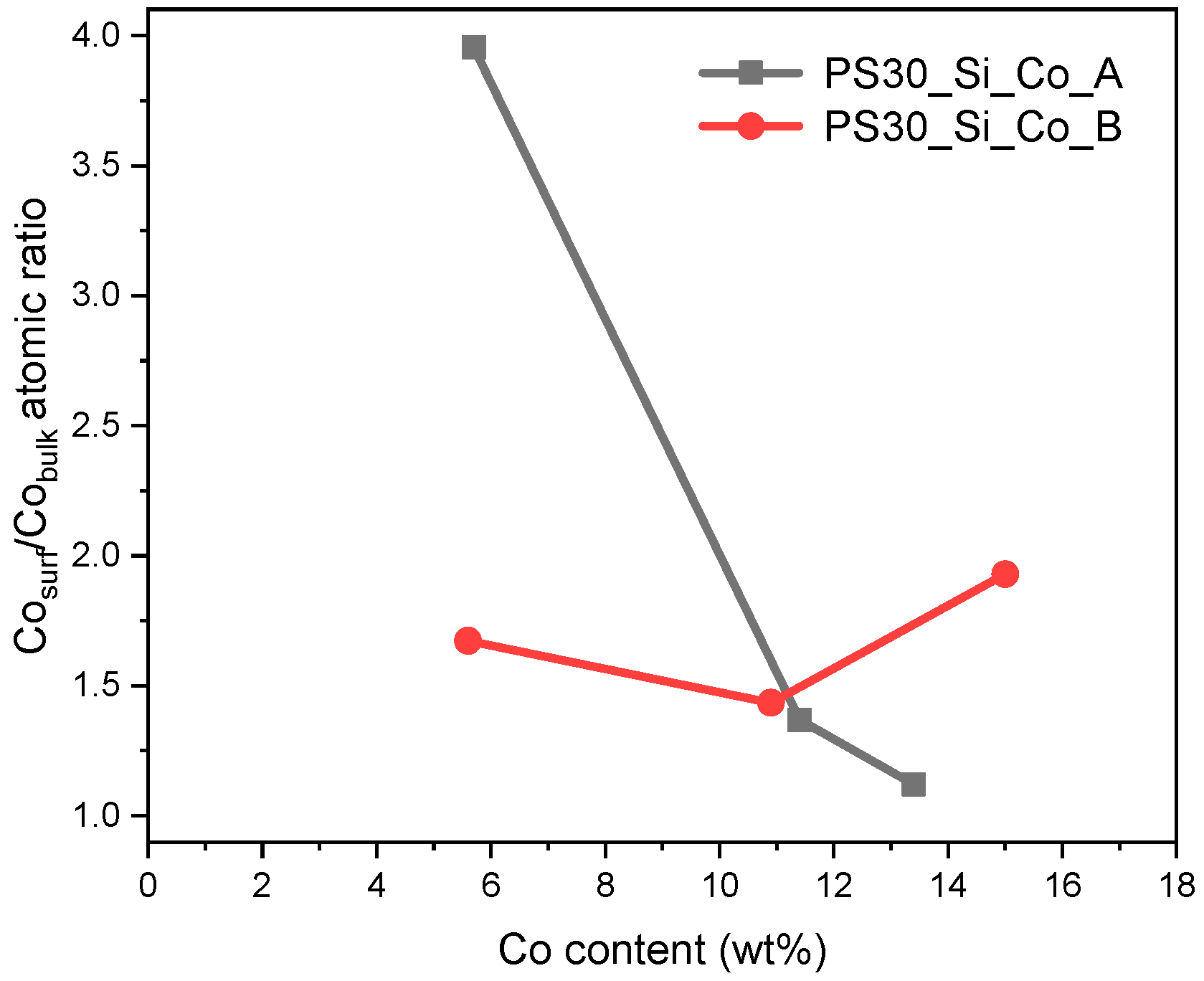
2 spheres (
Figure 7). For the PS30_Si_Co_B catalysts, the enrichment of the surface with cobalt ranges from 40% to 90%. In the case of the catalysts obtained in path A, there is a clear tendency to change the distribution along with the Co content. The PS30_Si_Co5_B sample has almost four times more Co on the surface than in the volume, while for higher Co contents, the enrichment of the surface with the active phase significantly decreases (for PS30_Si_Co15_B, the Co content on the surface is only 10% higher than in the entire volume).
The activity of oxide catalysts in the total oxidation of VOCs is often directly related to their redox properties, as the reaction involves lattice oxygen from the subsurface layers and even the interior of the solid phase [
43]. Susceptibility to reduction oxides more easily creates surface defects, which promotes catalytic activity [
44]. Therefore, we decided to use the temperature-programmed reduction (H
2-TPR) technique to study the reducibility of the developed catalysts (
Figure 8a,b).
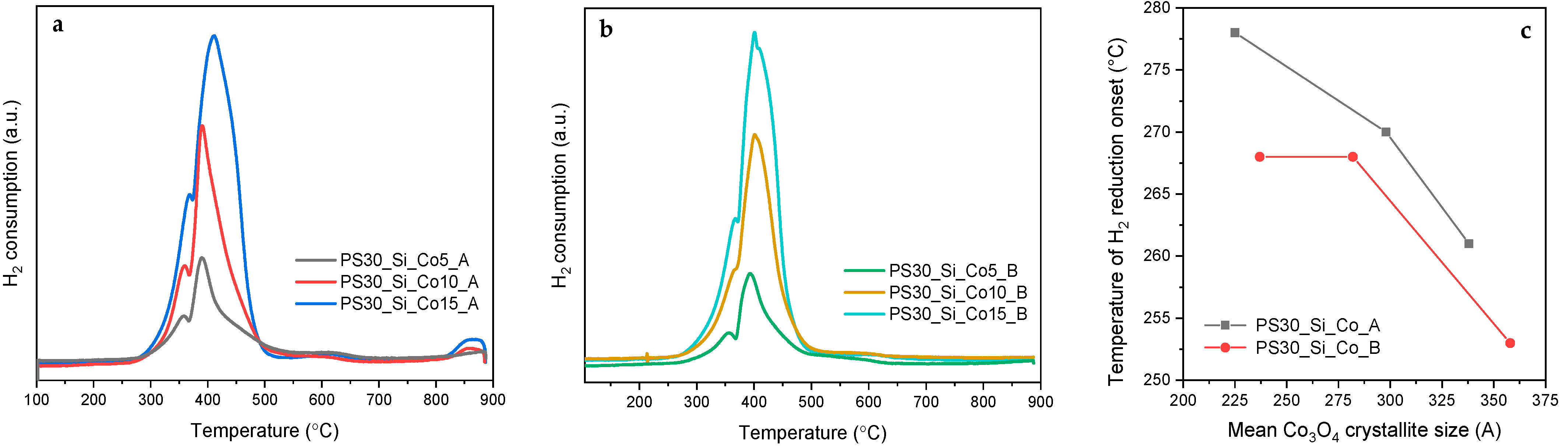
Mandal et al. [
45] postulated that large Co
3O
4 particles are typically reduced directly to metallic cobalt in one step, while the reduction of Co
3O
4 nanoparticles is often a two-step process. In the case of the tested materials, two main reduction peaks in the H
2-TPR profiles are observed, which confirm the nanometric dimensions of the active phase particles. The reduction of Co
3O
4 spinel proceeds through two steps described by the following reaction equations:
The first reduction step begins in the temperature range of 250–280 °C and reaches its maximum rate at about 360–370 °C. A deeper reduction in the active phase occurs at higher temperatures (a maximum of greater intensity at approximately 390–410 °C). For both series, an increase in the cobalt content in the studied catalysts results in an increase in the intensity of the reduction maxima and their shift towards higher temperatures. These effects are rather expected, since the higher content of the component to be reduced requires the consumption of more hydrogen, and on the other hand, kinetic circumstances lead to the achievement of the maximum rate of the polythermal reduction process at increasingly higher temperatures.
However, very interesting conclusions can be deduced by analyzing the temperatures of reduction onset determined for the individual catalysts (
Figure 8c). These values were determined based on an increase in a detector signal to a value exceeding by 1% a baseline value determined in the temperature range of 200–220 °C, in which no reaction resulting in the consumption of H
2 occurs. It is clearly visible that the reduction process begins at lower temperatures in the case of larger Co
3O
4 crystallites, the formation of which is favored by the higher Co content. Nevertheless, it is not the most important parameter responsible for easier reducibility. Clearly, the PS30_Si_Co_A catalysts require higher temperatures to initialize the reduction process than the analogous PS30_Si_Co_B materials, containing crystallites of similar sizes and comparable Co loadings. It should therefore be assumed that the observed differences result from a different distribution of Co
3O
4 nanoparticles, which in turn are varied by the synthesis procedure. In the case of PS30_Si_Co_A, the infiltration of the Co(NO
3)
2 solution leads to the aggregation of more active phase nanoparticles on the inner walls of the SiO
2 spheres, which closely adhere to the surface of the support stabilizing them. Differently, the introduction of the Co(NO
3)
2 solution into the hollow SiO
2 spheres provides a larger space for crystallization and the subsequent transformation of the active phase precursor to the oxide form. Hence, in this case, the Co
3O
4 crystallites are loosely bonded to the silica support and thus are easier to be reduced. The shift of the reduction onset towards lower temperatures with the activation of lattice oxygen should result in higher catalytic activity in the combustion of VOCs [
38,
46].
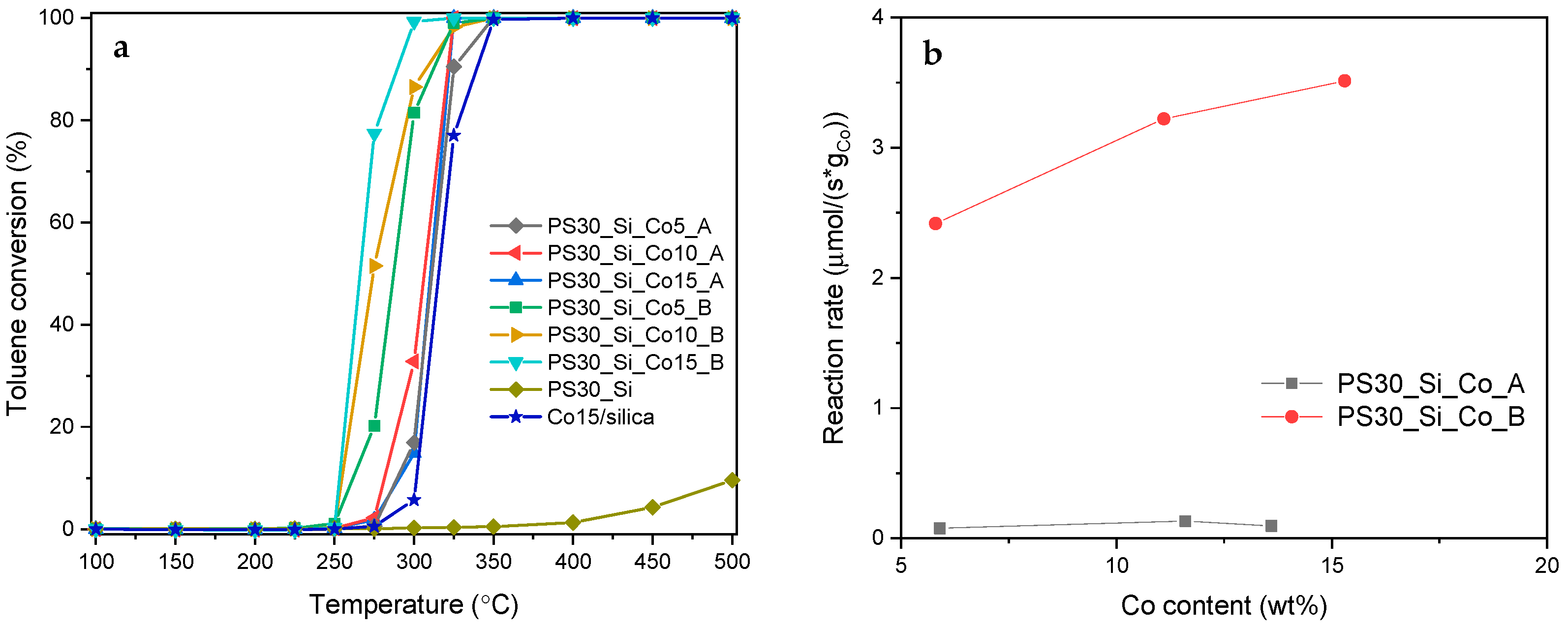
The Co
3O
4-containing catalysts synthesized on the basis of the PS30_Si composite template were tested for the catalytic activity in the total oxidation of toluene (
Figure 9a). The catalysts of both series A and B showed high activity in this reaction, the main products of which were CO
2 and H
2O. Selectivity to CO
2 remained at an extremely high level. For each material, regardless of the reaction temperature, it was > 99.9%. In only selected cases, the formation of traces of benzene, as a by-product, was detected. It is observed that the PS30_Si_Co_A catalysts demonstrate very similar activity, while the materials calcined prior to the inclusion of the cobalt precursor (PS30_Si_Co_B) exhibit apparent differences in the catalytic performance. Nevertheless, it should be noted that for the PS30_Si_Co_B catalysts, which are characterized by easier reducibility as previously shown by the H
2-TPR measurements, significantly better catalytic results were achieved. The toluene conversion at the level of 50% for the most active catalysts of both series was attained at temperatures of 306 °C and 266 °C for PS30_Si_Co10_A and PS30_Si_Co15_B, respectively (
Table 2). The extraordinary catalytic activity of the PS30_Si_Co_B catalysts can be found even better when the reaction rate (r) is compared:
where
Ftoluene represents the flow rate of toluene (mol/s),
Ctoluene is the toluene conversion, and
mCo is the content of Co in the working catalyst. The results collected for the experimental points measured at 275 °C are shown in
Figure 9b.
It should therefore be emphasized that the procedure used to introduce the active phase is of crucial importance in profiling the catalytic activity of Co3O4@SiO2 nanorattles. Due to the larger available nanospace for the arrangement of the active phase grains, much more favorable effects are obtained by the deposition of the Co3O4 precursor in the empty SiO2 spheres. In this case, the absence of strong interactions between the spinel phase crystallites and the silica support is confirmed even for the content of 15 wt.% of Co. Furthermore, the oxide particles do not aggregate significantly and finally the highest reaction rate is reached.
In order to verify the real usefulness of the developed catalysts in the process of toluene combustion, the catalytic test was also carried out for the material prepared using commercial spherical silica gel, which was modified by introducing 15 wt.% of Co with an analogous procedure as for the PS30_Si_Co samples. As can be seen from
Figure 9a, the reference Co15/silica sample shows a lower activity than all developed yolk–shell catalysts. Compared to the most active PS30_Si_Co15_B material, this difference is huge—the T
50 value is reached at a temperature approx. 50 °C lower.
On the other hand, additional measurements for the most active PS30_Si_Co15_B catalyst were performed to show its stability and behavior at much higher toluene content (the toluene content in the feed was increased from 1000 ppm to 2500 ppm). After the catalytic run in the temperature range of 100–500 °C in the presence of 1000 ppm of toluene in the flowing air (cycle 1), the reactor was cooled down to 100 °C and the measurement was repeated (cycle 2). After cycle 2, the toluene concentration in the feed was increased to 2500 ppm and two subsequent polythermal runs (cycle 3 and 4) were carried out. The results presented in
Figure 10 clearly confirm high stability of the developed material in the repeated runs. Only a slight decrease in the toluene conversion at 275 °C is found. Moreover, an increase in the toluene concentration results in a noticeable decrease in its conversion from 77.4% (cycle 1 at 1000 ppm) to 38.4% (cycle 3 at 2500 ppm) at this temperature. At 300 °C these differences are not so important, and starting from 325 °C, practically complete conversion is achieved regardless of the toluene concentration.
To conclude, the presented results clearly indicate the enormous potential resulting from the location of the oxide phase inside the SiO2 shell, where properly protected, can actively, selectively and stably act in the total oxidation of VOCs.
3. Materials and Methods
3.1. Chemicals
Styrene (Sigma-Aldrich, Germany, >99.0%), acrylic acid (Acros Organics, France, 98%, extra pure), potassium persulfate (Chempur, Piekary Śląskie, Poland, pure p.a.), tetraethyl orthosilicate (TEOS, Sigma-Aldrich, China, ≥99.0%), ammonia solution (28–30%, J.T. Baker, Philipsburg, NJ, USA), isopropanol (Stanlab, Lublin, Poland, pure), cobalt(II) nitrate(V) hexahydrate (Honeywell, Seelze, Germany, 98%), toluene (Honeywell, Seelze, Germany, puriss p.a.) and nitrogen (Air Products, Świerzewo, Poland, grade 5.2).
3.2. Synthesis
Poly(styrene-
co-acrylic acid) latex spheres were prepared by emulsion polymerization [
47]. Briefly, 360 mL of distilled water was introduced into a 1000 mL three-neck round-bottom flask equipped with a mechanical stirrer (300 rpm), a reflux condenser and a N
2 dosing tube. Then, acrylic acid (1.6 g) and styrene (6.4 g) were added slowly by dropping it into the flask, and the mixture was deoxygenated by bubbling with nitrogen gas for 30 min. The temperature was raised to 70 °C and an aqueous solution of potassium persulfate (0.24 g in 40 g water) was poured to initialize the polymerization reaction at a constant mixing speed of 300 rpm. After 24 h the reaction mixture was allowed to cool down to room temperature.
In the next step, the surface of the obtained poly(styrene-co-acrylic acid) spheres (coded as PS30) was coated with a SiO2 layer. For this purpose, 5 mL of the mixture containing the synthesized latex was placed along with 35 mL of distilled water and 200 mL of isopropanol in a 1000 mL vessel. After 15 min, 5 mL of ammonia solution and 4.5 mL of TEOS were added dropwise while vigorously stirring the mixture using a mechanical stirrer (250 rpm). The mixing was continued for another 3 h. Subsequently, the solid was centrifuged, rinsed twice with distilled water and once with isopropanol, and finally dried at 80 °C. The formed core–shell material is named as PS30_Si.
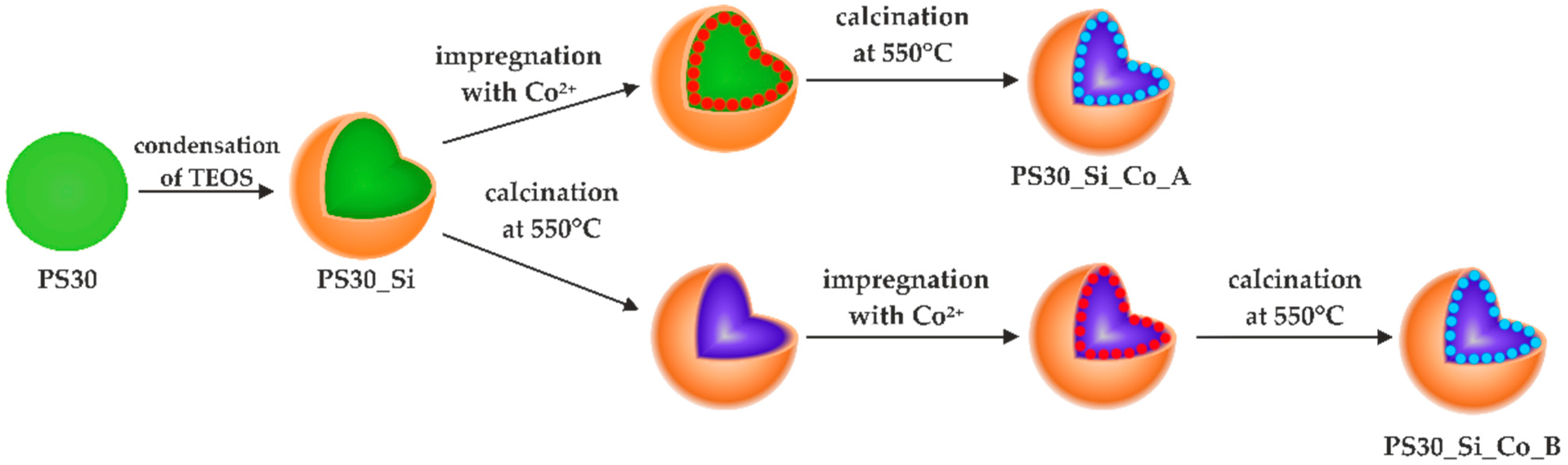
The synthesized composite was modified with Co using two different procedures (path A and B) shown schematically in
Scheme 1. The series A was prepared by infiltrating the active phase precursor into the previously obtained PS30_Si material, drying and then calcinating. In the case of the series B, the copolymer core was first removed by calcination, then the active phase precursor was introduced. The resulting material was dried and calcined again.
The preparations of both series were impregnated with aqueous solutions of Co(NO3)2 with concentrations that allowed Co loadings of 5, 10 and 15 wt.% in the final catalysts. Appropriate amounts of Co(NO3)2·6H2O were dissolved in distilled water and added to 1 g of the support placed in crucibles. After thorough mixing, the samples were allowed to dry at 80 °C. Finally, the materials were calcined at 550 °C for 4 h (heating rate of 1 °C/min) in order to remove the polymer core and form the Co3O4 spinel phase.
3.3. Characterization
Surface morphology was investigated by SEM imaging using a Hitachi S-4700 field emission scanning electron microscope at an accelerating voltage of 20 kV. Samples were mounted on sticky carbon discs and coated with a gold layer. Secondary electron (SE) signal was used for observations.
Particle size distribution and average ζ-potential were determined by DLS measurements in a Zetasizer Nano ZS instrument (Malvern Instruments) equipped with a He-Ne laser source (λ = 633 nm). Prior to analysis, a suspension containing 0.1 wt.% of a material was prepared using deionized water and sonicated in an ultrasonic bath for 30 min.
Chemical composition was studied by XRF using an ARL Quant’x (Thermo Scientific) spectrometer, whereas polymer and inorganic contents in the PS30_Si composite were determined by thermal analysis. About 10 mg of the sample was tested in a TA Instruments SDT Q600 thermobalance in the range of 20–1000 °C at a linear heating rate of 20 °C/min in flowing air flow (100 mL/min).
The structure of samples was examined by XRD. The XRD patterns were collected on a Bruker D2 Phaser instrument using Cu Kα radiation (λ = 1.54184° Å) and a LYNXEYE detector within a 2θ range of 5–70° at a step of 0.02°.
Textural properties were calculated from low-temperature N2 adsorption isotherms measured at −196 °C using a Micromeritics ASAP 2020 sorptometer. Samples were initially outgassed at 250 °C for 5 h under vacuum.
UV-Vis spectra were collected in a Thermo Scientific UV-VIS Evolution 200 double-beam spectrometer equipped with a xenon lamp and a diffuse reflectance accessory, enabling analyses of solids. The spectra were recorded in the wavelength range of 190–1100 nm, collecting 60 scans per min.
Surface analyses were performed by XPS in an ultra-high vacuum system manufactured by Prevac. The XPS spectra were collected using a monochromatized aluminum source AlKα (E = 1486.6 eV) and a hemispherical analyzer (VG SCIENTA R3000). The binding energy scale for the non-conductive samples was calibrated by referring to a position of Si 2p (Eb = 103.0 eV). The Shirley background and fitting with the mixed Gauss–Lorentz function (GL = 30) were used during interpretation of the spectra in the CasaXPS software.
H2-TPR measurements were performed in a self-made equipment. An amount of 50 mg of a dried sample was placed in a quartz reactor and heated from room temperature to 900 °C at a linear temperature increase of 10 °C/min in a mixture of Ar/H2 (95/5 vol.%), which was passed through the bed of the analyzed sample at a total flow rate of 40 mL/min. Hydrogen concentration in the gas mixture was analyzed by a thermal conductivity detector (TCD) and recalculated into consumption by the reduction reaction based on calibration with CuO as a reference.
3.4. Catalytic Activity
Catalytic combustion of toluene was studied as a model reaction in a quartz microreactor with an inner diameter of 8.0 mm. Before starting a measurement, a catalyst (0.1 g, particle size 160–315 μm) was degassed at 500 °C for 30 min in a stream of air (flow rate = 100 mL/min). After that time, the reactor was cooled to 100 °C and toluene dosing (at concentration of 1000 ppm) to flowing air was started. The organic substrate was introduced into the feed using a thermostated scrubber filled with the liquid compound kept at −7.5 °C ensuring its vapor pressure at the assumed level. The catalytic tests were carried out in the temperature range of 100–500 °C. The reactor was held at each temperature step for 80 min during which three analyses of the reaction products were performed. The reaction products were analyzed using a Bruker 450 gas chromatograph equipped with two capillary columns (Porapak S and Chromosorb WAW-DMCS), two flame ionization detectors, a thermal conductivity detector and a methanizer.


 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}