

Pd-Catalyzed Hirao P–C Coupling Reactions with Dihalogenobenzenes without the Usual P-Ligands under MW Conditions

 ,
,  and
and 
Abstract
:
1. Introduction

{kind=link}
{kind=link}
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | ArX1X2 | P-Reagent | Catalyst (Precursor)/Ligand | Additional Conditions | Base | Solvent | T (°C) | Isolated Yield (%) | Ref. |
| 1 | 1-Br-4-IC6H4 | Ph2P(O)H | Pd(PPh3)4 | – | NEt3 | PhMe | 110 | 85 | [21] |
| 2 | 1-Br-4-IC6H4 | (EtO)2P(O)H | Pd(PPh3)4 | MW | Cs2CO3 | THF | 120 | 19 | [22] |
| 3 | 1-Br-4-IC6H4 | (EtO)2P(O)H | Pd(OAc)2/dppf | KOAc additive | NEt3 | THF | 68 | 48 | [15,23] |
| 4 | 1-Br-2-IC6H4 | Ph2P(O)H | Pd(dba)2/dppp | – | DIPEA | PhMe | 120 | 65 | [24] |
| 5 | 1-Br-2-IC6H4 | Ph2P(O)H | Pd2(dba)3/dppp | – | DIPEA | PhMe | 90 | 64 | [25] |
| 6 | 1-Br-2-IC6H4 | (EtO)2P(O)H | Pd(OAc)2/PPh3 | – | DIPEA | EtOH | reflux | 74 | [26] |
| 7 | 1-Br-4-IC6H4, | Ph2P(O)H | Ni(cod)2/dtbbpy, Ru(bpy)3Cl2·6H2O | blue LED | Cs2CO3 | MeOH | 26 | 81 | [29] |
| 8 | 1-Br-4-IC6H4 | (EtO)2P(O)H | CuI/phen | – | Cs2CO3 | PhMe | 100 | 84 | [31] |
| 9 | 1-Br-4-IC6H4 | (EtO)2P(O)H | CuI/proline or pipecolonic acid | – | Cs2CO3 | PhMe | 110 | 86/85 | [32] |
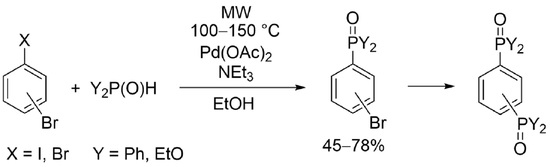
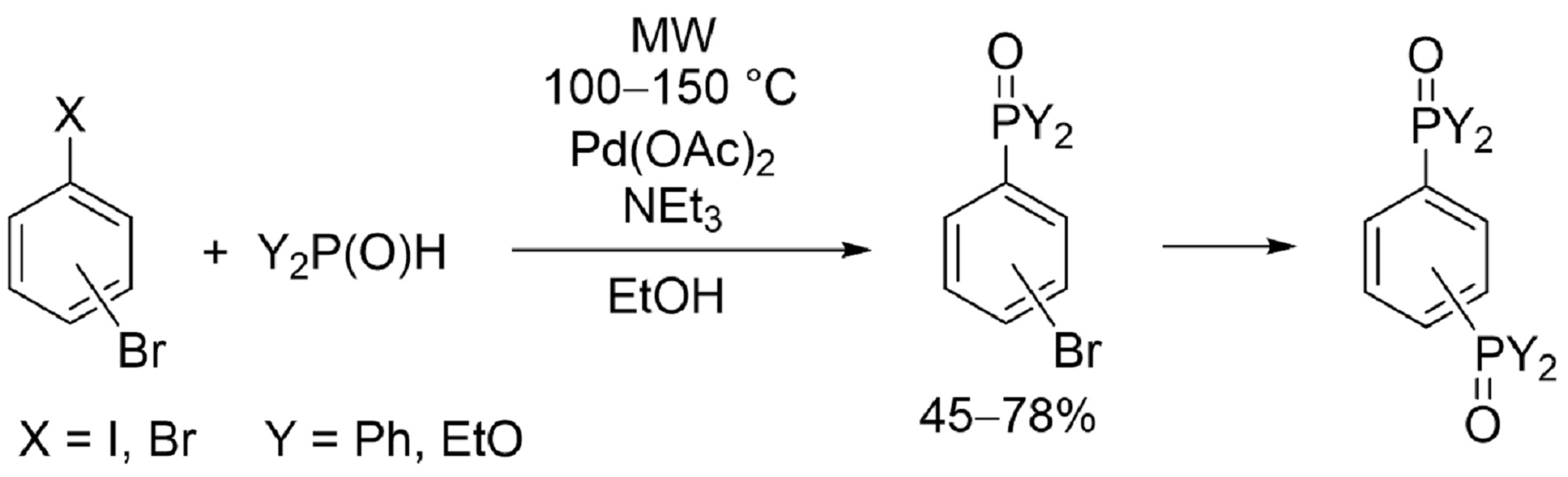
2. Results and Discussion
3. Experimental
3.1. General Information
3.2. Procedures for the P–C Coupling of 1,4- or 1,3-Dibromobenzene and Diphenylphosphine Oxide or Diethyl Phosphite (Table 2, Entries 1 and 3; Table 4, Entries 1, 2 and 4)
3.3. Procedures for the P–C Coupling of 4-Bromo- or 3-Bromo-Iodobenzenes and Diphenylphosphine Oxide or Diethyl Phosphite (Table 3, Entries 2, 4 and 5; Table 5, Entries 1, 3 and 4)
3.4. Procedure for the P–C Coupling of 3-Chloro-Bromobenzene and Diphenylpshosphine Oxide or Diethyl Phosphite (Table 6, Entry 1 and 2)
3.5. The Preparation of Diethyl 2-Bromophenylphosphonate 6b (Table 8, Entry 4)
3.6. The Synthesis of 4-Diethylphosphonoylphenyl-Diphenylphosphine Oxide 8 (Scheme 1)
3.7. The Procedure for the P–C Coupling of 3-Bromophenyl-Diphenylphosphine Oxide 3a with Diphenylphosphine Oxide and Diethyl Phosphite (Table 9, Entry 1 and 2)
3.8. Spectral Data for the Compounds Isolated
- (4-Bromophenyl)-diphenylphosphine Oxide (1a) (Table 2, Entry 1 and Table 3, Entry 2). Appearance: white crystals, mp. 157–158 °C; 31P NMR (CDCl3, 202.4 MHz) δ 28.5, δP [29] (CDCl3, 162 MHz) 25.2, δP [35] (CDCl3, 162 MHz) 28.7; 13C NMR (CDCl3, 125.7 MHz) δ 133.6 (d, J = 10.6, C2)a, 132.2 (d, J = 2.8, C4′), 132.1 (d, J = 104.9, C1′), 132.0 (d, J = 9.9, C2′)b, 131.84 (d, J = 12.5, C3)a, 131.76 (d, J = 104.3, C1), 128.7 (d, J = 12.2, C3′)b, 127.2 (d, J = 3.4, C4), a,b may be reversed, δC [29] (CDCl3, 100 MHz) 133.5 (d, J = 10.5), 132.1 (d, J = 2.8), 131.9 (d, J = 104.4), 131.9 (d, J = 10.0), 131.7 (d, J = 12.4), 131.6 (d, J = 107.1), 128.6 (d, J = 12.1), 128.1 (d, J = 3.4), δC [35] (CDCl3, 100 MHz) 133.6 (d, J = 10.6), 132.1 (d, J = 2.7), 132.0 (d, J = 10.0), 131.9 (d, J = 104.6), 131.8 (d, J = 12.4), 131.3 (d, J = 45.6), 128.6 (d, J = 12.2), 127.2 (d, J = 3.3); 1H NMR (CDCl3, 500 MHz) δ 7.66–7.59 (m, 6H), 7.57–7.51 (m, 4H), 7.48–7.45 (m, 4H), δH [29] (CDCl3, 600 MHz) 7.72–7.59 (m, 6H), 7.58–7.52 (m, 4H), 7.49–7.46 (m, 4H), δH [35] (CDCl3, 400 MHz) 7.68–7.65 (m, 3H), 7.63–7.62 (m, 2H), 7.60–7.58 (m, 2H), 7.56–7.51 (m, 4H), 7.49–7.47 (m, 3H); [M + H]+ = 357.0045 C18H15OPBr requires 357.0044.
- Diethyl 4-Bromophenylphosphonate (1b) (Table 2, Entry 3 and Table 3, Entry 4). Appearance: colorless oil; 31P NMR (CDCl3, 202.4 MHz) δ 17.8, δP [32] (CDCl3, 121 MHz) 18.3, δP [36] (CDCl3, 121 MHz) δ 16.7; 13C NMR (CDCl3, 125.7 MHz) δ 133.3 (d, J = 10.7, C2)a, 131.8 (d, J = 15.5, C3)a, 127.54 (d, J = 190.5, C1), 127.53 (d, J = 4.2, C4), 62.3, (d, J = 5.5,CH2), 16.3 (d, J = 6.4, CH3), a may be reversed, δC [32] (CDCl3, 75 MHz) 133.4, 133.3, 132.0, 131.8, 127.7 (J = 188.59), 127.6, 62.4 (J = 5.02), 16.3 (J = 6.46), δC [36] (CDCl3, 75 MHz) 133.3 (J = 10.1), 131.7 (J = 16.5), 129.8 (J = 185.0), 127.5 (J = 3.9), 61.8 (J = 5.7), 16.3 (J = 5.7); 1H NMR (CDCl3, 500 MHz) δ 7.72–7.68 (m, 2H), 7.65–7.62 (m, 2H), 4.21–4.06 (m, 4H, CH2), 1.34 (t, J = 7.0, 6H, CH3), δH [32] (CDCl3, 300 MHz) 7.60–7.72 (m, 4H), 4.10–4.20 (m, 4H), 1.32 (t, J = 7.2, 6H), δH [36] (CDCl3, 300 MHz) 7.62 (d, J = 8.94, 2H), 7.20 (d, J = 8.94, 2H), 3.84–3.86 (m, 4H), 1.17 (t, J = 7.2, 6H); [M + H]+ = 292.9939 C10H15O3PBr requires 292.9942.
- Tetraethyl 1,4-Phenylenebisphosphonate (2b) (Table 3, Entry 5). Appearance: colorless oil; 31P NMR (CDCl3, 202.4 MHz) δ 16.8, δP [37] (121 MHz, CDCl3) 17.5; 13C NMR (CDCl3, 125 MHz) δ 132.9 (dd, J1 = 189.9, J2 = 2.7, C1), 131.7–131.5 (m, C2), 62.5 (d, J = 5.4, CH2), 16.3 (d, J = 6.4, CH3), δC [37] (75 MHz, CDCl3) 134.1 (d, J = 3.1), 131.8–131.4 (m), 63.3–62.3 (m), 16.5–16.2 (m); 1H NMR (CDCl3, 500 MHz) δ 7.95–7.91 (m, 4H), 4.24–4.09 (m, 8H, CH2), 1.36 (t, J = 7.1, 12H, CH3), δH [37] (300 MHz, CDCl3) 7.97–7.84 (m, 4H), 4.24–4.01 (m, 8H), 1.39–1.28 (m, 12H); [M + H]+ = 351.1121 C14H25O6P2 requires 351.1126.
- 3-Bromophenyl-diphenylphosphine Oxide (3a) (Table 4, Entry 1 and Table 5, Entry 1). Appearance: white crystals, mp. 92–93 °C; 31P NMR (CDCl3, 202.4 MHz) δ 28.0; 13C NMR (CDCl3, 125.7 MHz) δ 135.4 (d, J = 100.8, C1), 135.0 (d, J = 2.5, C4), 134.6 (d, J = 10.5, C2), 132.3 (d, J = 2.8, C4′), 132.0 (d, J = 10.0, C2′)a, 131.8 (d, J = 105.0, C1′), 130.5 (d, J = 9.5, C6), 130.2 (d, J = 12.6, C5), 128.7 (d, J = 12.3, C3′)a, 123.2 (d, J = 15.2, C3), a may be reversed; 1H NMR (CDCl3, 500 MHz) δ 7.84–7.81 (m, 1H), 7.69–7.64 (m, 5H), 7.60–7.56 (m, 3H), 7.51–7.47 (m, 4H), 7.36–7.32 (m, 1H); [M + H]+ = 357.0046 C18H15OPBr requires 357.0044.
- 1,3-Phenylenebis(diphenylphosphine Oxide) (4a) (Table 5, Entry 3). Appearance: colorless oil; 31P NMR (CDCl3, 202.4 MHz) δ 28.5, δP [27] (CDCl3,162 MHz) 30.5; 13C NMR (CDCl3, 125.7 MHz) δ 135.5 (dd, J1 = 10.1, J2 = 3.1, C3), 135.4 (t, J = 11.2, C1), 133.6 (dd, J1 = 101.8, J2 = 10.7, C2), 132.2 (d, J = 2.3, C4′)a, 131.95 (d, J = 10.2, C2′)a, 131.7 (d, J = 105.1, C1′), 128.95 (t, J = 11.3, C4), 128.6 (d, J = 12.5, C3′)a, a may be reversed, δC [27] (CDCl3, 100 MHz) 135.2–135.4 (m, 2C), 135.1, 133.5 (dd, J1 = 101.7, J2 = 10.7), 132.0, 131.8 (d, J = 10.3), 131.5 (d, J = 104.9), 128.8 (t, J = 11.2), 128.4 (d, J = 12.6), 127.1; 1H NMR (CDCl3, 500 MHz) δ 7.96 (ddm, J1 = 12.5, J2 = 7.7, 2H), 7.69 (tt, J1 = 11.7, J2 = 1.5, 1H), 7.62 (tt, J1 = 7.7, J2 = 2.5, 1H), 7.58 (dd, J1 = 12.1, J2 = 7.9, 8H), 7.53 (t, J = 7.4, 4H), 7.41 (td, J1 = 7.7, J2 = 2.8, 8H), δH [27] (CDCl3, 400 MHz) 7.93–7.98 (m, 2H), 7.71 (t, J = 11.7, 1H), 7.50–7.63 (m, 13H), 7.38–7.43 (m, 8H); [M + H]+ = 479.1327 C30H25O2P2 requires 479.1330.
- Diethyl 3-Bromophenylphosphonate (3b) (Table 4, Entry 4 and Table 5, Entry 4). Appearance: colorless oil; 31P NMR (CDCl3, 202.4 MHz) δ 16.2; 13C NMR (CDCl3, 125.7 MHz) δ 135.6 (d, J = 2.8, C4), 134.7 (d, J = 10.7, C2)a, 131.2 (d, J = 187.3, C1), 130.4 (d, J = 6.0, C6), 130.3 (d, J = 12.8, C5)a, 123.0 (d, J = 19.7, C3), 62.6 (d, J = 5.5, CH2), 16.5 (d, J = 6.4, CH3), a may be reversed, δC [33] (101 MHz, CDCl3) 135.4 (d, J = 3.0), 134.5 (d, J = 10.6), 131.1 (d, J = 186), 130.2 (d, J = 4.9), 130.1 (d, J = 11.7), 122.9 (d, J = 19.8), 62.4 (d, J = 5.5), 16.3 (d, J = 6.4); 1H NMR (CDCl3, 500 MHz) δ 7.98–7.95 (m, 1H), 7.79–7.74 (m, 1H), 7.71–7.69 (m, 1H), 7.39–7.35 (m, 1H), 4.23–4.08 (m, 4H, CH2), 1.36 (t, J = 7.1, 6H, CH3), δH [33] (400 MHz, CDCl3) 7.93 (d, J = 13.6, 1H), 7.72 (dd, J1 = 12.9, J2 = 7.6, 1H), 7.66 (d, J = 8.0, 1H), 7.33 (td, J1 = 7.8, J2 = 4.8, 1H), 4.21–4.00 (m, 4H), 1.32 (t, J = 7.1, 6H); [M + H]+ = 292.9937 C10H15O3PBr requires 292.9942.
- Diphenyl-3-chlorophenylphosphine Oxide (5a) (Table 6, Entry 1). Appearance: white crystals, mp. 75–76 °C; 31P NMR (CDCl3, 202.4 MHz) δ 28.1; 13C NMR (CDCl3, 125.7 MHz) δ 135.1 (d, J = 101.3, C1), 135.0 (d, J = 15.6, C3), 132.3 (d, J = 2.8, C4′), 132.1 (d, J = 2.7, C4), 132.0 (d, J = 10.0, C2′)a, 131.84 (d, J = 10.7, C2)b, 131.82 (d, J = 105.1, C1′), 130.1 (d, J = 9.5, C6)b, 129.9 (d, J = 12.9, C5), 128.7 (d, J = 12.3, C3′)a, a,b may be reversed; 1H NMR (CDCl3, 500 MHz) δ 7.68–7.64 (m, 5H), 7.59–7.55 (m, 3H), 7.53–7.47 (m, 5H), 7.43–7.39 (m, 1H); [M + H]+ = 313.0547 C18H15OPCl requires 313.0549.
- Diethyl 3-Chlorophenylphosphonate (5b) (Table 6, Entry 2). Appearance: colorless oil; 31P NMR (CDCl3, 202.4 MHz) δ 16.5, δP [17] (CDCl3) 17.5; 13C NMR (CDCl3, 125.7 MHz) δ 134.8 (d, J = 20.3, C3), 132.5 (d, J = 3.0, C4), 131.6 (d, J = 10.7, C2)a, 130.8 (d, J = 187.8, C1), 129.9 (d, J = 16.5, C5), 129.8 (d, J = 9.2, C6)a, 62.4 (d, J = 5.5, CH2), 16.3 (d, J = 6.4, CH3), a may be reversed, δC [17] (CDCl3) 134.7 (d, J = 20.3, C2), 132.4 (d, J = 3.0, C4), 131.6 (d, J = 10.7, C3), 130.7 (d, J = 187.9, C1), 129.74 (d, J = 16.3, C6), 129.69 (d, J = 9.2, C5), 62.3 (d, J = 5.5, CH2), 16.2 (d, J = 6.4, CH3); 1H NMR (CDCl3, 500 MHz) δ 7.83–7.68 (m, 2H), 7.56–7.53 (m, 1H), 7.46–7.39 (m, 1H), 4.25–4.05 (m, 4H, CH2), 1.36 (t, J = 7.1, 6H, CH3), δH [17] (CDCl3) 7.81–7.59 (m, 2H, ArH), 7.51–7.43 (m, 1H, ArH), 7.40–7.31 (m, 1H, ArH), 4.20–3.96 (m, 4H, OCH2), 1.30 (t, J = 7.1, 6H, CH3); [M + H]+ = 249.0448 C10H15O3PCl requires 249.0447.
- (2-Bromophenyl)-diphenylphosphine Oxide (6a) (Table 8, Entry 1). Appearance: white crystals; 31P NMR (CDCl3, 202.4 MHz) δ 30.5, δP [25] (CDCl3, 200 MHz) 30.6, δP [24] (CDCl3, 162 MHz) 32.2; 13C NMR (CDCl3, 125.7 MHz) δ 136.0 (d, J = 10.5, C3)a, 134.9, (d, J = 7.5, C6)a, 133.4 (d, J = 2.4, C4), 133.0 (d, J = 104.6, C1), 132.1 (d, J = 10.0, C2′)b, 132.0 (d, J = 2.8, C4′), 131.7 (d, J = 108.0, C1′), 128.6 (d, J = 12.5, C3′)b, 127.0 (d, J = 11.1, C5)a 126.9 (d, J = 4.7, C2), a,b may be reversed, δC [25] (CDCl3, 75 MHz) 136.1 (d, J = 10.4), 135.0 (d, J = 7.7), 133.6 (d, J = 2.2), 133.2 (d, J = 104.3), 132.3 (d, J = 9.9), 132.1 (d, J = 2.7), 131.9 (d, J = 107.6), 128.7 (d, J = 12.6), 127.1 (d, J = 11.5), 127.1 (d, J = 4.9), δC [24] (CDCl3, 100 MHz) 136.3 (d, J = 10.4), 135.2 (d, J = 8.0), 133.7 (d, J = 2.4), 133.5 (d, J = 104.7), 132.5 (d, J = 9.6), 132.3 (d, J = 2.4), 132.2 (d, J = 107.9), 128.9 (d, J = 12.0), 127.3 (d, J = 11.2), 127.3; 1H NMR (CDCl3, 500 MHz) δ 7.74–7.71 (m, 4H), 7.70–7.67 (m, 1H), 7.59–7.55 (m, 2H), 7.50–7.46 (m, 4H), 7.41–7.32 (m, 3H,) δH [25] (CDCl3, 500 MHz) 7.75–7.65 (m, 5H), 7.58–7.53 (m, 2H), 7.60–7.45 (m, 4H), 7.41–7.30 (m, 3H), δH [24] (CDCl3, 400 MHz) 7.75–7.65 (m, 5H), 7.58–7.31 (m, 9H); [M + H]+ = 357.0045 C18H15OPBr requires 357.0044.
- Diethyl 2-Bromophenylphosphonate (6b) (Table 8, Entry 4). Appearance: colorless oil; 31P NMR (CDCl3, 202.4 MHz) δ 14.8, δP [26] (CDCl3, 121 MHz) 15.4; 13C NMR (CDCl3, 125.7 MHz) δ 136.3 (d, J = 8.3, C6)a, 134.3 (d, J = 11.2, C3)a, 133.6 (d, J = 2.7, C4), 129.5 (d, J = 192.0, C1), 126.9 (d, J = 13.6, C5)a, 125.2 (d, J = 3.8, C2), 62.6 (d, J = 5.6, CH2), 16.3 (d, J = 6.5, CH3), a may be reversed, δC [26] (CDCl3, 75 MHz) 136.2 (d, J = 8.3, Ar-CH), 133.5 (d, J = 2.7, Ar-CH), 129.3 (d, J = 192.0, Ar-qC), 126.8 (d, J = 13.6, Ar-CH), 125.1 (d, J = 3.9, Ar-qC), 62.5 (d, J = 5.6 OCH2CH3), 16.2 (d, J = 6.6 OCH2CH3); 1H NMR (CDCl3, 500 MHz) δ 8.07–8.02 (m, 1H), 7.71–7.68 (m, 1H), 7.45–7.38 (m, 2H), 4.27–4.12 (m, 4H, CH2), 1.39 (t, J = 7.1, 6H, CH3), δH [26] (CDCl3, 300 MHz) 8.05–7.96 (m, 1H, ArH), 7.70–7.62 (m, 1H, ArH), 7.43–7.33 (m, 2H, ArH), 4.28–4.04 (m, 4H, OCH2CH3), 1.35 (td, J1 = 7.1, J2 = 0.5) 6H, OCH2CH3); [M + Na]+ = 314.9761 C10H14O3PBrNa requires 314.9762.
- 4-Diethylphosphonoylphenyl-diphenylphosphine Oxide (8) (Scheme 1). Appearance: colorless oil; 31P NMR (CDCl3, 202.4 MHz) δ 28.4 (m, P(C6H5)2), 16.8 (d; J = 3.7, P(OCH2CH3)2); 13C NMR (CDCl3, 125.7 MHz) δ 137.3 (dd, J1 = 100.5, J2 = 3.0, C1), 132.6 (dd, J1 = 186.9, J2 = 2.9, C4), 132.3 (d, J = 2.8, C4′), 132.1 (d, J = 10.0, C2′)a, 132.0 (dd, J = 14.9, J = 9.8, C2), 131.7 (d, J ~ 105, C1′), 131.6 (dd, J1 = 11.8, J2 = 10.0, C3), 128.7 (d, J = 12.2, C3′)a, 62.5 (d, J = 5.6, CH2), 16.4 (d, J = 6.4, CH3), a may be reversed; 1H NMR (CDCl3, 500 MHz) 7.90 (ddd, J1 = 12.8, J2 = 8.3, J3 = 2.5, 2H), 7.78 (ddd, J1 = 11.6, J2 = 8.1, J3 = 3.9, 2H), 7.67 (ddd, J1 = 12.1, J2 = 7.8, J3 = 1,3, 4H), 7.58 (tm, J = 7.5, 2H), 7.49 (td, J1 = 7.6, J2 = 2.9, 4H), 4.18 (m) 4.10 (m, 4H, CH2), 1.34 (t, J = 7.1, 6H, CH3); [M + H]+ = 415.1228 C22H25O4P2 requires 415.1228.
- 3-Diethylphosphonoylphenyl-diphenylphosphine Oxide (9) (Table 9, Entry 2). Appearance: colorless oil; 31P NMR (CDCl3, 202.4 MHz) δ 28.3 (m, P(C6H5)2), 16.7 (d, J = 5.0, P(OCH2CH3)2); 13C NMR (CDCl3, 125.7 MHz) δ 135.8 (dd, J1 = 9.7, J2 = 2.8, C6), 135.2 (dd, J1 = 10.0, J2 = 2.6, C4), 135.0 (t, J = 10.6, C2), 133.6 (dd, J1 = 102.1, J2 = 13.7, C1), 132.3 (d, J = 2.9, C4′), 132.1 (d, J = 10.0, C2′)a, 132.0 (d, J = 104.8, C1′), 129.5 (dd, J1 = 188.6, J2 = 11.3, C3), 128.75 (dd, J1 = 14.2, J2 = 11.2, C), 128.7 (d, J = 12.3, C3′)a, 62.5 (d, J = 5.7, CH2), 16.3 (d, J = 6.4, CH3), a may be reversed; 1H NMR (CDCl3, 500 MHz) 8.06 (t, J = 12.6, 1H), 8.01 (ddq, J1 = 13.1, J2 = 7.7, J3 = 1.5, 1H), 7.89 (ddq, J1 = 11.7, J2 = 7.8, J3 = 1.5, 1H), 7.67 (ddd, J1 = 12.1, J2 = 8.0, J3 = 1.4, 4H), 7.59 (tt, J1b, J2 = 3.3, 1H), 7.57 (tq, J1 = 7.5, J2 = 1.5, 2H), 7.48 (td, J1 = 7.6, J2 = 2.9, 4H), 4.12 (m) 4.05 (m, 4H, CH2), 1.27 (t, J = 7.1, 6H, CH3), b the coupling could not be detected due to overlapping signals; [M + Na]+ = 437.1042 C22H24O4P2Na requires 437.1048.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hirao, T.; Masunaga, T.; Ohshiro, Y.; Agawa, T. Stereoselective synthesis of vinylphosphonate. Tetrahedron Lett. 1980, 21, 3595–3598. [Google Scholar] [CrossRef]
- Hirao, T.; Masunaga, T.; Yamada, N.; Ohshiro, Y.; Agawa, T. Palladium-catalyzed new carbon-phosphorus bond formation. Bull. Chem. Soc. Jpn. 1982, 55, 909–913. [Google Scholar] [CrossRef]
- Hirao, T.; Masunaga, T.; Ohshiro, Y.; Agawa, T. A Novel Synthesis of Dialkyl Arenephosphonates. Synthesis 1981, 1981, 56–57. [Google Scholar] [CrossRef]
- Lu, X.; Zhu, J. Palladium-catalyzed reaction of aryl polyfluoroalkanesulfonates with O,O-dialkyl phosphonates. Synthesis 1987, 1987, 726–727. [Google Scholar] [CrossRef]
- Holt, D.A.; Erb, J.M. Palladium-catalyzed phosphorylation of alkenyl triflates. Tetrahedron Lett. 1989, 30, 5393–5396. [Google Scholar] [CrossRef]
- Kazankova, M.A.; Trostyanskaya, I.G.; Lutsenko, S.V.; Beletskaya, I.P. Nickel- and palladium-catalyzed cross-coupling as a route to 1- and 2-alkoxy- or dialkylaminovinylphosphonates. Tetrahedron Lett. 1999, 40, 569–572. [Google Scholar] [CrossRef]
- Zhong, P.; Xiong, Z.X.; Huang, X. A facile regio- and stereocontrolled synthesis of (E)-vinylphosphonates VIA cross coupling of (E)-vinyl iodides with dialkyl phosphites. Synth. Commun. 2000, 30, 273–278. [Google Scholar] [CrossRef]
- Kobayashi, Y.; William, A.D. Palladium- and nickel-catalyzed coupling reactions of α-bromoalkenylphosphonates with arylboronic acids and lithium alkenylborates. Adv. Synth. Catal. 2004, 346, 1749–1757. [Google Scholar] [CrossRef]
- Jablonkai, E.; Keglevich, G. P–C bond formation by coupling reaction utilizing >P(O)H species as the reagents. Curr. Org. Synth. 2014, 11, 429–453. [Google Scholar] [CrossRef]
- Jablonkai, E.; Keglevich, G. Advances and new variations of the Hirao reaction. Org. Prep. Proc. Int. 2014, 46, 281–316. [Google Scholar] [CrossRef]
- Henyecz, R.; Keglevich, G. New developments on the Hirao reactions, especially from “green” point of view. Curr. Org. Synth. 2019, 16, 523–545. [Google Scholar] [CrossRef] [PubMed]
- Kalek, M.; Stawinski, J. Pd(0)-catalyzed phosphorus−carbon bond formation. Mechanistic and synthetic studies on the role of the palladium sources and anionic additives. Organometallics 2007, 26, 5840–5847. [Google Scholar] [CrossRef]
- Belabassi, Y.; Alzghari, S.; Montchamp, J.-L. Revisiting the Hirao cross-coupling: Improved synthesis of aryl and heteroaryl phosphonates. J. Organomet. Chem. 2008, 693, 3171–3178. [Google Scholar] [CrossRef] [PubMed]
- Deal, E.L.; Petit, C.; Montchamp, J.-L. Palladium-catalyzed cross-coupling of H-phosphinate esters with chloroarenes. Org. Lett. 2011, 13, 3270–3273. [Google Scholar] [CrossRef]
- Kalek, M.; Jezowska, M.; Stawinski, J. Preparation of arylphosphonates by palladium(0)-catalyzed cross-coupling in the presence of acetate additives: Synthetic and mechanistic studies. Adv. Synth. Catal. 2009, 351, 3207–3216. [Google Scholar] [CrossRef]
- Jablonkai, E.; Keglevich, G. P-ligand-free, microwave-assisted variation of the Hirao reaction under solvent-free conditions; the P–C coupling reaction of >P(O)H species and bromoarenes. Tetrahedron Lett. 2013, 54, 4185–4188. [Google Scholar] [CrossRef]
- Keglevich, G.; Jablonkai, E.; Balázs, L.B. A “green” variation of the Hirao reaction: The P–C coupling of diethyl phosphite, alkyl phenyl-H-phosphinates and secondary phosphine oxides with bromoarenes using a P-ligand-free Pd(OAc)2 catalyst under microwave and solvent-free conditions. RSC Adv. 2014, 4, 22808–22816. [Google Scholar] [CrossRef]
- Keglevich, G.; Henyecz, R.; Mucsi, Z.; Kiss, N.Z. The palladium acetate-catalyzed microwave-assisted Hirao reaction without an added phosphorus ligand as a “green” protocol: A quantum chemical study on the mechanism. Adv. Synth. Catal. 2017, 359, 4322–4331. [Google Scholar] [CrossRef]
- Henyecz, R.; Mucsi, Z.; Keglevich, G. Palladium-catalyzed microwave-assisted Hirao reaction utilizing the excess of the diarylphosphine oxide reagent as the P-ligand; a study on the activity and formation of the "PdP2" catalyst. Pure Appl. Chem. 2019, 91, 121–134. [Google Scholar] [CrossRef]
- Keglevich, G.; Henyecz, R.; Mucsi, Z. Focusing on the catalysts of the Pd- and Ni- catalyzed Hirao reactions. Molecules 2020, 25, 3897. [Google Scholar] [CrossRef]
- Ueoka, K.; Tanaka, D.; Arai, T.; Ichihashi, Y. Phosphine oxide derivative and light-emitting element provided with same. WO2014057873A1, 17 April 2014. [Google Scholar]
- Crawford, J.; Zak, M.; Kellar, T.; Cheng, Y.X.; Li, W.; Romero, A.F.; Gibbons, P.; Zhao, G.; Hamilton, G.; Goodacre, S.C. Pyrazole derivatives, compositions and therapeutic use thereof. WO2017191098A1, 9 November 2017. [Google Scholar]
- Marx, M.A.Z.; Lee, M.R.; Bobinski, T.P.; Burns, A.C.; Arora, N.; Christensen, J.G.; Ketcham, J.N. PRC2 inhibitors. WO2019152419A1, 8 August 2019. [Google Scholar]
- Baillie, C.; Xiao, J. Palladium-catalysed synthesis of biaryl phosphines. Tetrahedron 2004, 60, 4159–4168. [Google Scholar] [CrossRef]
- Zhang, Z.; Smal, V.; Retailleau, P.; Voituriez, A.; Frison, G.; Marinetti, A.; Guinchard, X. Tethered counterion-directed catalysis: Merging the chiral ion-pairing and bifunctional ligand strategies in enantioselective gold(I) catalysis. J. Am. Chem. Soc. 2020, 142, 3797–3805. [Google Scholar] [CrossRef]
- Bonnaventure, I.; Charette, A.B. Probing the importance of the hemilabile site of bis(phosphine) monoxide ligands in the copper-catalyzed addition of diethylzinc to N-phosphinoylimines: Discovery of new effective chiral ligands. J. Org. Chem. 2008, 73, 6330–6340. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, H.; Hu, X.; Tang, G.; Zhu, J.; Zhao, Y. Ni(II)/Zn catalyzed reductive coupling of aryl halides with diphenylphosphine oxide in water. Org. Lett. 2011, 13, 3478–3481. [Google Scholar] [CrossRef] [PubMed]
- Keglevich, G.; Henyecz, R.; Mucsi, Z. Experimental and theoretical study on the “2,2′-bipiridyl-Ni-catalyzed” Hirao reaction of >P(O)H reagents and halobenzenes: A Ni(0) → Ni(II) or a Ni(II) → Ni(IV) mechanism? J. Org. Chem. 2020, 85, 14486–14495. [Google Scholar] [CrossRef]
- Xuan, J.; Zeng, T.T.; Chen, J.; Lu, L.Q.; Xiao, W.J. Room temperature C–P bond formation enabled by merging nickel catalysis and visible-light-induced photoredox catalysis. Chem. Eur. J. 2015, 21, 4962–4965. [Google Scholar] [CrossRef] [PubMed]
- Stankevic, M.; Wlodarczyk, A. Efficient copper(I)-catalyzed coupling of secondary phosphine oxides with aryl halides. Tetrahedron 2013, 69, 73–81. [Google Scholar] [CrossRef]
- Karlstedt, N.B.; Beletskaya, I.P. Copper-catalyzed cross-coupling of diethyl phosphonate with aryl iodides. Russ. J. Organ. Chem. 2011, 47, 1011–1014. [Google Scholar] [CrossRef]
- Huang, C.; Tang, X.; Fu, H.; Jiang, Y.; Zhao, Y. Proline/pipecolinic acid-promoted copper-catalyzed P-arylation. J. Org. Chem. 2006, 71, 5020–5022. [Google Scholar] [CrossRef]
- Pan, L.; Kelley, A.S.; Cooke, V.M.; Deckert, M.M.; Laulhé, S. Transition-metal-free photoredox phosphonation of aryl C–N and C–X bonds in aqueous solvent mixtures. ACS Sustainable Chem. Eng. 2022, 10, 691–695. [Google Scholar] [CrossRef]
- Zöllner, M.; Rothe, C. Semiconducting material comprising a phosphine oxide matrix and metal salt. WO2015052284A1, 16 April 2015. [Google Scholar]
- Dong, J.; Liu, L.; Ji, X.; Shang, Q.; Liu, L.; Su, L.; Chen, B.; Kan, R.; Zhou, Y.; Yin, S.-F.; et al. General oxidative aryl C–P bond formation through palladium-catalyzed decarbonylative coupling of aroylhydrazides with P(O)H compounds. Org. Lett. 2019, 21, 3198–3203. [Google Scholar] [CrossRef] [PubMed]
- Rao, H.; Jin, Y.; Fu, H.; Jiang, Y.; Zhao, Y. A versatile and efficient ligand for copper-catalyzed formation of C–N, C–O, and P–C bonds: Pyrrolidine-2-phosphonnic acid phenyl monoester. Chem. Eur. J. 2006, 12, 3636–3646. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, R.S.; Düsel, S.J.S.; Koenig, B. Visible-light photo-Arbuzov reaction of aryl bromides and trialkyl phosphites yielding aryl phosphonates. ACS Catal. 2016, 6, 8410–8414. [Google Scholar] [CrossRef]

 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Y | P-Reagent (equiv.) | t (min) | Conversion (%) a | Product Composition (%) a | Isolated Yield of 1 (%) | |||
| 1 | 2 | Ph2(EtO)P(O) (A) or Ph(EtO)2P(O) (B) | Ph3P(O) | ||||||
| 1 | Ph (a) | 1.15 | 30 | 100 | 85 | 6 | 7 (A) | 2 | 63 (1a) |
| 2 | Ph (a) | 2.15 | 60 | 98 | 15 | 41 b | 16 (A) c | 26 d | – |
| 3 | EtO (b) | 1.15 | 60 | 99 | 69 | 18 e | 12 (B) f | n. r. | 48 (1b) |
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Y | P-Reagent (equiv.) | T (°C) | t (min) | Conversion (%) a | Product Composition (%) a | Isolated Yield of 1 (%) | |||
| 1 | 2 | Ph2(EtO)P(O) (A) or Ph(EtO)2P(O) (B) | Ph3P(O) | |||||||
| 1 | Ph (a) | 1.15 b | 100 | 60 | 100 | 96 | – | 2 (A) | 2 | 74 (1a) |
| 2 | Ph (a) | 1.15 b | 120 | 30 | 100 | 97 | – | 2 (A) | 1 | 75 (1a) |
| 3 | Ph (a) | 2.15 b | 120 | 60 | 100 | 19 | 42 | 8 (A) | 31 | – |
| 4 | EtO (b) | 1.15 b | 120 | 30 | 98 | 88 | 7 | 3 (B) | n. r. | 63 (1b) |
| 5 | EtO (b) | 2.3 c | 120 | 30 | 100 | 13 | 87 | – | n. r. | 65 (2b) |
 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Y | P-Reagent (equiv.) | T (°C) | t (min) | Solvent | Conversion (%) a | Product Composition (%) a | Isolated Yield (%) | |||
| 3 | 4 | Ph2(EtO)P(O) (A) or Ph(EtO)2P(O) (B) | Ph3P(O) | ||||||||
| 1 | Ph (a) | 1.15 | 120 | 25 | EtOH | 100 | 85 | 6 | 4 (A) | 5 | 68 (3a) |
| 2 | Ph (a) | 2.15 | 120 | 30 | EtOH | 100 | 2 | 92 | 2 (A) | 4 | 75 (4a) |
| 3 | EtO (b) | 1.15 | 120 | 60 | EtOH | 94 | 57 | 27 b | 10 (B) | n. r. | – |
| 4 | EtO (b) | 1.15 | 150 | 30 | – | 97 | 63 | 24 b | 10 (B) | n. r. | 45 (3b) |
 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Y | P-Reagent (equiv.) | T (°C) | t (min) | Solvent | Conversion (%) a | Product Composition (%) a | Isolated Yield (%) | |||
| 3 | 4 | Ph2(EtO)P(O) (A) or Ph(EtO)2P(O) (B) | Ph3P(O) | ||||||||
| 1 | Ph (a) | 1.15 | 100 | 60 | EtOH | 100 | 94 | – | 1 (A) | 5 | 78 (3a) |
| 2 | Ph (a) | 1.15 | 120 | 30 | EtOH | 100 | 95 | 3 | 1 (A) | 2 | 75 (3a) |
| 3 | Ph (a) | 2.15 | 120 | 35 | EtOH | 97 | – | 88 | 3 (A) | 6 | 70 (4a) |
| 4 | EtO (b) | 1.15 | 120 | 30 | – | 98 | 81 | 13 | 4 (B) | n. r. | 66 (3b) |
 | |||
|---|---|---|---|
| Entry | Y | t (min) | Isolated Yield of 5 (%) |
| 1 | Ph (a) | 60 | 80 (5a) |
| 2 | EtO (b) | 45 | 82 (5b) |
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Y | P-Reagent (equiv.) | t (min) | Solvent | Conversion (%) a | Product Composition (%) a | Isolated Yield of 6 (%) | |||
| 6 | 7 | Ph2(EtO)P(O) (A) or Ph(EtO)2P(O) (B) | Ph3P(O) | |||||||
| 1 | Ph (a) | 1.15 b | 60 | EtOH | 100 c | 20 | 19 d | – | 48 | – |
| 2 | Ph (a) | 1.3 e | 60 | EtOH | 100 | 20 | 20 d | 2 (A) | 58 | – |
| 3 | EtO (b) | 1.15 b | 30 | EtOH | 96 | 30 | – | 66 (B) | n. r. | – |
| 4 | EtO (b) | 1.15 b | 30 | – | 81 | 69 | – | 12 (B) | n. r. | 41 (6b) |
| 5 | EtO (b) | 1.3 e | 45 | – | 100 | 74 | – | 16 (B) | n. r. | 60 (6b) |
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Y | P-Reagent (equiv.) | t (min) | Solvent | Conversion (%) a | Product Composition (%) a | Isolated Yield (%) | |||
| 6 | Ph2(EtO)P(O) (A) or Ph(EtO)2P(O) (B) | Ph3P(O) | Ph2P(O)OH | |||||||
| 1 | Ph (a) | 1.15 b | 60 | EtOH | 100 | 65 | 9 (A) | 11 | 15 c | 45 (6a) |
| 2 | Ph (a) | 1.3 d | 60 | EtOH | 100 | 66 | 11 (A) | 11 | 12 c | – |
| 3 | EtO (b) | 1.3 d | 45 | EtOH | 95 | 36 | 36 (B) | n. r. | n. r. | – |
| 4 | EtO (b) | 1.3 d | 45 | – | 100 | 91 | 9 (B) | n. r. | n. r. | 75 (6b) |
| 5 | EtO (b) | 1.15 b | 30 | – | 85 | 69 | 16 (B) | n. r. | n. r. | 43 (6b) |
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Y | Product Composition (%) a | Isolated Yield (%) | |||
| 4a or 9 | (EtO)Ph2P(O) | Ph3P(O) | Ph2P(O)OH | |||
| 1 | Ph (a) | 91 (4a) | 3 | 4 | 2 | 67 (4a) |
| 2 | EtO (b) | 87 (9) | n. r. | 13 | n. r. | 64 (9) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huszár, B.; Varga, P.R.; Szűcs, N.Á.; Simon, A.; Drahos, L.; Keglevich, G. Pd-Catalyzed Hirao P–C Coupling Reactions with Dihalogenobenzenes without the Usual P-Ligands under MW Conditions. Catalysts 2022, 12, 1080. https://doi.org/10.3390/catal12101080
Huszár B, Varga PR, Szűcs NÁ, Simon A, Drahos L, Keglevich G. Pd-Catalyzed Hirao P–C Coupling Reactions with Dihalogenobenzenes without the Usual P-Ligands under MW Conditions. Catalysts. 2022; 12(10):1080. https://doi.org/10.3390/catal12101080
Chicago/Turabian StyleHuszár, Bianka, Petra Regina Varga, Nóra Á. Szűcs, András Simon, László Drahos, and György Keglevich. 2022. "Pd-Catalyzed Hirao P–C Coupling Reactions with Dihalogenobenzenes without the Usual P-Ligands under MW Conditions" Catalysts 12, no. 10: 1080. https://doi.org/10.3390/catal12101080
APA StyleHuszár, B., Varga, P. R., Szűcs, N. Á., Simon, A., Drahos, L., & Keglevich, G. (2022). Pd-Catalyzed Hirao P–C Coupling Reactions with Dihalogenobenzenes without the Usual P-Ligands under MW Conditions. Catalysts, 12(10), 1080. https://doi.org/10.3390/catal12101080