


Computational Insights into Ru, Pd and Pt fcc Nano-Catalysts from Density Functional Theory Calculations: The Influence of Long-Range Dispersion Corrections

Abstract
:1. Introduction
2. Computational Methods
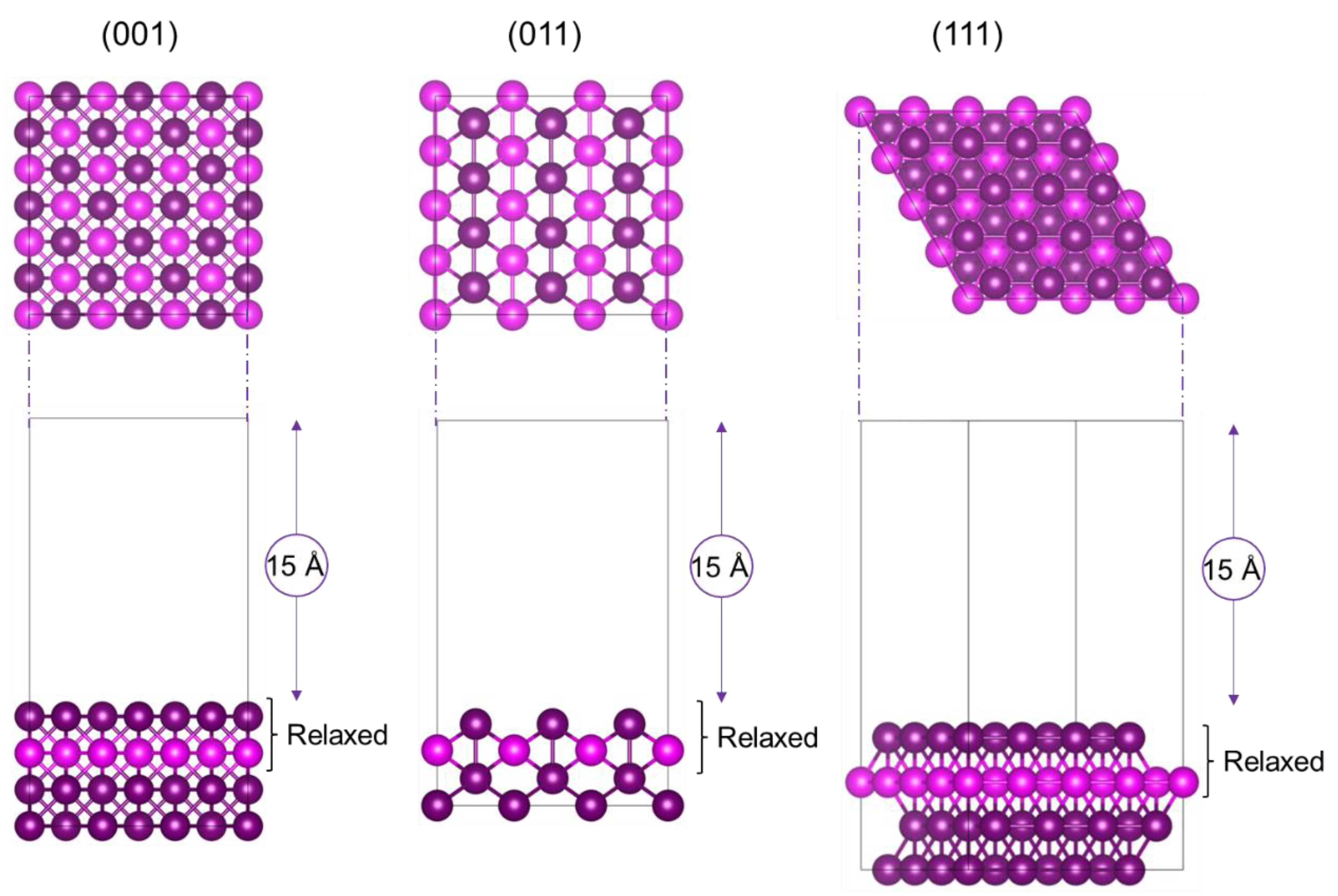
2.1. Calculation of Surfaces
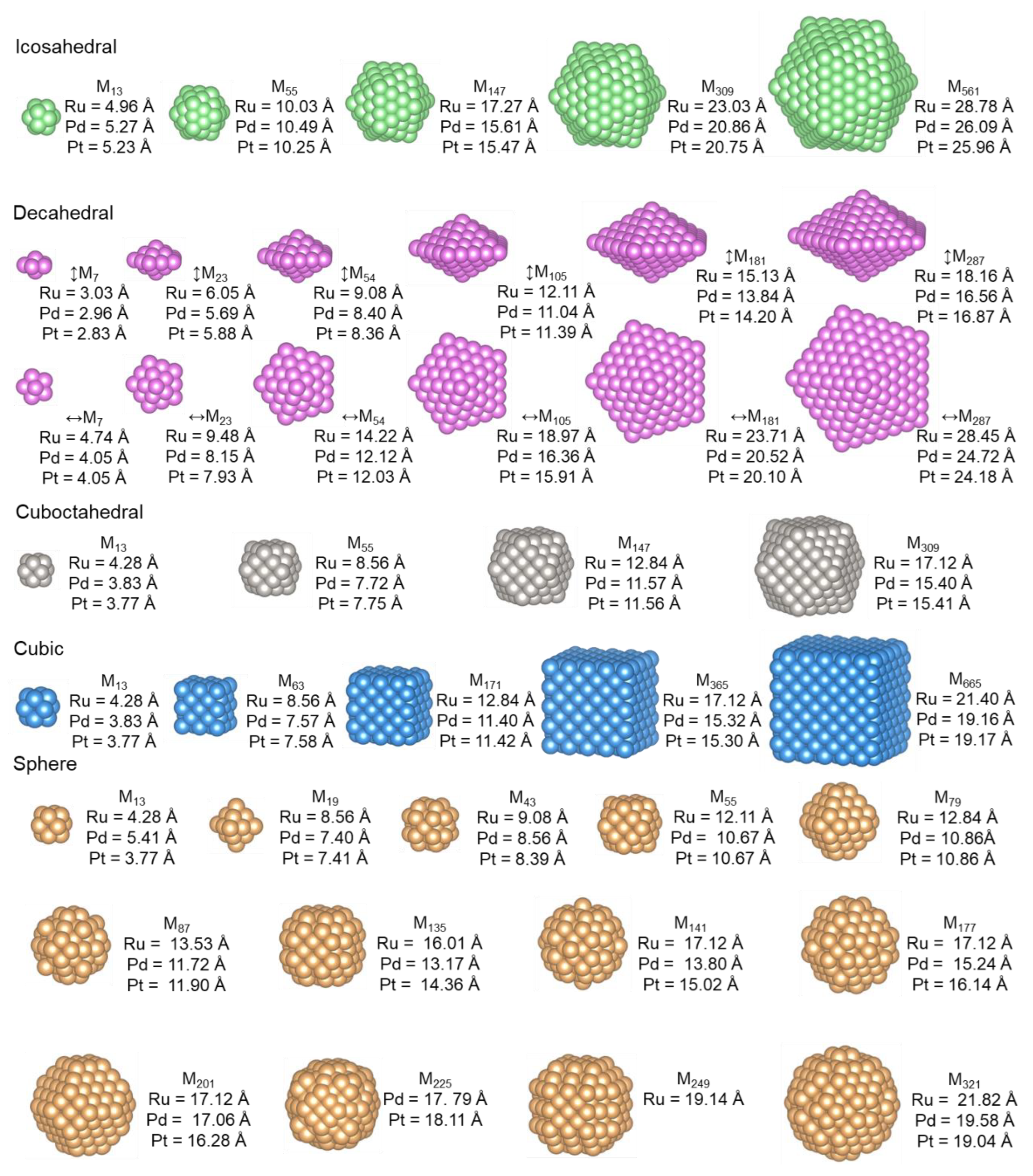
2.2. Calculation of Nanoparticles
3. Results and Discussion
3.1. Ruthenium, Palladium and Platinum Surfaces
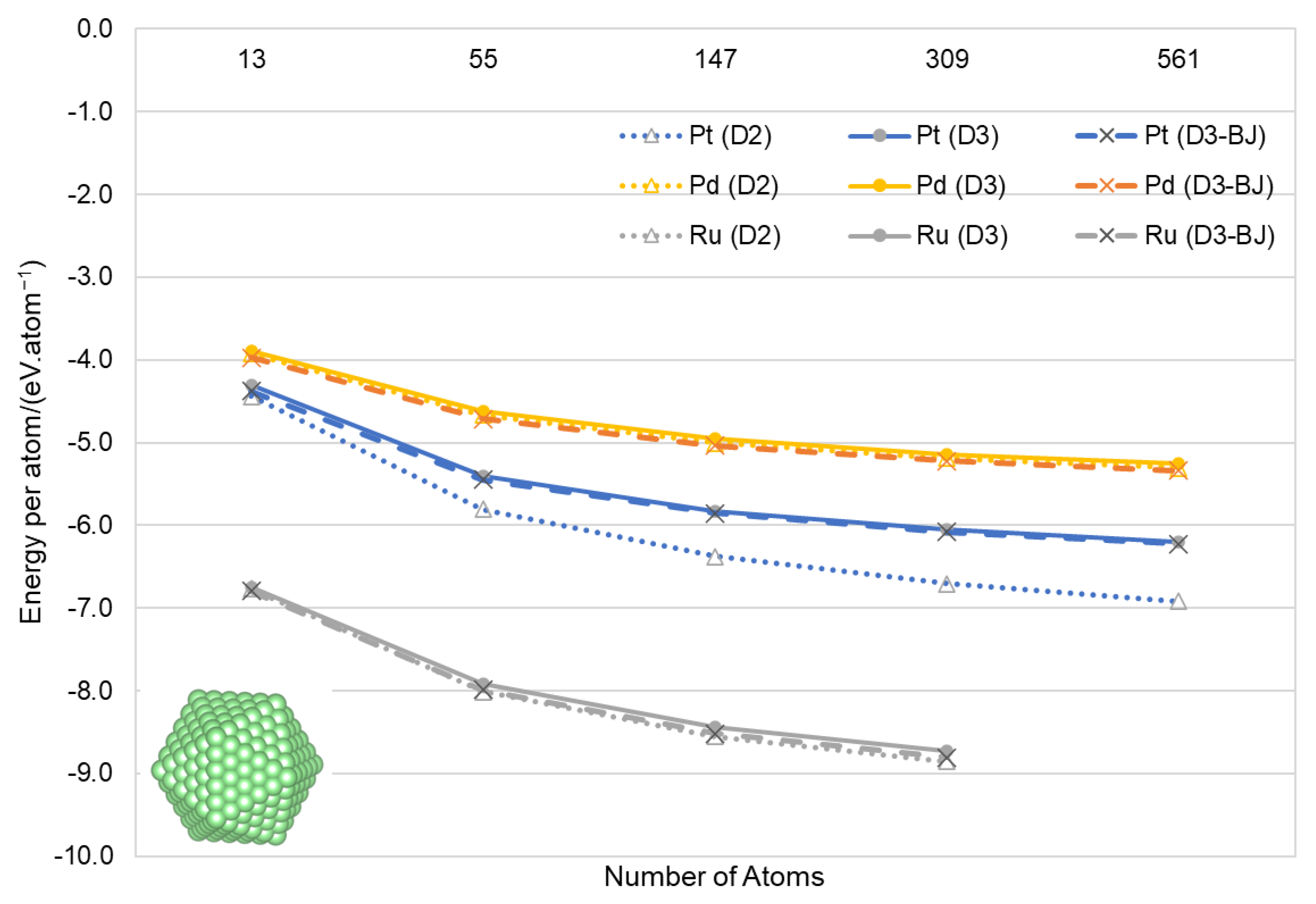
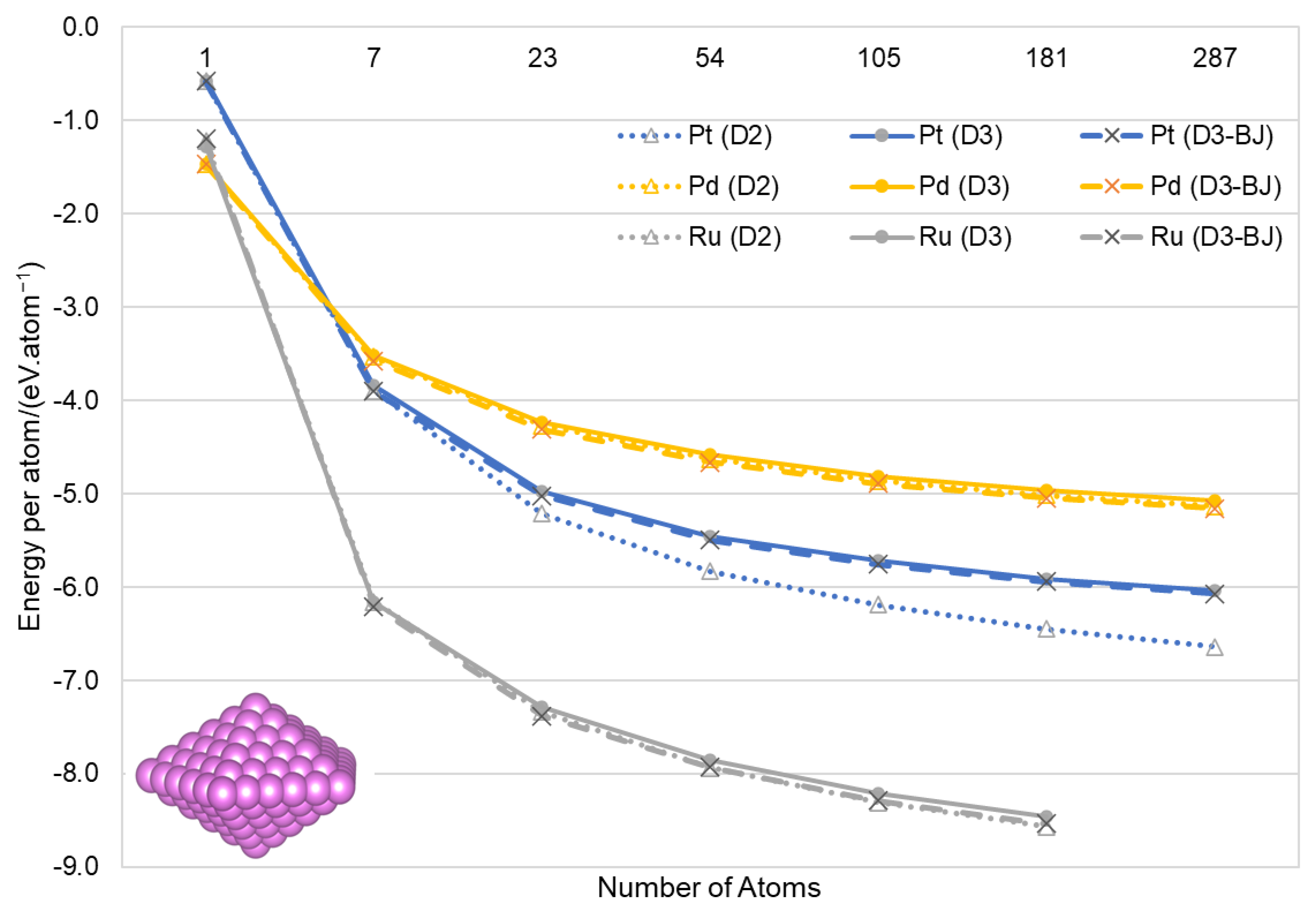
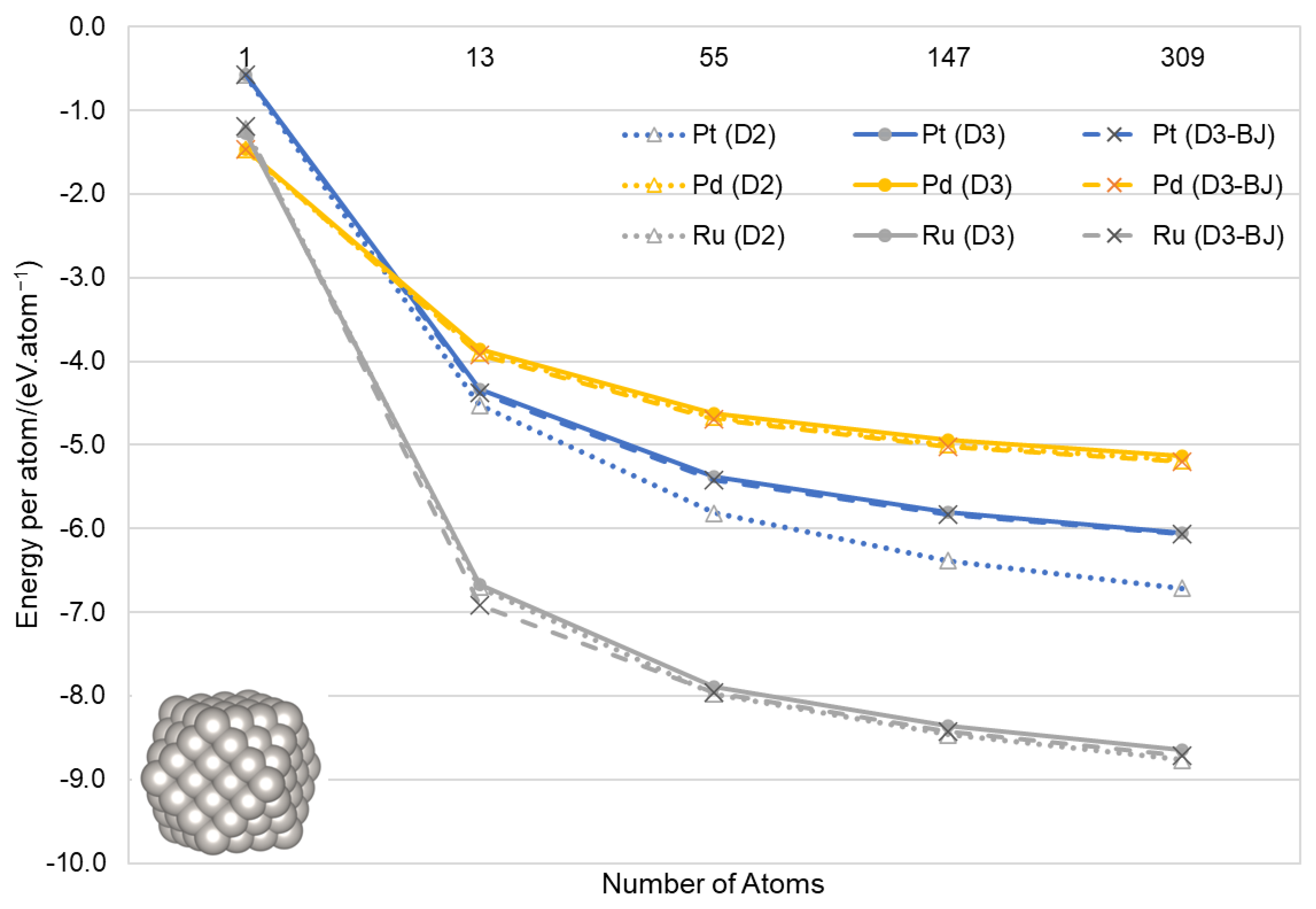
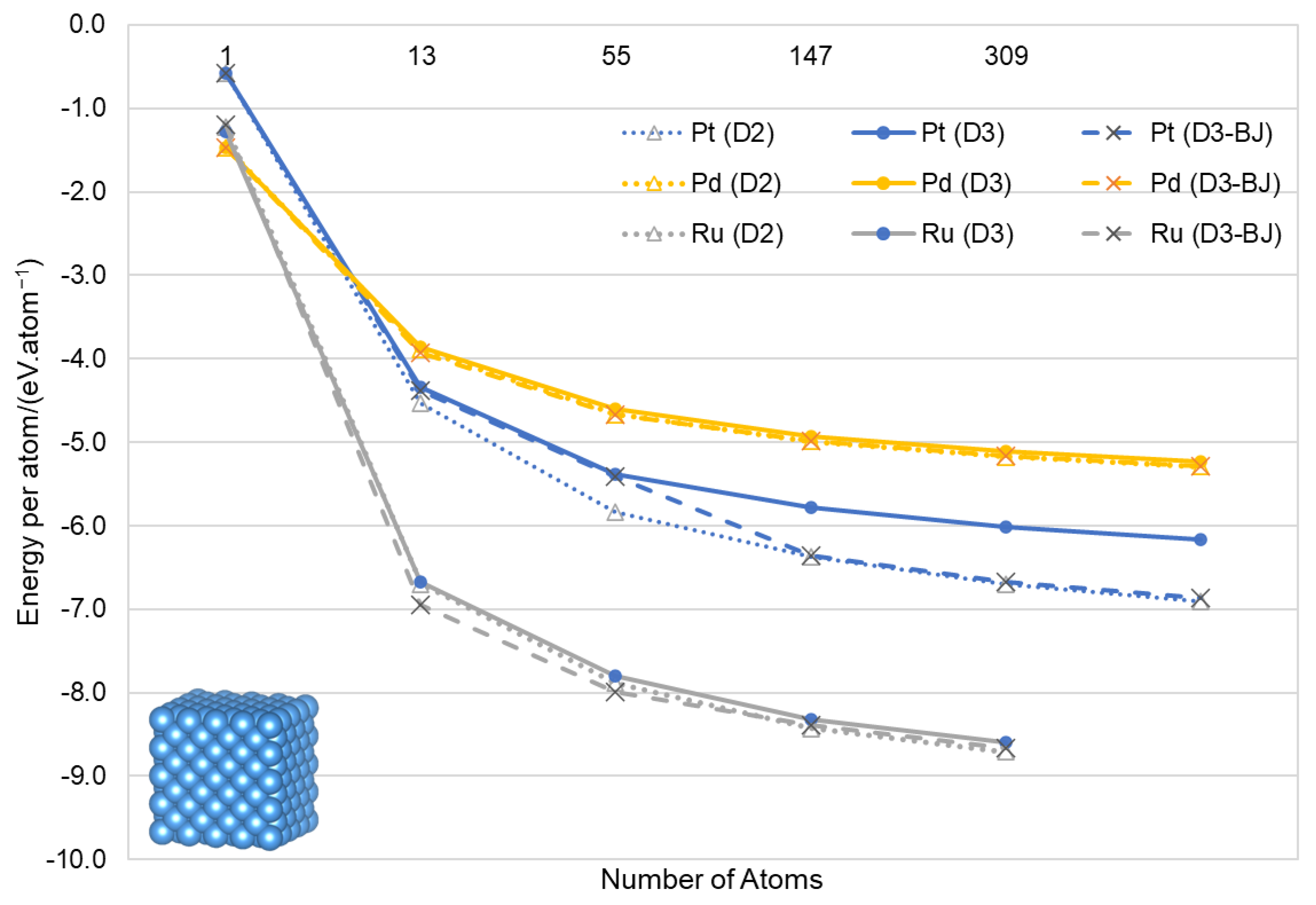
3.2. Nanoparticles
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Petitpas, G.; Aceves, S. Hydrogen Storage in Pressure Vessels: Liquid, Cryogenic, and Compressed Gas. In Hydrogen Storage Technology: Materials and Applications; Klebanoff, L., Ed.; CRC Press: Boca Raton, FL, USA, 2016; pp. 91–108. ISBN 9781439841082. [Google Scholar]
- Taube, M.; Rippin, D.; Cresswell, D.; Knecht, W. A System of Hydrogen-Powered Vehicles with Liquid Organic Hydrides. Int. J. Hydrog. Energy 1983, 8, 213–225. [Google Scholar] [CrossRef]
- Amende, M.; Gleichweit, C.; Schernich, S.; Höfert, O.; Lorenz, M.P.A.; Zhao, W.; Koch, M.; Obesser, K.; Papp, C.; Wasserscheid, P.; et al. Size and Structure Effects Controlling the Stability of the Liquid Organic Hydrogen Carrier Dodecahydro-N-Ethylcarbazole during Dehydrogenation over Pt Model Catalysts. J. Phys. Chem. Lett. 2014, 5, 1498–1504. [Google Scholar] [CrossRef]
- Schuster, R.; Bertram, M.; Runge, H.; Geile, S.; Chung, S.; Vonk, V.; Noei, H.; Poulain, A.; Lykhach, Y.; Stierle, A.; et al. Metastability of Palladium Carbide Nanoparticles during Hydrogen Release from Liquid Organic Hydrogen Carriers. Phys. Chem. Chem. Phys. 2021, 23, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Yang, M.; Chen, X.; Dong, Y.; Zhang, Z.; Cheng, H. A Highly Active Bifunctional Ru–Pd Catalyst for Hydrogenation and Dehydrogenation of Liquid Organic Hydrogen Carriers. J. Catal. 2019, 378, 382–391. [Google Scholar] [CrossRef]
- Modisha, P.M.; Ouma, C.N.M.; Garidzirai, R.; Wasserscheid, P.; Bessarabov, D. The Prospect of Hydrogen Storage Using Liquid Organic Hydrogen Carriers. Energy Fuels 2019, 33, 2778–2796. [Google Scholar] [CrossRef]
- Gall, D. Electron Mean Free Path in Elemental Metals. J. Appl. Phys. 2016, 119, 085101. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Xia, Y. Crystal-Phase and Surface-Structure Engineering of Ruthenium Nanocrystals. Nat. Rev. Mater. 2020, 5, 440–459. [Google Scholar] [CrossRef]
- Kusada, K.; Kobayashi, H.; Yamamoto, T.; Matsumura, S.; Sumi, N.; Sato, K.; Nagaoka, K.; Kubota, Y.; Kitagawa, H. Discovery of Face-Centered-Cubic Ruthenium Nanoparticles: Facile Size-Controlled Synthesis Using the Chemical Reduction Method. J. Am. Chem. Soc. 2013, 135, 5493–5496. [Google Scholar] [CrossRef]
- Liu, L.; Corma, A. Metal Catalysts for Heterogeneous Catalysis: From Single Atoms to Nanoclusters and Nanoparticles. Chem. Rev. 2018, 118, 4981–5079. [Google Scholar] [CrossRef] [Green Version]
- Cramer, C.J.; Truhlar, D.G. Density Functional Theory for Transition Metals and Transition Metal Chemistry. Phys. Chem. Chem. Phys. 2009, 11, 10757–10816. [Google Scholar] [CrossRef]
- Viñ, F.; Gomes, J.R.B.; Illas, F. Chem Soc Rev Chemical Society Reviews Understanding the Reactivity of Metallic Nanoparticles: Beyond the Extended Surface Model for Catalysis. Chem. Soc. Rev. 2014, 43, 4922–4939. [Google Scholar] [CrossRef]
- Nanda, K.K.; Maisels, A.; Kruis, F.E.; Fissan, H.; Stappert, S. Higher Surface Energy of Free Nanoparticles. Phys. Rev. Lett. 2003, 91, 106102. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. Ab Initio Molecular Dynamics for Liquid Metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab Initio Molecular-Dynamics Simulation of the Liquid-Metalamorphous- Semiconductor Transition in Germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S. Semiempirical GGA-Type Density Functional Constructed with a Long-Range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Methfessel, M.; Paxton, A.T. High-Precision Sampling for Brillouin-Zone Integration in Metals. Phys. Rev. B 1989, 40, 3616–3621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blöchl, P.E.; Jepsen, O.; Andersen, O.K. Improved Tetrahedron Method for Brillouin-Zone Integrations. Phys. Rev. B 1994, 49, 16223–16233. [Google Scholar] [CrossRef]
- Huang, B.; Kobayashi, H.; Yamamoto, T.; Toriyama, T.; Matsumura, S.; Nishida, Y.; Sato, K.; Nagaoka, K.; Haneda, M.; Xie, W.; et al. A CO Adsorption Site Change Induced by Copper Substitution in a Ruthenium Catalyst for Enhanced CO Oxidation Activity. Angew. Chem. 2019, 58, 2230–2235. [Google Scholar] [CrossRef]
- Hull, A.W. X-ray Crystal Analysis of Thirteen Common Metals. Phys. Rev. 1921, 17, 571–588. [Google Scholar] [CrossRef] [Green Version]
- Corbel, G.; Topić, M.; Gibaud, A.; Lang, C.I. Selective Dry Oxidation of the Ordered Pt-11.1 at.% v Alloy Surface Evidenced by in Situ Temperature-Controlled X-ray Diffraction. J. Alloy. Compd. 2011, 509, 6532–6538. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zon Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Song, C.; Sakata, O.; Kumara, L.S.R.; Kohara, S.; Yang, A.; Kusada, K.; Kobayashi, H.; Kitagawa, H. Size Dependence of Structural Parameters in Fcc and Hcp Ru Nanoparticles, Revealed by Rietveld Refinement Analysis of High-Energy X-ray Diffraction Data. Sci. Rep. 2016, 6, 31400. [Google Scholar] [CrossRef]
- Arblaster, J.W. Crystallographic Properties of Platinum. Platin. Met. Rev. 1997, 41, 12–21. [Google Scholar] [CrossRef]
- Arblaster, J.W. Crystallographic Properties of Platinum (Errata). Platin. Met. Rev. 2006, 50, 118–119. [Google Scholar] [CrossRef]
- Watson, G.W.; Kelsey, E.T.; de Leeuw, N.H.; Harris, D.J.; Parker, S.C. Atomistic Simulation of Dislocations, Surfaces and Interfaces in MgO. J. Chem. Soc. Faraday Trans. 1996, 92, 433. [Google Scholar] [CrossRef]
- Wulff, G., XXV. Zur Frage der Geschwindigkeit des Wachsthums und der Auflösung der Krystallflächen. Z. Krist. 1901, 34, 449–530. [Google Scholar] [CrossRef]
- Rahm, J.; Erhart, P. WulffPack: A Python Package for Wulff Constructions. J. Open Source Softw. 2020, 5, 1944. [Google Scholar] [CrossRef] [Green Version]
- Bhattarai, H.; Drisko, C.; Duraes, A.D.S.; Lin, T.; Charles, F.V., II; Christopher, J.; Fennell, M.A.M.; Louden, P.; Neidhart, S.; Kuang, S.; et al. OpenMD-2.6: Molecular Dynamics in the Open; University of Notre Dame: Notre Dame, Indiana, 2019. [Google Scholar]
- Sutton, A.P.; Chen, J. Long-Range Finnis–Sinclair Potentials. Philos. Mag. Lett. 1990, 61, 139–146. [Google Scholar] [CrossRef]
- Qi, Y.; Çağın, T.; Kimura, Y.; Goddard, W.A. Molecular-Dynamics Simulations of Glass Formation and Crystallization in Binary Liquid Metals: Cu-Ag and Cu-Ni. Phys. Rev. B 1999, 59, 3527–3533. [Google Scholar] [CrossRef] [Green Version]
- Prasai, B.; Ren, Y.; Shan, S.; Zhao, Y.; Cronk, H.; Luo, J.; Zhong, C.J.; Petkov, V. Synthesis-Atomic Structure-Properties Relationships in Metallic Nanoparticles by Total Scattering Experiments and 3D Computer Simulations: Case of Pt–Ru Nanoalloy Catalysts. Nanoscale 2015, 7, 8122–8134. [Google Scholar] [CrossRef]
- Prasai, B.; Ren, Y.; Shan, S.; Zhao, Y.; Cronk, H.; Luo, J.; Zhong, C.J.; Petkov, V. Correction: Synthesis-Atomic Structure–Properties Relationships in Metallic Nanoparticles by Total Scattering Experiments and 3D Computer Simulations: Case of Pt–Ru Nanoalloy Catalysts. Nanoscale 2015, 7, 10279. [Google Scholar] [CrossRef] [Green Version]
- Momma, K.; Izumi, F. VESTA 3 for Three-Dimensional Visualization of Crystal, Volumetric and Morphology Data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Ungerer, M.J.; Santos-Carballal, D.; Cadi-Essadek, A.; van Sittert, C.G.C.E.; de Leeuw, N.H. Interaction of H 2 O with the Platinum Pt (001), (011), and (111) Surfaces: A Density Functional Theory Study with Long-Range Dispersion Corrections. J. Phys. Chem. C 2019, 123, 27465–27476. [Google Scholar] [CrossRef]
- Posada-Pérez, S.; Santos-Carballal, D.; Terranova, U.; Roldan, A.; Illas, F.; de Leeuw, N.H. CO2 Interaction with Violarite (FeNi2S4) Surfaces: A Dispersion-Corrected DFT Study. Phys. Chem. Chem. Phys. 2018, 20, 20439–20446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.; Liu, J.X.; Fan, H.; Li, W.X. Compensation between Surface Energy and Hcp/Fcc Phase Energy of Late Transition Metals from First-Principles Calculations. J. Phys. Chem. C 2020, 124, 11005–11014. [Google Scholar] [CrossRef]
- Wang, J.; Wang, S.Q. Surface Energy and Work Function of Fcc and Bcc Crystals: Density Functional Study. Surf. Sci. 2014, 630, 216–224. [Google Scholar] [CrossRef]
- Singh-Miller, N.E.; Marzari, N. Surface Energies, Work Functions, and Surface Relaxations of Low-Index Metallic Surfaces from First Principles. Phys. Rev. B Condens. Matter Mater. Phys. 2009, 80, 235407. [Google Scholar] [CrossRef] [Green Version]
- Tran, R.; Xu, Z.; Radhakrishnan, B.; Winston, D.; Sun, W.; Persson, K.A.; Ong, S.P. Surface Energies of Elemental Crystals. Sci. Data 2016, 3, 160080. [Google Scholar] [CrossRef] [Green Version]
- Hulse, J.; Küppers, J.; Wandelt, K.; Ertl, G. UV-Photoelectron Spectroscopy from Xenon Adsorbed on Heterogeneous Metal Surfaces. Appl. Surf. Sci. 1980, 6, 453–463. [Google Scholar] [CrossRef]
- Tyson, W.R. Surface Energies of Solid Metals. Can. Metall. Q. 1975, 14, 307–314. [Google Scholar] [CrossRef]
- De Boer, F.R.; Boom, R.; Mattens, W.C.M.; Miedema, A.R.; Niessen, A.K. Cohesion in Metals; Elsevier Science Publishers: Amsterdam, The Netherlands, 1988. [Google Scholar]
- Tyson, W.R.; Miller, W.A. Surface Free Energies of Solid Metals: Estimation from Liquid Surface Tension Measurements. Surf. Sci. 1977, 62, 267–276. [Google Scholar] [CrossRef]
- Michaelson, H.B. The Work Function of the Elements and Its Periodicity. J. Appl. Phys. 1977, 48, 4729–4733. [Google Scholar] [CrossRef] [Green Version]
- Caglar, B.; Kizilkaya, A.C.; Niemantsverdriet, J.W.; Weststrate, C.J. Application of Work Function Measurements in the Study of Surface Catalyzed Reactions on Rh(1 0 0). Catal. Struct. React. 2018, 4, 1–11. [Google Scholar] [CrossRef]
- Tran, R.; Xu, Z.; Radhakrishnan, B.; Winston, D.; Sun, W.; Persson, K.A.; Ong, S.P.; Characteristic, S. The Inhibition of Epidermal Growth Factor Receptor Signaling by Hexagonal Selenium Nanoparticles Modified by SiRNA. Cancer Gene Ther. 2016, 23, 321–325. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.; Morales, R.; Albiter, M.A.; Zaera, F. Synthesis of Heterogeneous Catalysts with Well Shaped Platinum Particles to Control Reaction Selectivity. Proc. Natl. Acad. Sci. USA 2008, 105, 15241–15246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shevchenko, V.Y.; Madison, A.E. Structure of Nanoparticles: I. Generalized Crystallography of Nanoparticles and Magic Numbers. Glass Phys. Chem. 2002, 28, 40–43. [Google Scholar] [CrossRef]
- Mackay, A.L. A Dense Non-Crystallographic Packing of Equal Spheres. Acta Crystallogr. 1962, 15, 916–918. [Google Scholar] [CrossRef]
- Ungerer, M.J.; Santos-Carballal, D.; Cadi-Essadek, A.; van Sittert, C.G.C.E.; de Leeuw, N.H. Interaction of SO2 with the Platinum (001), (011), and (111) Surfaces: A DFT Study. Catalysts 2020, 10, 558. [Google Scholar] [CrossRef]
- Anderson, S.E.; McKay, B. Counting Polyhedra. Available online: http://www.numericana.com/data/polyhedra.htm (accessed on 1 June 2022).
- Johnson, N.W. Convex Polyhedra with Regular Faces. Can. J. Math. 1966, 18, 169–200. [Google Scholar] [CrossRef]
- Coxeter, H.S.M. 2.3 Quasi-Regular Polyhedra. In Regular Polytopes; Dover Publications: Dover, UK, 1973; pp. 18–19. ISBN 0-486-61480-8. [Google Scholar]
- Farges, J.; De Feraudy, M.F.; Raoult, B.; Torchet, G. Noncrystalline Structure of Argon Clusters. I. Polyicosahedral Structure of ArN Clusters, 20 < N < 50. J. Chem. Phys. 1983, 78, 5067–5080. [Google Scholar] [CrossRef]
- Farges, J.; de Feraudy, M.F.; Raoult, B.; Torchet, G. Noncrystalline Structure of Argon Clusters. II. Multilayer Icosahedral Structure of Ar N Clusters 50 < N < 750. J. Chem. Phys. 1986, 84, 3491–3501. [Google Scholar] [CrossRef]
- Sloane, N.J.A.; Teo, B.K. Theta Series and Magic Numbers for Close-Packed Spherical Clusters. J. Chem. Phys. 1985, 83, 6520–6534. [Google Scholar] [CrossRef]
- Vollath, D.; Fischer, F.D.; Holec, D. Surface Energy of Nanoparticles—Influence of Particle Size and Structure. Beilstein J. Nanotechnol. 2018, 9, 2265–2276. [Google Scholar] [CrossRef]
- Kerber, T.; Sierka, M.; Sauer, J. Application of Semiempirical Long-Range Dispersion Corrections to Periodic Systems in Density Functional Theory. J. Comput. Chem. 2008, 29, 2088–2097. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ruthenium | Palladium | Platinum | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DFT- D2 | DFT- D3 | DFT-D3(BJ) | Other Work | DFT- D2 | DFT- D3 | DFT-D3(BJ) | Other Work | DFT- D2 | DFT- D3 | DFT-D3(BJ) | Other Work | |
| Ebulk (eV/atom) | −9.83 | −9.64 | −9.74 | −5.90 | −5.85 | −5.93 | −8.05 | −7.01 | −7.01 | |||
| a (Å) | 3.763 | 3.777 | 3.778 | 3.818 [44], 3.87 [30] | 3.889 | 3.886 | 3.886 | 3.949 [44], 3.89 [27] | 3.837 | 3.917 | 3.924 | 3.924 [31,32] |
| (001) | ||||||||||||
| γu (J.m−2) | 3.77 | 3.75 | 3.65 | 2.30 | 2.31 | 2.28 | 4.52 | 2.79 | 2.73 | |||
| γr (J.m−2) | 3.71 | 3.67 | 3.58 | 2.99 [44] | 2.30 | 2.30 | 2.32 | 2.15 [45] | 4.19 | 2.79 | 2.73 | 1.81 [46] |
| A (Å2) | 127.48 | 128.40 | 128.46 | 135.94 | 135.93 | 135.93 | 132.50 | 138.10 | 138.60 | |||
| Φ (eV) | 5.18 | 5.05 | 5.08 | 4.85 [47] | 5.16 | 5.06 | 5.06 | 5.08 [45], 5.65 [48] | 5.81 | 5.77 | 5.58 | 5.66 [46] |
| (011) | ||||||||||||
| γu (J.m−2) | 3.95 | 3.71 | 3.83 | 2.44 | 2.42 | 2.36 | 4.31 | 2.77 | 2.86 | |||
| γr (J.m−2) | 3.81 | 3.55 | 3.67 | 2.77 [44] | 2.41 | 2.41 | 2.34 | 2.30 [45] | 4.19 | 2.70 | 2.79 | 1.85 [46] |
| A (Å2) | 120.18 | 121.05 | 121.12 | 128.16 | 128.16 | 128.16 | 124.92 | 130.20 | 130.67 | |||
| Φ (eV) | 4.21 | 4.20 | 4.14 | 4.28 [47] | 5.08 | 5.07 | 5.06 | 5.13 [45], 5.20 [48] | 5.45 | 5.53 | 5.55 | 5.26 [46] |
| (111) | ||||||||||||
| γu (J.m−2) | 3.30 | 3.20 | 3.11 | 2.17 | 2.15 | 2.14 | 3.96 | 2.18 | 2.34 | |||
| γr (J.m−2) | 3.28 | 3.18 | 3.08 | 2.37 [44], 3.04 [49] | 2.15 | 2.14 | 2.13 | 1.90 [45], 2.05 [50] | 3.92 | 2.18 | 2.33 | 1.49 [46], 2.49 [51] |
| A (Å2) | 98.13 | 98.83 | 98.89 | 104.64 | 104.64 | 104.64 | 102.00 | 106.31 | 106.69 | |||
| Φ (eV) | 5.19 | 5.19 | 5.23 | 4.94 [47], 4.71 [52] | 5.18 | 5.18 | 5.18 | 5.53 [45], 5.90 [48] | 5.84 | 5.77 | 5.71 | 5.69 [46] |
| DFT-D2 | % | DFT-D3 | % | DFT-D3(BJ) | % | |
|---|---|---|---|---|---|---|
| Ru | ||||||
 |  | 64.9 |  | 57.2 |  | 72.5 |
| 24.1 | 20.4 | 21.7 | ||||
| 10.9 | 22.4 | 5.8 | ||||
| Pd | ||||||
 |  | 54.6 |  | 55.0 |  | 58.4 |
| 29.4 | 28.9 | 27.8 | ||||
| 16.0 | 16.1 | 13.7 | ||||
| Pt | ||||||
 |  | 45.9 |  | 87.1 |  | 74.3 |
| 18.9 | 12.9 | 20.9 | ||||
| 35.2 | - | 4.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ungerer, M.J.; De Leeuw, N.H. Computational Insights into Ru, Pd and Pt fcc Nano-Catalysts from Density Functional Theory Calculations: The Influence of Long-Range Dispersion Corrections. Catalysts 2022, 12, 1287. https://doi.org/10.3390/catal12101287
Ungerer MJ, De Leeuw NH. Computational Insights into Ru, Pd and Pt fcc Nano-Catalysts from Density Functional Theory Calculations: The Influence of Long-Range Dispersion Corrections. Catalysts. 2022; 12(10):1287. https://doi.org/10.3390/catal12101287
Chicago/Turabian StyleUngerer, Marietjie J., and Nora H. De Leeuw. 2022. "Computational Insights into Ru, Pd and Pt fcc Nano-Catalysts from Density Functional Theory Calculations: The Influence of Long-Range Dispersion Corrections" Catalysts 12, no. 10: 1287. https://doi.org/10.3390/catal12101287
APA StyleUngerer, M. J., & De Leeuw, N. H. (2022). Computational Insights into Ru, Pd and Pt fcc Nano-Catalysts from Density Functional Theory Calculations: The Influence of Long-Range Dispersion Corrections. Catalysts, 12(10), 1287. https://doi.org/10.3390/catal12101287