
Cofactor and Process Engineering for Nicotinamide Recycling and Retention in Intensified Biocatalysis

 ,
, {kind=link}
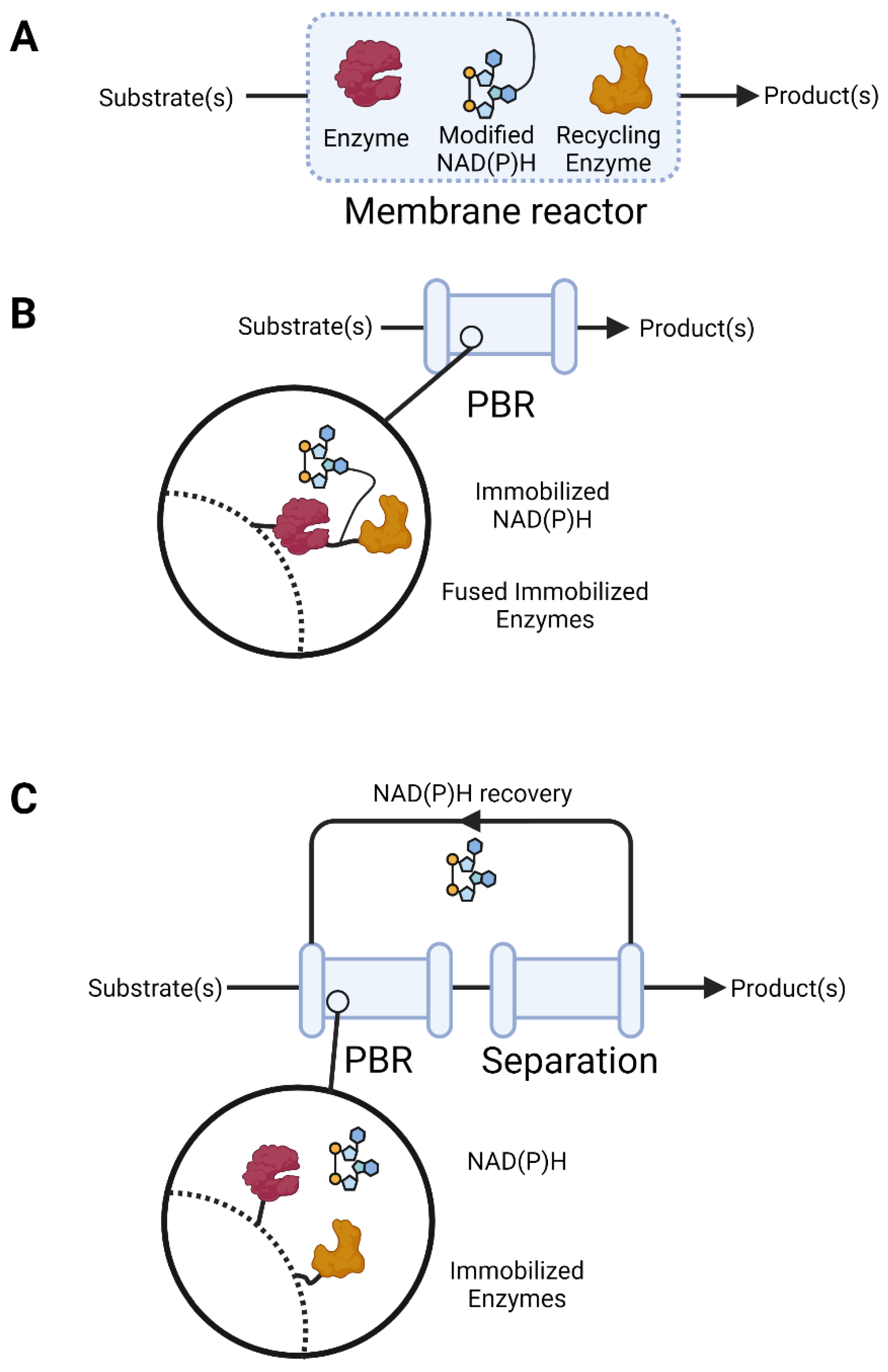{kind=link}
{kind=link}
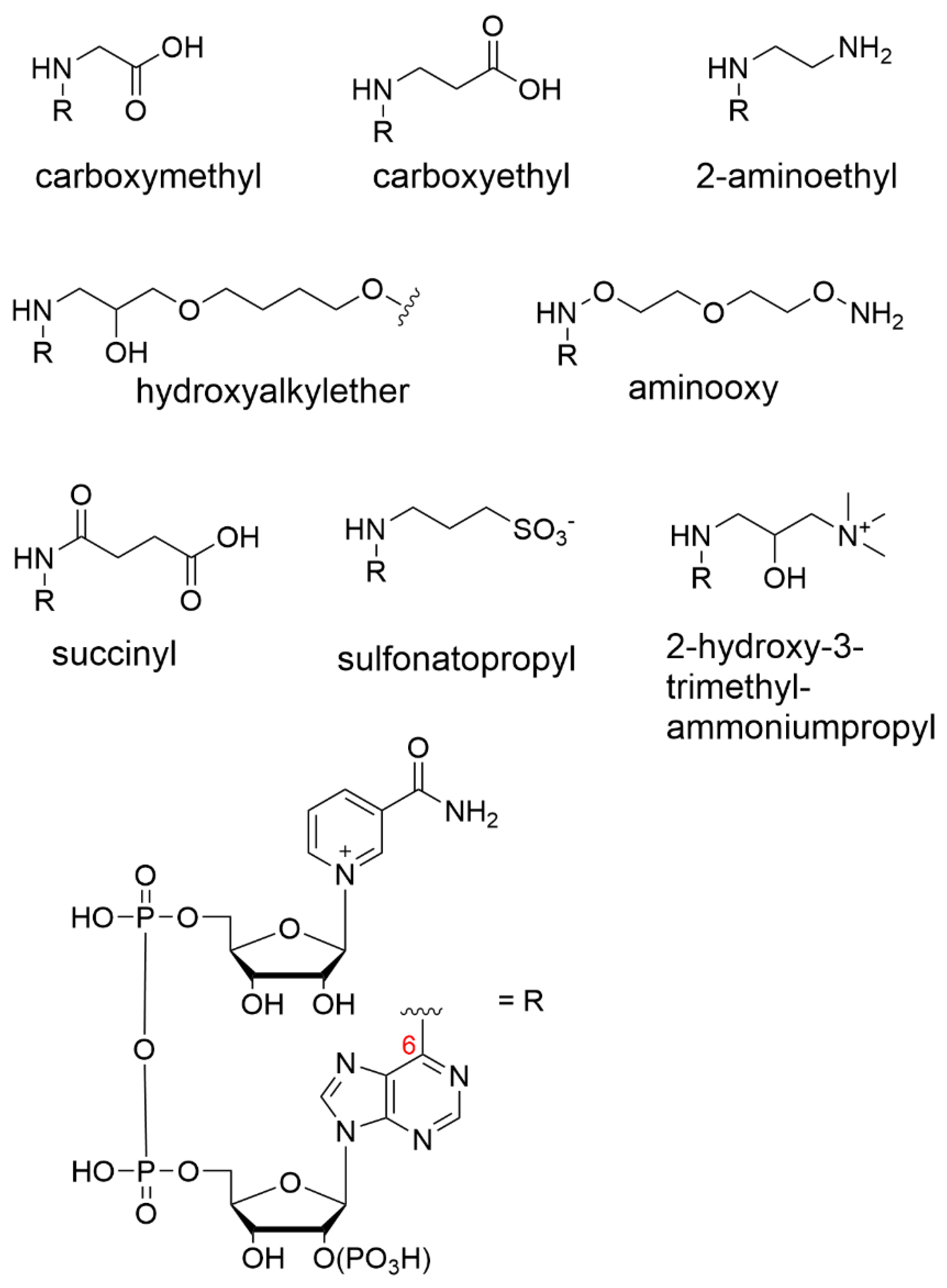{kind=link}
{kind=link}
Abstract
:1. Introduction
2. NAD(P)H Use in Continuous-Flow Biocatalysis
3. Engineering Nicotinamide Cofactors for Continuous-Flow Biocatalysis
3.1. The Dimroth Rearrangement Route
3.2. Phosphate Coupling
4. Nicotinamide Cofactor Biomimetics
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Adams, J.P.; Brown, M.J.B.; Diaz-Rodriguez, A.; Lloyd, R.C.; Roiban, G.D. Biocatalysis: A pharma perspective. Adv. Synth. Catal. 2019, 361, 2421–2432. [Google Scholar] [CrossRef] [Green Version]
- Bornscheuer, U.T.; Huisman, G.W.; Kazlauskas, R.J.; Lutz, S.; Moore, J.C.; Robins, K. Engineering the third wave of biocatalysis. Nature 2012, 485, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Faber, K.; Fessner, W.D.; Turner, N.J. Biocatalysis: Ready to master increasing complexity. Adv. Synth. Catal. 2019, 361, 2373–2376. [Google Scholar] [CrossRef] [Green Version]
- Winkler, C.K.; Schrittwieser, J.H.; Kroutil, W. Power of biocatalysis for organic synthesis. ACS Cent. Sci. 2021, 7, 55–71. [Google Scholar] [CrossRef] [PubMed]
- García-Junceda, E.; Lavandera, I.; Rother, D.; Schrittwieser, J.H. (Chemo) enzymatic cascades—Nature’s synthetic strategy transferred to the laboratory. J. Mol. Catal. B Enzym. 2015, 114, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Reetz, M.T. Biocatalysis in organic chemistry and biotechnology: Past, present, and future. J. Am. Chem. Soc. 2013, 135, 12480–12496. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Brady, D. The limits to biocatalysis: Pushing the envelope. Chem. Commun. 2018, 54, 6088–6104. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Pereira, P.C. Biocatalysis engineering: The big picture. Chem. Soc. Rev. 2017, 46, 2678–2691. [Google Scholar] [CrossRef]
- Faber, K. Biocatalytic applications. In Biotransformations in Organic Chemistry; Springer: Cham, Switzerland, 2011; pp. 31–313. [Google Scholar]
- Grogan, G. Synthesis of chiral amines using redox biocatalysis. Curr. Opin. Chem. Biol. 2018, 43, 15–22. [Google Scholar] [CrossRef]
- Toogood, H.S.; Scrutton, N.S. Discovery, characterization, engineering, and applications of ene-reductases for industrial biocatalysis. ACS Catal. 2018, 8, 3532–3549. [Google Scholar] [CrossRef]
- Rodriguez-Abetxuko, A.; Reifs, A.; Sanchez-deAlcazar, D.; Beloqui, A. A Versatile Chemoenzymatic Nanoreactor that Mimics NAD(P)H Oxidase for the In Situ Regeneration of Cofactors. Angew. Chem. Int. Ed. 2022, 61, e202206926. [Google Scholar]
- Kroutil, W.; Mang, H.; Edegger, K.; Faber, K. Recent advances in the biocatalytic reduction of ketones and oxidation of sec-alcohols. Curr. Opin. Chem. Biol. 2004, 8, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.; Scott, C.; Grogan, G. F420-dependent enzymes-potential for applications in biotechnology. Trends Biotechnol. 2013, 31, 63–64. [Google Scholar] [CrossRef] [PubMed]
- Chenault, H.K.; Whitesides, G.M. Regeneration of nicotinamide cofactors for use in organic synthesis. Appl. Biochem. Biotechnol. 1987, 14, 147–197. [Google Scholar] [CrossRef]
- Fukuzumi, S.; Lee, Y.-M.; Nam, W. Catalytic recycling of NAD(P)H. J. Inorg. Biochem. 2019, 199, 110777. [Google Scholar] [CrossRef]
- Hummel, W.; Gröger, H. Strategies for regeneration of nicotinamide coenzymes emphasizing self-sufficient closed-loop recycling systems. J. Biotechnol. 2014, 191, 22–31. [Google Scholar] [CrossRef]
- Weckbecker, A.; Gröger, H.; Hummel, W. Regeneration of nicotinamide coenzymes: Principles and applications for the synthesis of chiral compounds. In Biosystems Engineering I; Springer: Berlin/Heidelberg, Germany, 2010; pp. 195–242. [Google Scholar]
- Wu, H.; Tian, C.; Song, X.; Liu, C.; Yang, D.; Jiang, Z. Methods for the regeneration of nicotinamide coenzymes. Green Chem. 2013, 15, 1773–1789. [Google Scholar] [CrossRef]
- Zhao, H.; Van Der Donk, W.A. Regeneration of cofactors for use in biocatalysis. Curr. Opin. Biotechnol. 2003, 14, 583–589. [Google Scholar] [CrossRef]
- Rodriguez, C.; Lavandera, I.; Gotor, V. Recent advances in cofactor regeneration systems applied to biocatalyzed oxidative processes. Current Org. Chem. 2012, 16, 2525–2541. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Saba, T.; Yiu, H.H.P.; Howe, R.F.; Anderson, J.A.; Shi, J. Cofactor NAD(P)H regeneration inspired by heterogeneous pathways. Chem 2017, 2, 621–654. [Google Scholar] [CrossRef] [Green Version]
- Mordhorst, S.; Andexer, J.N. Round, round we go–strategies for enzymatic cofactor regeneration. Nat. Prod. Rep. 2020, 37, 1316–1333. [Google Scholar] [CrossRef] [PubMed]
- Tassano, E.; Hall, M. Enzymatic self-sufficient hydride transfer processes. Chem. Soc. Rev. 2019, 48, 5596–5615. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.Q.; Feng, J.Q.; Cui, J.W.; Jiang, G.D.; Harrison, W.; Zang, X.; Zhou, J.H.; Wang, B.J.; Zhao, H.M. Photoinduced chemomimetic biocatalysis for enantioselective intermolecular radical conjugate addition. Nat. Catal. 2022, 5, 586–593. [Google Scholar] [CrossRef]
- De Santis, P.; Meyer, L.E.; Kara, S. The rise of continuous flow biocatalysis—Fundamentals, very recent developments and future perspectives. React. Chem. Eng. 2020, 5, 2155–2184. [Google Scholar] [CrossRef]
- Tamborini, L.; Fernandes, P.; Paradisi, F.; Molinari, F. Flow bioreactors as complementary tools for biocatalytic process intensification. Trends Biotechnol. 2018, 36, 73–88. [Google Scholar] [CrossRef]
- Woltinger, J.; Karau, A.; Leuchtenberger, W.; Drauz, K. Membrane reactors at Degussa. In Technology Transfer in Biotechnology: From Lab to Industry to Production; Springer: Berlin, Germany, 2005; Volume 92, pp. 289–316. [Google Scholar]
- Bitterwolf, P.; Ott, F.; Rabe, K.S.; Niemeyer, C.M. Imine Reductase Based All-Enzyme Hydrogel with Intrinsic Cofactor Regeneration for Flow Biocatalysis. Micromachines 2019, 10, 783. [Google Scholar] [CrossRef] [Green Version]
- Gutmann, B.; Cantillo, D.; Kappe, C.O. Continuous-flow technology—A tool for the safe manufacturing of active pharmaceutical ingredients. Angew. Chem. Int. Ed. 2015, 54, 6688–6728. [Google Scholar] [CrossRef]
- Hessel, V.; Kralisch, D.; Kockmann, N.; Noël, T.; Wang, Q. Novel process windows for enabling, accelerating, and uplifting flow chemistry. ChemSusChem 2013, 6, 746–789. [Google Scholar] [CrossRef]
- Newman, S.G.; Jensen, K.F. The role of flow in green chemistry and engineering. Green Chem. 2013, 15, 1456–1472. [Google Scholar] [CrossRef] [Green Version]
- Thompson, M.P.; Peñafiel, I.; Cosgrove, S.C.; Turner, N.J. Biocatalysis using immobilized enzymes in continuous flow for the synthesis of fine chemicals. Org. Process Res. Dev. 2018, 23, 9–18. [Google Scholar] [CrossRef]
- Hartley, C.J.; Williams, C.C.; Scoble, J.A.; Churches, Q.I.; North, A.; French, N.G.; Nebl, T.; Coia, G.; Warden, A.C.; Simpson, G. Engineered enzymes that retain and regenerate their cofactors enable continuous-flow biocatalysis. Nat. Catal. 2019, 2, 1006–1015. [Google Scholar] [CrossRef]
- Velasco-Lozano, S.; Benítez-Mateos, A.I.; López-Gallego, F. Co-immobilized phosphorylated cofactors and enzymes as self-sufficient heterogeneous biocatalysts for chemical processes. Angew. Chem. Int. Ed. 2017, 56, 771–775. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.P.; Derrington, S.R.; Heath, R.S.; Porter, J.L.; Mangas-Sanchez, J.; Devine, P.N.; Truppo, M.D.; Turner, N.J. A generic platform for the immobilisation of engineered biocatalysts. Tetrahedron 2019, 75, 327–334. [Google Scholar] [CrossRef]
- Mattey, A.P.; Ford, G.J.; Citoler, J.; Baldwin, C.; Marshall, J.R.; Palmer, R.B.; Thompson, M.; Turner, N.J.; Cosgrove, S.C.; Flitsch, S.L. Development of Continuous Flow Systems to Access Secondary Amines Through Previously Incompatible Biocatalytic Cascades. Angew. Chem. Int. Ed. 2021, 60, 18660–18665. [Google Scholar] [CrossRef]
- Roura Padrosa, D.; Nisar, Z.; Paradisi, F. Efficient amino donor recycling in amination reactions: Development of a new alanine dehydrogenase in continuous flow and dialysis membrane reactors. Catalysts 2021, 11, 520. [Google Scholar] [CrossRef]
- Peschke, T.; Skoupi, M.; Burgahn, T.; Gallus, S.; Ahmed, I.; Rabe, K.S.; Niemeyer, C.M. Self-immobilizing fusion enzymes for compartmentalized biocatalysis. ACS Catal. 2017, 7, 7866–7872. [Google Scholar] [CrossRef]
- Contente, M.L.; Paradisi, F. Self-sustaining closed-loop multienzyme-mediated conversion of amines into alcohols in continuous reactions. Nat. Catal. 2018, 1, 452–459. [Google Scholar] [CrossRef]
- Padrosa, D.R.; Benítez-Mateos, A.I.; Calvey, L.; Paradisi, F. Cell-free biocatalytic syntheses of L-pipecolic acid: A dual strategy approach and process intensification in flow. Green Chem. 2020, 22, 5310–5316. [Google Scholar] [CrossRef]
- Baumer, B.; Classen, T.; Pohl, M.; Pietruszka, J. Efficient Nicotinamide Adenine Dinucleotide Phosphate [NADP (H)] Recycling in Closed-Loop Continuous Flow Biocatalysis. Adv. Synth. Catal. 2020, 362, 2894–2901. [Google Scholar] [CrossRef] [Green Version]
- Kula, M.-R.; Wandrey, C. [2] Continuous enzymatic transformation in an enzyme-membrane reactor with simultaneous NADH regeneration. Methods Enzymol. 1987, 136, 9–21. [Google Scholar]
- Månsson, M.-O.; Mosbach, K. Immobilized active coenzymes. Meth. Enzymol. 1987, 136, 3–9. [Google Scholar]
- Mosbach, K.; Larsson, P.-O.; Lowe, C. Immobilized coenzymes. Methods Enzymol. 1976, 44, 859–887. [Google Scholar] [PubMed]
- Wichmann, R.; Vasic-Racki, D. Cofactor regeneration at the lab scale. Technol. Transf. Biotechnol. 2005, 92, 225–260. [Google Scholar]
- Wichmann, R.; Wandrey, C.; Bückmann, A.F.; Kula, M.R. Continuous enzymatic transformation in an enzyme membrane reactor with simultaneous NAD(H) regeneration. Biotechnol. Bioeng. 1981, 23, 2789–2802. [Google Scholar] [CrossRef]
- Wandrey, C.; Wichmann, R. Production of l-amino acids in the membrane reactor. Biotech. 1987, 1, 85–92. [Google Scholar]
- Katz, E.; Bückmann, A.F.; Willner, I. Self-powered enzyme-based biosensors. J. Am. Chem. Soc. 2001, 123, 10752–10753. [Google Scholar] [CrossRef]
- Fu, J.; Yang, Y.R.; Johnson-Buck, A.; Liu, M.; Liu, Y.; Walter, N.G.; Woodbury, N.W.; Yan, H. Multi-enzyme complexes on DNA scaffolds capable of substrate channelling with an artificial swinging arm. Nat. Nanotechnol. 2014, 9, 531–536. [Google Scholar] [CrossRef]
- Kim, S.; Kwon, K.; Tae, G.; Kwon, I. Nano-Entrapping Multiple Oxidoreductases and Cofactor for All-In-One Nanoreactors. ACS Sustain. Chem. Eng. 2021, 9, 6741–6747. [Google Scholar] [CrossRef]
- Lindberg, M.; Larsson, P.O.; Mosbach, K. A new immobilized NAD+ analogue, its application in affinity chromatography and as a functioning coenzyme. Eur. J. Biochem. 1973, 40, 187–193. [Google Scholar] [CrossRef]
- Schmidt, H.L.; Grenner, G. Coenzyme Properties of NAD+ Bound to Different Matrices through the Amino Group in the 6-Position. Eur. J. Biochem. 1976, 67, 295–302. [Google Scholar] [CrossRef]
- Wykes, J.R.; Dunnill, P.; Lilly, M.D. The preparation of soluble high molecular weight NAD derivative active as a cofactor. Biochem. Biophys. Acta Gen. Subj. 1972, 286, 260–268. [Google Scholar] [CrossRef]
- Mosbach, K. Immobilised enzymes. FEBS Lett. 1976, 62, E80–E95. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Mullens, C.; Gorski, W. Coimmobilization of dehydrogenases and their cofactors in electrochemical biosensors. Anal. Chem. 2007, 79, 2446–2450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radoi, A.; Compagnone, D. Recent advances in NADH electrochemical sensing design. Bioelectrochemistry 2009, 76, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.J.; Buehner, M.; Chandrasekhar, K.; Ford, G.C.; Hackert, M.L.; Liljas, A.; Rossmann, M.G.; Smiley, I.E.; Allison, W.S.; Everse, J. Structure-function relationships in lactate dehydrogenase. Proc. Natl. Acad. Sci. USA 1973, 70, 1968–1972. [Google Scholar] [CrossRef]
- Hendle, J.; BÜCkmann, A.F.; Aehle, W.; Schomburg, D.; Schmid, R.D. Structure/activity relationship of adenine-modified NAD derivatives with respect to porcine heart lactate dehydrogenase isozyme H4 simulated with molecular mechanics. Eur. J. Biochem. 1993, 213, 947–956. [Google Scholar] [CrossRef]
- Grenner, G.; Schmidt, H.L.; Voelkl, W. Coenzym-Eigenschaften einiger in l- und N6-stellung des Adeninringes substituierterN AD+-Derivate. Z. Physiol. Chem. 1976, 357, 887–902. [Google Scholar]
- Ottolina, G.; Carrea, G.; Riva, S.; Bückmann, A.F. Coenzymatic properties of low molecular-weight and macromolecular N6-derivatives of NAD+ and NADP+ with dehydrogenases of interest for organic synthesis. Enzym. Microb. Technol. 1990, 12, 596–602. [Google Scholar] [CrossRef]
- Zappelli, P.; Pappa, R.; Rossodivita, A.; Re, L. Preparation and Coenzymic Activity of Soluble Polyethyleneimine-Bound NADP+ Derivatives. Eur. J. Biochem. 1977, 72, 309–315. [Google Scholar] [CrossRef]
- Engel, J.D. Mechanism of the Dimroth rearrangement in adenosine. Biochem. Biophys. Res. Commun. 1975, 64, 581–586. [Google Scholar] [CrossRef]
- Lowe, C.R.; Mosbach, K. The synthesis of adenine-substituted derivatives of NADP+ and their potential as active coenzymes and affinity adsorbents. Eur. J. Biochem. 1974, 49, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, J.; Vieille, C. Activity of select dehydrogenases with Sepharose-immobilized N6-carboxymethyl-NAD. Bioengineered 2015, 6, 106–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishimoto, T.; Itami, M.; Yomo, T.; Urabe, I.; Yamada, Y.; Okada, H. Improved methods for the preparation of N6-(2-carboxyethyl)-NAD and poly (ethylene glycol)-bound NAD (H). J. Ferment. Bioeng. 1991, 71, 447–449. [Google Scholar] [CrossRef]
- Muramatsu, M.; Urabe, I.; Yamada, Y.; Okada, H. Synthesis and kinetic properties of a new NAD+ derivative carrying a vinyl group. Eur. J. Biochem. 1977, 80, 111–117. [Google Scholar] [CrossRef]
- Fuller, C.W.; Rubin, J.R.; Bright, H.J. A simple procedure for covalent immobilization of NADH in a soluble and enzymically active form. Eur. J. Biochem. 1980, 103, 421–430. [Google Scholar] [CrossRef]
- Carrea, G.; Ottolina, G.; Riva, S.; Danieli, B.; Lesma, G.; Palmisano, G. Alkylation of Adenine, Adenosine, and NAD+ with 1, 3-Propanesultone. Synthesis of N6-(3-sulfonatopropyl)-NAD+, a new NAD+ derivative with substantial coenzyme activity. Helv. Chim. Acta 1988, 71, 762–772. [Google Scholar] [CrossRef]
- Okuda, K.; Suntinanalerts, P.; Miyoshi, S.; Urabe, I.; Yamada, Y.; Okada, H. Preparation and characterization of NADP derivatives alkylated at 2′-phosphate and 6-amino groups. Eur. J. Biochem. 1985, 147, 241–247. [Google Scholar] [CrossRef]
- Sogin, D.C. 2′, 3′-Cyclic NADP as a substrate for 2′, 3′-cyclic nucleotide 3′-phosphohydrolase. J. Neurochem. 1976, 27, 1333–1337. [Google Scholar] [CrossRef]
- Reiss, J.R.; Moffatt, J.G. Dismutation Reactions of Nucleoside Polyphosphates. III. The Synthesis of α, ι-Dinucleoside 5′-Polyphosphates1. J. Org. Chem. 1965, 30, 3381–3387. [Google Scholar] [CrossRef]
- Appy, L.; Chardet, C.; Peyrottes, S.; Roy, B. Synthetic Strategies for Dinucleotides Synthesis. Molecules 2019, 24, 4334. [Google Scholar] [CrossRef] [Green Version]
- Buntz, A.; Wallrodt, S.; Gwosch, E.; Schmalz, M.; Beneke, S.; Ferrando-May, E.; Marx, A.; Zumbusch, A. Real-time cellular imaging of protein poly (ADP-ribos) ylation. Angew. Chem. Int. Ed. 2016, 55, 11256–11260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, N.A.; Kenner, G.W.; Todd, A. Codehydrogenases. Part III. A synthesis of diphosphopyridine nucleotide (cozymase), and some observations on the synthesis of triphosphopyridine nucleotide. J. Chem. Soc. 1957, 3733–3738. [Google Scholar] [CrossRef]
- Wallrodt, S.; Buntz, A.; Wang, Y.; Zumbusch, A.; Marx, A. Bioorthogonally Functionalized NAD+ Analogues for In-Cell Visualization of Poly (ADP-Ribose) Formation. Angew. Chem. Int. Ed. 2016, 55, 7660–7664. [Google Scholar] [CrossRef] [PubMed]
- Cen, Y.; Falco, J.N.; Xu, P.; Youn, D.Y.; Sauve, A.A. Mechanism-based affinity capture of sirtuins. Org. Biomol. Chem. 2011, 9, 987–993. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.T.; Zhang, X.-N.; Courouble, V.V.; Strutzenberg, T.S.; Pei, H.; Stiles, B.L.; Louie, S.G.; Griffin, P.R.; Zhang, Y. A Bifunctional NAD+ for Profiling Poly-ADP-Ribosylation-Dependent Interacting Proteins. ACS Chem. Biol. 2021, 16, 389–396. [Google Scholar] [CrossRef]
- Wang, Y.; Rösner, D.; Grzywa, M.; Marx, A. Chain-Terminating and Clickable NAD+ Analogues for Labeling the Target Proteins of ADP-Ribosyltransferases. Angew. Chem. Int. Ed. 2014, 53, 8159–8162. [Google Scholar] [CrossRef]
- Maeda, M.; Patel, A.D.; Hampton, A. Formation of ribonucleotide 2′, 3′-cyclic carbonates during conversion of ribonucleoside 5′-phosphates to diphosphates and triphosphates by the phosphorimidazolidate procedure. Nucleic Acids Res. 1977, 4, 2843–2853. [Google Scholar] [CrossRef] [Green Version]
- Zatorski, A.; Goldstein, B.M.; Colby, T.D.; Jones, J.P.; Pankiewicz, K.W. Potent inhibitors of human inosine monophosphate dehydrogenase type II. fluorine-substituted analogs of thiazole-4-carboxamide adenine dinucleotide. J. Med. Chem. 1995, 38, 1098–1105. [Google Scholar] [CrossRef]
- Lynch, J.; áP Volante, R.; Reider, P. A chemical synthesis of nicotinamide adenine dinucleotide (NAD+). Chem. Commun. 1999, 729–730. [Google Scholar] [CrossRef]
- Pergolizzi, G.; Butt, J.N.; Bowater, R.P.; Wagner, G.K. A novel fluorescent probe for NAD-consuming enzymes. Chem. Commun. 2011, 47, 12655–12657. [Google Scholar] [CrossRef] [Green Version]
- Pergolizzi, G.; Cominetti, M.M.D.; Butt, J.N.; Field, R.A.; Bowater, R.P.; Wagner, G.K. Base-modified NAD and AMP derivatives and their activity against bacterial DNA ligases. Org. Biomol. Chem. 2015, 13, 6380–6398. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wagner, G.K.; Weber, K.; Garnham, C.; Morgan, A.J.; Galione, A.; Guse, A.H.; Potter, B.V.L. 2′-Deoxy cyclic adenosine 5′-diphosphate ribose derivatives: Importance of the 2′-hydroxyl motif for the antagonistic activity of 8-substituted cADPR derivatives. J. Med. Chem. 2008, 51, 1623–1636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansell, R.J.; Lowe, C.R. Artificial redox coenzymes: Biomimetic analogues of NAD+. Appl. Microbiol. Biotechnol. 1999, 51, 703–710. [Google Scholar] [CrossRef]
- Paul, C.E.; Arends, I.W.C.E.; Hollmann, F. Is simpler better? Synthetic nicotinamide cofactor analogues for redox chemistry. ACS Catal. 2014, 4, 788–797. [Google Scholar] [CrossRef]
- Paul, C.E.; Hollmann, F. A survey of synthetic nicotinamide cofactors in enzymatic processes. Appl. Microbiol. Biotechnol. 2016, 100, 4773–4778. [Google Scholar] [CrossRef] [Green Version]
- Zachos, I.; Nowak, C.; Sieber, V. Biomimetic cofactors and methods for their recycling. Curr. Opin. Chem. Biol. 2019, 49, 59–66. [Google Scholar] [CrossRef]
- Geddes, A.; Paul, C.E.; Hay, S.; Hollmann, F.; Scrutton, N.S. Donor–acceptor distance sampling enhances the performance of “better than nature” nicotinamide coenzyme biomimetics. J. Am. Chem. Soc. 2016, 138, 11089–11092. [Google Scholar] [CrossRef] [Green Version]
- Knaus, T.; Paul, C.E.; Levy, C.W.; De Vries, S.; Mutti, F.G.; Hollmann, F.; Scrutton, N.S. Better than nature: Nicotinamide biomimetics that outperform natural coenzymes. J. Am. Chem. Soc. 2016, 138, 1033–1039. [Google Scholar] [CrossRef] [Green Version]
- Lutz, J.; Hollmann, F.; Ho, T.V.; Schnyder, A.; Fish, R.H.; Schmid, A. Bioorganometallic chemistry: Biocatalytic oxidation reactions with biomimetic NAD+/NADH co-factors and [Cp* Rh (bpy) H]+ for selective organic synthesis. J. Organomet. Chem. 2004, 689, 4783–4790. [Google Scholar] [CrossRef] [Green Version]
- Paul, C.E.; Gargiulo, S.; Opperman, D.J.; Lavandera, I.; Gotor-Fernández, V.; Gotor, V.; Taglieber, A.; Arends, I.W.C.E.; Hollmann, F. Mimicking Nature: Synthetic Nicotinamide Cofactors for C=C Bioreduction Using Enoate Reductases. Org. Lett. 2013, 15, 180–183. [Google Scholar] [CrossRef] [Green Version]
- Campbell, E.; Meredith, M.; Minteer, S.D.; Banta, S. Enzymatic biofuel cells utilizing a biomimetic cofactor. Chem. Commun. 2012, 48, 1898–1900. [Google Scholar] [CrossRef] [PubMed]
- Kara, S.; Schrittwieser, J.H.; Hollmann, F.; Ansorge-Schumacher, M.B. Recent trends and novel concepts in cofactor-dependent biotransformations. Appl. Microbiol. Biotechnol. 2014, 98, 1517–1529. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Danishefsky, S.J. The total synthesis of proteasome inhibitors TMC-95A and TMC-95B: Discovery of a new method to generate cis-propenyl amides. Angew. Chem. Int. Ed. 2002, 114, 530–533. [Google Scholar] [CrossRef]
- Huang, R.; Chen, H.; Upp, D.M.; Lewis, J.C.; Zhang, Y.-H.P.J. A high-throughput method for directed evolution of NAD(P)+-dependent dehydrogenases for the reduction of biomimetic nicotinamide analogues. ACS Catal. 2019, 9, 11709–11719. [Google Scholar] [CrossRef] [PubMed]
- Drenth, J.; Yang, G.; Paul, C.E.; Fraaije, M.W. A Tailor-Made Deazaflavin-Mediated Recycling System for Artificial Nicotinamide Cofactor Biomimetics. ACS Catal. 2021, 11, 11561–11569. [Google Scholar] [CrossRef]
- Nowak, C.; Beer, B.C.; Pick, A.; Roth, T.; Lommes, P.; Sieber, V. A water-forming NADH oxidase from Lactobacillus pentosus suitable for the regeneration of synthetic biomimetic cofactors. Front. Microbiol. 2015, 6, 957. [Google Scholar] [CrossRef]
- Nowak, C.; Pick, A.; Csepei, L.I.; Sieber, V. Characterization of biomimetic cofactors according to stability, redox potentials, and enzymatic conversion by NADH oxidase from Lactobacillus pentosus. ChemBioChem 2017, 18, 1944–1949. [Google Scholar] [CrossRef]
- Nowak, C.; Pick, A.; Lommes, P.; Sieber, V. Enzymatic reduction of nicotinamide biomimetic cofactors using an engineered glucose dehydrogenase: Providing a regeneration system for artificial cofactors. ACS Catal. 2017, 7, 5202–5208. [Google Scholar] [CrossRef]
- Bashiri, G.; Rehan, A.M.; Greenwood, D.R.; Dickson, J.M.J.; Baker, E.N. Metabolic engineering of cofactor F420 production in Mycobacterium smegmatis. PLoS ONE 2010, 5, e15803. [Google Scholar] [CrossRef] [Green Version]
- Shah, M.V.; Antoney, J.; Kang, S.W.; Warden, A.C.; Hartley, C.J.; Nazem-Bokaee, H.; Jackson, C.J.; Scott, C. Cofactor F420-dependent enzymes: An under-explored resource for asymmetric redox biocatalysis. Catalysts 2019, 9, 868. [Google Scholar] [CrossRef] [Green Version]
- Shah, M.V.; Nazem-Bokaee, H.; Antoney, J.; Kang, S.W.; Jackson, C.J.; Scott, C. Improved production of the non-native cofactor F420 in Escherichia coli. Sci. Rep. 2021, 11, 21774. [Google Scholar] [CrossRef] [PubMed]
- Voss, C.V.; Gruber, C.C.; Faber, K.; Knaus, T.; Macheroux, P.; Kroutil, W. Orchestration of concurrent oxidation and reduction cycles for stereoinversion and deracemisation of sec-alcohols. J. Am. Chem. Soc. 2008, 130, 13969–13972. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rocha, R.A.; North, A.J.; Speight, R.E.; Williams, C.C.; Scott, C. Cofactor and Process Engineering for Nicotinamide Recycling and Retention in Intensified Biocatalysis. Catalysts 2022, 12, 1454. https://doi.org/10.3390/catal12111454
Rocha RA, North AJ, Speight RE, Williams CC, Scott C. Cofactor and Process Engineering for Nicotinamide Recycling and Retention in Intensified Biocatalysis. Catalysts. 2022; 12(11):1454. https://doi.org/10.3390/catal12111454
Chicago/Turabian StyleRocha, Raquel A., Andrea J. North, Robert E. Speight, Charlotte C. Williams, and Colin Scott. 2022. "Cofactor and Process Engineering for Nicotinamide Recycling and Retention in Intensified Biocatalysis" Catalysts 12, no. 11: 1454. https://doi.org/10.3390/catal12111454
APA StyleRocha, R. A., North, A. J., Speight, R. E., Williams, C. C., & Scott, C. (2022). Cofactor and Process Engineering for Nicotinamide Recycling and Retention in Intensified Biocatalysis. Catalysts, 12(11), 1454. https://doi.org/10.3390/catal12111454