
Electrocatalytic Isomerization of Allylic Alcohols: Straightforward Preparation of β-Aryl-Ketones


Abstract
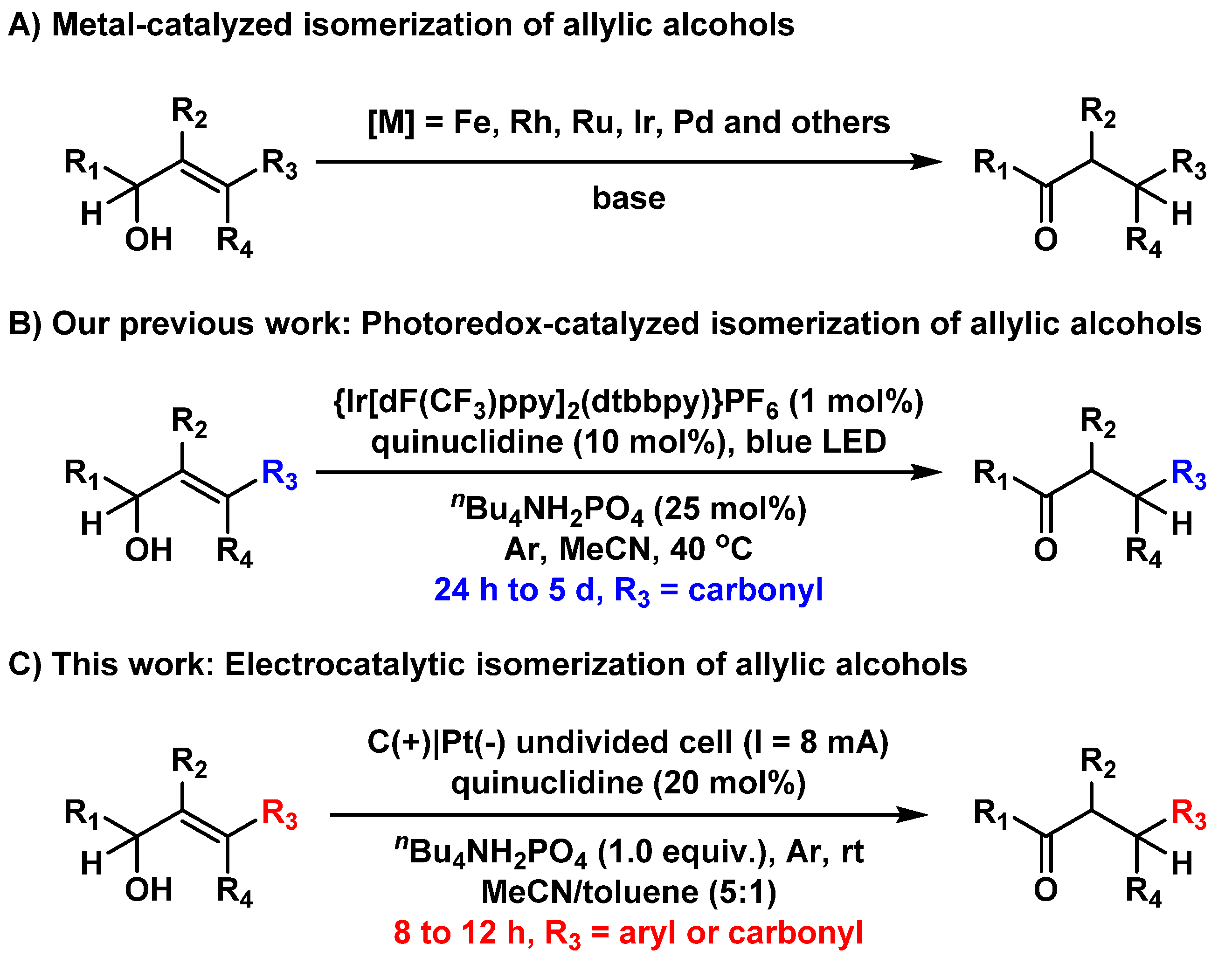
:1. Introduction
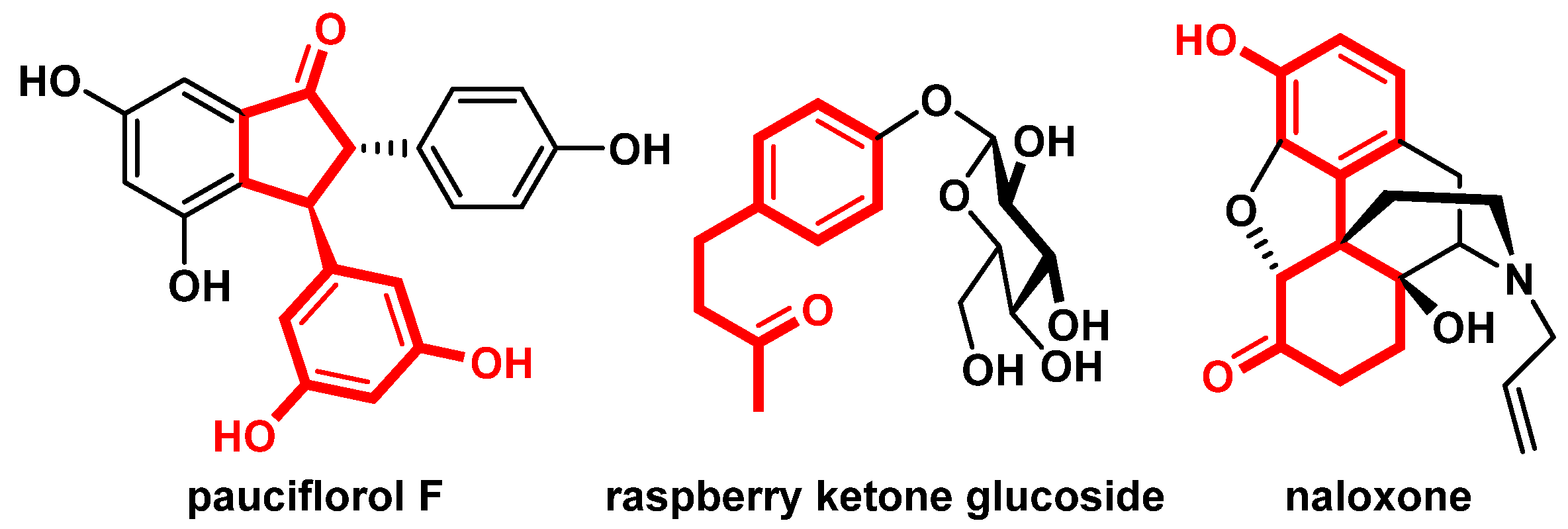
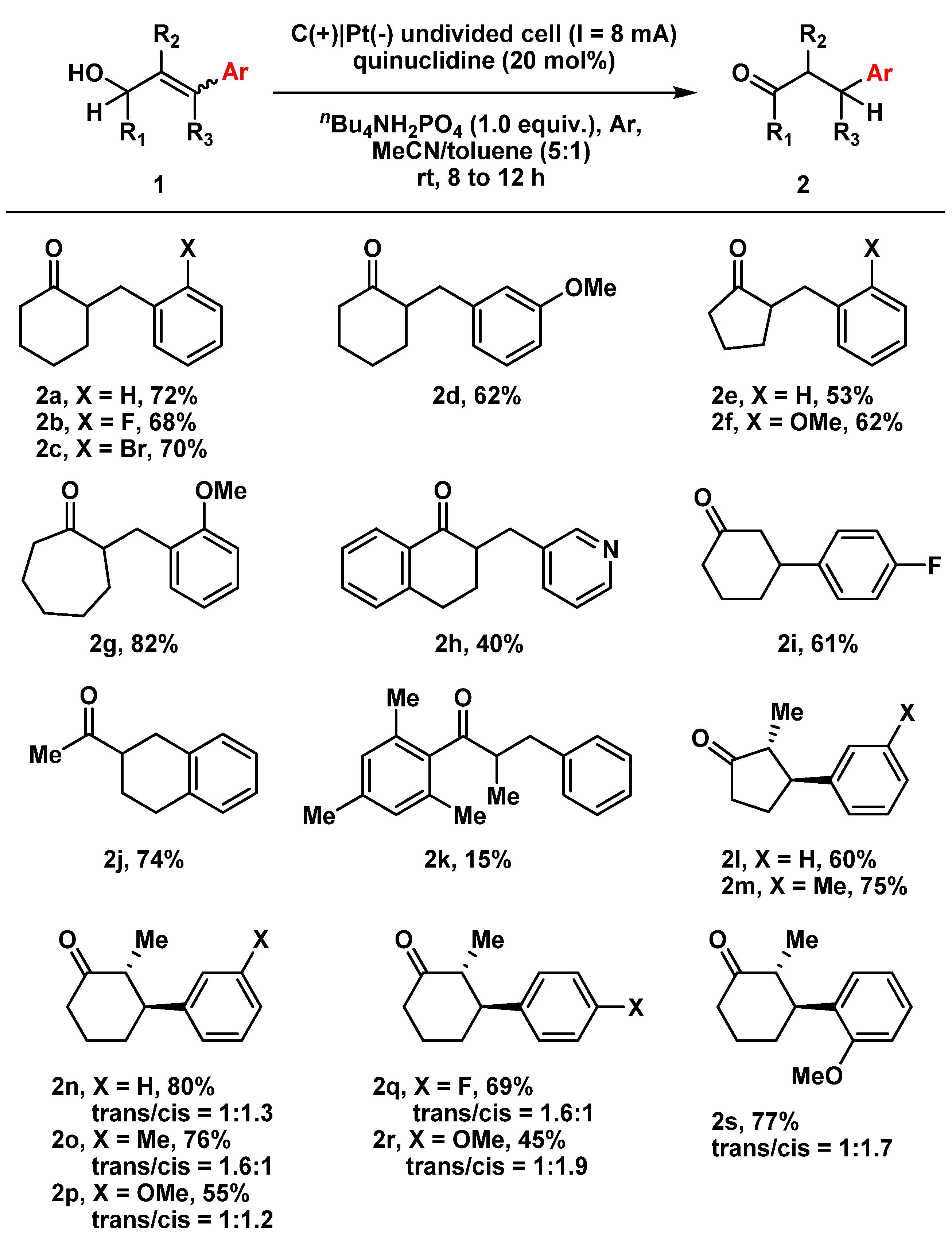
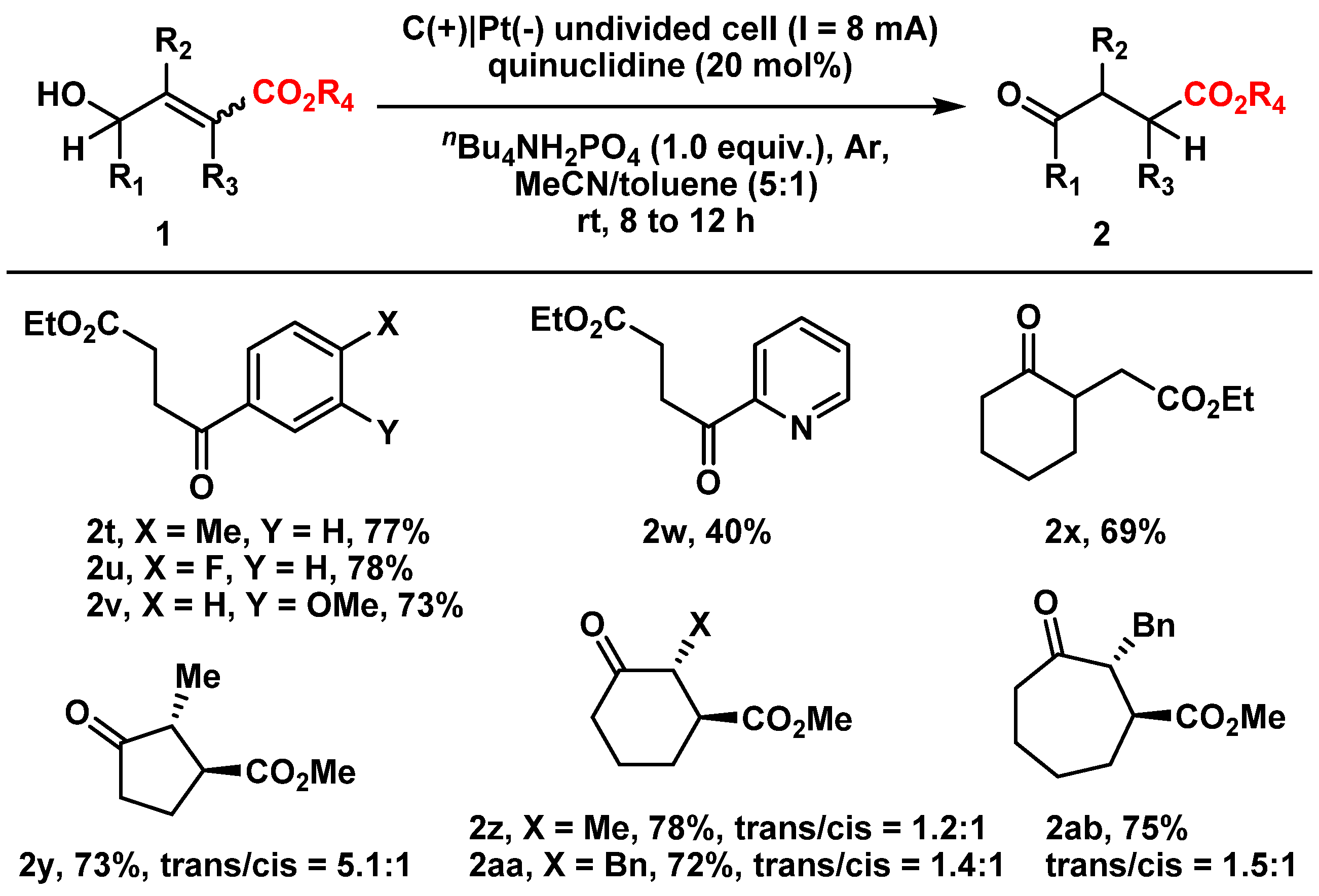
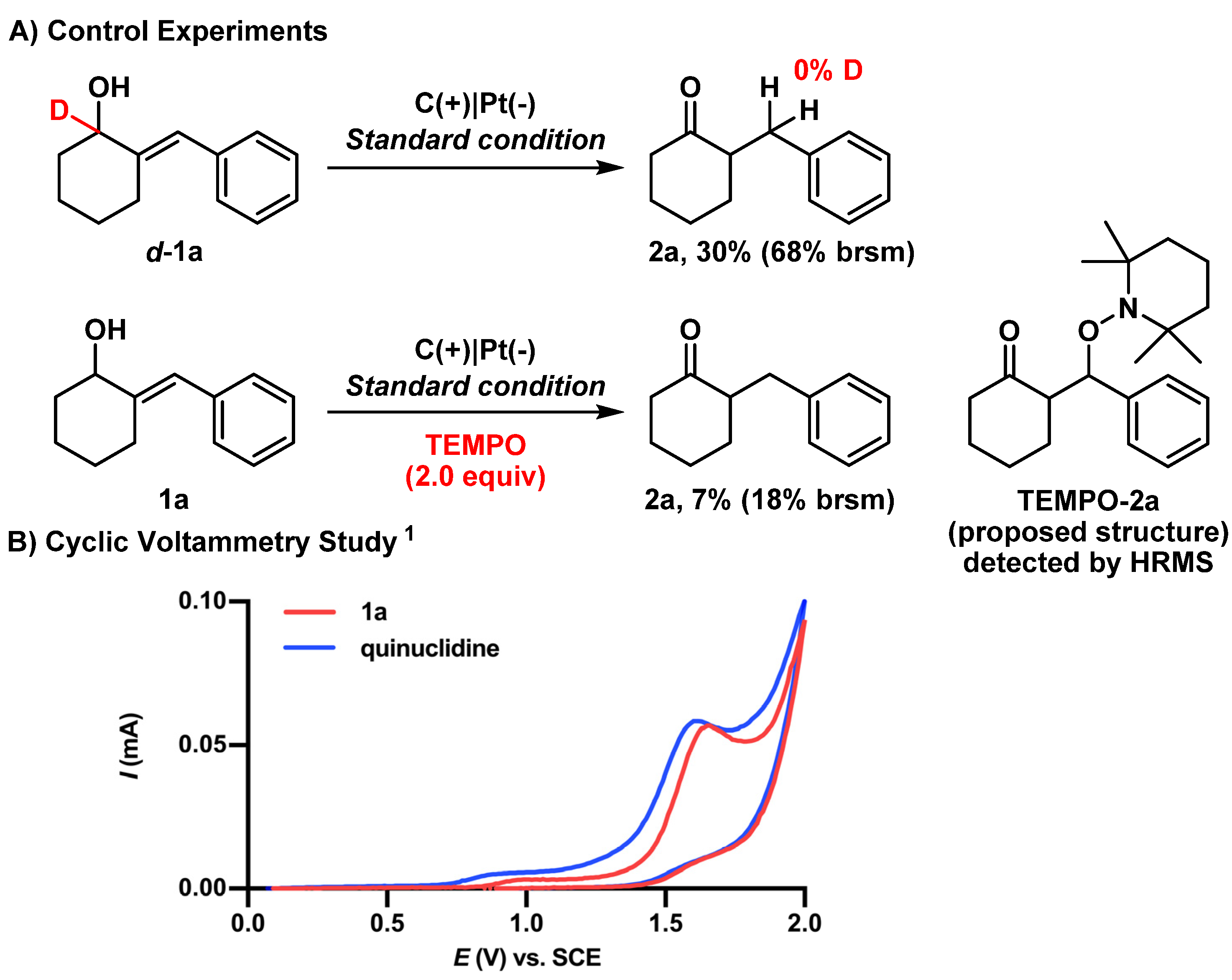
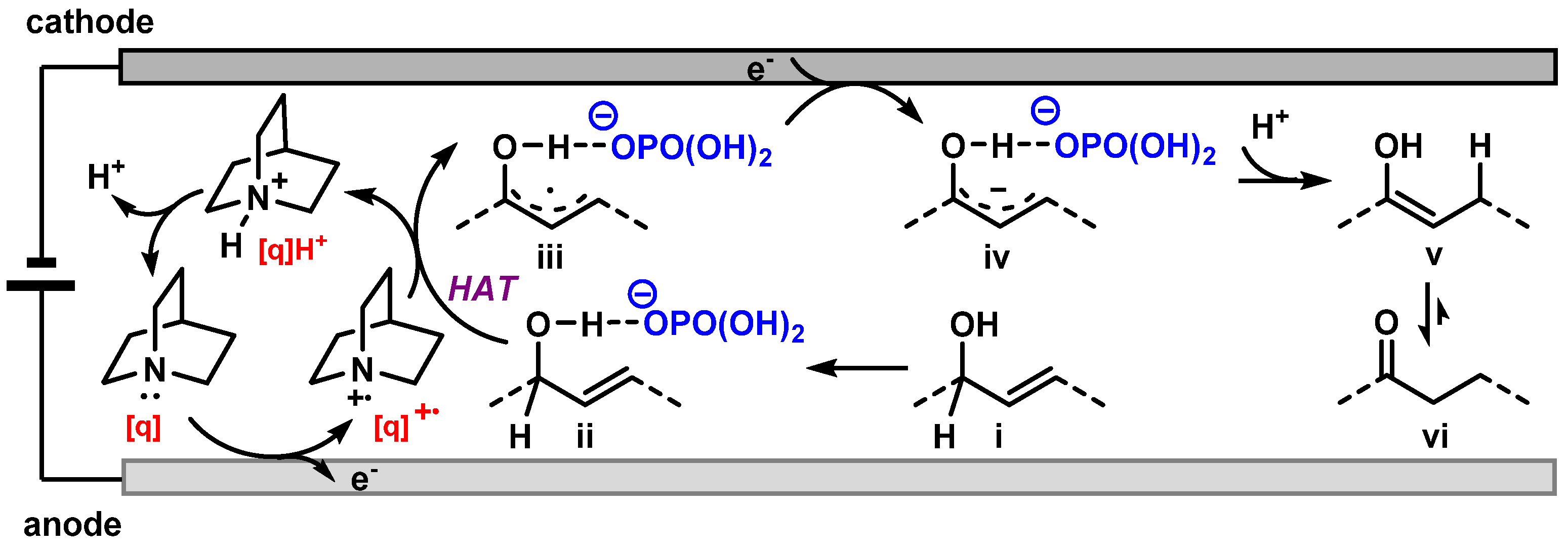
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. General Experimental Procedure for Electrocatalytic Isomerization of Allylic Alcohols
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shi, F.; Wang, H.; Dai, X. Carbonyl Compounds: Reactants, Catalysts and Products; Wiley-VCH: Hoboken, NJ, USA, 2021. [Google Scholar] [CrossRef]
- Van der Drift, R.C.; Bouwman, E.; Drent, E. Homogeneously catalysed isomerisation of allylic alcohols to carbonyl compounds. J. Organomet. Chem. 2002, 650, 1–24. [Google Scholar] [CrossRef]
- Luca, M.; Clément, M. Platinum metals in the catalytic asymmetric isomerization of allylic alcohols. Chem. Lett. 2011, 40, 341–344. [Google Scholar]
- Ahlsten, N.; Bartoszewicz, A.; Martín-Matute, B. Allylic alcohols as synthetic enolate equivalents: Isomerisation and tandem reactions catalysed by transition metal complexes. Dalton Trans. 2012, 41, 1660–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzo-Luis, P.; Romerosa, A.; Serrano-Ruiz, M. Catalytic isomerization of allylic alcohols in water. ACS Catal. 2012, 2, 1079–1086. [Google Scholar] [CrossRef]
- Li, H.; Mazet, C. Iridium-catalyzed selective isomerization of primary allylic alcohols. Acc. Chem. Res. 2016, 49, 1232–1241. [Google Scholar] [CrossRef]
- Trost, B.M. The atom economy—A search for synthetic efficiency. Science 1991, 254, 1471–1477. [Google Scholar] [CrossRef]
- Sheldon, R.A. The E factor: Fifteen years on. Green Chem. 2007, 9, 1273–1283. [Google Scholar] [CrossRef]
- Newhouse, T.; Baran, P.S.; Hoffmann, R.W. The economies of synthesis. Chem. Soc. Rev. 2009, 38, 3010–3021. [Google Scholar] [CrossRef]
- Anastas, P.; Eghbali, N. Green chemistry: Principles and practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef]
- Branchadell, V.; Crévisy, C.; Grée, R. Theoretical study on the mechanism of iron carbonyls mediated isomerization of allylic alcohols to saturated carbonyls. Chem. Eur. J. 2003, 9, 2062–2067. [Google Scholar] [CrossRef]
- Ahlsten, N.; Lundberg, H.; Martín-Matute, B. Rhodium-catalysed isomerisation of allylic alcohols in water at ambient temperature. Green Chem. 2010, 12, 1628–1633. [Google Scholar] [CrossRef]
- Liu, T.-L.; Ng, T.W.; Zhao, Y. Rhodium-catalyzed enantioselective isomerization of secondary allylic alcohols. J. Am. Chem. Soc. 2017, 139, 3643–3646. [Google Scholar] [CrossRef] [PubMed]
- Cadierno, V.; García-Garrido, S.E.; Gimeno, J.; Varela-Álvarez, A.; Sordo, J.A. Bis(allyl)-ruthenium(IV) complexes as highly efficient catalysts for the redox isomerization of allylic alcohols into carbonyl compounds in organic and aqueous media: Scope, limitations, and theoretical analysis of the mechanism. J. Am. Chem. Soc. 2006, 128, 1360–1370. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Beauchamps, M.G.; Laquidara, J.M.; Sowa, J.R., Jr. Ruthenium-catalyzed asymmetric transfer hydrogenation of allylic alcohols by an enantioselective isomerization/transfer hydrogenation mechanism. Angew. Chem. Int. Ed. 2012, 51, 2106–2110. [Google Scholar] [CrossRef] [PubMed]
- Manzini, S.; Poater, A.; Nelson, D.J.; Cavallo, L.; Nolan, S.P. How phenyl makes a difference: Mechanistic insights into the ruthenium(ii)-catalysed isomerisation of allylic alcohols. Chem. Sci. 2014, 5, 180–188. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Mazet, C. Catalyst-directed diastereoselective isomerization of allylic alcohols for the stereoselective construction of C(20) in steroid side chains: Scope and topological diversification. J. Am. Chem. Soc. 2015, 137, 10720–10727. [Google Scholar] [CrossRef]
- Mantilli, L.; Gérard, D.; Torche, S.; Besnard, C.; Mazet, C. Iridium-catalyzed asymmetric isomerization of primary allylic alcohols. Angew. Chem. Int. Ed. 2009, 48, 5143–5147. [Google Scholar] [CrossRef]
- Martinez-Erro, S.; Sanz-Marco, A.; Gómez, A.B.; Vάzquez-Romero, A.; Ahlquist, M.S.G.; Martín-Matute, B. Base-catalyzed stereospecific isomerization of electron-eeficient allylic alcohols and ethers through ion-pairing. J. Am. Chem. Soc. 2016, 138, 13408–13414. [Google Scholar] [CrossRef]
- Sutar, R.L.; Sen, S.; Eivgi, O.; Segalovich, G.; Schapiro, I.; Reany, O.; Lemcoff, N.G. Guiding a divergent reaction by photochemical control: Bichromatic selective access to levulinates and butenolides. Chem. Sci. 2018, 9, 1368–1374. [Google Scholar] [CrossRef] [Green Version]
- Lai, L.; Li, A.N.; Zhou, J.; Guo, Y.; Lin, L.; Chen, W.; Wang, R. Mg(OMe)2 promoted allylic isomerization of γ-hydroxy-α,β-alkenoic esters to synthesize γ-ketone esters. Org. Biomol. Chem. 2017, 15, 2185–2190. [Google Scholar] [CrossRef]
- Mantilli, L.; Mazet, C. Iridium-catalyzed isomerization of primary allylic alcohols under mild reaction conditions. Tetrahedron Lett. 2009, 50, 4141–4144. [Google Scholar] [CrossRef]
- Larionov, E.; Lin, L.; Guénée, L.; Mazet, C. Scope and mechanism in palladium-catalyzed isomerizations of highly substituted allylic, homoallylic, and alkenyl Alcohols. J. Am. Chem. Soc. 2014, 136, 16882–16894. [Google Scholar] [CrossRef] [PubMed]
- Romero, N.A.; Nicewicz, D.A. Organic photoredox catalysis. Chem. Rev. 2016, 116, 10075–10166. [Google Scholar] [CrossRef] [PubMed]
- Skubi, K.L.; Blum, T.R.; Yoon, T.P. Dual catalysis strategies in photochemical synthesis. Chem. Rev. 2016, 116, 10035–10074. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.Y.; Perry, I.B.; Bissonnette, N.B.; Buksh, B.F.; Edwards, G.A.; Frye, L.I.; Garry, O.L.; Lavagnino, M.N.; Li, B.X.; Liang, Y.; et al. Metallaphotoredox: The merger of photoredox and transition metal catalysis. Chem. Rev. 2022, 122, 1485–1542. [Google Scholar] [CrossRef]
- Jeffrey, J.L.; Terrett, J.A.; MacMillan, D.W.C. O–H hydrogen bonding promotes H-atom transfer from α C-H bonds for C-alkylation of alcohols. Science 2015, 349, 1532–1536. [Google Scholar] [CrossRef] [Green Version]
- Le, C.; Liang, Y.; Evans, R.W.; Li, X.; MacMillan, D.W.C. Selective sp3 C-H alkylation via polarity-match-based cross-coupling. Nature 2017, 547, 7983. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Kalvet, I.; Schoenebeck, F.; Rovis, T. Direct α-alkylation of primary aliphatic amines enabled by CO2 and electrostatics. Nat. Chem. 2018, 10, 1037–1041. [Google Scholar] [CrossRef]
- Yang, H.-B.; Feceu, A.; Martin, D.B.C. Catalyst-controlled C-H functionalization of adamantanes using selective H-atom transfer. ACS Catal. 2019, 9, 5708–5715. [Google Scholar] [CrossRef] [Green Version]
- Ashley, M.A.; Yamauchi, C.; Chu, J.C.K.; Otsuka, S.; Yorimitsu, H.; Rovis, T. Photoredox-catalyzed site-selective α-C(sp3)-H alkylation of primary amine derivatives. Angew. Chem. Int. Ed. 2019, 58, 4002–4006. [Google Scholar] [CrossRef]
- Dimakos, V.; Su, H.Y.; Garrett, G.E.; Taylor, M.S. Site-selective and stereoselective C-H alkylations of aarbohydrates via combined siarylborinic acid and photoredox catalysis. J. Am. Chem. Soc. 2019, 141, 5149–5153. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.; Zhang, Z.; Li, A.; Li, Y.; Huang, J.; Yang, Z. Photoredox-catalyzed isomerization of highly substituted allylic alcohols by C-H bond activation. Angew. Chem. Int. Ed. 2020, 59, 11660–11668. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Kawamata, Y.; Baran, P.S. Synthetic organic electrochemical methods since 2000: On the verge of a renaissance. Chem. Rev. 2017, 117, 13230–13319. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Xu, K.; Zeng, C. Use of electrochemistry in the synthesis of heterocyclic structures. Chem. Rev. 2018, 118, 4485–4540. [Google Scholar] [CrossRef]
- Heard, D.M.; Lennox, A.J.J. Electrode materials in modern organic electrochemistry. Angew. Chem. Int. Ed. 2020, 59, 18866–18884. [Google Scholar] [CrossRef]
- Murray, P.R.D.; Cox, J.H.; Chiappini, N.D.; Roos, C.B.; McLoughlin, E.A.; Hejna, B.G.; Nguyen, S.T.; Ripberger, H.H.; Ganley, J.M.; Tsui, E.; et al. Photochemical and electrochemical applications of proton-coupled electron transfer in organic synthesis. Chem. Rev. 2022, 122, 2017–2291. [Google Scholar] [CrossRef]
- Munda, M.; Niyogi, S.; Shaw, K.; Kundu, S.; Nandia, R.; Bisai, A. Electrocatalysis as a key strategy for the total synthesis of natural products. Org. Biomol. Chem. 2022, 20, 727–748. [Google Scholar] [CrossRef]
- Kawamata, Y.; Yan, M.; Liu, Z.; Bao, D.-H.; Chen, J.; Starr, J.T.; Baran, P.S. Scalable, electrochemical oxidation of unactivated C-H bonds. J. Am. Chem. Soc. 2017, 139, 7448–7451. [Google Scholar] [CrossRef] [Green Version]
- Peters, B.K.; Rodriguez, K.X.; Reisberg, S.H.; Beil, S.B.; Hickey, D.P.; Kawamata, Y.; Collins, M.; Starr, J.; Chen, L.; Udyavara, S.; et al. Scalable and safe synthetic organic electroreduction inspired by Li-ion battery chemistry. Science 2019, 363, 838–845. [Google Scholar] [CrossRef]
- Gnaim, S.; Takahira, Y.; Wilke, H.R.; Yao, Z.; Li, J.; Delbrayelle, D.; Echeverria, P.-G.; Vantourout, J.C.; Baran, P.S. Electrochemically driven desaturation of carbonyl compounds. Nat. Chem. 2021, 13, 367–372. [Google Scholar] [CrossRef]
- Huang, C.; Qian, X.-Y.; Xu, H.-C. Continuous-flow electrosynthesis of benzofused S-heterocycles by dehydrogenative C−S cross-coupling. Angew. Chem. Int. Ed. 2019, 58, 6650–6653. [Google Scholar] [CrossRef] [PubMed]
- Fu, N.; Song, L.; Liu, J.; Shen, Y.; Siu, J.C.; Lin, S. New bisoxazoline ligands enable enantioselective electrocatalytic cyanofunctionalization of vinylarenes. J. Am. Chem. Soc. 2019, 141, 14480–14485. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Shi, S.-H.; Jin, R.; Qiu, X.; Wei, J.; Tan, H.; Jiang, X.; Shi, X.; Song, S.; Jiao, N. Electrochemically induced nickel catalysis for oxygenation reactions with water. Nat. Catal. 2021, 4, 116–123. [Google Scholar] [CrossRef]
- Hou, Z.-W.; Liu, D.-J.; Xiong, P.; Lai, X.-L.; Song, J.; Xu, H.-C. Site-selective electrochemical benzylic C-H amination. Angew. Chem. Int. Ed. 2021, 60, 2943–2947. [Google Scholar] [CrossRef]
- Qin, J.-H.; Luo, M.-J.; An, D.-L.; Li, J.-H. Electrochemical 1,2-diarylation of alkenes enabled by direct dual C-H functionalizations of electron-rich aromatic hydrocarbons. Angew. Chem. Int. Ed. 2021, 60, 1861–1868. [Google Scholar] [CrossRef]
- Zhu, J.; Chen, Z.; He, M.; Wang, D.; Li, L.; Qi, J.; Shi, R.; Lei, A. Metal-free electrochemical C3-sulfonylation of imidazo[1,2-a]pyridines. Org. Chem. Front. 2021, 8, 3815–3819. [Google Scholar] [CrossRef]
- Saito, M.; Kawamata, Y.; Meanwell, M.; Navratil, R.; Chiodi, D.; Carlson, E.; Hu, P.; Chen, L.; Udyavara, S.; Kingston, C.; et al. N-ammonium ylide mediators for electrochemical C-H oxidation. J. Am. Chem. Soc. 2021, 143, 7859–7867. [Google Scholar] [CrossRef]
- Wu, Z.-J.; Li, S.-R.; Xu, H.-C. Synthesis of N-heterocycles by dehydrogenative annulation of N-allyl amides with 1,3-dicarbonyl compounds. Angew. Chem. Int. Ed. 2018, 57, 14070–14074. [Google Scholar] [CrossRef]
- Jie, L.-H.; Guo, B.; Song, J.; Xu, H.-C. Organoelectrocatalysis enables direct cyclopropanation of methylene compounds. J. Am. Chem. Soc. 2022, 144, 2343–2350. [Google Scholar] [CrossRef]
- Liu, Y.; Shi, B.; Liu, Z.; Gao, R.; Huang, C.; Alhumade, H.; Wang, S.; Qi, X.; Lei, A. Time-resolved EPR revealed the formation, structure, and reactivity of N-centered radicals in an electrochemical C(sp3)-H arylation reaction. J. Am. Chem. Soc. 2021, 143, 20863–20872. [Google Scholar] [CrossRef]
- Liang, H.; Wang, L.-J.; Ji, Y.-X.; Wang, H.; Zhang, B. Selective electrochemical hydrolysis of hydrosilanes to silanols via anodically generated silyl cations. Angew. Chem. Int. Ed. 2021, 60, 1839–1844. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-R.; Zhang, S.-Q.; Meyer, T.H.; Yang, C.-H.; Zhang, Q.-H.; Liu, J.-R.; Xu, H.-J.; Cao, F.-H.; Ackermann, L.; Hong, X. Carboxylate breaks the arene C-H bond via a hydrogen-atom-transfer mechanism in electrochemical cobalt catalysis. Chem. Sci. 2020, 11, 5790–5796. [Google Scholar] [CrossRef] [PubMed]
- Kurimoto, Y.; Yamashita, J.; Mitsudo, K.; Sato, E.; Suga, S. Electrosynthesis of phosphacycles via dehydrogenative C-P bond formation using DABCO as a mediator. Org. Lett. 2021, 23, 3120–3124. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Farmer, L.A.; Pratt, D.A.; Maldonado, S.; Stephenson, C.R.J. Mechanism of electrochemical generation and decomposition of phthalimide-N-oxyl. J. Am. Chem. Soc. 2021, 143, 10324–10332. [Google Scholar] [CrossRef]
- Huang, H.; Strater, Z.M.; Lambert, T.H. Electrophotocatalytic C-H functionalization of ethers with high regioselectivity. J. Am. Chem. Soc. 2020, 142, 1698–1703. [Google Scholar] [CrossRef]
- Ghorbani, F.; Harry, S.A.; Capilato, J.N.; Pitts, C.R.; Joram, J.; Peters, G.N.; Tovar, J.D.; Smajlagic, I.; Siegler, M.A.; Dudding, T.; et al. Carbonyl-directed aliphatic fluorination: A special type of hydrogen atom transfer beats out Norrish II. J. Am. Chem. Soc. 2020, 142, 14710–14724. [Google Scholar] [CrossRef]
- Wu, J.; Zhou, Y.; Zhou, Y.; Chiang, C.-W.; Lei, A. Electro-oxidative C(sp3)-H amination of azoles via intermolecular oxidative C(sp3)-H/N−H cross-coupling. ACS Catal. 2017, 7, 8320–8323. [Google Scholar] [CrossRef]
- Hu, X.; Zhang, G.; Bu, F.; Nie, L.; Lei, A. Electrochemical-oxidation-induced site-selective intramolecular C(sp3)-H amination. ACS Catal. 2018, 8, 9370–9375. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
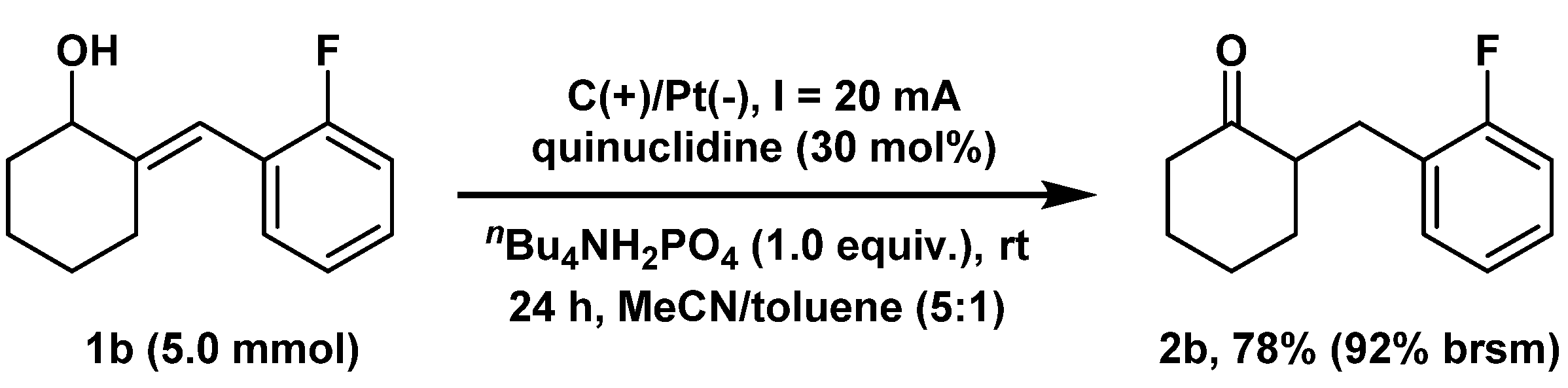{kind=link}

| Entry | Variation from the Standard Conditions | Yield (%) 2 |
|---|---|---|
| 1 | none | 72 |
| 2 | no electricity | n.d. (100) |
| 3 | no quinuclidine | n.d. (90) |
| 4 | 5 mA | 15 (59) |
| 5 | 12 mA | n.d. |
| 6 | DABCO instead of quinuclidine | n.d. (95) |
| 7 | MeCN as solvent | 62 |
| 8 | DMF as solvent | trace (80) |
| 9 | HIFP as solvent | trace (42) |
| 10 | solvent containing 0.5 mL H2O | n.d. (98) |
| 11 | nBu4NBF4 instead of nBu4NH2PO4 | 31 (50) |
| 12 | nBu4NPF6 instead of nBu4NH2PO4 | n.d. (95) |
| 13 | under Air | trace (55) |
| 14 | Zn(-) instead of Pt(-) | 29 |
| 15 | Glassy carbon(-) instead of Pt(-) | trace |
| 16 | reaction time to 24 h | 70 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, A.; Zheng, N.; Guo, K.; Zhang, Z.; Yang, Z. Electrocatalytic Isomerization of Allylic Alcohols: Straightforward Preparation of β-Aryl-Ketones. Catalysts 2022, 12, 333. https://doi.org/10.3390/catal12030333
Li A, Zheng N, Guo K, Zhang Z, Yang Z. Electrocatalytic Isomerization of Allylic Alcohols: Straightforward Preparation of β-Aryl-Ketones. Catalysts. 2022; 12(3):333. https://doi.org/10.3390/catal12030333
Chicago/Turabian StyleLi, Anding, Nan Zheng, Kai Guo, Zhongchao Zhang, and Zhen Yang. 2022. "Electrocatalytic Isomerization of Allylic Alcohols: Straightforward Preparation of β-Aryl-Ketones" Catalysts 12, no. 3: 333. https://doi.org/10.3390/catal12030333
APA StyleLi, A., Zheng, N., Guo, K., Zhang, Z., & Yang, Z. (2022). Electrocatalytic Isomerization of Allylic Alcohols: Straightforward Preparation of β-Aryl-Ketones. Catalysts, 12(3), 333. https://doi.org/10.3390/catal12030333