
Whole-Genome Sequence and Comparative Analysis of Trichoderma asperellum ND-1 Reveal Its Unique Enzymatic System for Efficient Biomass Degradation

Abstract
:
1. Introduction
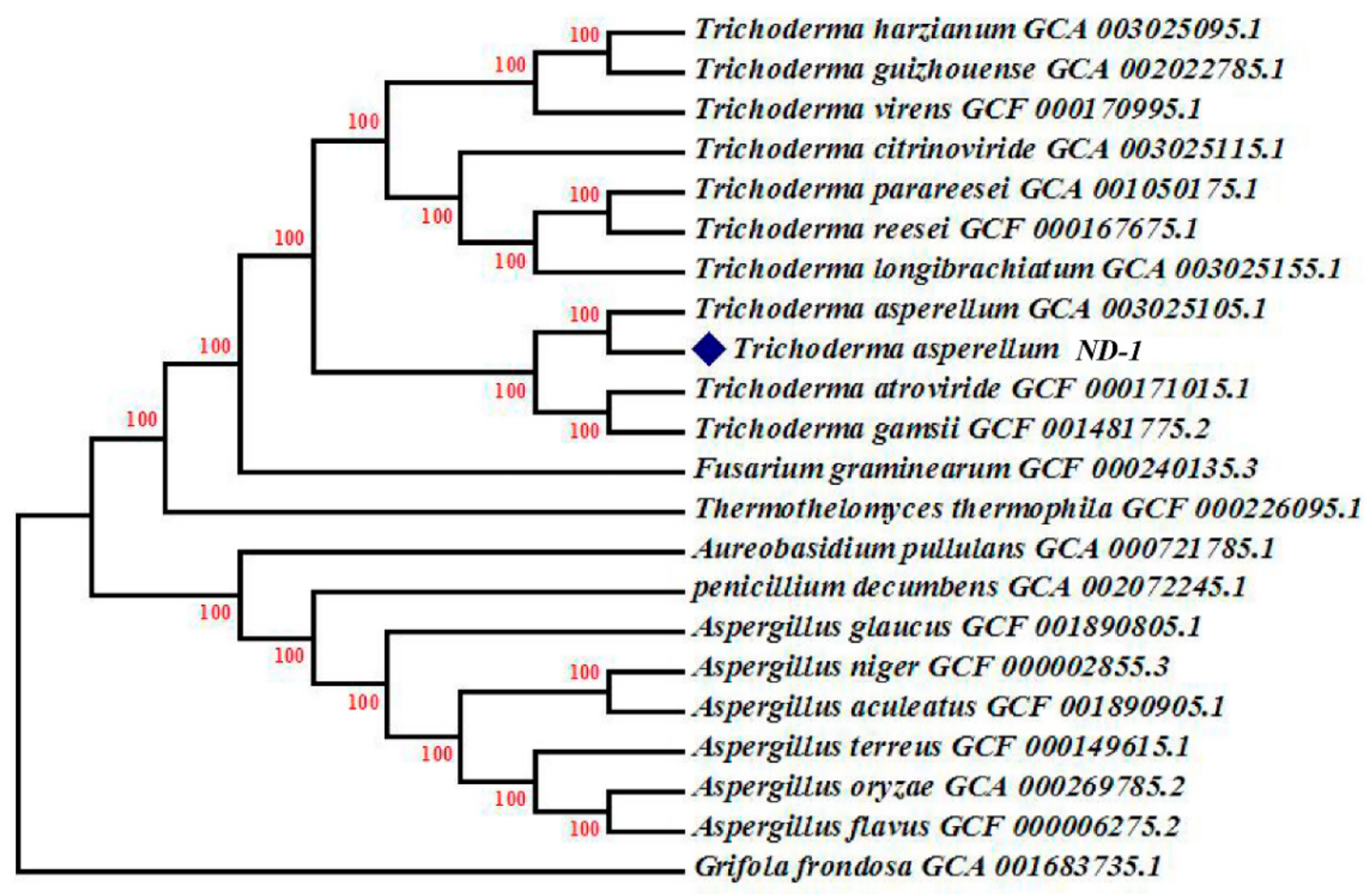
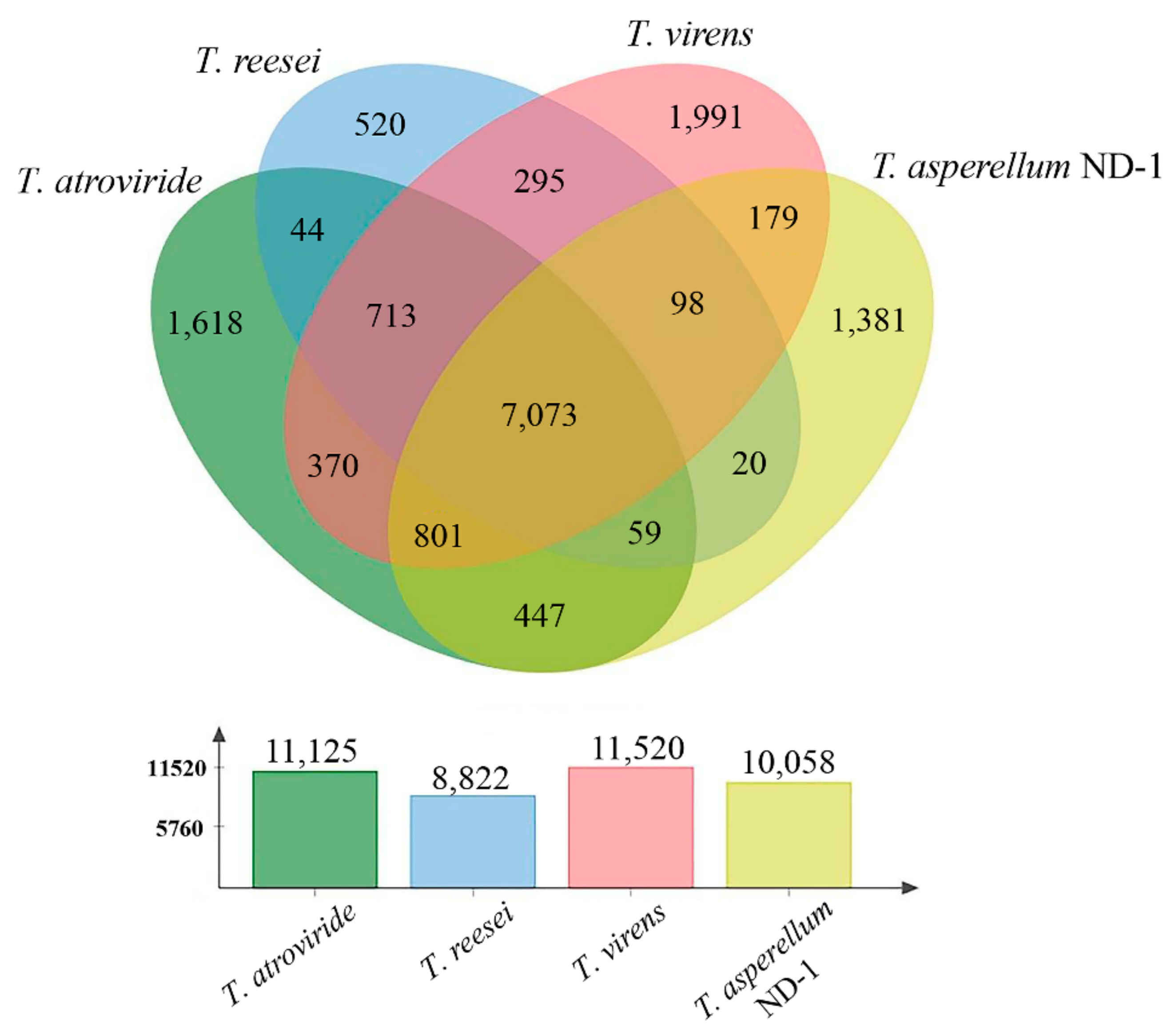
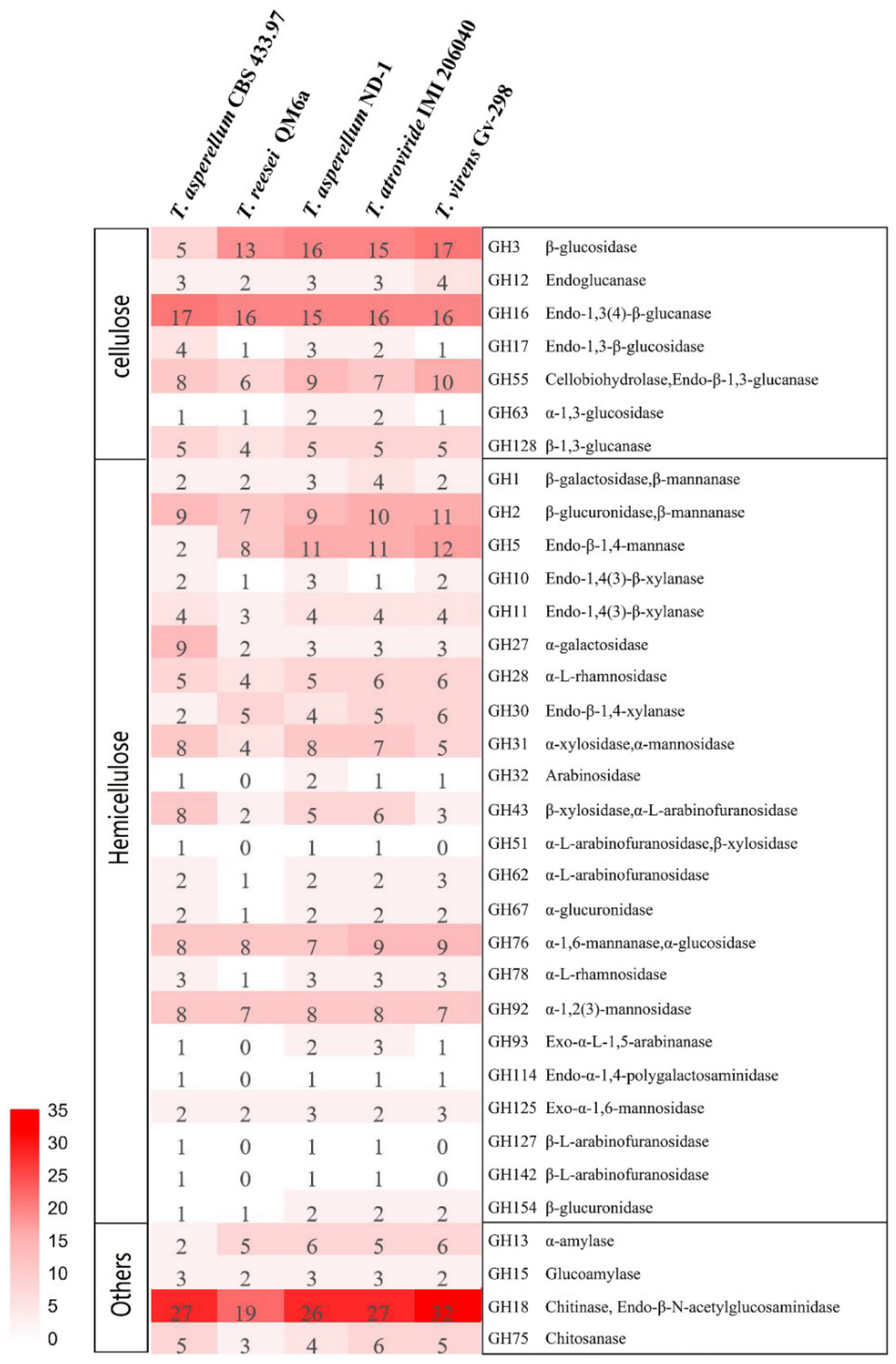
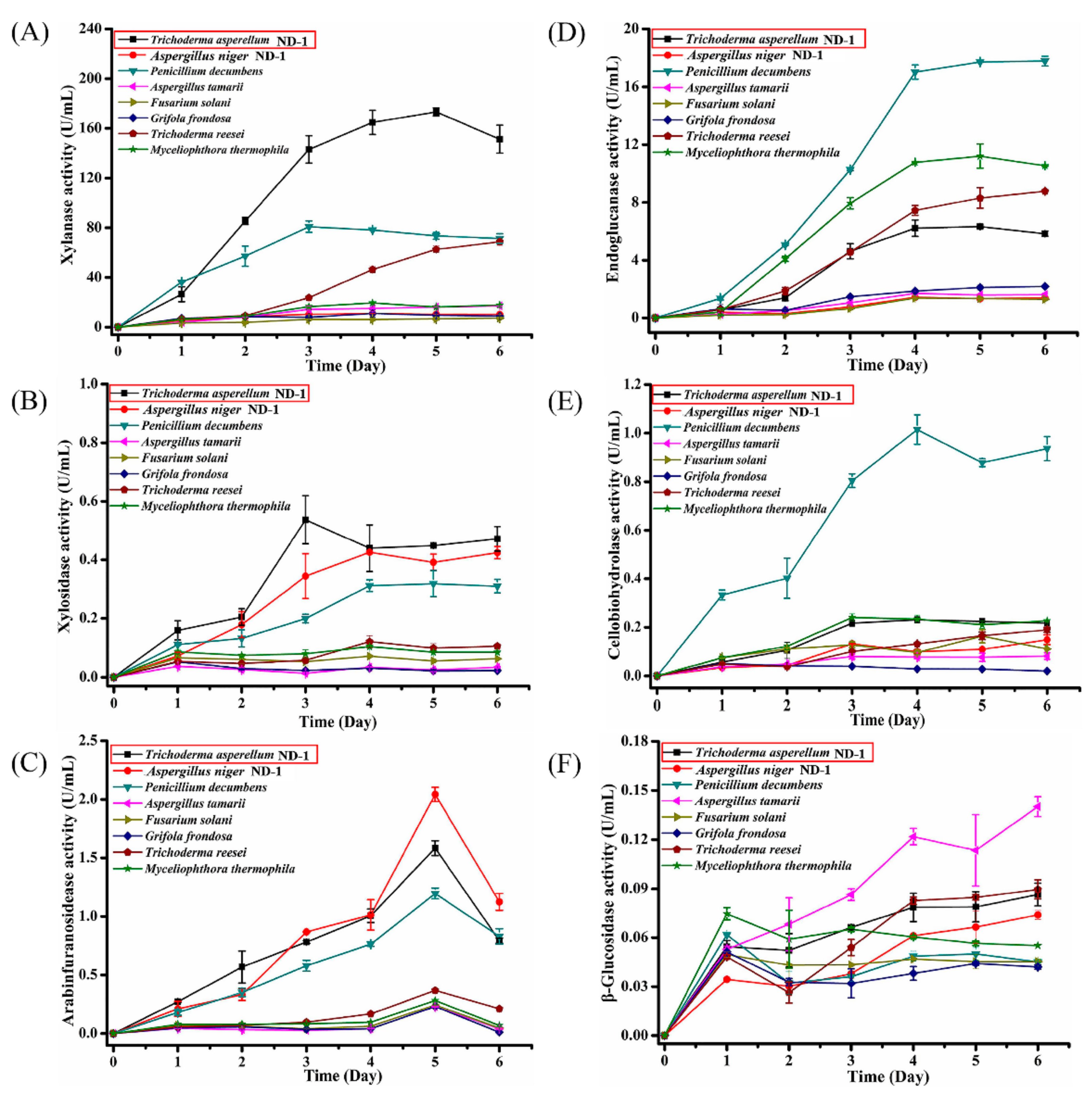
2. Results and Discussion
3. Conclusions
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Jiang, L.; Wu, Y.-X.; Wang, X.-B.; Zheng, A.-Q.; Zhao, Z.-L.; Li, H.-B.; Feng, X. Crude glycerol pretreatment for selective saccharification of lignocellulose via fast pyrolysis and enzyme hydrolysis. Energy Convers. Manag. 2019, 199, 111894. [Google Scholar] [CrossRef]
- Straathof, A.J.J. Transformation of biomass into commodity chemicals using enzymes or cells. Chem. Rev. 2013, 114, 1871–1908. [Google Scholar] [CrossRef] [PubMed]
- Sudarsanam, P.; Zhong, R.; Bosch, S.V.D.; Coman, S.M.; Parvulescu, V.I.; Sels, B.F. Functionalised heterogeneous catalysts for sustainable biomass valorisation. Chem. Soc. Rev. 2018, 47, 8349–8402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidi, L.; Moraïs, S.; Artzi, L.; Knop, D.; Hadar, Y.; Arfi, Y.; Bayer, E.A. Toward combined delignification and saccharification of wheat straw by a laccase-containing designer cellulosome. Proc. Natl. Acad. Sci. USA 2016, 113, 10854–10859. [Google Scholar] [CrossRef] [Green Version]
- Nitsos, C.; Lazaridis, P.A.; Mach-Aigner, A.; Matis, K.A.; Triantafyllidis, K.S. Enhancing lignocellulosic biomass hydrolysis by hydrothermal pretreatment, extraction of surface lignin, wet milling and production of cellulolytic enzymes. ChemSusChem 2019, 12, 1179–1195. [Google Scholar] [CrossRef]
- Somerville, C.; Bauer, S.; Brininstool, G.; Facette, M.; Hamann, T.; Milne, J.; Osborne, E.; Paredez, A.; Persson, S.; Raab, T.; et al. Toward a systems approach to understanding plant cell walls. Science 2004, 306, 2206–2211. [Google Scholar] [CrossRef] [Green Version]
- Finore, I.; Poli, A.; Di Donato, P.; Lama, L.; Trincone, A.; Fagnano, M.; Mori, M.; Nicolaus, B.; Tramice, A. The hemicellulose extract from Cynara cardunculus: A source of value-added biomolecules produced by xylanolytic thermozymes. Green Chem. 2015, 18, 2460–2472. [Google Scholar] [CrossRef]
- Gaurav, N.; Sivasankari, S.; Kiran, G.; Ninawe, G.; Selvin, J. Utilization of bioresources for sustainable biofuels: A Review. Renew. Sustain. Energy Rev. 2017, 73, 205–214. [Google Scholar] [CrossRef]
- Zhou, F.; Olman, V.; Xu, Y. Large-scale analyses of glycosylation in cellulases. Genom. Proteom. Bioinform. 2009, 7, 194–199. [Google Scholar] [CrossRef] [Green Version]
- Basit, A.; Miao, T.; Liu, J.; Wen, J.; Song, L.; Zheng, F.; Lou, H.; Jiang, W. Highly efficient degradation of xylan into xylose by a single enzyme. ACS Sustain. Chem. Eng. 2019, 7, 11360–11368. [Google Scholar] [CrossRef]
- Zheng, F.; Song, L.; Basit, A.; Liu, J.; Miao, T.; Wen, J.; Cao, Y.; Jiang, W. An endoxylanase rapidly hydrolyzes xylan into major product xylobiose via transglycosylation of xylose to xylotriose or xylotetraose. Carbohydr. Polym. 2020, 237, 116121. [Google Scholar] [CrossRef] [PubMed]
- Rubin, E.M. Genomics of cellulosic biofuels. Nature 2008, 454, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Himmel, M.E.; Ding, S.-Y.; Johnson, D.K.; Adney, W.S.; Nimlos, M.R.; Brady, J.W.; Foust, T.D. Biomass recalcitrance: Engineering plants and enzymes for biofuels production. Science 2007, 315, 804–807. [Google Scholar] [CrossRef] [Green Version]
- Wahlström, R.M.; Suurnäkki, A. Enzymatic hydrolysis of lignocellulosic polysaccharides in the presence of ionic liquids. Green Chem. 2014, 17, 694–714. [Google Scholar] [CrossRef] [Green Version]
- Zheng, F.; Liu, J.; Basit, A.; Miao, T.; Jiang, W. Insight to improve α-L-arabinofuranosidase productivity in Pichia pastoris and its application on corn stover degradation. Front. Microbiol. 2018, 9, 3016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, L.E., 2nd; Knott, B.C.; Baker, J.O.; Alahuhta, P.M.; Hobdey, S.E.; Linger, J.G.; Lunin, V.V.; Amore, A.; Subramanian, V.; Podkaminer, K.; et al. Engineering enhanced cellobiohydrolase activity. Nat. Commun. 2018, 9, 1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, D.; Uppugundla, N.; Chundawat, S.P.; Yu, X.; Hermanson, S.; Gowda, K.; Brumm, P.; Mead, D.; Balan, V.; Dale, B.E. Hemicellulases and auxiliary enzymes for improved conversion of lignocellulosic biomass to monosaccharides. Biotechnol. Biofuels 2011, 4, 5. [Google Scholar] [CrossRef] [Green Version]
- Lagaert, S.; Pollet, A.; Courtin, C.M.; Volckaert, G. β-Xylosidases and α-l-arabinofuranosidases: Accessory enzymes for arabinoxylan degradation. Biotechnol. Adv. 2014, 32, 316–332. [Google Scholar] [CrossRef]
- Laothanachareon, T.; Bunterngsook, B.; Suwannarangsee, S.; Eurwilaichitr, L.; Champreda, V. Synergistic action of recombinant accessory hemicellulolytic and pectinolytic enzymes to Trichoderma reesei cellulase on rice straw degradation. Bioresour. Technol. 2015, 198, 682–690. [Google Scholar] [CrossRef]
- McKee, L.S.; Peña, M.J.; Rogowski, A.; Jackson, A.; Lewis, R.J.; York, W.S.; Krogh, K.B.R.M.; Viksø-Nielsen, A.; Skjøt, M.; Gilbert, H.J.; et al. Introducing endo-xylanase activity into an exo-acting arabinofuranosidase that targets side chains. Proc. Natl. Acad. Sci. USA 2012, 109, 6537–6542. [Google Scholar] [CrossRef] [Green Version]
- Eijsink, V.G.H.; Petrovic, D.; Forsberg, Z.; Mekasha, S.; Røhr, Å.K.; Várnai, A.; Bissaro, B.; Vaaje-Kolstad, G. On the functional characterization of lytic polysaccharide monooxygenases (LPMOs). Biotechnol. Biofuels 2019, 12, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filiatrault-Chastel, C.; Navarro, D.; Haon, M.; Grisel, S.; Herpoël-Gimbert, I.; Chevret, D.; Fanuel, M.; Henrissat, B.; Heiss-Blanquet, S.; Margeot, A.; et al. AA16, a new lytic polysaccharide monooxygenase family identified in fungal secretomes. Biotechnol. Biofuels 2019, 12, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levasseur, A.; Drula, E.; Lombard, V.; Coutinho, P.M.; Henrissat, B. Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol. Biofuels 2013, 6, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabbadin, F.; Hemsworth, G.R.; Ciano, L.; Henrissat, B.; DuPree, P.; Tryfona, T.; Marques, R.D.S.; Sweeney, S.T.; Besser, K.; Elias, L.; et al. An ancient family of lytic polysaccharide monooxygenases with roles in arthropod development and biomass digestion. Nat. Commun. 2018, 9, 756. [Google Scholar] [CrossRef] [PubMed]
- Vu, V.V.; Beeson, W.T.; Span, E.A.; Farquhar, E.R.; Marletta, M.A. A family of starch-active polysaccharide monooxygenases. Proc. Natl. Acad. Sci. USA 2014, 111, 13822–13827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, E.; Hijnen, M.; Bunker, R.D.; Boudes, M.; Rajendran, C.; Aizel, K.; Olieric, V.; Schulze-Briese, C.; Mitsuhashi, W.; Young, V.; et al. Structural basis for the enhancement of virulence by viral spindles and their in vivo crystallization. Proc. Natl. Acad. Sci. USA 2015, 112, 3973–3978. [Google Scholar] [CrossRef] [Green Version]
- Couturier, M.; Ladevèze, S.; Sulzenbacher, G.; Ciano, L.; Fanuel, M.; Moreau, C.; Villares, A.; Cathala, B.; Chaspoul, F.; Frandsen, K.E.; et al. Lytic xylan oxidases from wood-decay fungi unlock biomass degradation. Nature 2018, 14, 306–310. [Google Scholar] [CrossRef] [Green Version]
- Rani Singhania, R.; Dixit, P.; Kumar Patel, A.; Shekher Giri, B.; Kuo, C.H.; Chen, C.W.; Di Dong, C. Role and significance of lytic polysaccharide monooxygenases (LPMOs) in lignocellulose deconstruction. Bioresour. Technol. 2021, 335, 125261. [Google Scholar] [CrossRef]
- Li, J.; Solhi, L.; Goddard-Borger, E.D.; Mathieu, Y.; Wakarchuk, W.W.; Withers, S.G.; Brumer, H. Four cellulose-active lytic polysaccharide monooxygenases from Cellulomonas species. Biotechnol. Biofuels 2021, 14, 29. [Google Scholar] [CrossRef]
- Gaber, Y.; Rashad, B.; Hussein, R.; Abdelgawad, M.; Ali, N.S.; Dishisha, T.; Várnai, A. Heterologous expression of lytic polysaccharide monooxygenases (LPMOs). Biotechnol. Adv. 2020, 43, 107583. [Google Scholar] [CrossRef]
- Chahal, D.S. Solid-state fermentation with Trichoderma reesei for cellulase production. Appl. Environ. Microbiol. 1985, 49, 205–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latifian, M.; Hamidi-Esfahani, Z.; Barzegar, M. Evaluation of culture conditions for cellulase production by two Trichoderma reesei mutants under solid-state fermentation conditions. Bioresour. Technol. 2007, 98, 3634–3637. [Google Scholar] [CrossRef] [PubMed]
- Mekala, N.; Singhania, R.; Sukumaran, R.; Pandey, A. Cellulase production under solid-state fermentation by Trichoderma reesei RUT C30: Statistical optimization of process parameters. Appl. Biochem. Biotechnol. 2008, 151, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Shen, X. High-yield cellulase production by Trichoderma reesei ZU-02 on corn cob residue. Bioresour. Technol. 2003, 91, 259–262. [Google Scholar] [CrossRef]
- Singhania, R.; Sukumaran, R.; Pandey, A. Improved cellulase production by Trichoderma reesei RUT C30 under SSF through process optimization. Appl. Biochem. Biotechnol. 2007, 142, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Li, W.C.; Lin, T.C.; Chen, C.L.; Liu, H.C.; Lin, H.N.; Chao, J.L.; Hsieh, C.H.; Ni, H.F.; Chen, R.S.; Wang, T.F. Complete genome sequences and genome-wide characterization of Trichoderma biocontrol agents provide new insights into their evolution and variation in genome organization, sexual development, and fungal-plant interactions. Microbiol. Spectr. 2021, 9, e0066321. [Google Scholar] [CrossRef]
- Ravalason, H.; Grisel, S.; Chevret, D.; Favel, A.; Berrin, J.-G.; Sigoillot, J.-C.; Herpoël-Gimbert, I. Fusarium verticillioides secretome as a source of auxiliary enzymes to enhance saccharification of wheat straw. Bioresour. Technol. 2012, 114, 589–596. [Google Scholar] [CrossRef]
- Marx, I.J.; van Wyk, N.; Smit, S.; Jacobson, D.; Viljoen-Bloom, M.; Volschenk, H. Comparative secretome analysis of Trichoderma asperellum S4F8 and Trichoderma reesei Rut C30 during solid-state fermentation on sugarcane bagasse. Biotechnol. Biofuels 2013, 6, 172. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, H.; Sakagawa, E.; Toyoda, R.; Motomura, T.; Murase, M.; Takahashi, A.; Fukuyoshi, S.; Kanamasa, S. Draft genome sequence of glycoside hydrolase-producing Trichoderma asperellum strain IC-1. Microbiol. Resour. Announc. 2020, 9, e00958-20. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, L.; Yu, D.; Lin, H.; Shen, Q.; Zhao, Y. Excellent waste biomass-degrading performance of Trichoderma asperellum T-1 during submerged fermentation. Sci. Total Environ. 2017, 609, 1329–1339. [Google Scholar] [CrossRef]
- Da Silva Aires, R.; Steindorff, A.S.; Ramada, M.H.S.; de Siqueira, S.J.L.; Ulhoa, C.J. Biochemical characterization of a 27 kDa 1,3-β-D-glucanase from Trichoderma asperellum induced by cell wall of Rhizoctonia solani. Carbohydr. Polym. 2012, 87, 1219–1223. [Google Scholar] [CrossRef] [Green Version]
- Subbaiyan, G.K.; Waters, D.L.E.; Katiyar, S.K.; Sadananda, A.R.; Satyadev, V.; Henry, R. Genome-wide DNA polymorphisms in elite indica rice inbreds discovered by whole-genome sequencing. Plant Biotechnol. J. 2012, 10, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Kubicek, C.P.; Herrera-Estrella, A.; Seidl-Seiboth, V.; Martinez, D.A.; Druzhinina, I.S.; Thon, M.; Zeilinger, S.; Casas-Flores, S.; Horwitz, B.A.; Mukherjee, P.K.; et al. Comparative genome sequence analysis underscores mycoparasitism as the ancestral life style of Trichoderma. Genome Biol. 2011, 12, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, D.; Berka, R.M.; Henrissat, B.; Saloheimo, M.; Arvas, M.; Baker, S.E.; Chapman, J.; Chertkov, O.; Coutinho, P.M.; Cullen, D.; et al. Genome sequencing and analysis of the biomass-degrading fungus Trichoderma reesei (syn. Hypocrea jecorina). Nat. Biotechnol. 2008, 26, 553–560. [Google Scholar] [CrossRef] [Green Version]
- Teng, J.L.L.; Yeung, M.L.; Chan, E.; Jia, L.; Lin, C.H.; Huang, Y.; Tse, H.; Wong, S.S.Y.; Sham, P.C.; Lau, S.K.P.; et al. PacBio but not Illumina technology can achieve fast, accurate and complete closure of the high GC, complex Burkholderia pseudomallei two-chromosome genome. Front. Microbiol. 2017, 8, 1448. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Li, Y.; Chen, Y.; Xu, S.; Du, G.; Shi, H.; Zhou, J.; Chen, J. Complete genome sequence and analysis of the industrial Saccharomyces cerevisiae strain N85 used in Chinese rice wine production. DNA Res. 2018, 25, 297–306. [Google Scholar] [CrossRef] [Green Version]
- Chin, C.-S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B.; et al. Real-time DNA sequencing from single polymerase molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef]
- Nakano, K.; Shiroma, A.; Shimoji, M.; Tamotsu, H.; Ashimine, N.; Ohki, S.; Shinzato, M.; Minami, M.; Nakanishi, T.; Teruya, K.; et al. Advantages of genome sequencing by long-read sequencer using SMRT technology in medical area. Hum. Cell 2017, 30, 149–161. [Google Scholar] [CrossRef] [Green Version]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 1, 686–689. [Google Scholar] [CrossRef] [PubMed]
- Bodega, B.; Orlando, V. Repetitive elements dynamics in cell identity programming, maintenance and disease. Curr. Opin. Cell Biol. 2014, 31, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Adav, S.S.; Chao, L.T.; Sze, S.K. Quantitative secretomic analysis of Trichoderma reesei strains reveals enzymatic composition for lignocellulosic biomass degratation. Mol. Cell Proteom. 2012, 11, M111.012419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barriuso, J.; Nguyen, D.T.; Li, J.W.-H.; Roberts, J.N.; MacNevin, G.; Chaytor, J.L.; Marcus, S.L.; Vederas, J.C.; Ro, D.-K. Double oxidation of the cyclic nonaketide dihydromonacolin L to monacolin J by a single cytochrome P450 monooxygenase, LovA. J. Am. Chem. Soc. 2011, 133, 8078–8081. [Google Scholar] [CrossRef] [PubMed]
- Kelkar, H.S.; Skloss, T.W.; Haw, J.F.; Keller, N.P.; Adams, T.H. Aspergillus nidulans stcL encodes a putative cytochrome P-450 monooxygenase required for bisfuran desaturation during aflatoxin/sterigmatocystin biosynthesis. J. Biol. Chem. 1997, 272, 1589–1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moktali, V.; Park, J.; Fedorova-Abrams, N.D.; Park, B.; Choi, J.; Lee, Y.-H.; Kang, S. Systematic and searchable classification of cytochrome P450 proteins encoded by fungal and oomycete genomes. BMC Genom. 2012, 13, 525. [Google Scholar] [CrossRef] [Green Version]
- Aachmann, F.L.; Sørlie, M.; Skjåk-Bræk, G.; Eijsink, V.G.H.; Vaaje-Kolstad, G. NMR structure of a lytic polysaccharide monooxygenase provides insight into copper binding, protein dynamics, and substrate interactions. Proc. Natl. Acad. Sci. USA 2012, 109, 18779–18784. [Google Scholar] [CrossRef] [Green Version]
- Kont, R.; Bissaro, B.; Eijsink, V.G.H.; Väljamäe, P. Kinetic insights into the peroxygenase activity of cellulose-active lytic polysaccharide monooxygenases (LPMOs). Nat. Commun. 2020, 11, 5786. [Google Scholar] [CrossRef]
- Grondona, I.; Hermosa, R.; Tejada, M.; Gomis, M.D.; Mateos, P.; Bridge, P.D.; Monte, E.; Garcia-Acha, I. Physiological and biochemical characterization of Trichoderma harzianum, a biological control agent against soilborne fungal plant pathogens. Appl. Environ. Microbiol. 1997, 63, 3189–3198. [Google Scholar] [CrossRef] [Green Version]
- Harman, G.E.; Howell, C.R.; Viterbo, A.; Chet, I.; Lorito, M. Trichoderma species-opportunistic, avirulent plant symbionts. Nat. Rev. Microbiol. 2004, 2, 43–56. [Google Scholar] [CrossRef]
- Seidl, V.; Huemer, B.; Seiboth, B.; Kubicek, C.P. A complete survey of Trichoderma chitinases reveals three distinct subgroups of family 18 chitinases. FEBS J. 2005, 272, 5923–5939. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Liu, H.; Wang, C.; Xu, J.-R. Comparative analysis of fungal genomes reveals different plant cell wall degrading capacity in fungi. BMC Genom. 2013, 14, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viterbo, A.; Landau, U.; Kim, S.; Chernin, L.; Chet, I. Characterization of ACC deaminase from the biocontrol and plant growth-promoting agent Trichoderma asperellum T203. FEMS Microbiol. Lett. 2010, 305, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, Y.; Ichikawa, H.; Naznin, H.A.; Kogure, A.; Hyakumachi, M. Systemic resistance induced in Arabidopsis thaliana by Trichoderma asperellum SKT-1, a microbial pesticide of seedborne diseases of rice. Pest Manag. Sci. 2011, 68, 60–66. [Google Scholar] [CrossRef]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.G.; Arantes, V.; Saddler, J.N. The enhancement of enzymatic hydrolysis of lignocellulosic substrates by the addition of accessory enzymes such as xylanase: Is it an additive or synergistic effect? Biotechnol. Biofuels 2011, 4, 36. [Google Scholar] [CrossRef] [Green Version]
- Zhu, N.; Yang, J.; Ji, L.; Liu, J.; Yang, Y.; Yuan, H. Metagenomic and metaproteomic analyses of a corn stover-adapted microbial consortium EMSD5 reveal its taxonomic and enzymatic basis for degrading lignocellulose. Biotechnol. Biofuels 2016, 9, 243. [Google Scholar] [CrossRef] [Green Version]
- Guo, B.; Sato, N.; Biely, P.; Amano, Y.; Nozaki, K. Comparison of catalytic properties of multiple β-glucosidases of Trichoderma reesei. Appl. Microbiol. Biotechnol. 2016, 100, 4959–4968. [Google Scholar] [CrossRef]
- Horn, S.J.; Vaaje-Kolstad, G.; Westereng, B.; Eijsink, V.G. Novel enzymes for the degradation of cellulose. Biotechnol. Biofuels 2012, 5, 45. [Google Scholar] [CrossRef] [Green Version]
- Kostylev, M.; Wilson, D. Synergistic interactions in cellulose hydrolysis. Biofuels 2012, 3, 61–70. [Google Scholar] [CrossRef]
- Häkkinen, M.; Arvas, M.; Oja, M.; Aro, N.; Penttilä, M.; Saloheimo, M.; Pakula, T.M. Re-annotation of the CAZy genes of Trichoderma reesei and transcription in the presence of lignocellulosic substrates. Microb. Cell Fact 2012, 11, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, A.-P.; Wang, H.; Luo, Y.; Yang, Z.; Liu, Z.; Wang, Z.; Li, B.; Yang, S.; Zhou, Z.; Lu, X.; et al. Dissecting Cellular Function and Distribution of β-Glucosidases in Trichoderma reesei. mBio 2021, 12, e03671-20. [Google Scholar] [CrossRef] [PubMed]
- Kubicek, C.P.; Steindorff, A.S.; Chenthamara, K.; Manganiello, G.; Henrissat, B.; Zhang, J.; Cai, F.; Kopchinskiy, A.G.; Kubicek, E.M.; Kuo, A.; et al. Evolution and comparative genomics of the most common Trichoderma species. BMC Genom. 2019, 20, 485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berlin, K.; Koren, S.; Chin, C.-S.; Drake, J.P.; Landolin, J.M.; Phillippy, A.M. Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat. Biotechnol. 2015, 33, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 2012, 1, 18. [Google Scholar] [CrossRef]
- Ter-Hovhannisyan, V.; Lomsadze, A.; Chernoff, Y.O.; Borodovsky, M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 2008, 18, 1979–1990. [Google Scholar] [CrossRef] [Green Version]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef] [Green Version]
- UniProt, C. Reorganizing the protein space at the universal protein resource (UniProt). Nucleic Acids Res. 2012, 40, 71–75. [Google Scholar]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, 277–280. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, 222–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eddy, S.R. Profile hidden Markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Götz, S.; Arnold, R.; Sebastián-León, P.; Martín-Rodríguez, S.; Tischler, P.; Jehl, M.A.; Dopazo, J.; Rattei, T.; Conesa, A. B2G-FAR, a species-centered GO annotation repository. Bioinformatics 2011, 27, 919–924. [Google Scholar] [CrossRef] [PubMed]
- Urban, M.; Cuzick, A.; Rutherford, K.; Irvine, A.; Pedro, H.; Pant, R.; Sadanadan, V.; Khamari, L.; Billal, S.; Mohanty, S.; et al. PHI-base: A new interface and further additions for the multi-species pathogen–host interactions database. Nucleic Acids Res. 2016, 45, D604–D610. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Nordberg, H.; Cantor, M.; Dusheyko, S.; Hua, S.; Poliakov, A.; Shabalov, I.; Smirnova, T.; Grigoriev, I.; Dubchak, I. The genome portal of the Department of Energy Joint Genome Institute: 2014 updates. Nucleic Acids Res. 2013, 42, D26–D31. [Google Scholar] [CrossRef]
- Enright, A.J.; Van Dongen, S.; Ouzounis, C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar] [CrossRef]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, 121. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Mao, X.; Yang, J.; Chen, X.; Mao, F.; Xu, Y. dbCAN: A web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2012, 40, W445–W451. [Google Scholar] [CrossRef]
- Saha, B.C.; Bothast, R.J. Production, purification, and characterization of a highly glucose-tolerant novel β-glucosidase from Candida peltata. Appl. Environ. Microb. 1996, 62, 3165–3170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, G.L. Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal. Chem. 1959, 31, 426–428. [Google Scholar] [CrossRef]
- National Genomics Data Center Members and Partners. Database Resources of the National Genomics Data Center in 2020. Nucleic Acids Res. 2020, 48, 24–33. [Google Scholar]








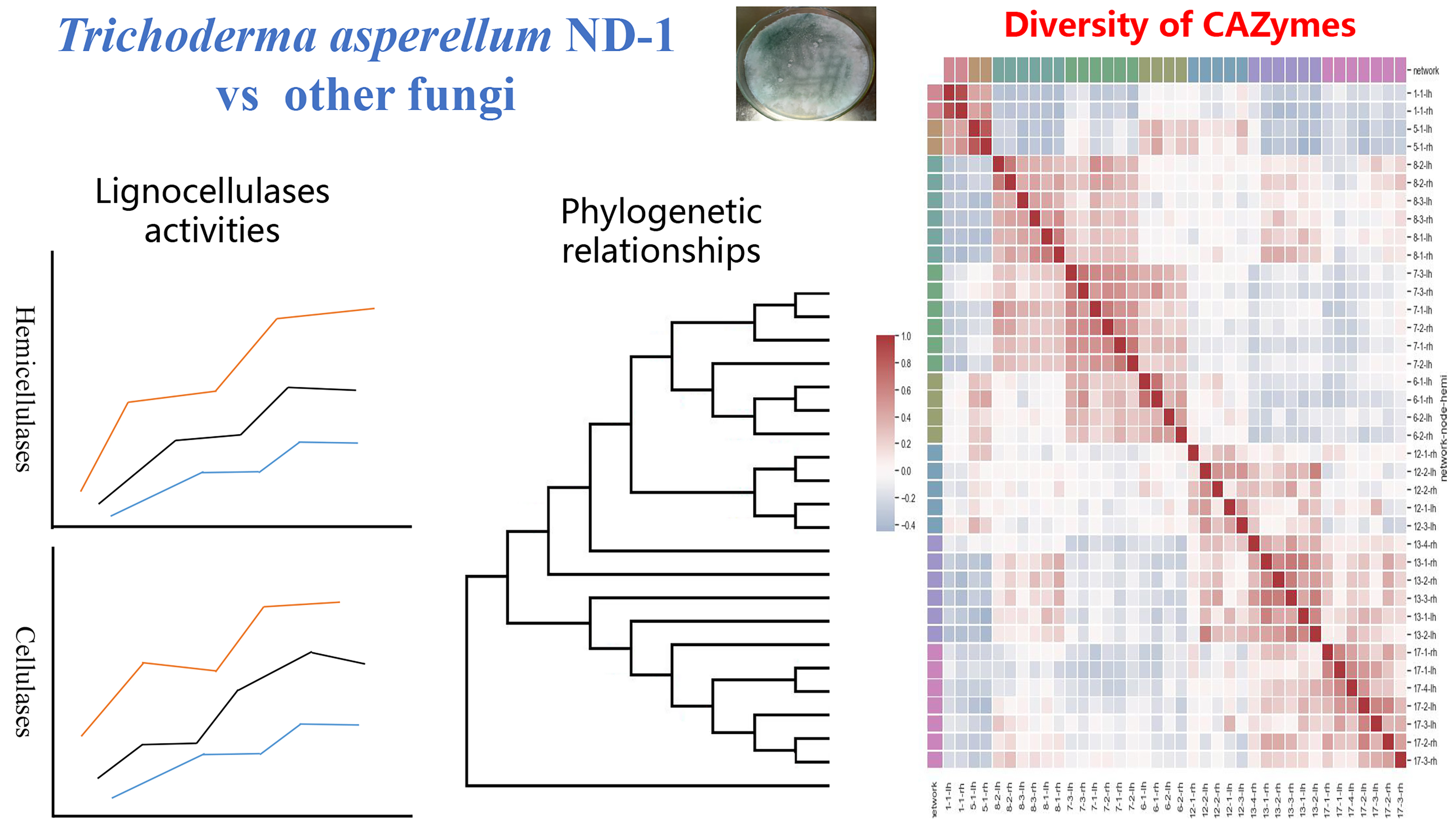{kind=link}
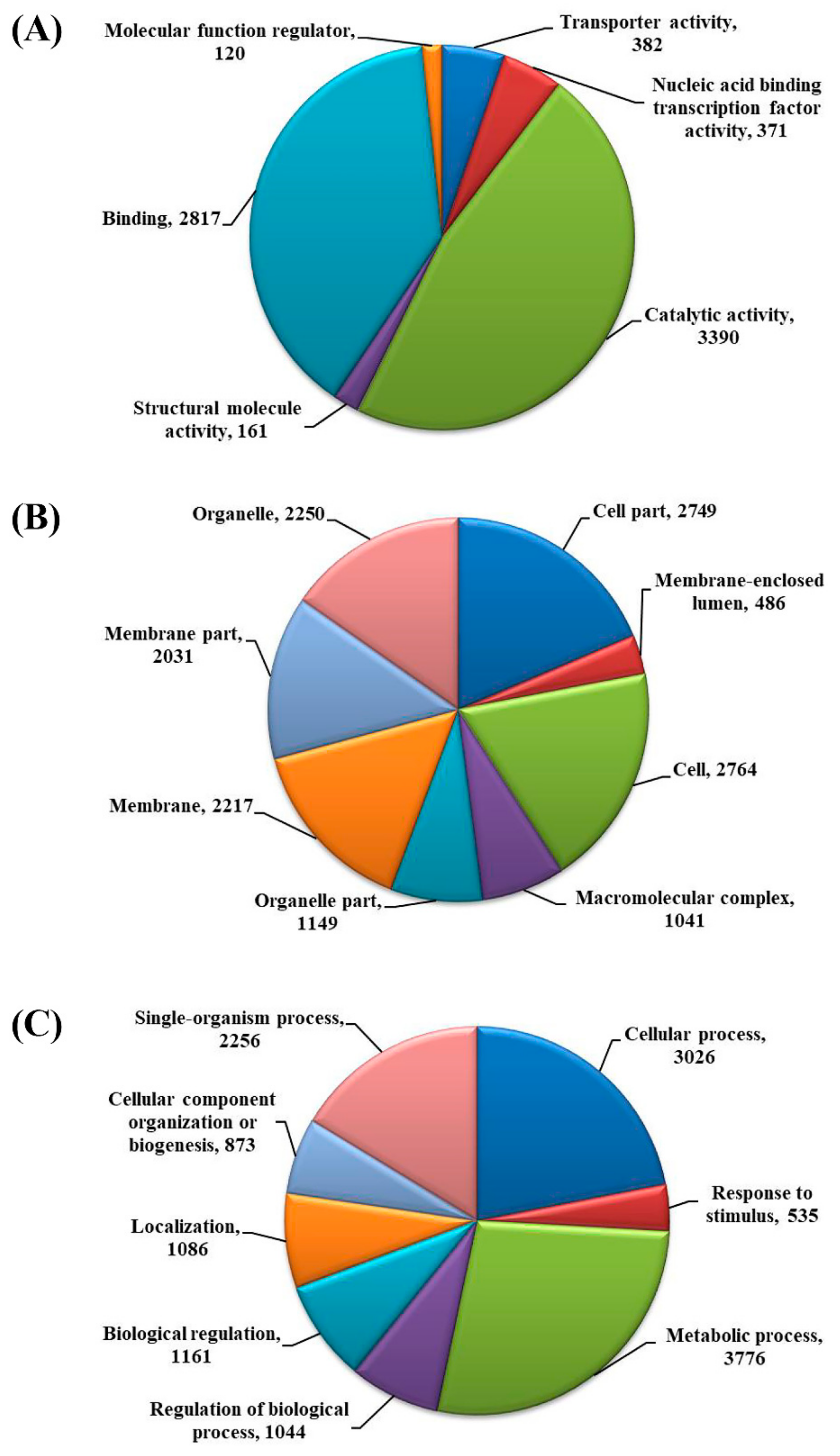{kind=link}
{kind=link}
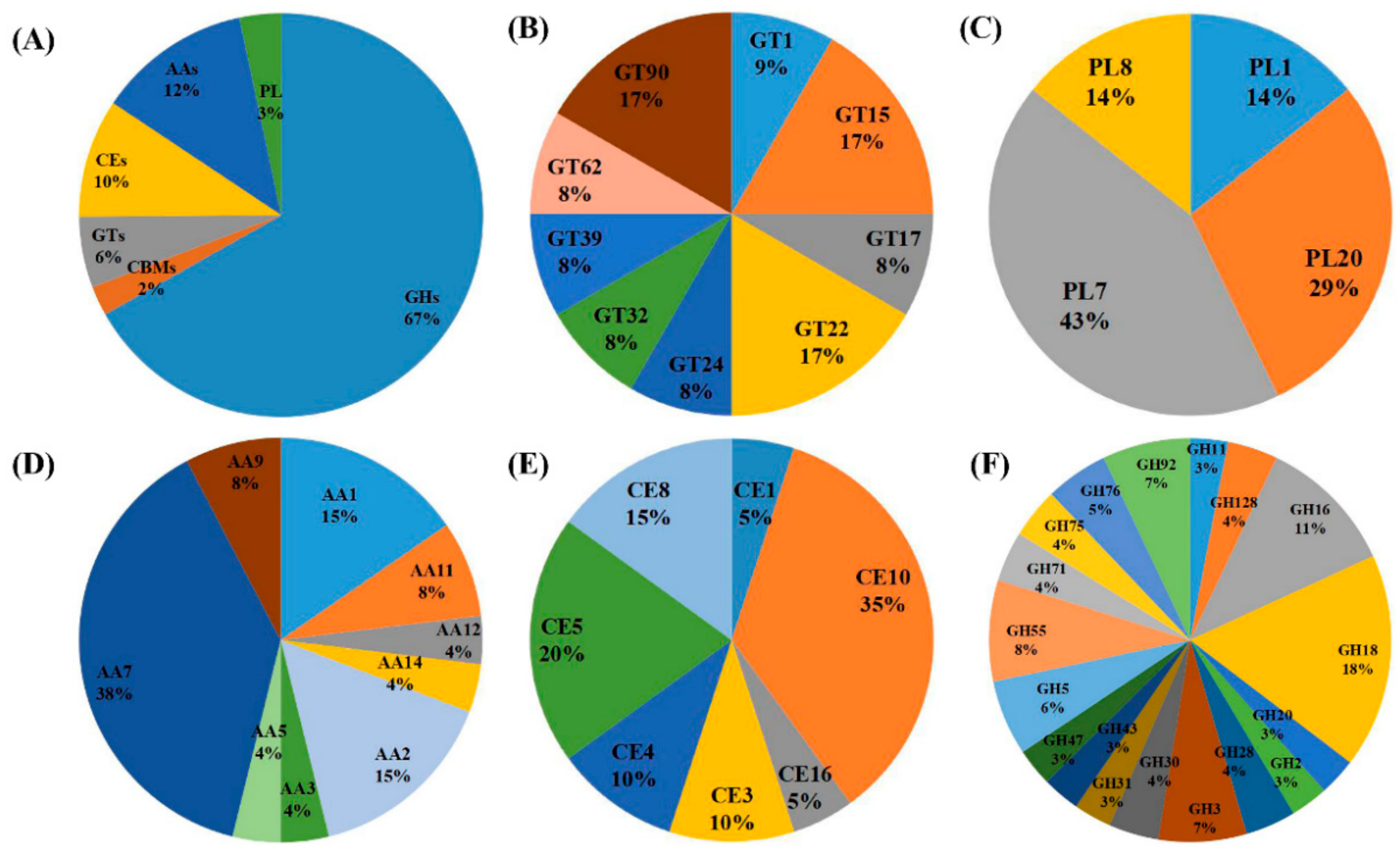{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Featuers | Trichoderma asperellum ND-1 |
|---|---|
| Coverage | 99.3% |
| Protein length, amino acids | 516.18 |
| Avg. Gene Density (genes/kb) | 0.29 |
| Avg. Gene length (bp) | 1.86 kb |
| Repeat Content % | 1.66 |
| tRNAs | 246 |
| Secreted Proteins | 895 |
| PHI genes | 2340 |
| Proteases | 82 |
| Average exons per gene | 2.98 |
| Average exon length (bp) | 0.52 kb |
| Average introns per gene | 1.98 |
| Average intron length (bp) | 0.16 kb |
| Supported by homology, Swissprot | 6746 (64%) |
| Supported by homology, NR | 9496 (90%) |
| Has PFAM domain | 7281 (69%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, F.; Han, T.; Basit, A.; Liu, J.; Miao, T.; Jiang, W. Whole-Genome Sequence and Comparative Analysis of Trichoderma asperellum ND-1 Reveal Its Unique Enzymatic System for Efficient Biomass Degradation. Catalysts 2022, 12, 437. https://doi.org/10.3390/catal12040437
Zheng F, Han T, Basit A, Liu J, Miao T, Jiang W. Whole-Genome Sequence and Comparative Analysis of Trichoderma asperellum ND-1 Reveal Its Unique Enzymatic System for Efficient Biomass Degradation. Catalysts. 2022; 12(4):437. https://doi.org/10.3390/catal12040437
Chicago/Turabian StyleZheng, Fengzhen, Tianshuo Han, Abdul Basit, Junquan Liu, Ting Miao, and Wei Jiang. 2022. "Whole-Genome Sequence and Comparative Analysis of Trichoderma asperellum ND-1 Reveal Its Unique Enzymatic System for Efficient Biomass Degradation" Catalysts 12, no. 4: 437. https://doi.org/10.3390/catal12040437
APA StyleZheng, F., Han, T., Basit, A., Liu, J., Miao, T., & Jiang, W. (2022). Whole-Genome Sequence and Comparative Analysis of Trichoderma asperellum ND-1 Reveal Its Unique Enzymatic System for Efficient Biomass Degradation. Catalysts, 12(4), 437. https://doi.org/10.3390/catal12040437