2.1. Characterization
Figure 1 shows the SEM images of natural bentonite (B), the bentonite nanocomposite (CB), and the activated bentonite nanocomposite (CBA). As can be observed, natural bentonite B (
Figure 1a) consists of particles that seem to be formed by several flaky particles stacked together in the form of agglomerates [
31]. On the other hand, solid CB (
Figure 1b) has smaller particles than B, and they seem to have a sponge-type structure, indicating that the condensation process took place [
32]. Finally, the acid treatment generated the disaggregation and decrease in size of the particles, as can be observed in
Figure 1c (solid CBA).
Table 1 shows the composition of solids B, CB, and CBA determined by EDS analysis. As observed, the Si content was higher in solids CB and CBA due to the incorporation of TEOS. However, in solid CBA (treated with nitric acid) the sodium content decreased, indicating that the Na
+ ions were replaced by the protons from the acid solution.
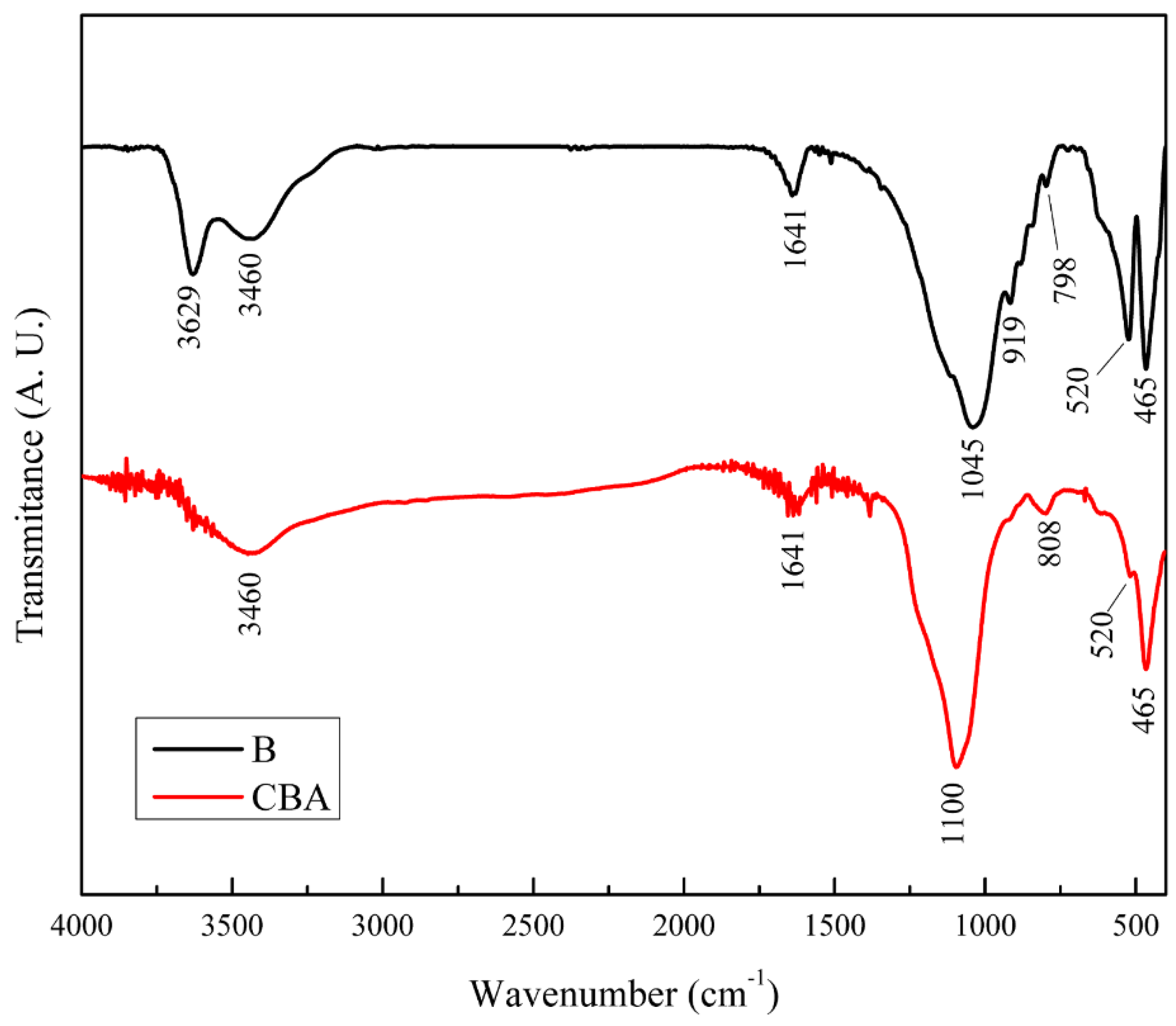
Figure 2 shows the infrared spectra of solids CBA and B. The spectra of both solids presented signals at 465 and 520 cm
−1, which corresponded to the Si–O vibrations in the tetrahedral layer. In addition, the signal observed at 798 cm
−1 in solid B was related to the Si–O vibration, which is characteristic of quartz, and the signals at 919 cm
−1 and 3629 cm
−1 were attributed to Al–OH vibrations in the octahedral layers of the bentonite [
33]. The bands observed at 1641 and 3460 cm
−1 were attributed to the H–OH vibrations of the water adsorbed on the interlayer structure and were observed in both solids [
34].
On the other hand, solid CB presented two bands at 808 and 1100 cm
−1, which could be attributed to the Si–O–Si unions from the condensation of TEOS [
33]. As can be observed, the acid treatment did not generate important changes in the solid structure.
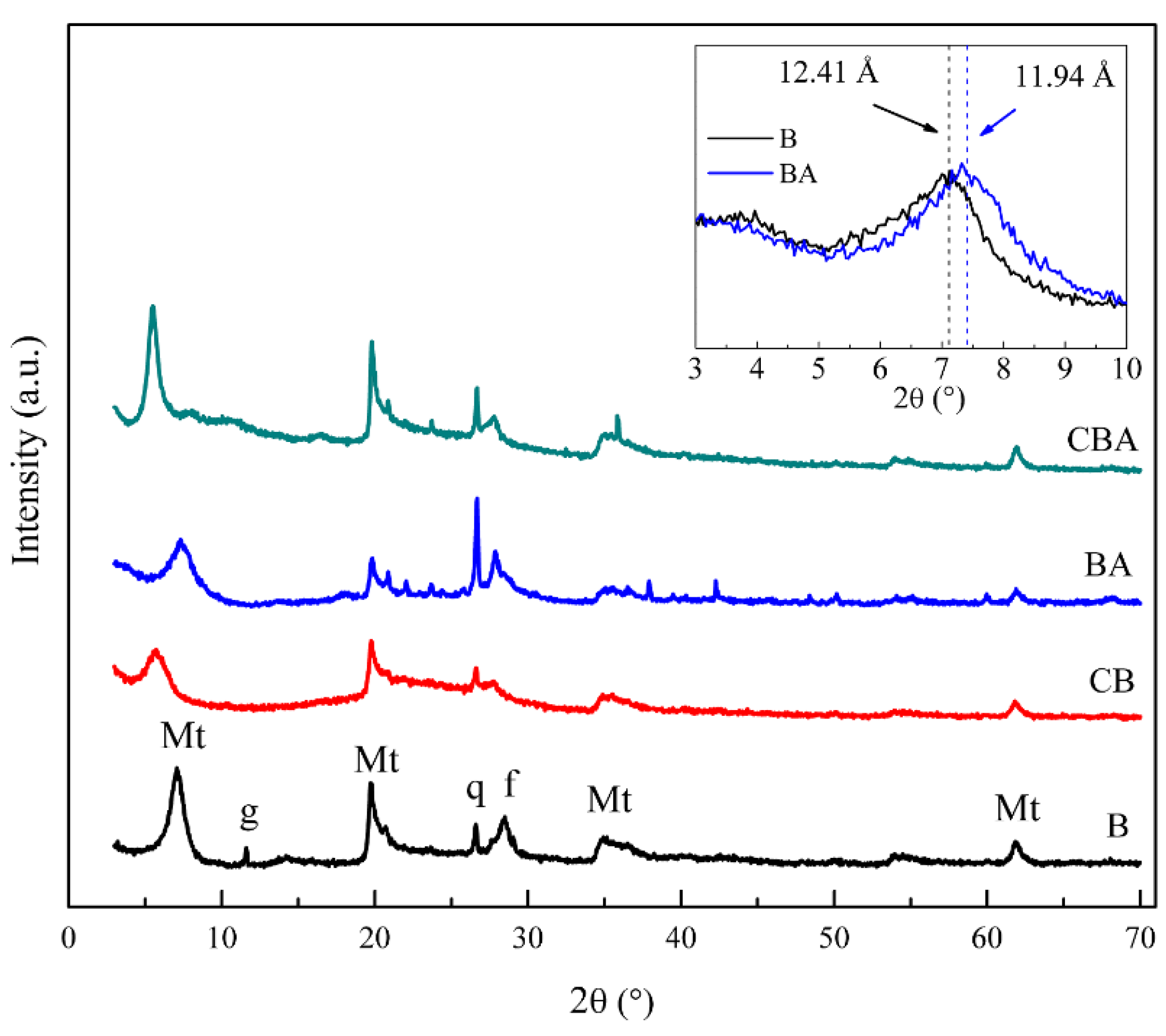
The X-ray diffraction technique was used to study the structure of the bentonite in the composite and to evaluate the effect of acidification on the bentonite’s structure.
The diffraction patterns of natural bentonite B (
Figure 3) were analyzed and compared with the shown reflections of montmorillonite and the accompanying minerals, such as quartz, at positions 2θ = 20.8° and 2θ = 26.8° (PDF card No. 00-046-1045), gypsum at 2θ = 11.0° (PDF card No. 00-021-0816), and feldspar at 2θ = 28.8° (PDF card No. 00-020-0548). The rest of the peaks assigned to montmorillonite corresponded to PDF card No. 00-029-1498, and the reflection d001 at position 2θ = 7.1° was characteristic of dry montmorillonite [
35].
The diffractogram of bentonite composite CB (
Figure 3) shows that the d001 peak shifted to lower angles, from 2θ = 7.1° to 2θ = 5.6°, indicating the expansion of the interlaminar space from 12.41 to 15.8 Å, respectively. There were numerous sol-gel reactions involved in the formation of the hybrid composite, such as the complete hydrolysis of tetraethyl orthosilicate (TEOS) and the condensation of the hydroxyl groups (–OH) in the organic and inorganic precursors with the hydroxyl groups (–OH) of montmorillonite layer edges. This generated a continuous network and, as it grew, it probably penetrated the interlayer and expanded the basal space. Then, after the aging and curing stages, the structure remained swollen, pillaring the montmorillonite. It was also observed that the peaks corresponding to the bentonite, such as montmorillonite and other minerals, preserved their structure in the composite, although they overlapped the amorphous silicon band.
As can be observed, the activated bentonite (BA) and the activated bentonite composite (CBA) conserved the peaks of the crystalline structure of the bentonite layers, showing that the acid treatment did not modify the crystalline structure of the bentonite. Additionally, the activation of the B solid produced a shift of the d001 peak at 2θ = 7.1° to 2θ = 7.4° (Figure inserted in
Figure 1), whereas this effect could not be observed in the solid CBA due to the expansion of the basal space mentioned above.
The use of nitrogen to calculate the specific surface area only allows the determination of the external specific surface area (ESSA) [
36,
37], a value that can be around 10% of the total specific surface area (TSSA) [
38].
The results obtained are shown in
Table 2, employing the BET technique for ESSA and water adsorption for TSSA. It can be observed that the ESSA of solid B was approximately 110 m
2 g
−1, a value that is in agreement with those reported in the literature [
36].
As can be observed in
Table 2, the composites CB and CBA presented a remarkably lower TSSA than the bentonites due to the inclusion of the silica–resin structure in the interlaminar spaces of the bentonites, as discussed above in the XRD diffractograms. In addition, the ESSA values of CB and CBA were similar, which is in agreement with Timofeeva and coworkers, who did not observe any differences in the ESSA when weak conditions were employed for the activation of bentonites [
21].
Furthermore,
Table 2 shows the results of the potentiometric titration technique. The acid strength of sites can be studied through the potentiometric titration technique with n-butylamine, in which the initial electrode potential (Ei) indicates the maximum acid strength according to the following scale: very strong, Ei > 100 mV; strong, 0 < Ei < 100 mV; weak, −100 < Ei < 0 mV; and very weak, Ei < −100 mV [
39]. As observed, the acid treatment of the solids generated an increment in the Ei value from −62.7 mV to ~ 490 mV, indicating the generation of acid sites stronger than those present in solids B and CB. Furthermore, the values of Ei for BA and CBA were similar (~ 490 mV), indicating that the acid strength generated was comparable in both solids.
To study the distribution of these acid sites, the ammonia temperature-programmed desorption (NH
3-TPD) and FTIR spectroscopy of pyridine adsorption techniques were used. However, the latter did not produce good results because the signal corresponding to the silica–resin structure overlapped the signal of adsorbed pyridine.
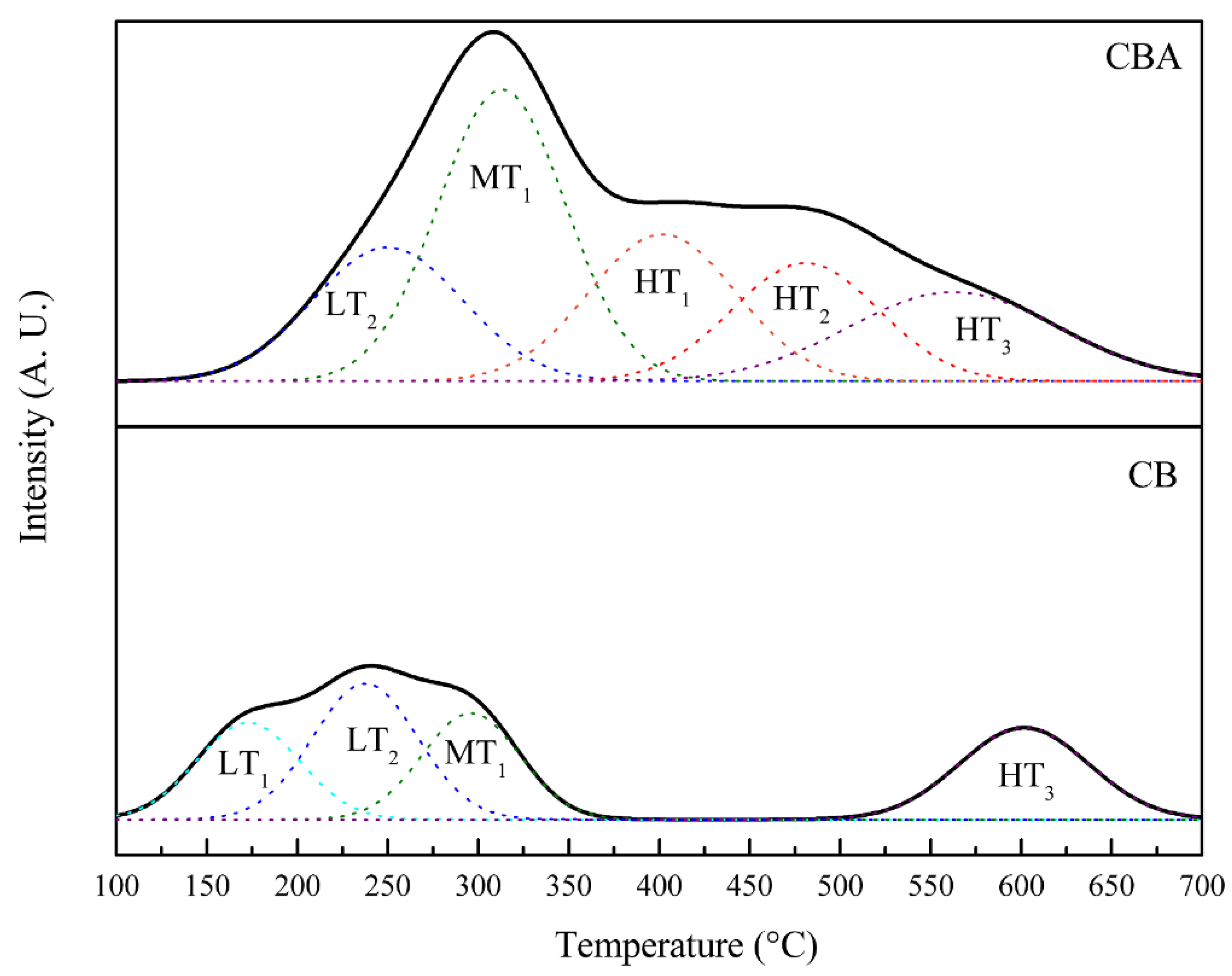
Figure 4 shows the NH
3 desorption spectrum and the peak deconvolution. The measurements were carried out between 50 °C and 700 °C, identifying three desorption zones: the first at low temperatures (150–250 °C), identified as LT, which corresponded to weak acid sites, the second at medium temperatures (250–350 °C), identified as MT, which corresponded to medium acid sites, and the third zone at high temperatures (>350 °C), identified as HT, which corresponded to strong acid sites [
25]. Signals observed below 150 °C were not considered because NH
3 desorbed at such low temperatures corresponds to physisorbed molecules [
40].
The CB solid presented two desorption peaks with maximums at 160 °C (denoted as “LT
1”) and 240 °C (denoted as “LT
2”), which corresponded to acid sites of weak strength that proceeded from Si-OH groups on the surface. Furthermore, it presented a peak with a maximum at 300 °C (denoted as “MT
1”), attributed to medium acid sites, which proceeded from the polarized water molecules in the interlayer space. The region at ~600 °C (HT
3) could be attributed to the desorption of NH
3 from the framework Al sites [
41].
On the other hand, the CBA solid conserved the peaks at LT
2 and MT
1 and presented an increase in the desorption peaks in the region at temperatures higher than 400 °C, attributed to the generation of sites with strong acid strength. This solid presented a desorption peak with a maximum at 400 °C (denoted as “HT
1”), which corresponded to acid sites generated by the protonation of Si–OH groups, forming terminal Si–OH
2+ species. In addition, it presented a desorption peak with a maximum at 500 °C (denoted as “HT
2”) attributed to the presence of protons in the interlaminar space [
42] as a result of the acid treatment with HNO
3. This result is in agreement with the EDS results, which indicated the reduction in the Na
+ content due to the incorporation of protons from the acid solution.
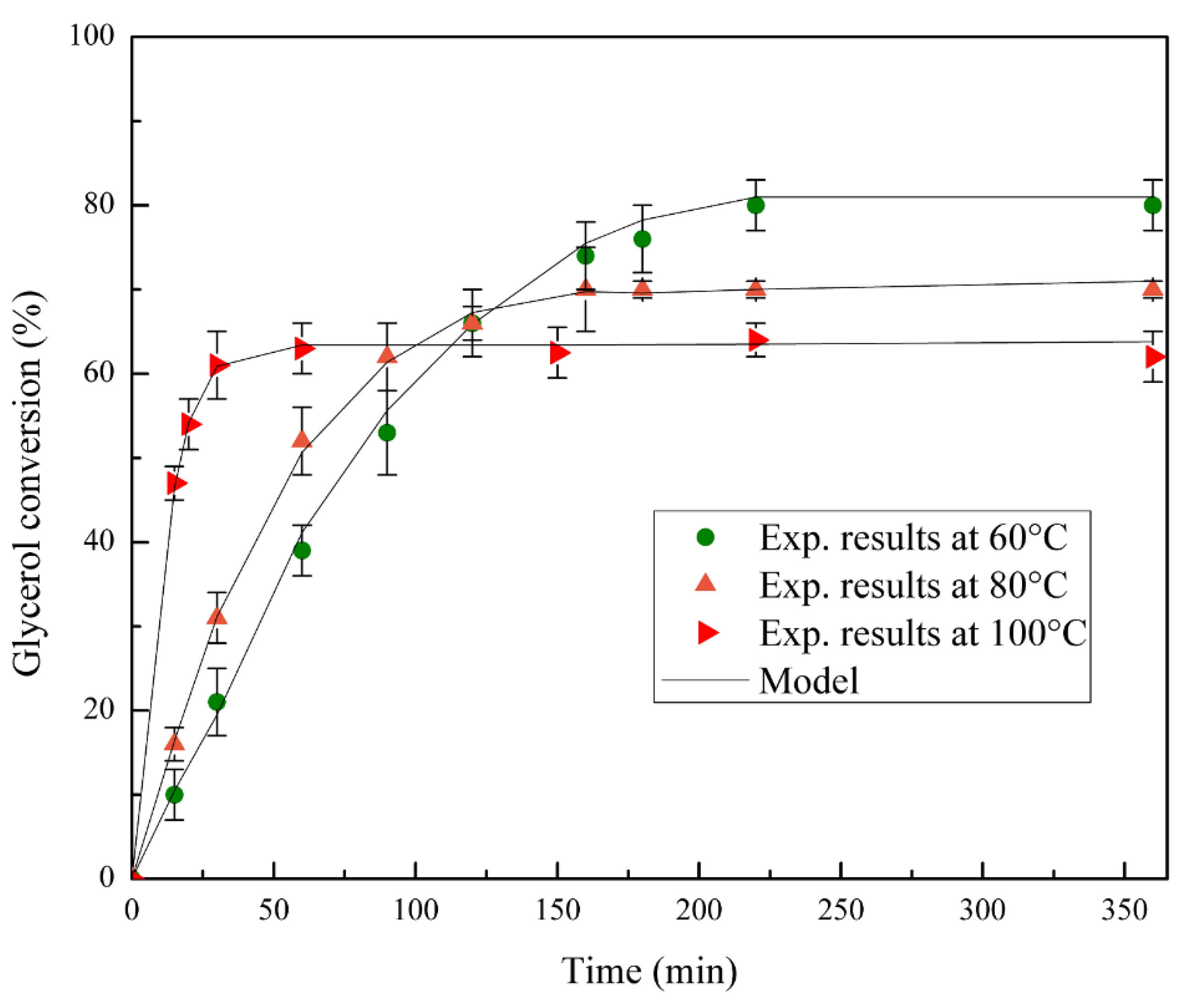
The acid site density, obtained by NH3-TPD, was about 4 mmol per gram of bentonite for solids BA and CBA, and it was corroborated by the neutralization back titration method with NaOH. This result confirmed that the acid sites were generated on the clay and not on the silica–resin structure.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}