
Substituent’s Effects of PNP Ligands in Ru(II)-Catalyzed CO2 Hydrogenation to Formate: Theoretical Analysis Considering Steric Hindrance and Promotion of Hydrogen Bonding


Abstract
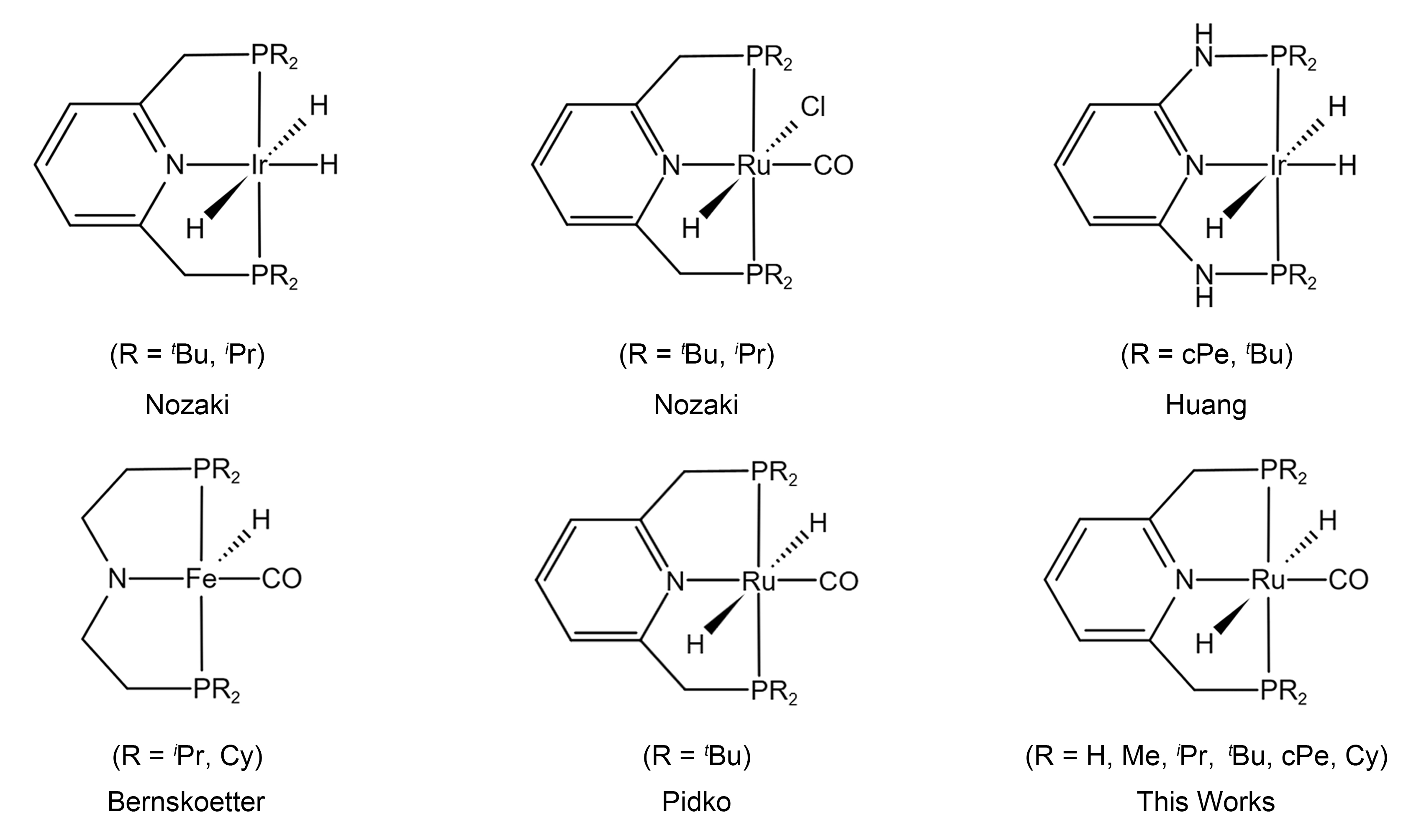
:1. Introduction
2. Results and Discussion
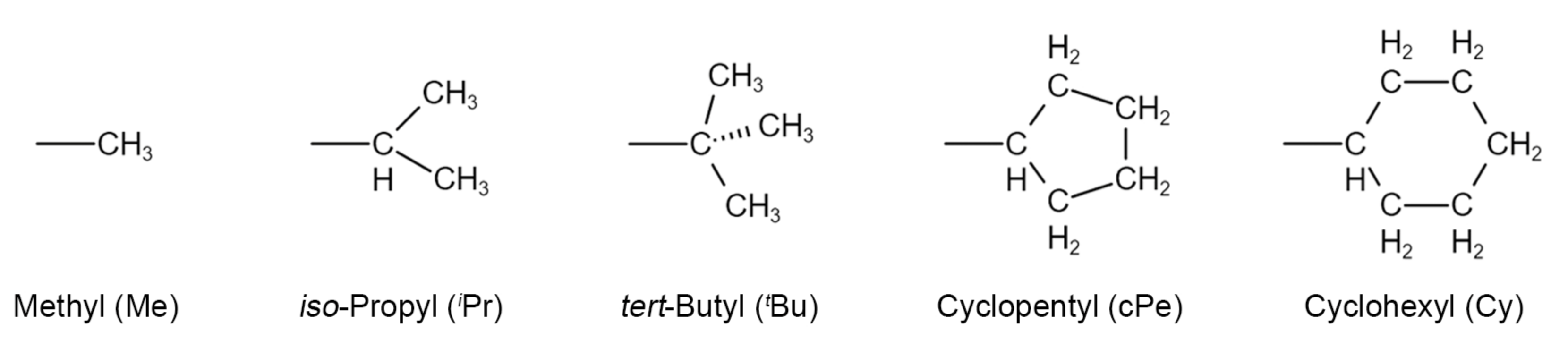
2.1. Substituent’s Effects on CO2 Coordination
2.2. Substituent’s Effects on Hydride Addition to CO2
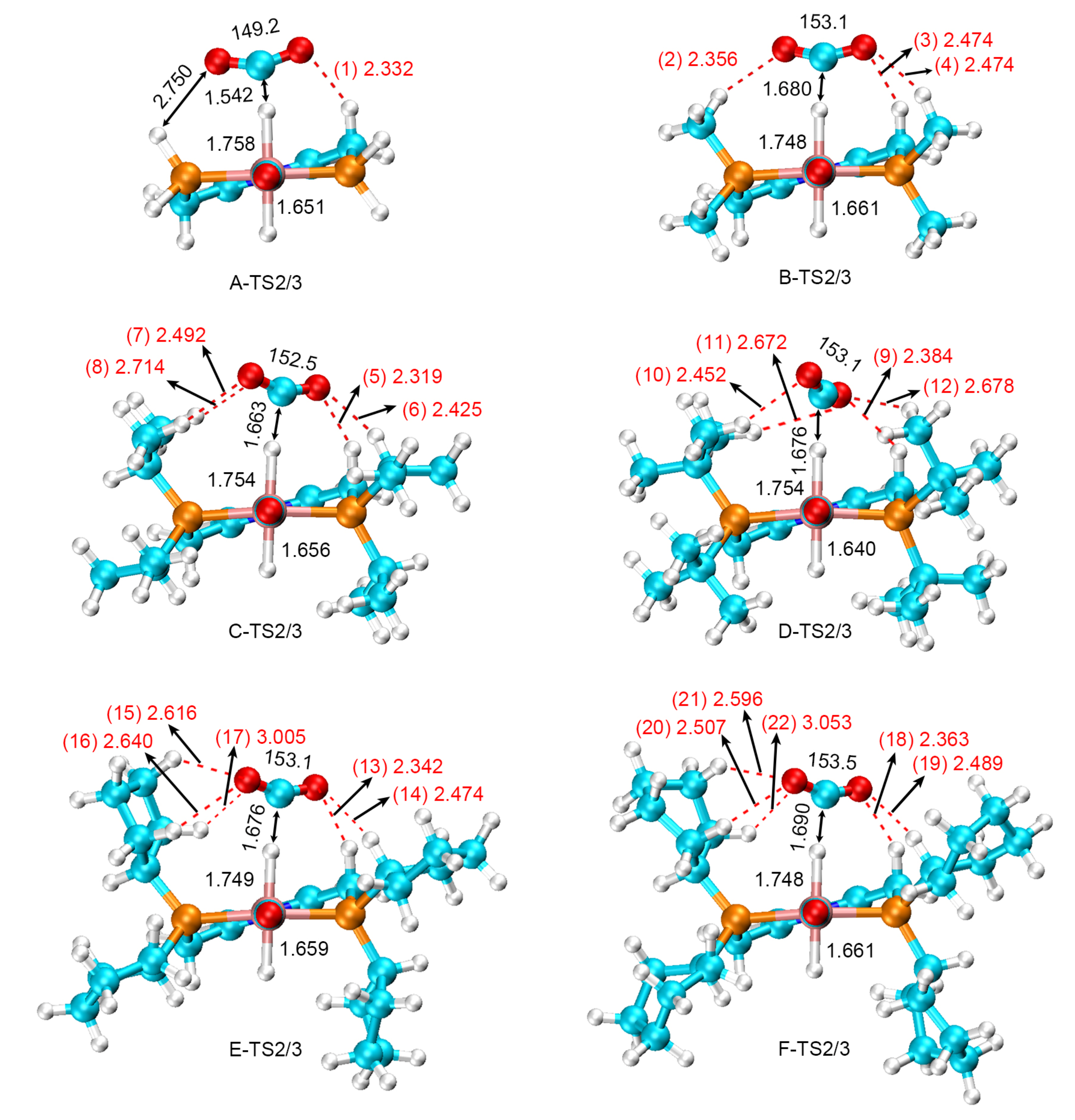
2.3. Substituent’s Effects on HCOO− Rotation
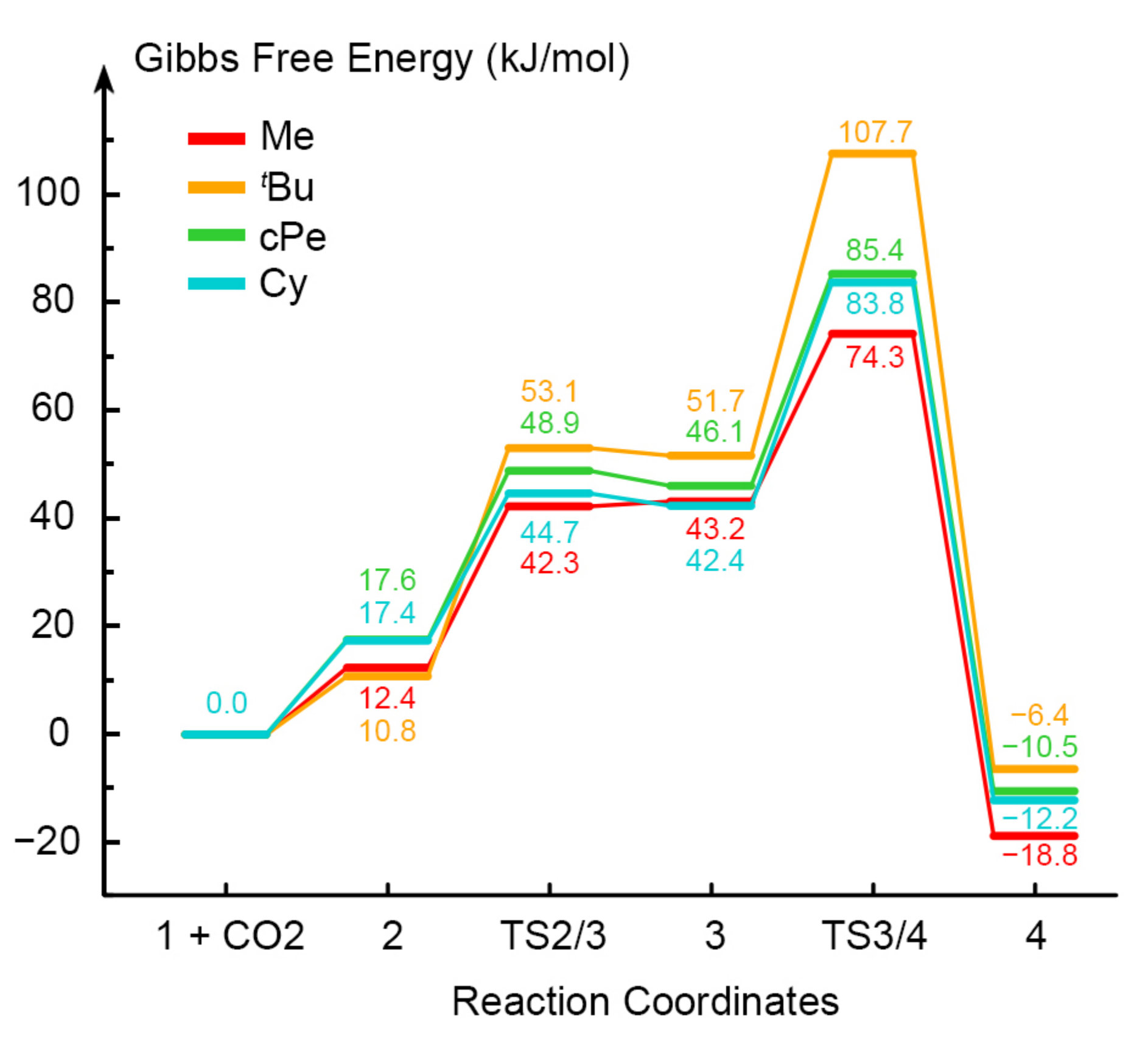
2.4. Substituent’s Effects on Total Reaction
2.5. Impact of Solvents on Substituent’s Effects
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gasser, T.; Guivarch, C.; Tachiiri, K.; Jones, C.D.; Ciais, P. Negative Emissions Physically Needed to Keep Global Warming Below 2 °C. Nat. Commun. 2015, 6, 7958–7964. [Google Scholar] [CrossRef] [PubMed]
- Sakakura, T.; Choi, J.; Yasuda, H. Transformation of Carbon Dioxide. Chem. Rev. 2007, 107, 2365–2387. [Google Scholar] [CrossRef] [PubMed]
- Aresta, M.; Dibenedetto, A.; Angelini, A. Catalysis for the Valorization of Exhaust Carbon: From CO2 to Chemicals, Materials, and Fuels. Technological Use of CO2. Chem. Rev. 2014, 114, 1709–1742. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, M.; Zhang, J.; Tu, J.; Ge, J.; Jiao, S. Green and Sustainable Molten Salt Electrochemistry for the Conversion of Secondary Carbon Pollutants to Advanced Carbon Materials. J. Mater. Chem. A 2021, 9, 14119–14146. [Google Scholar] [CrossRef]
- Moret, S.; Dyson, P.J.; Laurenczy, G. Direct Synthesis of Formic Acid from Carbon Dioxide by Hydrogenation in Acidic Media. Nat. Commun. 2014, 5, 4017–4023. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Himeda, Y.; Muckerman, J.T.; Manbeck, G.F.; Fujita, E. CO2 Hydrogenation to Formate and Methanol as an Alternative to Photo- and Electrochemical CO2 Reduction. Chem. Rev. 2015, 115, 12936–12973. [Google Scholar] [CrossRef]
- Dong, K.; Razzaq, R.; Hu, Y.; Ding, K. Homogeneous Reduction of Carbon Dioxide with Hydrogen. Top. Curr. Chem. 2017, 375, 23–48. [Google Scholar] [CrossRef]
- Langer, R.; Diskin-Posner, Y.; Leitus, G.; Shimon, L.J.; Ben-David, Y.; Milstein, D. Low-Pressure Hydrogenation of Carbon Dioxide Catalyzed by an Iron Pincer Complex Exhibiting Noble Metal Activity. Angew. Chem. Int. Ed. 2011, 50, 9948–9952. [Google Scholar] [CrossRef]
- Huff, C.A.; Sanford, M.S. Catalytic CO2 Hydrogenation to Formate by a Ruthenium Pincer Complex. ACS Catal. 2013, 3, 2412–2416. [Google Scholar] [CrossRef]
- Filonenko, G.A.; Smykowski, D.; Szyja, B.M.; Li, G.; Szczygiel, J.; Hensen, E.J.M.; Pidko, E.A. Catalytic Hydrogenation of CO2 to Formates by a Lutidine-Derived Ru–CNC Pincer Complex: Theoretical Insight into the Unrealized Potential. ACS Catal. 2015, 5, 1145–1154. [Google Scholar] [CrossRef]
- Rawat, K.S.; Pathak, B. Aliphatic Mn–PNP Complexes for the CO2 Hydrogenation Reaction: A Base Free Mechanism. Catal. Sci. Technol. 2017, 7, 3234–3242. [Google Scholar] [CrossRef]
- Takaoka, S.; Eizawa, A.; Kusumoto, S.; Nakajima, K.; Nishibayashi, Y.; Nozaki, K. Hydrogenation of Carbon Dioxide with Organic Base by PCIIP-Ir Catalysts. Organometallics 2018, 37, 3001–3009. [Google Scholar] [CrossRef]
- Tanaka, R.; Yamashita, M.; Nozaki, K. Catalytic Hydrogenation of Carbon Dioxide Using Ir(III)-Pincer Complexes. J. Am. Chem. Soc. 2009, 131, 14168–14169. [Google Scholar] [CrossRef]
- Aoki, W.; Wattanavinin, N.; Kusumoto, S.; Nozaki, K. Development of Highly Active Ir-PNP Catalysts for Hydrogenation of Carbon Dioxide with Organic Bases. Bull. Chem. Soc. Jpn. 2016, 89, 113–124. [Google Scholar] [CrossRef]
- Pan, Y.; Guan, C.; Li, H.; Chakraborty, P.; Zhou, C.; Huang, K. CO2 Hydrogenation by Phosphorus–Nitrogen PN3P-Pincer Iridium Hydride Complexes: Elucidation of the Deactivation Pathway. Dalton Trans. 2019, 48, 12812–12816. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Y.; MacIntosh, A.D.; Wong, J.L.; Bielinski, E.A.; Williard, P.G.; Mercado, B.Q.; Hazari, N.; Bernskoetter, W.H. Iron Catalyzed CO2 Hydrogenation to Formate Enhanced by Lewis acid co-catalysts. Chem. Sci. 2015, 6, 4291–4299. [Google Scholar] [CrossRef] [Green Version]
- Filonenko, G.A.; Putten, R.; Schulpen, E.N.; Hensen, E.J.M.; Pidko, E.A. Highly Efficient Reversible Hydrogenation of Carbon Dioxide to Formates Using a Ruthenium PNP-Pincer Catalyst. ChemCatChem 2014, 6, 1526–1530. [Google Scholar] [CrossRef]
- Himeda, Y.; Miyazawa, S.; Hirose, T. Interconversion Between Formic Acid and H2/CO2 Using Rhodium and Ruthenium Catalysts for CO2 Fixation and H2 Storage. ChemSusChem 2011, 4, 487–493. [Google Scholar] [CrossRef]
- Li, Y.; He, L.; Liu, A.; Lang, X.; Yang, Z.; Yu, B.; Luan, C. In Situ Hydrogenation of Captured CO2 to Formate with Polyethyleneimine and Rh/Monophosphine System. Green Chem. 2013, 15, 2825–2829. [Google Scholar] [CrossRef]
- Rohmann, K.; Kothe, J.; Haenel, M.W.; Englert, U.; Holscher, M.; Leitner, W. Hydrogenation of CO2 to Formic Acid with a highly Active Ruthenium Acriphos Complex in DMSO and DMSO/Water. Angew. Chem.-Int. Edit. 2016, 55, 8966–8969. [Google Scholar] [CrossRef]
- Gunasekar, G.H.; Shin, J.; Jung, K.; Park, K.; Yoon, S. Design Strategy Toward Recyclable and Highly Efficient Heterogeneous Catalysts for the Hydrogenation of CO2 to Formate. ACS Catal. 2018, 8, 4346–4353. [Google Scholar] [CrossRef] [Green Version]
- Guan, C.; Pan, Y.; Ang, E.P.L.; Hu, J.; Yao, C.; Huang, M.; Li, H.; Lai, Z.; Huang, K. Conversion of CO2 from Air into Formate Using Amines and Phosphorus-Nitrogen PN3P-Ru(II) Pincer Complexes. Green Chem. 2018, 20, 4201–4205. [Google Scholar] [CrossRef]
- Filonenko, G.A.; Hensen, E.J.M.; Pidko, E.A. Mechanism of CO2 Hydrogenation to Formates by Homogeneous Ru-PNP Pincer Catalyst: From a Theoretical Description to Performance Optimization. Catal. Sci. Technol. 2014, 4, 3474–3485. [Google Scholar] [CrossRef] [Green Version]
- Ohnishi, Y.; Matsunaga, T.; Nakao, Y.; Sato, H.; Sakaki, S. Ruthenium(II)-Catalyzed Hydrogenation of Carbon Dioxide to Formic Acid. Theoretical Study of Real Catalyst, Ligand Effects, and Solvation Effects. J. Am. Chem. Soc. 2005, 127, 4021–4032. [Google Scholar] [CrossRef]
- Ogo, S.; Kabe, R.; Hayashi, H.; Harada, R.; Fukuzumi, S. Mechanistic Investigation of CO2 Hydrogenation by Ru(II) and Ir(III) Aqua Complexes under Acidic Conditions: Two Catalytic Systems Differing in the Nature of the Rate Determining Step. Dalton Trans. 2006, 39, 4657–4663. [Google Scholar] [CrossRef]
- Li, J.; Yoshizawa, K. Catalytic Hydrogenation of Carbon Dioxide with a Highly Active Hydride on Ir(III)-Pincer Complex: Mechanism for CO2 Insertion and Nature of Metal-Hydride Bond. Bull. Chem. Soc. Jpn. 2011, 84, 1039–1048. [Google Scholar] [CrossRef]
- Li, J.; Liu, S.; Lu, X. Theoretical Study of the Mechanism for Direct Addition of Hydride to CO2 on Ruthenium Complexes: Nature of Ru-H Bond and Effect of Hydrogen Bonding. Bull. Chem. Soc. Jpn. 2016, 89, 905–910. [Google Scholar] [CrossRef]
- Fellr, M.; Gellrich, U.; Anaby, A.; Diskin-Posner, Y.; Milstein, D. Reductive Cleavage of CO2 by Metal–Ligand-Cooperation Mediated by an Iridium Pincer Complex. J. Am. Chem. Soc. 2016, 138, 6445–6454. [Google Scholar] [CrossRef] [PubMed]
- Filonenko, G.A.; Conley, M.P.; Coperet, C.; Lutz, M.; Hensen, E.J.M.; Pidko, E.A. The Impact of Metal–Ligand Cooperation in Hydrogenation of Carbon Dioxide Catalyzed by Ruthenium PNP Pincer. ACS Catal. 2013, 3, 2522–2526. [Google Scholar] [CrossRef]
- Liu, T.; Liu, Z.; Tang, L.; Li, J.; Yang, Z. Trans Influence of Boryl Ligands in CO2 Hydrogenation on Ruthenium Complexes: Theoretical Prediction of Highly Active Catalysts for CO2 Reduction. Catalysts 2021, 11, 1356. [Google Scholar] [CrossRef]
- Praveen, C.S.; Comas-Vives, A.; Coperet, C.; VandeVondel, J. Role of Water, CO2, and Noninnocent Ligands in the CO2 Hydrogenation to Formate by an Ir(III) PNP Pincer Catalyst Evaluated by Static-DFT and Ab Initio Molecular Dynamics under Reaction Conditions. Organometallics 2017, 36, 4908–4919. [Google Scholar] [CrossRef]
- Musashi, Y.; Sakaki, S. Theoretical Study of Rhodium(III)-Catalyzed Hydrogenation of Carbon Dioxide into Formic Acid. Significant Differences in Reactivity Among Rhodium(III), Rhodium(I), and Ruthenium(II) Complexes. J. Am. Chem. Soc. 2002, 124, 7588–7603. [Google Scholar] [CrossRef] [PubMed]
- Yang, X. Hydrogenation of Carbon Dioxide Catalyzed by PNP Pincer Iridium, Iron, and Cobalt Complexes: A Computational Design of Base Metal Catalysts. ACS Catal. 2011, 1, 849–854. [Google Scholar] [CrossRef]
- Tanaka, R.; Yamashita, M.; Chung, L.W.; Morokuma, K.; Nozaki, K. Mechanistic Studies on the Reversible Hydrogenation of Carbon Dioxide Catalyzed by an Ir-PNP Complex. Organometallics 2011, 30, 6742–6750. [Google Scholar] [CrossRef]
- Osadchuk, I.; Tamm, T.; Ahlquist, M.S.G. Theoretical Investigation of a Parallel Catalytic Cycle in CO2 Hydrogenation by (PNP)IrH3. Organometallics 2015, 34, 4932–4940. [Google Scholar] [CrossRef]
- Sawatlon, B.; Wodrich, M.D.; Corminboeuf, C. Unraveling Metal/Pincer Ligand Effects in the Catalytic Hydrogenation of Carbon Dioxide to Formate. Organometallics 2018, 37, 4568–4575. [Google Scholar] [CrossRef]
- Ahlquist, M.S.G. Iridium Catalyzed Hydrogenation of CO2 under Basic Conditions—Mechanistic Insight from Theory. J. Mol. Catal. A-Chem. 2010, 324, 3–8. [Google Scholar] [CrossRef]
- Schmeier, T.J.; Dobereiner, G.E.; Crabtree, R.H.; Hazari, N. Secondary Coordination Sphere Interactions Facilitate the Insertion Step in an Iridium(III) CO2 Reduction Catalyst. J. Am. Chem. Soc. 2011, 133, 9274–9277. [Google Scholar] [CrossRef]
- Zhang, P.; Ni, S.; Dang, L. Steric and Electronic Effects of Bidentate Phosphine Ligands on Ruthenium(II)-Catalyzed Hydrogenation of Carbon Dioxide. Chem. Asian J. 2016, 11, 2528–2536. [Google Scholar] [CrossRef]
- Ono, T.; Qu, S.; Gimbert-Surinñach, C.; Johnson, M.A.; Marell, D.J.; Benet-Buchholz, J.; Cramer, C.J.; Llobet, A. Hydrogenative Carbon Dioxide Reduction Catalyzed by Mononuclear Ruthenium Polypyridyl Complexes: Discerning between Electronic and Steric Effects. ACS Catal. 2017, 7, 5932–5940. [Google Scholar] [CrossRef] [Green Version]
- Matsubara, T. Promotion Effect of the Protonated Amine Arm of a Ruthenium Complex on Hydrido Migration to CO2: A Density Functional Study. Organometallics 2001, 20, 19–24. [Google Scholar] [CrossRef]
- Ohnishi, Y.; Nakao, Y.; Sato, H.; Sakaki, S. Ruthenium(II)-Catalyzed Hydrogenation of Carbon Dioxide to Formic Acid. Theoretical Study of Significant Acceleration by Water Molecules. Organometallics 2006, 25, 3352–3363. [Google Scholar] [CrossRef]
- Chapovetsky, A.; Welborn, M.; Luna, J.M.; Haiges, R.; Miller III, T.F.; Marinescu, S.C. Pendant Hydrogen-Bond Donors in Cobalt Catalysts Independently Enhance CO2 Reduction. ACS Cent. Sci. 2018, 4, 397–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, J.; Xie, R.; Xie, Z.; Chai, G.; Liu, T.; Chen, R.; Huang, Y.; Cao, R. Highly Selective CO2 Electroreduction to CH4 by in Situ Generated Cu2O Single-Type Sites on a Conductive MOF: Stabilizing Key Intermediates with Hydrogen Bonding. Angew. Chem. Int. Ed. 2020, 59, 23641–23648. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Sang, R.; Sponholz, P.; Beller, M. Reversible Hydrogenation of Carbon Dioxide to Formic Acid using a Mn-Pincer Complex in the Presence of Lysine. Nat. Energy 2022, 7, 438–447. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Dolg, M. Effective Core Potentials. In Modern Methods and Algorithms of Quantum Chemistry; Grotendorst, J., Ed.; John von Neumann Institute for Computing: Jülich, Germany, 2000; Volume 1, pp. 479–508. [Google Scholar]
- Martin, J.M.L.; Sundermann, A. Correlation Consistent Valence Basis Sets for Use with the Stuttgart-Dresden-Bonn Relativistic Effective Core Potentials: The Atoms Ga-Kr and In-Xe. J. Chem. Phys. 2001, 114, 3408–3420. [Google Scholar] [CrossRef] [Green Version]
- Dunning, T.H. Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic Interaction of a Solute with a Continuum. A Direct Utilizaion of AB initio Molecular Potentials for the Prevision of Solvent Effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Ab initio Study of Solvated Molecules: A New Implementation of the Polarizable Continuum Model. Chem. Phys. Lett. 1996, 255, 327–335. [Google Scholar] [CrossRef]
- Mennucci, B.; Tomasi, J. Continuum Solvation Models: A New Approach to the Problem of Solute’s Charge Distribution and Cavity Boundaries. J. Chem. Phys. 1997, 106, 5151–5158. [Google Scholar] [CrossRef]
- Cancès, E.; Mennucci, B.; Tomasi, J. A New Integral Equation Formalism for the Polarizable Continuum Model: Theoretical Background and Applications to Isotropic and Anisotropic Dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Molec. Graphics 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Falivene, L.; Cao, Z.; Petta, A.; Serra, L.; Poater, A.; Oliva, R.; Scarano, V.; Cavallo, L. Towards the online Computer-aided Design of Catalytic Pockets. Nat. Chem. 2019, 11, 872–879. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex Code | Substituent Type | Binding Energy (kJ/mol) | Binding Free Energy (kJ/mol) |
|---|---|---|---|
| A | Hydrogen (H) | −11.2 | 14.2 |
| B | Methyl (Me) | −13.0 | 12.4 |
| C | iso-Propyl (iPr) | −12.5 | 17.1 |
| D | tert-Butyl (tBu) | −10.0 | 10.8 |
| E | Cyclopentyl (cPe) | −12.6 | 17.6 |
| F | Cyclohexyl (Cy) | −14.9 | 17.4 |
| PR2 Group | %Vbur for Sphere Radius at 3.5 Å |
|---|---|
| PH2 | 17.7 |
| PMe2 | 20.1 |
| P(iPr)2 | 24.5 |
| P(tBu)2 | 27.5 |
| P(cPe)2 | 24.2 |
| P(Cy)2 | 24.4 |
| Serial Number | Distance of O···H | Angel of O···H−C | Serial Number | Distance of O···H | Angel of O···H−C |
|---|---|---|---|---|---|
| (1) | 2.566 | 146.8 | (11) | 3.029 | 119.3 |
| (2) | 2.612 | 155.8 | (12) | 3.048 | 112.3 |
| (3) | 2.841 | 142.2 | (13) | 2.644 | 153.1 |
| (4) | 2.849 | 145.6 | (14) | 2.670 | 157.8 |
| (5) | 2.611 | 165.3 | (15) | 2.736 | 132.4 |
| (6) | 2.645 | 157.0 | (16) | 2.504 | 161.6 |
| (7) | 2.924 | 139.8 | (17) | 2.553 | 161.5 |
| (8) | 2.983 | 135.0 | (18) | 2.803 | 151.6 |
| (9) | 2.670 | 160.9 | (19) | 3.009 | 123.7 |
| (10) | 2.687 | 152.5 |
| Serial Number | Distance of O···H | Angel of O···H−C | Serial Number | Distance of O···H | Angel of O···H−C |
|---|---|---|---|---|---|
| (1) | 2.332 | 141.3 | (12) | 2.678 | 140.0 |
| (2) | 2.356 | 147.2 | (13) | 2.342 | 147.9 |
| (3) | 2.474 | 141.9 | (14) | 2.474 | 148.9 |
| (4) | 2.474 | 143.1 | (15) | 2.616 | 137.4 |
| (5) | 2.319 | 145.0 | (16) | 2.640 | 126.5 |
| (6) | 2.425 | 153.5 | (17) | 3.005 | 133.7 |
| (7) | 2.492 | 149.8 | (18) | 2.363 | 146.3 |
| (8) | 2.714 | 141.2 | (19) | 2.489 | 148.5 |
| (9) | 2.384 | 134.9 | (20) | 2.507 | 155.1 |
| (10) | 2.452 | 151.2 | (21) | 2.596 | 140.5 |
| (11) | 2.672 | 162.1 | (22) | 3.053 | 113.0 |
| Serial Number | Distance of O···H | Angel of O···H−C | Serial Number | Distance of O···H | Angel of O···H−C |
|---|---|---|---|---|---|
| (1) | 2.092 | 136.1 | (10) | 2.161 | 152.5 |
| (2) | 2.167 | 140.5 | (11) | 2.189 | 172.1 |
| (3) | 2.207 | 140.6 | (12) | 2.113 | 146.1 |
| (4) | 2.222 | 138.8 | (13) | 2.169 | 147.4 |
| (5) | 2.136 | 151.6 | (14) | 2.494 | 144.1 |
| (6) | 2.161 | 142.6 | (15) | 2.753 | 130.8 |
| (7) | 2.236 | 142.8 | (16) | 2.105 | 146.5 |
| (8) | 2.437 | 137.7 | (17) | 2.173 | 146.8 |
| (9) | 2.054 | 152.9 | (18) | 2.300 | 163.1 |
| Complex Code | Substituent Type | Total Barrier (kJ/mol) | ||
|---|---|---|---|---|
| In Gas Phase | In Aqueous Phase | In THF Solution | ||
| A | H | 90.0 | 74.6 | 78.3 |
| B | Me | 74.3 | 64.0 | 65.8 |
| C | iPr | 80.2 | 61.9 | 66.5 |
| D | tBu | 107.7 | 67.6 | 76.0 |
| E | cPe | 85.4 | 61.2 | 61.8 |
| F | Cy | 83.8 | 60.8 | 64.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, X.; Li, J.; Yang, Z. Substituent’s Effects of PNP Ligands in Ru(II)-Catalyzed CO2 Hydrogenation to Formate: Theoretical Analysis Considering Steric Hindrance and Promotion of Hydrogen Bonding. Catalysts 2022, 12, 760. https://doi.org/10.3390/catal12070760
Feng X, Li J, Yang Z. Substituent’s Effects of PNP Ligands in Ru(II)-Catalyzed CO2 Hydrogenation to Formate: Theoretical Analysis Considering Steric Hindrance and Promotion of Hydrogen Bonding. Catalysts. 2022; 12(7):760. https://doi.org/10.3390/catal12070760
Chicago/Turabian StyleFeng, Xiangyang, Jun Li, and Zhuhong Yang. 2022. "Substituent’s Effects of PNP Ligands in Ru(II)-Catalyzed CO2 Hydrogenation to Formate: Theoretical Analysis Considering Steric Hindrance and Promotion of Hydrogen Bonding" Catalysts 12, no. 7: 760. https://doi.org/10.3390/catal12070760
APA StyleFeng, X., Li, J., & Yang, Z. (2022). Substituent’s Effects of PNP Ligands in Ru(II)-Catalyzed CO2 Hydrogenation to Formate: Theoretical Analysis Considering Steric Hindrance and Promotion of Hydrogen Bonding. Catalysts, 12(7), 760. https://doi.org/10.3390/catal12070760