
5-Hydroxymethylfurfural Oxidation to 2,5-Furandicarboxylic Acid on Noble Metal-Free Nanocrystalline Mixed Oxide Catalysts

 , ,
, , 
 ,
,  ,
,  ,
, 
 ,
,
Abstract
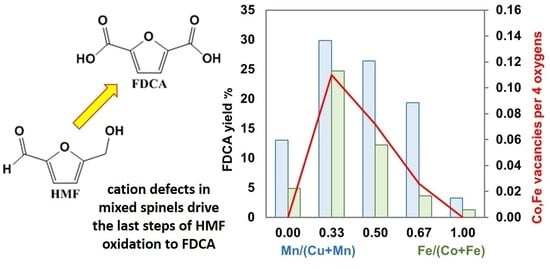
:
1. Introduction
2. Results
2.1. Characterisation of Catalysts
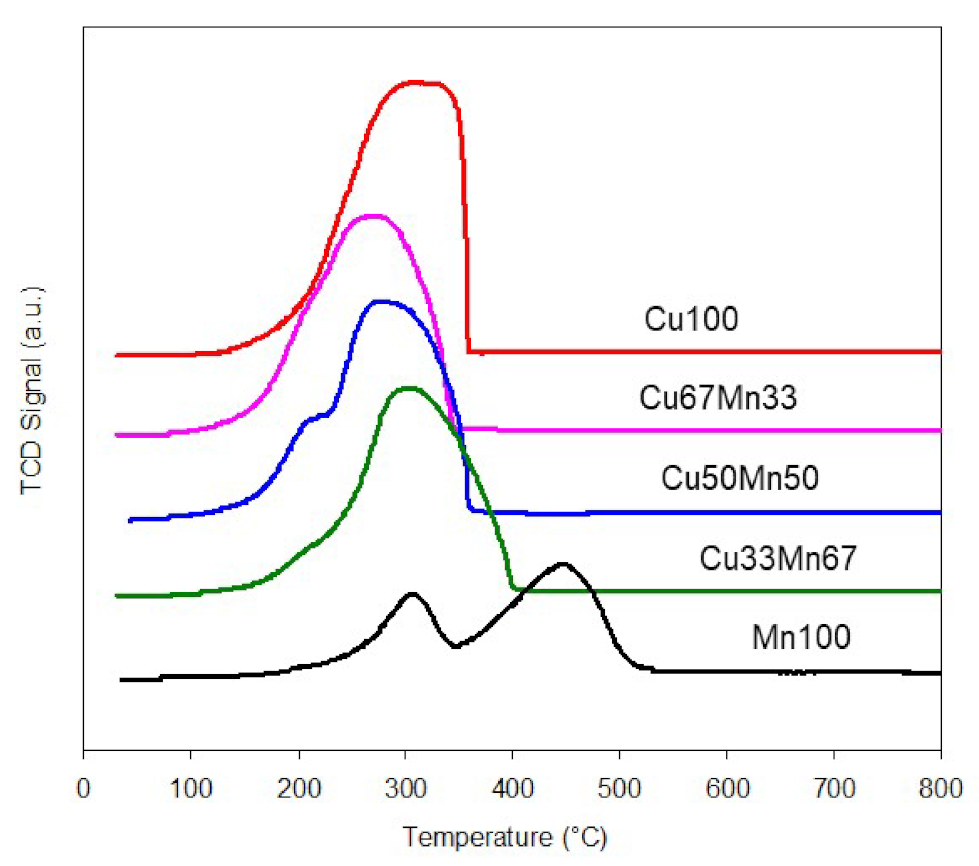
2.1.1. Copper-Manganese Oxides
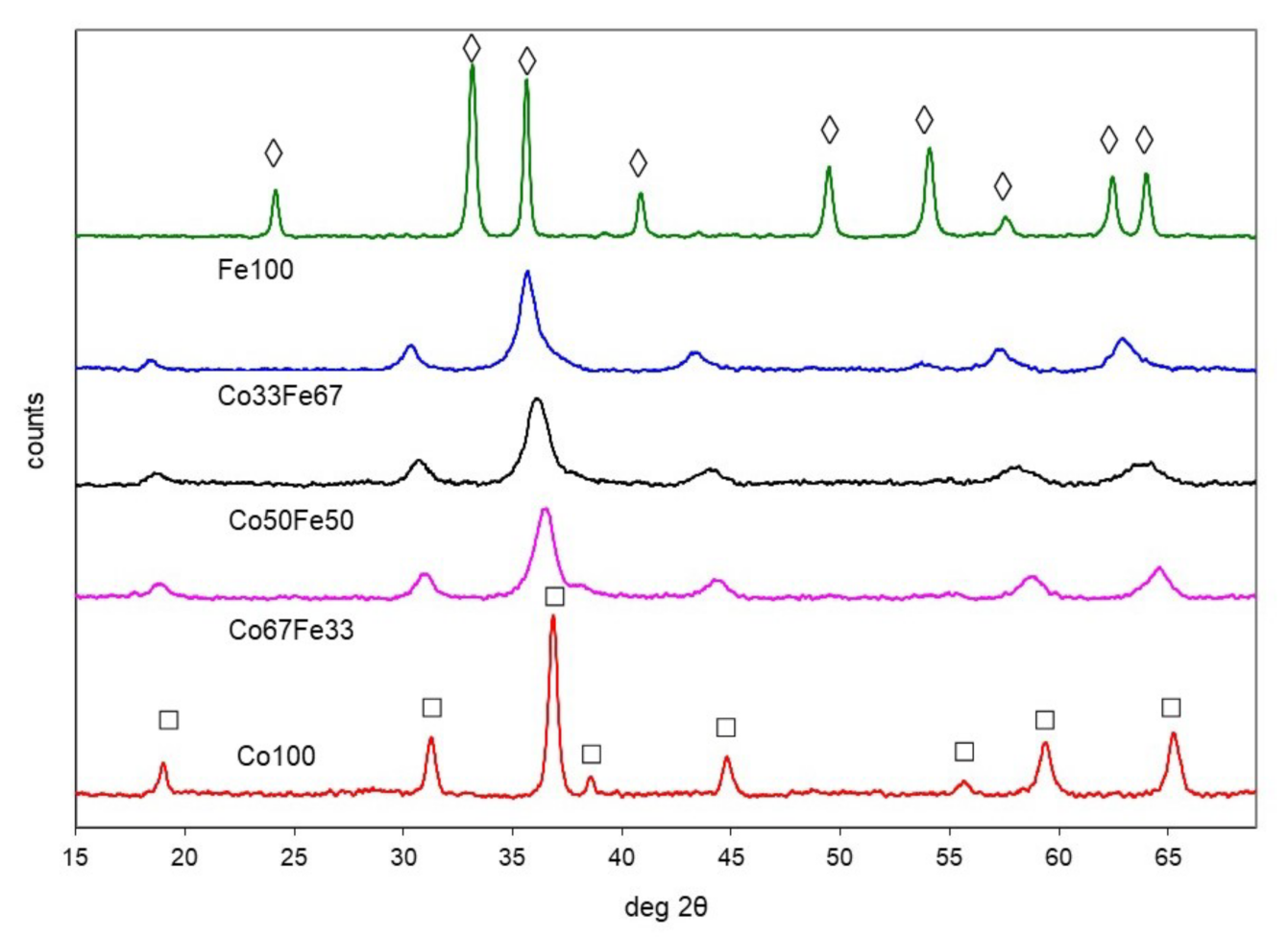
2.1.2. Cobalt-Iron Oxides
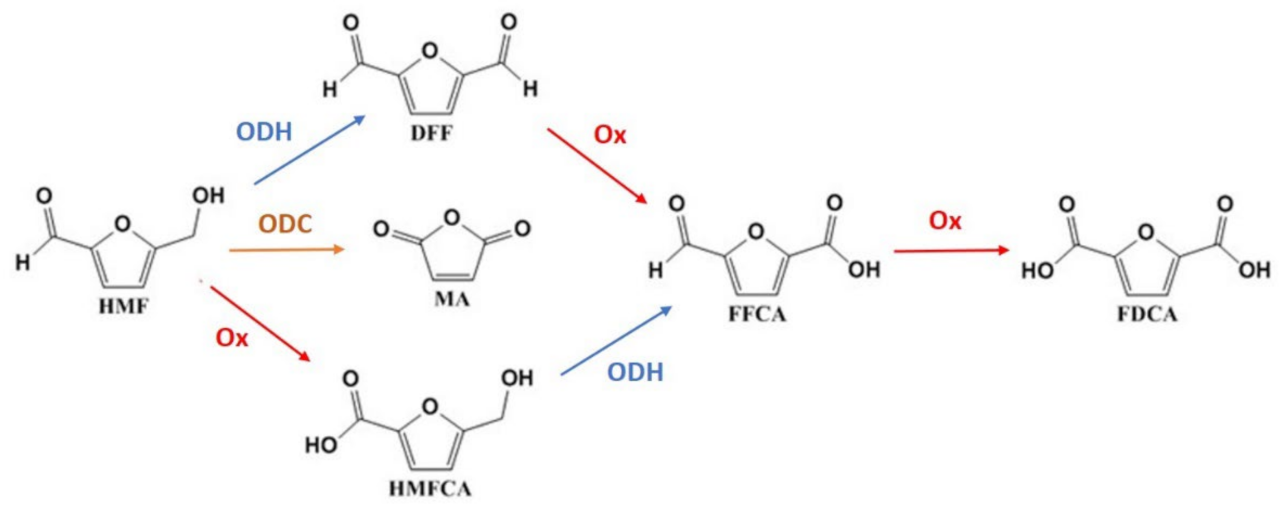
2.2. HMF Oxidation
2.2.1. Blank Tests in the Absence of Catalyst
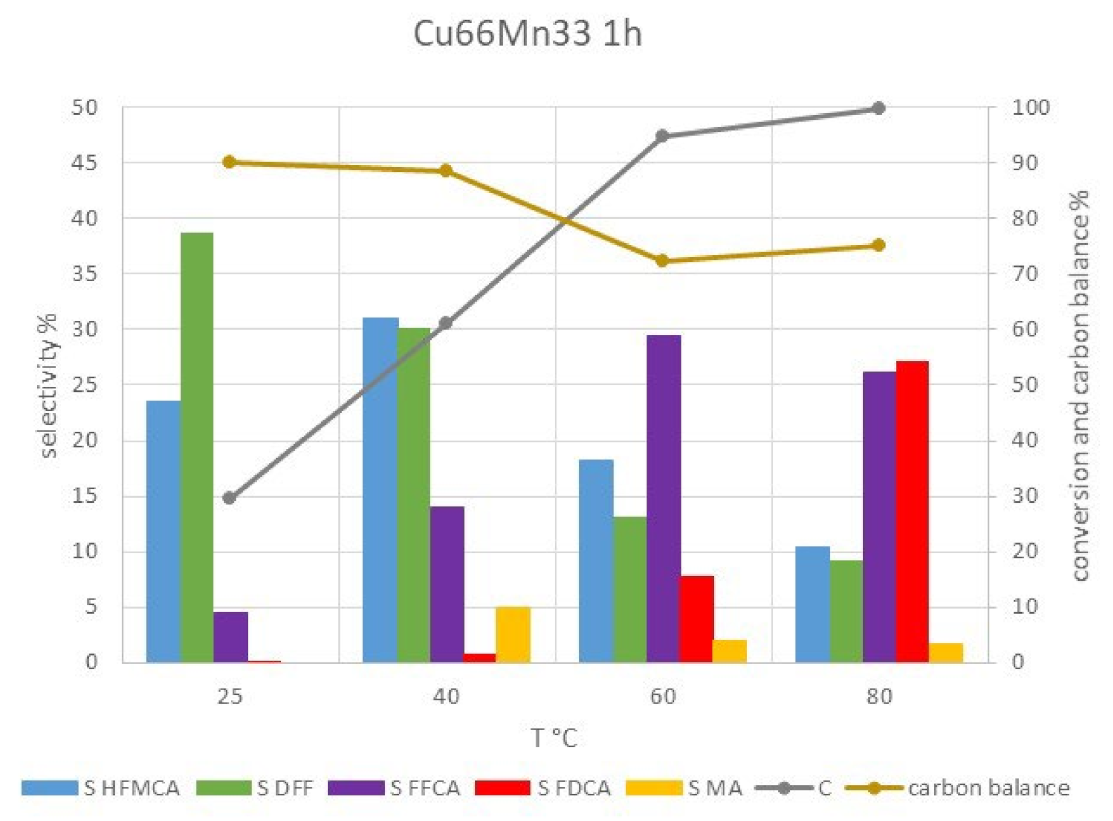
2.2.2. Catalytic Tests on Copper-Manganese Oxides
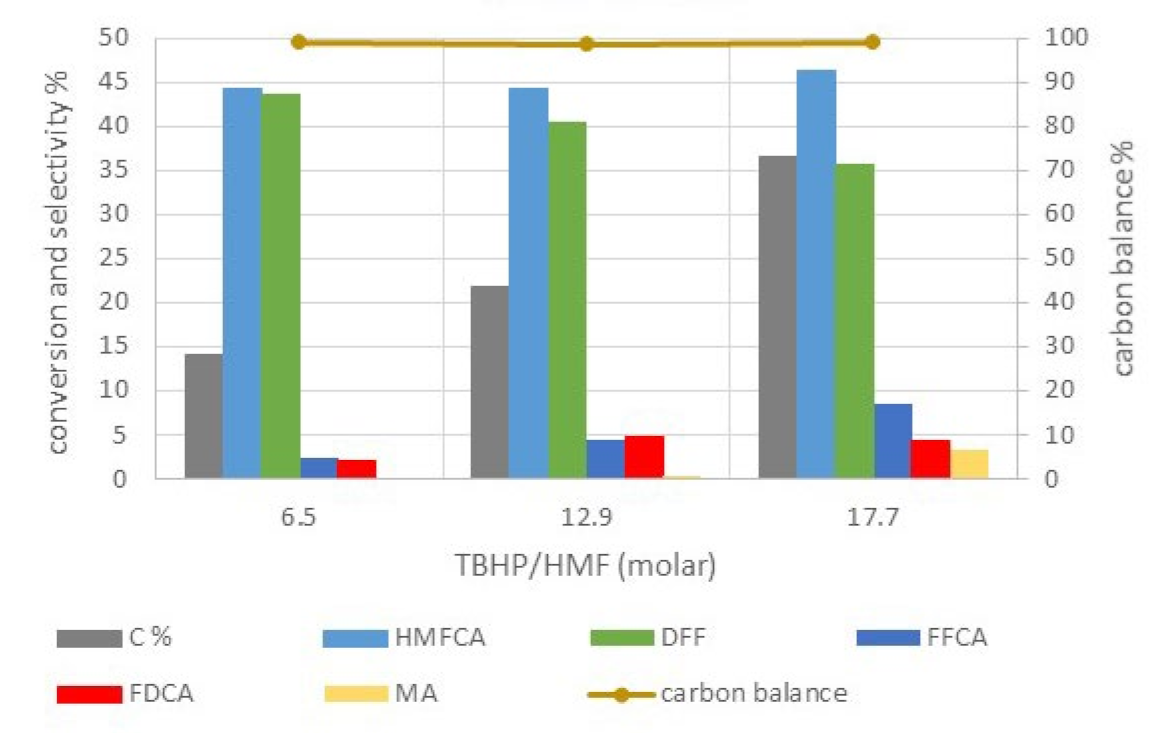
2.2.3. Catalytic Tests on Cobalt-Iron Oxides
3. Discussion
4. Materials and Methods
4.1. Synthesis of Catalysts
4.2. Characterisation Methods
4.3. Catalytic Tests
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bozell, J.J.; Petersen, G.R. Technology development for the production of biobased products from biorefinery carbohydrates-the US Department of Energy’s “Top 10” revisited. Green Chem. 2010, 12, 539–554. [Google Scholar] [CrossRef]
- Eerhart, A.J.J.E.; Faaij, A.P.C.; Patel, M.K. Replacing fossil based PET with biobased PEF; process analysis, energy and GHG balance. Energy Environ. Sci. 2012, 5, 6407–6422. [Google Scholar] [CrossRef]
- Service, R.F. Sugary Recipe Boosts Grow-Your-Own Plastics. Science 2006, 312, 1861. [Google Scholar] [CrossRef]
- Roman-Leshkov, Y.; Barrett, C.J.; Liu, Z.Y.; Dumesic, J.A. Production of dimethylfuran for liquid fuels from biomass-derived carbohydrates. Nature 2007, 447, 982–986. [Google Scholar] [CrossRef]
- van Putten, R.J.; van der Waal, J.C.; de Jong, E.; Rasdendra, C.B.; Heeres, H.J.; de Vries, J.G. Hydroxymethylfurfural, A Versatile Platform Chemical Made from Renewable Resources. Chem. Rev. 2013, 113, 1499–1597. [Google Scholar] [CrossRef]
- Gandini, A.; Lacerda, T.M.; Carvalho, A.J.F.; Trovatti, E. Progress of Polymers from Renewable Resources: Furans, Vegetable Oils, and Polysaccharides. Chem. Rev. 2016, 116, 1637–1669. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Romain, C.; Williams, C.K. Sustainable polymers from renewable resources. Nature 2016, 540, 354–362. [Google Scholar] [CrossRef]
- Huang, Y.T.; Wong, J.J.; Huang, C.J.; Li, C.L.; Jang, G.W.B. 2,5-Furandicarboxylic Acid Synthesis and Use. In Chemicals and Fuels from Bio-Based Building Blocks; Cavani, F., Albonetti, S., Basile, F., Gandini, A., Eds.; Wiley-VCH: Weinheim, Germany, 2016; pp. 191–215. [Google Scholar]
- Du, Z.; Ma, J.; Wang, F.; Liu, J.; Xu, J. Oxidation of 5-hydroxymethylfurfural to maleic anhydride with molecular oxygen. Green Chem. 2011, 13, 554–557. [Google Scholar] [CrossRef]
- Patil, S.K.R.; Lund, C.R.F. Formation and Growth of Humins via Aldol Addition and Condensation during Acid-Catalyzed Conversion of 5-Hydroxymethylfurfural. Energy Fuels 2011, 25, 4745–4755. [Google Scholar] [CrossRef]
- Tsilomelekis, G.; Orella, M.J.; Lin, Z.; Cheng, Z.; Zheng, W.; Nikolakis, V.; Vlachos, D.G. Molecular structure, morphology and growth mechanisms and rates of 5-hydroxymethyl furfural (HMF) derived humins. Green Chem. 2016, 18, 1983–1993. [Google Scholar] [CrossRef] [Green Version]
- Casanova, O.; Iborra, S.; Corma, A. Biomass into Chemicals: Aerobic Oxidation of 5-Hydroxymethyl-2-furfural into 2,5-Furandicarboxylic Acid with Gold Nanoparticle Catalysts. ChemSusChem 2009, 2, 1138–1144. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Zhou, C.; Chen, J.; Deng, W.; Zhang, Q.; Yang, Y.; Wang, Y. Base-Free Aerobic Oxidation of 5-Hydroxymethyl-furfural to 2,5-Furandicarboxylic Acid in Water Catalyzed by Functionalized Carbon Nanotube-Supported Au−Pd Alloy Nanoparticles. ACS Catal. 2014, 4, 2175–2185. [Google Scholar] [CrossRef]
- Wojcieszak, R.; Santarelli, F.; Paul, S.; Dumeignil, F.; Cavani, F.; Gonçalves, R.V. Recent Developments in maleic acid synthesis from bio-based chemicals. Sustain. Chem. Process. 2015, 3, 9. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Su, K.; Li, Z.; Cheng, B. Selective oxidation of 5-hydroxymethylfurfural with H2O2 catalyzed by a molybdenum complex. Green Chem. 2016, 18, 2122–2128. [Google Scholar] [CrossRef]
- Davis, S.E.; Houk, L.R.; Tamargo, E.R.; Datye, A.K.; Davis, R.J. Oxidation of 5-hydroxymethylfurfural over supported Pt, Pd and Au catalysts. Catal. Today 2011, 160, 55–60. [Google Scholar] [CrossRef]
- Xu, C.; Paone, E.; Rodriguez-Padron, D.; Luque, R.; Mauriello, F. Recent catalytic routes for the preparation and the upgrading of biomass derived furfural and 5-hydroxymethylfurfural. Chem. Soc. Rev. 2020, 49, 4273–4306. [Google Scholar] [CrossRef]
- Zhang, Z.; Deng, K. Recent Advances in the Catalytic Synthesis of 2,5-Furandicarboxylic Acid and Its Derivatives. ACS Catal. 2015, 5, 6529–6544. [Google Scholar] [CrossRef]
- Zhao, D.; Su, T.; Wang, Y.; Varma, R.S.; Len, C. Recent advances in catalytic oxidation of 5-hydroxymethylfurfural. Mol. Catal. 2020, 495, 111133. [Google Scholar] [CrossRef]
- Hameed, S.; Lin, L.; Wang, A.; Luo, W. Recent Developments in Metal-Based Catalysts for the Catalytic Aerobic Oxidation of 5-Hydroxymethyl-Furfural to 2,5-Furandicarboxylic Acid. Catalysts 2020, 10, 120. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Li, C.; Guo, Y.; Liu, X.; Zhang, Y.; Wang, Y. N-doped carbon supported Pt catalyst for base-free oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid. Appl. Catal. A Gen. 2016, 526, 1–8. [Google Scholar] [CrossRef]
- Zhang, Y.; Xue, Z.; Wang, J.; Zhao, X.; Deng, Y.; Zhao, W.; Mu, T. Controlled deposition of Pt nanoparticles on Fe3O4@carbon microspheres for efficient oxidation of 5-hydroxymethylfurfural. RSC Adv. 2016, 6, 51229–51237. [Google Scholar] [CrossRef]
- Han, X.; Geng, L.; Guo, Y.; Jia, R.; Liu, X.; Zhang, Y.; Wang, Y. Base-free aerobic oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid over a Pt/C–O–Mg catalyst. Green Chem. 2016, 18, 1597–1604. [Google Scholar] [CrossRef]
- Zhou, C.; Deng, W.; Wan, X.; Zhang, Q.; Yang, Y.; Wang, Y. Functionalized Carbon Nanotubes for Biomass Conversion: The Base-Free Aerobic Oxidation of 5-Hydroxymethyl-furfural to 2,5-Furandicarboxylic Acid over Platinum Supported on a Carbon Nanotube Catalyst. ChemCatChem 2015, 7, 2853–2863. [Google Scholar] [CrossRef]
- Ait Rass, H.; Essayem, N.; Besson, M. Selective Aerobic Oxidation of 5-HMF into 2,5-Furandicarboxylic Acid with Pt Catalysts Supported on TiO2- and ZrO2-Based Supports. ChemSusChem 2015, 8, 1206–1217. [Google Scholar] [CrossRef]
- Miao, Z.; Wu, T.; Li, J.; Yi, T.; Zhang, Y.; Yang, X. Aerobic oxidation of 5-hydroxymethylfurfural (HMF) effectively catalyzed by a Ce0.8Bi0.2O2-δ supported Pt catalyst at room temperature. RSC Adv. 2015, 5, 19823–19829. [Google Scholar] [CrossRef]
- Siankevich, S.; Savoglidis, G.; Fei, Z.; Laurenczy, G.; Alexander, D.T.L.; Yan, N.; Dyson, P.J. A novel platinum nanocatalyst for the oxidation of 5-Hydroxymethylfurfural into 2,5-Furandicarboxylic acid under mild conditions. J. Catal. 2014, 315, 67–74. [Google Scholar] [CrossRef]
- Ait Rass, H.; Essayem, N.; Besson, M. Selective aqueous phase oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid over Pt/C catalysts: Influence of the base and effect of bismuth promotion. Green Chem. 2013, 15, 2240–2251. [Google Scholar] [CrossRef]
- Lolli, A.; Maslova, V.; Bonincontro, D.; Basile, F.; Ortelli, S.; Albonetti, S. Selective Oxidation of HMF via Catalytic and Photocatalytic Processes Using Metal-Supported Catalysts. Molecules 2018, 23, 2792. [Google Scholar] [CrossRef] [Green Version]
- Qu, H.; Deng, J.; Wang, B.; Ouyang, L.; Tang, Y.; Yu, K.; Lou, L.-L.; Liu, S. Thermoresponsive block copolymer supported Pt nanocatalysts for base-free aerobic oxidation of 5-hydroxymethyl-2-furfural. Front. Chem. Sci. Eng. 2021, 15, 1514–1523. [Google Scholar] [CrossRef]
- Cheng, X.; Li, S.; Liu, S.; Xin, Y.; Yang, J.; Chen, B.; Liu, H. Highly efficient catalytic oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid using bimetallic Pt–Cu alloy nanoparticles as Catalysts. Chem. Commun. 2022, 58, 1183–1186. [Google Scholar] [CrossRef]
- Gui, Z.; Cao, W.; Saravanamurugan, S.; Riisager, A.; Chen, L.; Qi, Z. Efficient Aerobic Oxidation of 5-Hydroxymethylfurfural in Aqueous Media with Au–Pd Supported on Zinc Hydroxycarbonate. ChemCatChem 2016, 8, 3636–3643. [Google Scholar] [CrossRef]
- Gao, Z.; Xie, R.; Fan, G.; Yang, L.; Li, F. Highly Efficient and Stable Bimetallic AuPd over La-Doped Ca−Mg−Al Layered Double Hydroxide for Base-Free Aerobic Oxidation of 5-Hydroxymethylfurfural in Water. ACS Sustain. Chem. Eng. 2017, 5, 5852–5861. [Google Scholar] [CrossRef]
- Gupta, K.; Rai, R.K.; Dwivedi, A.D.; Singh, S.K. Catalytic Aerial Oxidation of Biomass-Derived Furans to Furan Carboxylic Acids in Water over Bimetallic Nickel–Palladium Alloy Nanoparticles. ChemCatChem 2017, 9, 2760–2767. [Google Scholar] [CrossRef]
- Choudhary, H.; Ebitani, K. Hydrotalcite-supported PdPt-catalyzed Aerobic Oxidation of 5-Hydroxymethylfurfural to 2,5-Furandicarboxylic Acid in Water. Chem. Lett. 2016, 45, 613–615. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, K.; Lei, D.; Si, W.; Feng, Y.; Lou, L.-L.; Liu, L. Basicity-Tuned Hydrotalcite-Supported Pd Catalysts for Aerobic Oxidation of 5-Hydroxymethyl-2-furfural under Mild Conditions. ACS Sustain. Chem. Eng. 2016, 4, 4752–4761. [Google Scholar] [CrossRef]
- Liu, B.; Ren, Y.; Zhang, Z. Aerobic oxidation of 5-hydroxymethylfurfural into 2,5-furandicarboxylic acid in water under mild conditions. Green Chem. 2015, 17, 1610–1617. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhen, J.; Liu, B.; Lv, K.; Deng, K. Selective aerobic oxidation of the biomass-derived precursor 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid under mild conditions over a magnetic palladium nanocatalyst. Green Chem. 2015, 17, 1308–1317. [Google Scholar] [CrossRef]
- Lolli, A.; Albonetti, S.; Utili, L.; Amadori, R.; Ospitali, F.; Lucarelli, C.; Cavani, F. Insights into the reaction mechanism for 5-hydroxymethylfurfural oxidation to FDCA on bimetallic Pd–Au nanoparticles. Appl. Catal. A Gen. 2015, 504, 408–419. [Google Scholar] [CrossRef]
- Siyo, B.; Schneider, M.; Radnik, J.; Pohl, M.-M.; Langer, P.; Steinfeldt, N. Influence of support on the aerobic oxidation of HMF into FDCA over preformed Pd nanoparticle based materials. Appl. Catal. A Gen. 2014, 478, 107–116. [Google Scholar] [CrossRef]
- Gupta, N.K.; Nishimura, S.; Takagaki, A.; Ebitani, K. Hydrotalcite-supported gold-nanoparticle-catalyzed highly efficient base-free aqueous oxidation of 5-hydroxymethylfurfural into 2,5-furandicarboxylic acid under atmospheric oxygen pressure. Green Chem. 2011, 13, 824–827. [Google Scholar] [CrossRef]
- Davis, S.E.; Zope, B.N.; Davis, R.J. On the mechanism of selective oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid over supported Pt and Au catalysts. Green Chem. 2012, 14, 143–147. [Google Scholar] [CrossRef]
- Albonetti, S.; Pasini, T.; Lolli, A.; Blosi, M.; Piccinini, M.; Dimitratos, N.; Lopez-Sanchez, J.A.; Morgan, D.J.; Carley, A.F.; Hutchings, G.J.; et al. Selective oxidation of 5-hydroxymethyl-2-furfural over TiO2–supported gold–copper catalysts prepared from preformed nanoparticles: Effect of Au/Cu ratio. Catal. Today 2012, 195, 120–126. [Google Scholar] [CrossRef]
- Villa, A.; Schiavoni, M.; Campisi, S.; Veith, G.M.; Prati, L. Pd-modified Au on Carbon as an Effective and Durable Catalyst for the Direct Oxidation of HMF to 2,5-Furandicarboxylic Acid. ChemSusChem 2013, 6, 609–612. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Ma, H.; Zhang, J.; Song, Q.; Du, Z.; Huang, Y.; Xu, J. Gold Nanoclusters Confined in a Supercage of Y Zeolite for Aerobic Oxidation of HMF under Mild Conditions. Chem. Eur. J. 2013, 19, 14215–14223. [Google Scholar] [CrossRef]
- Ardemani, L.; Cibin, G.; Dent, A.J.; Isaacs, M.A.; Kyriakou, G.; Lee, A.F.; Parlett, C.M.A.; Parry, S.A.; Wilson, K. Solid base catalysed 5-HMF oxidation to 2,5-FDCA over Au/hydrotalcites: Fact or fiction? Chem. Sci. 2015, 6, 4940–4945. [Google Scholar] [CrossRef] [Green Version]
- Bonincontro, D.; Lolli, A.; Villa, A.; Prati, L.; Dimitratos, N.; Veith, G.M.; Chinchilla, L.E.; Botton, G.A.; Cavani, F.; Albonetti, S. AuPd-nNiO as an effective catalyst for the base-free oxidation of HMF under mild reaction conditions. Green Chem. 2019, 21, 4090–4099. [Google Scholar] [CrossRef]
- Uzunidis, G.; Schade, O.; Schild, D.; Grunwaldt, J.-D.; Behrens, S. Design of bimetallic Au/Cu nanoparticles in ionic liquids: Synthesis and catalytic properties in 5-(hydroxymethyl)furfural oxidation. ChemNanoMat 2021, 7, 1108–1116. [Google Scholar] [CrossRef]
- Gorbanev, Y.Y.; Kegnæs, S.; Riisager, A. Selective Aerobic Oxidation of 5-Hydroxymethylfurfural in Water Over Solid Ruthenium Hydroxide Catalysts with Magnesium-Based Supports. Catal. Lett. 2011, 141, 1752–1760. [Google Scholar] [CrossRef]
- Yi, G.; Teong, S.P.; Zhang, Y. Base-free conversion of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid over a Ru/C catalyst. Green Chem. 2016, 18, 979–983. [Google Scholar] [CrossRef]
- Mishra, D.K.; Lee, H.J.; Kim, J.; Lee, H.-S.; Cho, J.K.; Suh, T.-W.; Yi, Y.; Kim, Y.J. MnCo2O4 spinel supported ruthenium catalyst for air-oxidation of HMF to FDCA under aqueous phase and base-free conditions. Green Chem. 2017, 19, 1619–1623. [Google Scholar] [CrossRef]
- Yang, Z.; Qi, W.; Su, R.; He, Z. Selective Synthesis of 2,5-Diformylfuran and 2,5-Furandicarboxylic Acid from 5-Hydroxymethylfurfural and Fructose Catalyzed by Magnetically Separable Catalysts. Energy Fuels 2017, 31, 533–541. [Google Scholar] [CrossRef]
- Zheng, L.; Zhao, J.; Du, Z.; Zong, B.; Liu, H. Efficient aerobic oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid on Ru/C catalysts. Sci. China Chem. 2017, 60, 950–957. [Google Scholar] [CrossRef]
- Zhao, D.; Rodriguez-Padron, D.; Triantafyllidis, K.S.; Wang, Y.; Luque, R.; Len, C. Microwave-Assisted Oxidation of Hydroxymethyl Furfural to Added-Value Compounds over a Ruthenium-Based Catalyst. ACS Sustain. Chem. Eng. 2020, 8, 3091–3102. [Google Scholar] [CrossRef]
- Kandasamy, P.; Gogoi, P.; Venugopalan, A.T.; Raja, T. A highly efficient and reusable Ru-NaY catalyst for the base free oxidation of 5-Hydroxymethylfurfural to 2,5-Furandicarboxylic acid. Catal. Today 2021, 375, 145–154. [Google Scholar] [CrossRef]
- Su, T.; Liu, Q.; Lü, H.; Alasmary, F.A.; Zhao, D.; Len, C. Selective oxidation of 5-hydroxymethylfurfural to 5-hydroxymethyl-2-furancarboxylic acid using silver oxide supported on calcium carbonate. Mol. Catal. 2021, 502, 111374. [Google Scholar] [CrossRef]
- Zhao, D.; Rodriguez-Padron, D.; Luque, R.; Len, C. Insights into the Selective Oxidation of 5-Hydroxymethylfurfural to 5-Hydroxymethyl-2-furancarboxylic Acid Using Silver Oxide. ACS Sustain. Chem. Eng. 2020, 8, 8486–8495. [Google Scholar] [CrossRef]
- Celaya, C.A.; Oukhrib, R.; Ait El Had, M.; Abdellaoui, Y.; Abou Oualid, H.; Bourzi, H.; Chahboun, R.; Zhao, D.; Osman, S.M.; Parnar, V.S.; et al. Density functional theory study of the selective oxidation of 5-Hydroxymethylfurfural (HMF) to 5-Hydroxymethyl-2-furancarboxylic acid (HMFCA) on the Silver oxide surface (001). Mol. Catal. 2022, 519, 112117. [Google Scholar] [CrossRef]
- Zope, B.N.; Davis, S.E.; Davis, R.J. Influence of Reaction Conditions on Diacid Formation During Au-Catalyzed Oxidation of Glycerol and Hydroxymethylfurfural. Top. Catal. 2012, 55, 24–32. [Google Scholar] [CrossRef]
- Pasini, T.; Piccinini, M.; Blosi, M.; Bonelli, R.; Albonetti, S.; Dimitratos, N.; Lopez-Sancheze, Q.; Kiely, C.J.; Hutchings, G.J.; Cavani, F. Selective oxidation of 5-hydroxymethyl-2-furfural using supported gold–copper nanoparticles. Green Chem. 2011, 13, 2091–2099. [Google Scholar] [CrossRef]
- Vuyyuru, R.R.; Strasser, P. Oxidation of biomass derived 5-hydroxymethylfurfural using heterogeneous and electrochemical catalysis. Catal. Today 2012, 195, 144–154. [Google Scholar] [CrossRef]
- Albonetti, S.; Lolli, A.; Morandi, V.; Migliori, A.; Lucarelli, C.; Cavani, F. Conversion of 5-hydroxymethylfurfural to 2,5-furandicarboxylicacid over Au-based catalysts: Optimization of active phase and metal–support interaction. Appl. Catal. B Environ. 2015, 163, 520–530. [Google Scholar] [CrossRef]
- Subbiah, S.; Simeonov, S.P.; Esperança, J.M.S.S.; Rebelo, L.P.N.; Afonso, C.A.M. Direct transformation of 5-hydroxymethylfurfural to the building blocks 2,5-dihydroxymethylfurfural (DHMF) and 5-hydroxymethyl furanoic acid (HMFA) via Cannizzaro reaction. Green Chem. 2013, 15, 2849–2853. [Google Scholar] [CrossRef]
- Kar, S.; Zhou, Q.-Q.; Ben-David, Y.; Milstein, D. Catalytic Furfural/5-Hydroxymethyl Furfural Oxidation to Furoic Acid/Furan-2,5-dicarboxylic Acid with H2 Production Using Alkaline Water as the Formal Oxidant. J. Am. Chem. Soc. 2022, 144, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.K.R.; Heltzel, J.; Lund, C.R.F. Comparison of Structural Features of Humins Formed Catalytically from Glucose, Fructose, and 5-Hydroxymethylfurfuraldehyde. Energy Fuels 2012, 26, 5281–5293. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, L. A facile and effective method for preparation of 2.5-furandicarboxylic acid via hydrogen peroxide direct oxidation of 5-hydroxymethylfurfural. Pol. J. Chem. Technol. 2017, 19, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Partenheimer, W.; Grushin, V.V. Synthesis of 2,5-Diformylfuran and Furan-2,5-Dicarboxylic Acid by Catalytic Air-Oxidation of 5-Hydroxymethylfurfural. Unexpectedly Selective Aerobic Oxidation of Benzyl Alcohol to Benzaldehyde with Metal/Bromide Catalysts. Adv. Synth. Catal. 2001, 343, 102–111. [Google Scholar] [CrossRef]
- Zuo, X.; Venkitasubramanian, P.; Busch, D.H.; Subramaniam, B. Optimization of Co/Mn/Br-Catalyzed Oxidation of 5-Hydroxymethylfurfural to Enhance 2,5-Furandicarboxylic Acid Yield and Minimize Substrate Burning. ACS Sustain. Chem. Eng. 2016, 4, 3659–3668. [Google Scholar] [CrossRef]
- Zuo, X.; Chaudhari, A.S.; Snavely, K.; Niu, F.; Zhu, H.; Martin, K.J.; Subramaniam, B. Kinetics of Homogeneous 5-Hydroxymethylfurfural Oxidation to 2,5-Furandicarboxylic Acid with Co/Mn/Br Catalyst. AIChE J. 2017, 63, 162–171. [Google Scholar] [CrossRef]
- Chen, S.; Guo, X.; Ban, H.; Pan, T.; Zheng, L.; Cheng, Y.; Wang, L.; Li, X. Reaction Mechanism and Kinetics of the Liquid-Phase Oxidation of 5-Hydroxymethylfurfural to 2,5-Furandicarboxylic Acid. Ind. Eng. Chem. Res. 2021, 60, 16887–16898. [Google Scholar] [CrossRef]
- Han, X.; Li, C.; Liu, X.; Xia, Q.; Wang, Y. Selective oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid over MnOx–CeO2 composite catalysts. Green Chem. 2017, 19, 996–1004. [Google Scholar] [CrossRef]
- Fu, M.; Yang, W.; Yang, C.; Zhang, Y.; Shen, C. Mechanistic insights into CoOx–Ag/CeO2 catalysts for the aerobic oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid. Catal. Sci. Technol. 2022, 12, 116–123. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, Z.; Liu, B. Catalytic Conversion of Fructose and 5-Hydroxymethylfurfural into 2,5-Furandicarboxylic Acid over a Recyclable Fe3O4−CoOx Magnetite Nanocatalyst. ACS Sustain. Chem. Eng. 2015, 3, 406–412. [Google Scholar] [CrossRef]
- Jain, A.; Jonnalagadda, S.C.; Ramanujachary, K.V.; Mugweru, A. Selective oxidation of 5-hydroxymethyl-2-furfural to furan-2,5-dicarboxylic acid over spinel mixed metal oxide catalyst. Catal. Commun. 2015, 58, 179–182. [Google Scholar] [CrossRef]
- Gawade, A.B.; Nakhate, A.V.; Yadav, G.D. Selective synthesis of 2, 5-furandicarboxylic acid by oxidation of 5-hydroxymethylfurfural over MnFe2O4 catalyst. Catal. Today 2018, 309, 119–125. [Google Scholar] [CrossRef]
- Wang, F.; Lai, J.; Liu, Z.; Wen, S.; Liu, X. Copper-manganese oxide for highly selective oxidation of 5-hydroxymethylfurfural to bio-monomer 2, 5-furandicarboxylic acid. Biomass Convers. Biorefin. 2022; in press. [Google Scholar] [CrossRef]
- Gao, L.; Deng, K.; Zheng, J.; Liu, B.; Zhang, Z. Efficient oxidation of biomass derived 5-hydroxymethylfurfural into 2,5-furandicarboxylic acid catalyzed by Merrifield resin supported cobalt porphyrin. Chem. Eng. J. 2015, 270, 444–449. [Google Scholar] [CrossRef]
- Vandarkuzhali, S.A.A.; Karthikeyan, G.; Pachamuthu, M.P. Efficient oxidation of 5-Hydroxymethylfurfural to 2,5-furandicarboxylic acid over FeNPs@NH2-SBA-15 catalyst in water. Mol. Catal. 2021, 516, 111951. [Google Scholar] [CrossRef]
- Saha, B.; Gupta, D.; Abu-Omar, M.M.; Modak, A.; Bhaumik, A. Porphyrin-based porous organic polymer-supported iron(III) catalyst for efficient aerobic oxidation of 5-hydroxymethyl-furfural into 2,5-furandicarboxylic acid. J. Catal. 2013, 299, 316–320. [Google Scholar] [CrossRef]
- Yang, C.; Li, X.; Zhang, Z.; Lv, B.; Li, J.; Liu, Z.; Zhu, W.; Tao, F.; Lv, G.; Yang, Y. High efficient catalytic oxidation of 5-hydroxymethylfurfural into 2,5-furandicarboxylic acid under benign conditions with nitrogen-doped graphene encapsulated Cu nanoparticles. J. Energy Chem. 2010, 50, 96–105. [Google Scholar] [CrossRef]
- Cheng, F.; Guo, D.; Lai, J.; Long, M.; Zhao, W.; Liu, X.; Yin, D. Efficient base-free oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid over copper-doped manganese oxide nanorods with tert-butanol as solvent. Front. Chem. Sci. Eng. 2021, 15, 960–968. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, B.; Lv, K.; Sun, J.; Deng, K. Aerobic oxidation of biomass derived 5-hydroxymethylfurfural into 5-hydroxymethyl-2-furancarboxylic acid catalyzed by a montmorillonite K-10 clay immobilized molybdenum acetylacetonate complex. Green Chem. 2014, 16, 2762–2770. [Google Scholar] [CrossRef]
- Neatu, F.; Marina, R.S.; Florea, M.; Petrea, N.; Pavel, O.D.; Parvulescu, V.I. Selective oxidation of 5-hydroxymethyl furfural over non-precious metal heterogeneous catalysts. Appl. Catal. B Environ. 2016, 180, 751–757. [Google Scholar] [CrossRef]
- Hayashi, E.; Komanoya, T.; Kamata, K.; Hara, M. Heterogeneously-Catalyzed Aerobic Oxidation of 5-Hydroxymethylfurfural to 2,5-Furandicarboxylic Acid with MnO2. ChemSusChem 2017, 10, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, G.; Lewkowski, J.; Skowronski, R. The electrochemical oxidation of 5-hydroxymethylfurfural with the nickel oxide/hydroxide electrode. Electrochim. Acta 1991, 36, 1995. [Google Scholar] [CrossRef]
- Zhang, J.; Yu, P.; Zeng, G.; Bao, F.; Yuan, Y.; Huang, H. Boosting HMF oxidation performance via decorating ultrathin nickel hydroxide nanosheets with amorphous copper hydroxide islands. J. Mater. Chem. A 2021, 9, 9685–9691. [Google Scholar] [CrossRef]
- Su, T.; Zhao, D.; Wang, Y.; Lu, H.; Varma, R.S.; Len, C. Innovative Protocols in the Catalytic Oxidation of 5-Hydroxymethylfurfural. ChemSusChem 2021, 14, 266–280. [Google Scholar] [CrossRef]
- Wei, P.; Bieringer, M.; Cranswick, L.M.D.; Petric, A. In situ high-temperature X-ray and neutron diffraction of Cu–Mn oxide phases. J. Mat. Sci. 2010, 45, 1056–1064. [Google Scholar] [CrossRef] [Green Version]
- Vandenberghe, R.E.; Robbrechr, G.G.; Brabers, V.A.M. Structure and ionic configuration of oxidic copper-manganese spinels (CuxMn3-xO4). Phys. Stat. Sol. A 1976, 34, 583–592. [Google Scholar] [CrossRef]
- Martin, B.E.; Petric, A. Electrical properties of copper–manganese spinel solutions and their cation valence and cation distribution. J. Phys. Chem. Solids 2007, 68, 2262–2270. [Google Scholar] [CrossRef]
- Maunders, C.; Martin, B.E.; Wei, P.; Petric, A.; Botton, G.A. Investigation of the electronic structure of the cubic spinel Cu1.2Mn1.8O4 using electron energy loss spectroscopy. Solid State Ion. 2008, 179, 718–724. [Google Scholar] [CrossRef]
- Broemme, A.D.D.; Brabers, V.A.M. Preparation and Properties of Copper and Manganese Containing Mixed Oxide. Solid State Ion. 1985, 16, 171–178. [Google Scholar] [CrossRef]
- Lavina, B.; Salviulo, G.; Della Giusta, A. Cation distribution and structure modelling of spinel solid solutions. Phys. Chem. Miner. 2002, 29, 10–18. [Google Scholar] [CrossRef]
- Vozniuk, O.; Bazzo, C.; Albonetti, S.; Tanchoux, N.; Bosselet, F.; Millet, J.-M.; Di Renzo, F.; Cavani, F. Structural Changes of Binary/Ternary Spinel Oxides During Ethanol Anaerobic Decomposition. ChemCatChem 2017, 9, 2219–2230. [Google Scholar] [CrossRef]
- Cornell, R.M.; Schwertmann, U. The Iron Oxides. Structure, Properties, Reactions, Occurrences and Uses; Wiley-VCH: Weinheim, Germany, 2003; pp. 158–159. [Google Scholar]
- Cedeño-Mattei, Y.; Perales-Pérez, O.; Uwakweh, O.N.C. Synthesis of high-coercivity non-stoichiometric cobalt ferrite nanocrystals: Structural and magnetic characterization. Mat. Chem. Phys. 2012, 132, 999–1006. [Google Scholar] [CrossRef]
- Murray, P.J.; Linnett, J.W. Cation Distribution in the Spinels CoxFe3-xO4. J. Phys. Chem. Solids 1976, 37, 1041–1042. [Google Scholar] [CrossRef]
- Li, K.Z.; Haneda, M.; Ozawa, M. The synthesis of iron oxides with different phases or exposure crystal planes and their catalytic property for propene oxidation. Adv. Mat. Res. 2012, 463–464, 189–193. [Google Scholar] [CrossRef]
- Tiernan, M.J.; Barnes, P.A.; Parkes, G.M.B. Reduction of Iron Oxide Catalysts: The Investigation of Kinetic Parameters Using Rate Perturbation and Linear Heating Thermoanalytical Techniques. J. Phys. Chem. B 2001, 105, 220–228. [Google Scholar] [CrossRef]
- Zhu, J.; Andersson, S.L.T. Reaction Network and Kinetics for the Catalytic Oxidation of Toluene over V2O5. J. Catal. 1990, 126, 92–100. [Google Scholar] [CrossRef]
- Horvat, J.; Klaic, B.; Metelko, B.; Sunjic, V. Mechanism of levulinic acid formation. Tetrahedron Lett. 1985, 26, 2111–2114. [Google Scholar] [CrossRef]
- van Zandvoort, I.; Koers, E.J.; Weingarth, M.; Bruijnincx, P.C.A.; Baldus, M.; Weckhuysen, B.M. Structural characterization of 13C-enriched humins and alkali-treated 13C humins by 2D solid-state NMR. Green Chem. 2015, 17, 4383–4392. [Google Scholar] [CrossRef] [Green Version]
- Nichela, D.A.; Berkovic, A.M.; Costante, M.R.; Juliarena, M.P.; Einschlag, F.S.G. Nitrobenzene degradation in Fenton-like systems using Cu(II) as catalyst. Comparison between Cu(II)- and Fe(III)-based systems. Chem. Eng. J. 2013, 228, 1148–1157. [Google Scholar] [CrossRef]
- Shen, Y.; Zhang, Z.; Xiao, K. Evaluation of cobalt oxide, copper oxide and their solid solutions as heterogeneous catalysts for Fenton-degradation of dye pollutants. RSC Adv. 2015, 5, 91846–91854. [Google Scholar] [CrossRef]
- Qi, L.; Qi, X.; Wang, L.; Feng, L.; Lu, S. Decomposition of tert-butyl hydroperoxide into tert-butyl alcohol and O2 catalyzed by birnessite-type manganese oxides: Kinetics and activity. Catal. Commun. 2014, 49, 6–9. [Google Scholar] [CrossRef] [Green Version]
- Debnath, B.; Roy, A.S.; Kapri, S.; Bhattacharyya, S. Efficient Dye Degradation Catalyzed by Manganese Oxide Nanoparticles and the Role of Cation Valence. ChemistrySelect 2016, 1, 4265–4273. [Google Scholar] [CrossRef]
- Weng, Z.; Li, J.; Weng, Y.; Feng, M.; Zhuang, Z.; Yu, Y. Surfactant-free porous nano-Mn3O4 as a recyclable Fenton-like reagent that can rapidly scavenge phenolics without H2O2. J. Mater. Chem. A 2017, 5, 15650–15660. [Google Scholar] [CrossRef]
- Lin, Z.-R.; Zhao, L.; Dong, Y.-H. Effects of low molecular weight organic acids and fulvic acid on 2,4,4’-trichlorobiphenyl degradation and hydroxyl radical formation in a goethite-catalyzed Fenton-like reaction. Chem. Eng. J. 2017, 326, 201–209. [Google Scholar] [CrossRef]
- Araujo, F.V.F.; Yokoyama, L.; Teixeira, L.A.C.; Campos, J.C. Heterogeneous Fenton process using the mineral hematite for the discolouration of a reactive dye solution. Braz. J. Chem. Eng. 2011, 28, 605–616. [Google Scholar] [CrossRef]
- Busca, G. The surface acidity of solid oxides and its characterization by IR spectroscopic methods. An attempt at systematization. Phys. Chem. Chem. Phys. 1999, 1, 723–736. [Google Scholar] [CrossRef]
- Biswas, S.; Dutta, B.; Mannodi-Kanakkithodi, A.; Clarke, R.; Song, W.; Ramprasad, R.; Suib, S.L. Heterogeneous mesoporous manganese/cobalt oxide catalysts for selective oxidation of 5-hydroxymethylfurfural to 2,5-diformylfuran. Chem. Commun. 2017, 53, 11751–11754. [Google Scholar] [CrossRef]
- Hanna, K.; Kone, T.; Medjahdi, G. Synthesis of the mixed oxides of iron and quartz and their catalytic activities for the Fenton-like oxidation. Catal. Commun. 2008, 9, 955–959. [Google Scholar] [CrossRef]
- Voinov, M.A.; Sosa Pagan, J.O.; Morrison, E.; Smirnova, T.I.; Smirnov, A.I. Surface-Mediated Production of Hydroxyl Radicals as a Mechanism of Iron Oxide Nanoparticle Biotoxicity. J. Am. Chem. Soc. 2011, 133, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Yin, J.J.; Zhou, Y.T.; Zhang, Y.; Song, L.; Song, M.; Hu, S.; Gu, N. Dual Enzyme-like Activities of Iron Oxide Nanoparticles and Their Implication for Diminishing Cytotoxicity. ACS Nano 2012, 6, 4001–4012. [Google Scholar] [CrossRef] [PubMed]
- Zasada, F.; Piskorz, W.; Janas, J.; Grybos, J.; Indyka, P.; Sojka, Z. Reactive Oxygen Species on the (100) Facet of Cobalt Spinel Nanocatalyst and their Relevance in 16O2/18O2 Isotopic Exchange, deN2O, and deCH4 Processes. A Theoretical and Experimental Account. ACS Catal. 2015, 5, 6879–6892. [Google Scholar] [CrossRef]
- Zasada, F.; Janas, J.; Piskorz, W.; Sojka, Z. Surface oxygen dynamics and H2 oxidation on cobalt spinel surface probed by 18O/16O isotopic exchange and accounted for by DFT molecular modeling: Facile interfacial oxygen atoms flipping through transient peroxy intermediate. Res. Chem. Intermed. 2017, 43, 2865–2880. [Google Scholar] [CrossRef] [Green Version]
- Brazdil, J.F. Scheelite: A versatile structural template for selective alkene oxidation catalysts. Catal. Sci. Technol. 2015, 5, 3452–3458. [Google Scholar] [CrossRef]
- Massa, M.; Häggblad, R.; Andersson, A. Cation Vacant Fe3-x-yVx□yO4 Spinel-Type Catalysts for the Oxidation of Methanol to Formaldehyde. Top. Catal. 2011, 54, 685–697. [Google Scholar] [CrossRef]
- Reddy, A.S.; Gopinath, C.S.; Chilukuri, S. Selective ortho-methylation of phenol with methanol over copper manganese mixed-oxide spinel catalysts. J. Catal. 2006, 243, 278–291. [Google Scholar] [CrossRef]
- Gurrala, L.; Nagpure, A.S.; Gurav, H.R.; Chilukuri, S. Spinel-Type Mixed Oxides for Stable and Selective Partial Oxidation of Benzyl Alcohol. ChemistrySelect 2018, 3, 3751–3761. [Google Scholar] [CrossRef]
- Tirsoaga, A.; El Fergani, M.; Nuns, N.; Simon, P.; Granger, P.; Parvulescu, V.; Comana, S.M. Multifunctional nanocomposites with non-precious metals and magnetic core for 5-HMF oxidation to FDCA. Appl. Catal. A Gen. 2020, 278, 119309. [Google Scholar] [CrossRef]
- Girisuta, B.; Janssen, L.P.B.M.; Heeres, H.J. A kinetic study on the decomposition of 5-hydroxymethylfurfural into levulinic acid. Green Chem. 2006, 8, 701–709. [Google Scholar] [CrossRef] [Green Version]
- van Zandvoort, I.; Wang, Y.; Rasrendra, C.B.; van Eck, E.R.H.; Bruijnincx, P.C.A.; Heeres, H.J.; Weckhuysen, B.M. Formation, Molecular Structure, and Morphology of Humins in Biomass Conversion: Influence of Feedstock and Processing Conditions. ChemSusChem 2013, 6, 1745–1758. [Google Scholar] [CrossRef] [PubMed]
- Pipitone, G.; Zoppi, G.; Frattini, A.; Bocchini, S.; Pirone, R.; Bensaid, S. Aqueous phase reforming of sugar-based biorefinery streams: From the simplicity of model compounds to the complexity of real feeds. Catal. Today 2020, 345, 267–279. [Google Scholar] [CrossRef]
- Dong, X.; Wang, X.; Song, H.; Zhang, Y.; Yuan, A.; Guo, Z.; Wang, Q.; Yang, F. Enabling Efficient Aerobic 5-Hydroxymethylfurfural Oxidation to 2,5-Furandicarboxylic Acid in Water by Interfacial Engineering Reinforced Cu−Mn Oxides Hollow Nanofiber. ChemSusChem 2022, 15, e202200076. [Google Scholar] [CrossRef]
- Megías-Sayago, C.; Chabarova, K.; Penkova, A.; Lolli, A.; Ivanova, S.; Albonetti, S.; Cavani, F.; Odriozola, J.A. Understanding the Role of the Acid Sites in 5-Hydroxymethylfurfural Oxidation to 2,5-Furandicarboxylic Acid Reaction over Gold Catalysts: Surface Investigation on CexZr1−xO2 Compounds. ACS Catal. 2018, 8, 11154–11164. [Google Scholar] [CrossRef]
- Sreekumar, K.; Sugunan, S. Ferrospinels based on Co and Ni prepared via a low temperature route as efficient catalysts for the selective synthesis of o-cresol and 2,6-xylenol from phenol and methanol. J. Mol. Catal. A Gen. 2002, 185, 259–268. [Google Scholar] [CrossRef]
- Ballarini, N.; Cavani, F.; Passeri, S.; Pesaresi, L.; Lee, A.F.; Wilson, K. Phenol methylation over nanoparticulate CoFe2O4 inverse spinel catalysts: The effect of morphology on catalytic performance. Appl. Catal. A Gen. 2009, 366, 184–192. [Google Scholar] [CrossRef]
- O’Neill, H.S.C.; Navrotsky, A. Cation distributions and thermodynamic properties of binary spinel solid solutions. Am. Miner. 1983, 68, 181–194. [Google Scholar]
- Ferreira, T.A.S.; Waerenborgh, J.C.; Mendonça, M.H.R.M.; Nunes, M.R.; Costa, F.M. Structural and morphological characterization of FeCo2O4 and CoFe2O4 spinels prepared by a coprecipitation method. Solid State Sci. 2003, 5, 383–392. [Google Scholar] [CrossRef]
- Hill, R.J.; Craig, J.R.; Gibbs, G.V. Systematics of the Spinel Structure Type. Phys. Chem. Miner. 1979, 4, 317–339. [Google Scholar] [CrossRef]
- Ustinov, E.A.; Do, D.D.; Jaroniec, M. Equilibrium Adsorption in Cylindrical Mesopores: A Modified Broekhoff and de Boer Theory versus Density Functional Theory. J. Phys. Chem. B 2005, 109, 1947–1958. [Google Scholar] [CrossRef]
- Payne, S.M.; Kerton, F.M. Solubility of bio-sourced feedstocks in ‘green’ solvents. Green Chem. 2010, 12, 1648–1653. [Google Scholar] [CrossRef] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T | Time | Conversion | Productivity | Selectivity % | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Catalyst | Oxidant | Solvent | °C | h | HMF % | mmol g−1 h−1 | DFF | HMFCA | FFCA | FDCA | Ref. |
| Pt (4.9%)/Mg-carbon | O2 (0.5 MPa) | H2O | 110 | 3 | 86 | 7.20 | 26 | 0 | 58 | 17 | [24] |
| 17 | 100 | 1.48 | 0 | 0 | 0 | 100 | |||||
| Pt (5%)/N-carbon | O2 (1 MPa) | H2O | 110 | 4 | 60 | 3.20 | 18 | 0 | 53 | 33 | [23] |
| 12 | 99 | 1.06 | 0 | 0 | 0 | 98 | |||||
| Pt (1%)/LDH | O2 (40 mL/min) | H2O | 95 | 2 | 74 | 2.60 | 0 | 0 | 54 | 31 | [35] |
| 8 | 100 | 0.89 | 0 | 0 | 7 | 87 | |||||
| Pt0.4Cu0.6 (0.74%)/C | O2 (1 MPa) | H2O | 150 | 1 | 100 | 7.50 | 16 | 0 | 74 | 9 | [31] |
| 6 | 100 | 1.25 | 0 | 0 | 0 | 99 | |||||
| Pt0.8Pd0.2 (1%)/LDH | O2 (40 mL/min) | H2O | 95 | 2 | 87 | 3.10 | 0 | 3 | 51 | 42 | [35] |
| 8 | 100 | 0.89 | 0 | 0 | 1 | 99 | |||||
| Pd (1%)/LDH | O2 (40 mL/min) | H2O | 95 | 2 | 91 | 3.20 | 0 | 20 | 44 | 16 | [35] |
| 8 | 98 | 0.87 | 0 | 8 | 30 | 53 | |||||
| Au (1%)/NiO | O2 (1 MPa) | H2O | 90 | 6 | 70 | 0.59 | 0 | 24 | 40 | 35 | [47] |
| 20 | 74 | 0.18 | 0 | 21 | 30 | 48 | |||||
| Au (1.92%)/LDH | O2 flowing | H2O | 95 | 3 | 97 | 1.20 | 0 | 53 | 8 | 37 | [41] |
| 7 | 100 | 0.55 | 0 | 0 | 0 | 100 | |||||
| Au0.5Pd0.5 (1%)/CNT | O2 (0.5 MPa) | H2O | 90 | 2 | 68 | 2.24 | 19 | 0 | 53 | 23 | [13] |
| 12 | 100 | 0.55 | 0 | 0 | 2 | 94 | |||||
| Au0.67Pd0.33 (1%)/NiO | O2 (1 MPa) | H2O | 90 | 2 | 76 | 1.93 | 2 | 7 | 57 | 33 | [47] |
| 14 | 100 | 0.32 | 0 | 0 | 0 | 100 | |||||
| Au0.56Pd0.44 (2.75%)/La-LDH | O2 (0.5 MPa) | H2O | 120 | 0.5 | 86 | 17.20 | 3 | 45 | 23 | 27 | [33] |
| 4 | 100 | 2.50 | 0 | 0 | 0 | 100 | |||||
| Ru (5%)/C | O2 (0.2 MPa) | H2O | 120 | 1 | 86 | 4.30 | 14 | 0 | 75 | 0 | [50] |
| 10 | 100 | 0.50 | 0 | 0 | 0 | 88 | |||||
| Ru (2.4%)/LDH | O2 (0.25 MPa) | H2O | 140 | 2 | 98 | 4.30 | 11 | 56 | n.a | 28 | [49] |
| 6 | 100 | 0.50 | 0 | 1 | n.a. | 99 | |||||
| T | Time | Conversion | Productivity | Selectivity % | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Catalyst | Oxidant | Base | °C | h | HMF % | mmol g−1 h−1 | DFF | HMFCA | FFCA | FDCA | Ref. |
| Pt (20%)/C | O2 (1 MPa) | NaOH/ HMF 20 | 50 | 1 | 99 | 10.20 | 0 | 31 | 15 | 47 | [61] |
| 8 | 100 | 1.30 | 0 | 13 | 1 | 83 | |||||
| Pt (3.4%) Bi (0.7%)/ TiO2 | air (4 MPa) | Na2CO3/ HMF 2 | 100 | 0.5 | 100 | 34.90 | 0 | 13 | 25 | 52 | [28] |
| 4 | 100 | 4.35 | 0 | 0 | 0 | 99 | |||||
| Au0.6Pt0.4 (1%)/C | O2 (0.3 MPa) | Na2CO3/ HMF 2 | 60 | 2 | 100 | 5.10 | n.a | 38 | n.a | 62 | [44] |
| 6 | 100 | 1.70 | n.a | 6 | n.a | 94 | |||||
| Au (2.6%)-CeO2 | air (1 MPa) | Na2OH/ HMF 4 | 65 | 1 | 100 | 19.80 | 0 | 98 | 0 | 1 | [12] |
| 8 | 100 | 2.48 | 0 | 0 | 0 | 99 | |||||
| Au (1.5%)-CeO2 | O2 (1 MPa) | Na2OH/ HMF 4 | 95 | 0.5 | 100 | 15.20 | 0 | 1.7 | 67 | 30 | [62] |
| 3 | 100 | 2.50 | 0 | 0 | 1.4 | 98 | |||||
| Au0.86Pd0.14 (1.5%)/ TiO2 | O2 (1 MPa) | Na2OH/ HMF 2 | 70 | 0.5 | 100 | 16.30 | 0 | 43 | 4 | 52 | [39] |
| 4 | 100 | 2.00 | 0 | 13 | 4 | 85 | |||||
| Au0.5Pd0.5 (2%)/ZOC | O2 (0.3 MPa) | Na2HCO3/HMF 4 | 80 | 0.5 | 74 | 19.50 | traces | 25 | 55 | 19 | [32] |
| 4 | 100 | 3.30 | traces | 0 | 0 | 100 | |||||
| Au0.5Cu0.5 (1.5%)/ TiO2 | O2 (1 MPa) | Na2OH/ HMF 4 | 95 | 1 | 100 | 11.50 | n.a. | 90 | 0 | 9 | [60] |
| 4 | 100 | 2.90 | n.a. | 2 | 0 | 98 | |||||
| T | Time | Conversion | Productivity | Selectivity % | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Catalyst | Oxidant | Solvent | °C | h | HMF % | mmol g−1 h−1 | DFF | HMFCA | FFCA | FDCA | Ref. |
| Co (7%)Ox/α-Fe3O4 | TBHP/ HMF 7 | DMSO | 80 | 2 | 39 | 1.08 | 33 | 12 | 0 | 51 | [73] |
| 12 | 97 | 0.45 | 8 | 0 | 0 | 70 | |||||
| O2 20 mL/min | DMSO | 80 | 12 | 13 | 0.06 | 50 | 0 | 0 | 32 | ||
| Cu (2.6%)-MnO2 | TBHP/ HMF 65 | TBA | 80 | 1 | 39 | 1.08 | 13 | 0 | 57 | 28 | [81] |
| 12 | 99 | 0.41 | 0.4 | 0 | 3 | 96 | |||||
| O2 (0.1 MPa) | TBA | 80 | 12 | 57 | 0.24 | 73 | 0 | 24 | 3 | ||
| CuMn2O4 | TBHP/ HMF 15 | CH3CN | 80 | 4 | 100 | 5 | 8 | 8 | 55 | 29 | [76] |
| 14 | 100 | 1.43 | 0 | 0 | 3 | 95 | |||||
| Co (0.12%)-Py-resin | TBHP/ HMF 9 | CH3CN | 110 | 3 | 40 | 1.48 | 51 | 3 | n.a | 39 | [77] |
| 24 | 78 | 0.36 | 15 | 2 | n.a | 78 | |||||
| FeNP@NH2-SBA-15 | O2 (0.6 MPa) | H2O | 120 | 6 | 86 | 2.4 | 0 | 37 | 14 | 47 | [78] |
| 14 | 100 | 1.2 | 0 | 5 | 5 | 88 | |||||
| Fe3+ (0.015%)/POP | O2 (1 MPa) | H2O | 100 | 2 | 85 | 75 | 42 | n.a | 17 | 42 | [79] |
| 10 | 100 | 26 | 3 | n.a | 3 | 85 | |||||
| Cu (0.62%)/ N-graphene | TBHP/ HMF 8 | CH3CN | 70 | 2 | 100 | 25 | 4 | 3 | 82 | 7 | [80] |
| 24 | 100 | 2.1 | 0 | 0 | 30 | 60 | |||||
| Catalyst | S(BET)/m2 g−1 | Grain Size/nm | Spinel Cell/Å | |||
|---|---|---|---|---|---|---|
| CuO | Spinel | Mn3O4 | Mn5O8 | |||
| Cu100 | 11 | 38 | ||||
| Cu67Mn33 | 106 | 11 | 11 | 8.286 (4) | ||
| Cu50Mn50 | 75 | 15 | 14 | 8.290 (0) | ||
| Cu33Mn67 | 51 | 15 | 8.291 (4) | |||
| Mn100 | 24 | 22 | 25 | |||
| Catalyst | Phase | Cell Size/Å | S(BET)/m2 g−1 | Grain Size/nm | |
|---|---|---|---|---|---|
| Spinel | Hematite | ||||
| Co100 | spinel | 8.086 (3) | 35 | 29 | |
| Co67Fe33 | spinel | 8.167 (4) | 81 | 7 | |
| Co50Fe50 | spinel | 8.263 (4) | 78 | 11 | |
| Co33Fe67 | spinel | 8.360 (1) | 84 | 11 | |
| Fe100 | α-hematite | 5.04, 13.76 | 43 | 25 | |
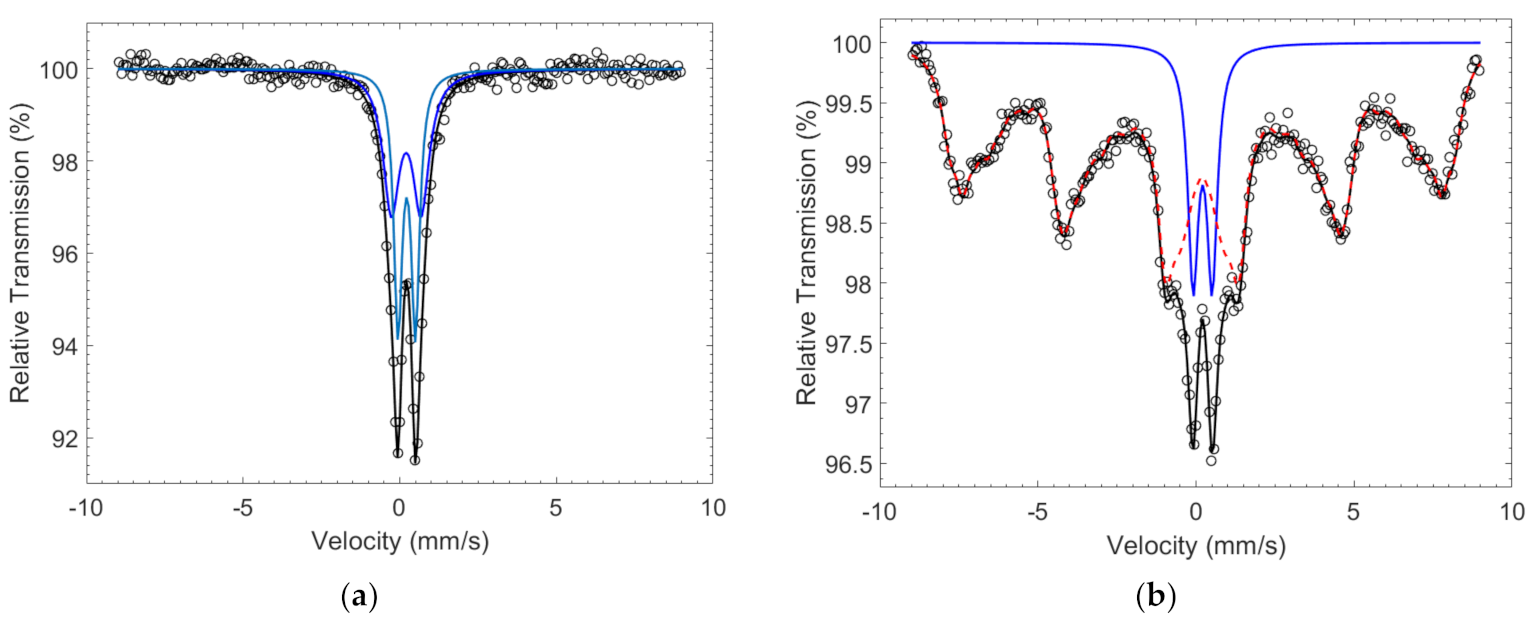
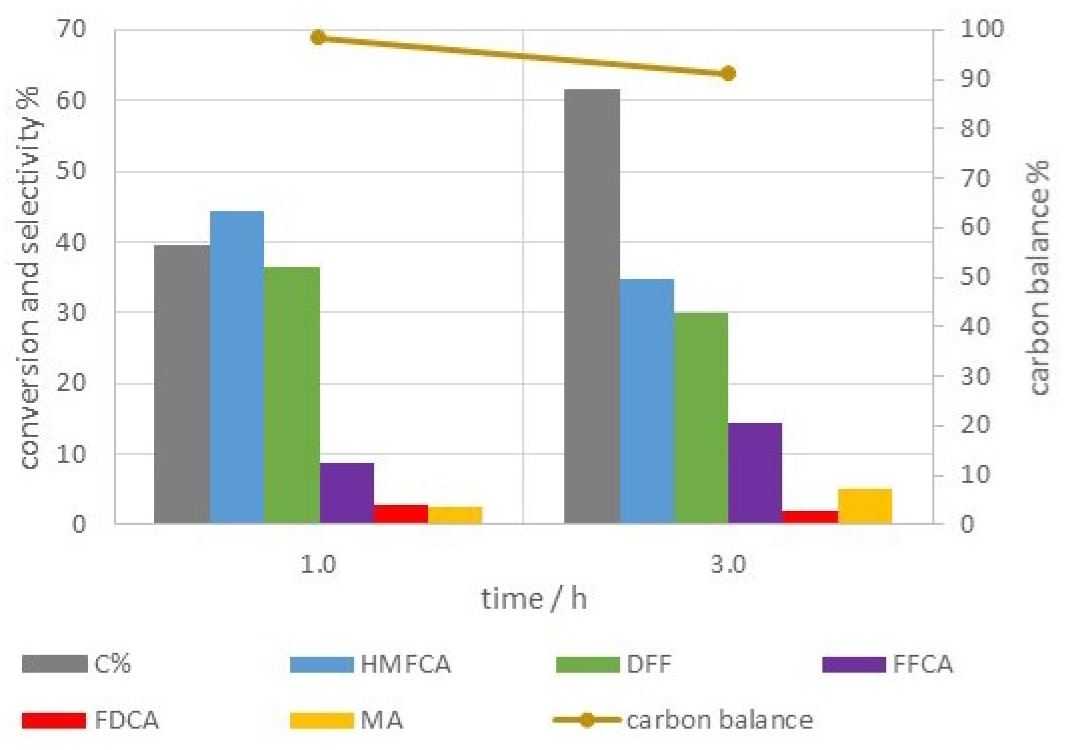
| Catalyst | BHF (T) | Δ (mm/s) | δ (mm/s) | Γ (mm/s) | Area (%) | Site |
|---|---|---|---|---|---|---|
| Co67Fe33 | - | 0.95 (8) | 0.31 (1) | 0.65 (3) | 51 (9) | Fe3+ |
| - | 0.53 (2) | 0.33 (1) | 0.32 (3) | 49 (9) | Fe3+ | |
| Co50Fe50 | 52-2 | 0.00 (1) | 0.30 (1) | 0.6 | 88 (1) | Fe3+ |
| - | 0.60 (2) | 0.32 (1) | 0.40 (3) | 12 (1) | Fe3+ |
| Catalyst | Co2+ Tetr | Fe3+ Tetr | Co2+ Oct | Fe3+ Oct | Co3+ Oct | Cation Vacancies |
|---|---|---|---|---|---|---|
| Co100 | 1 | 0 | 0 | 0 | 2 | 0 |
| Co67Fe33 | 0.50 | 0.50 | 0.17 | 0.46 | 1.26 | 0.110 |
| Co50Fe50 | 0.35 | 0.65 | 0.43 | 0.81 | 0.68 | 0.072 |
| Co33Fe67 | 1 | 0.90 | 0.82 | 1.08 | 0.07 | 0.026 |
| Catalyst | (FFCA + FDCA)/ (HMFCA + DFF) | FDCA/FFCA | HMF Reaction Rate (mmol g−1 h−1) |
|---|---|---|---|
| Cu100 | 2.00 | 0.40 | 10.1 |
| Cu67Mn33 | 23.30 | 1.26 | 10.2 |
| Cu50Mn50 | 10.05 | 0.91 | 11.4 |
| Cu33Mn67 | 3.78 | 0.60 | 11.2 |
| Mn100 | 0.52 | 0.18 | 8.9 |
| Co100 | 0.87 | 0.26 | 9.4 |
| Co67Fe33 | 8.70 | 0.79 | 11.2 |
| Co50Fe50 | 1.73 | 0.43 | 10.3 |
| Co33Fe67 | 0.53 | 0.20 | 9.6 |
| Fe100 | 0.25 | 0.18 | 5.5 |
| no catalyst | 0.26 | 0.12 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Demet, A.E.; Gimello, O.; Arletti, R.; Tanchoux, N.; Sougrati, M.T.; Stievano, L.; Quignard, F.; Centi, G.; Perathoner, S.; Di Renzo, F. 5-Hydroxymethylfurfural Oxidation to 2,5-Furandicarboxylic Acid on Noble Metal-Free Nanocrystalline Mixed Oxide Catalysts. Catalysts 2022, 12, 814. https://doi.org/10.3390/catal12080814
Demet AE, Gimello O, Arletti R, Tanchoux N, Sougrati MT, Stievano L, Quignard F, Centi G, Perathoner S, Di Renzo F. 5-Hydroxymethylfurfural Oxidation to 2,5-Furandicarboxylic Acid on Noble Metal-Free Nanocrystalline Mixed Oxide Catalysts. Catalysts. 2022; 12(8):814. https://doi.org/10.3390/catal12080814
Chicago/Turabian StyleDemet, Atif Emre, Olinda Gimello, Rossella Arletti, Nathalie Tanchoux, Moulay Tahar Sougrati, Lorenzo Stievano, Françoise Quignard, Gabriele Centi, Siglinda Perathoner, and Francesco Di Renzo. 2022. "5-Hydroxymethylfurfural Oxidation to 2,5-Furandicarboxylic Acid on Noble Metal-Free Nanocrystalline Mixed Oxide Catalysts" Catalysts 12, no. 8: 814. https://doi.org/10.3390/catal12080814
APA StyleDemet, A. E., Gimello, O., Arletti, R., Tanchoux, N., Sougrati, M. T., Stievano, L., Quignard, F., Centi, G., Perathoner, S., & Di Renzo, F. (2022). 5-Hydroxymethylfurfural Oxidation to 2,5-Furandicarboxylic Acid on Noble Metal-Free Nanocrystalline Mixed Oxide Catalysts. Catalysts, 12(8), 814. https://doi.org/10.3390/catal12080814