
On the Optimization of Ni/A and Ni/X Synthesis Procedure toward Active and Selective Catalysts for the Production of CH4 from CO2


 and
and
Abstract
:1. Introduction
2. Results and Discussion
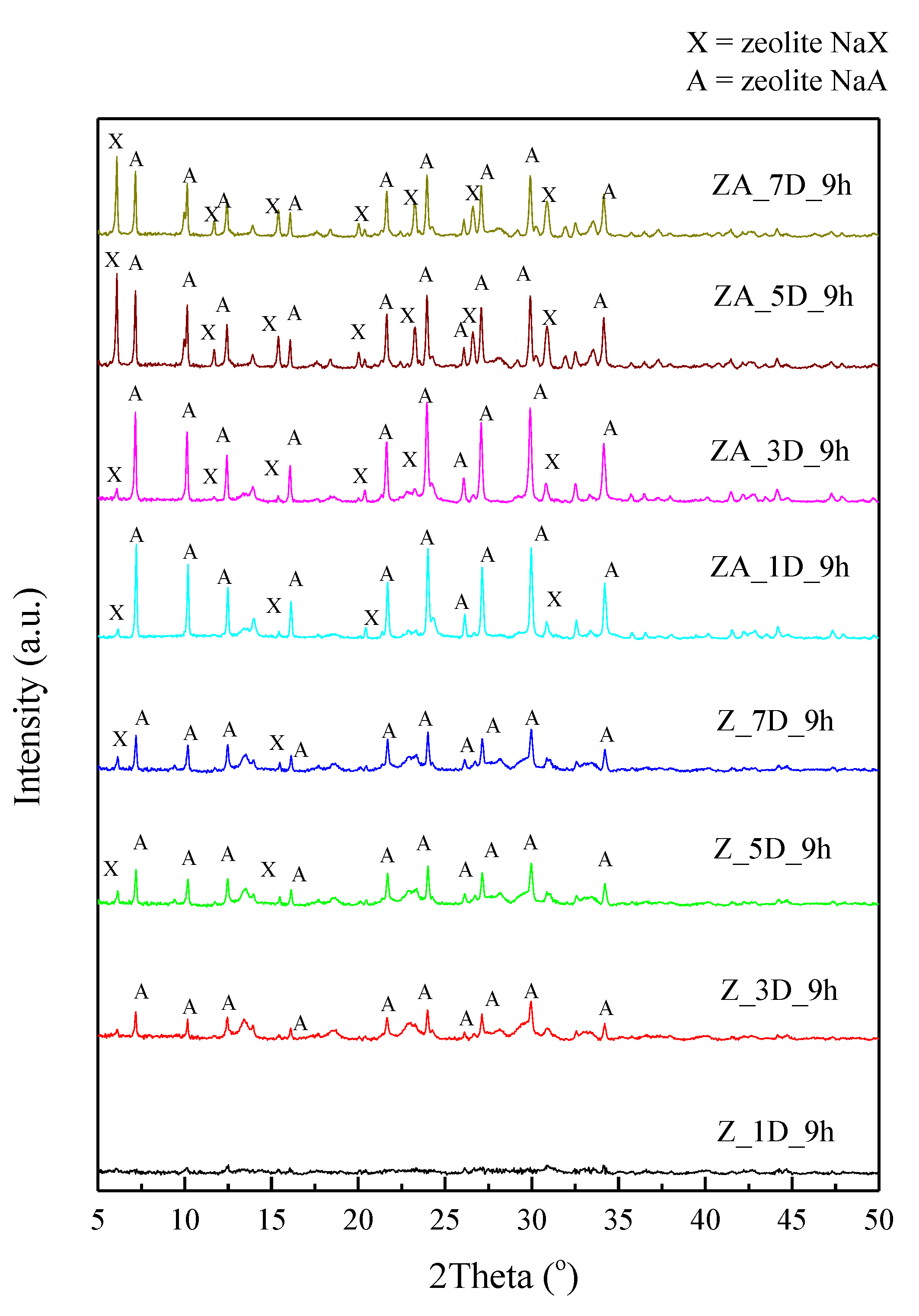
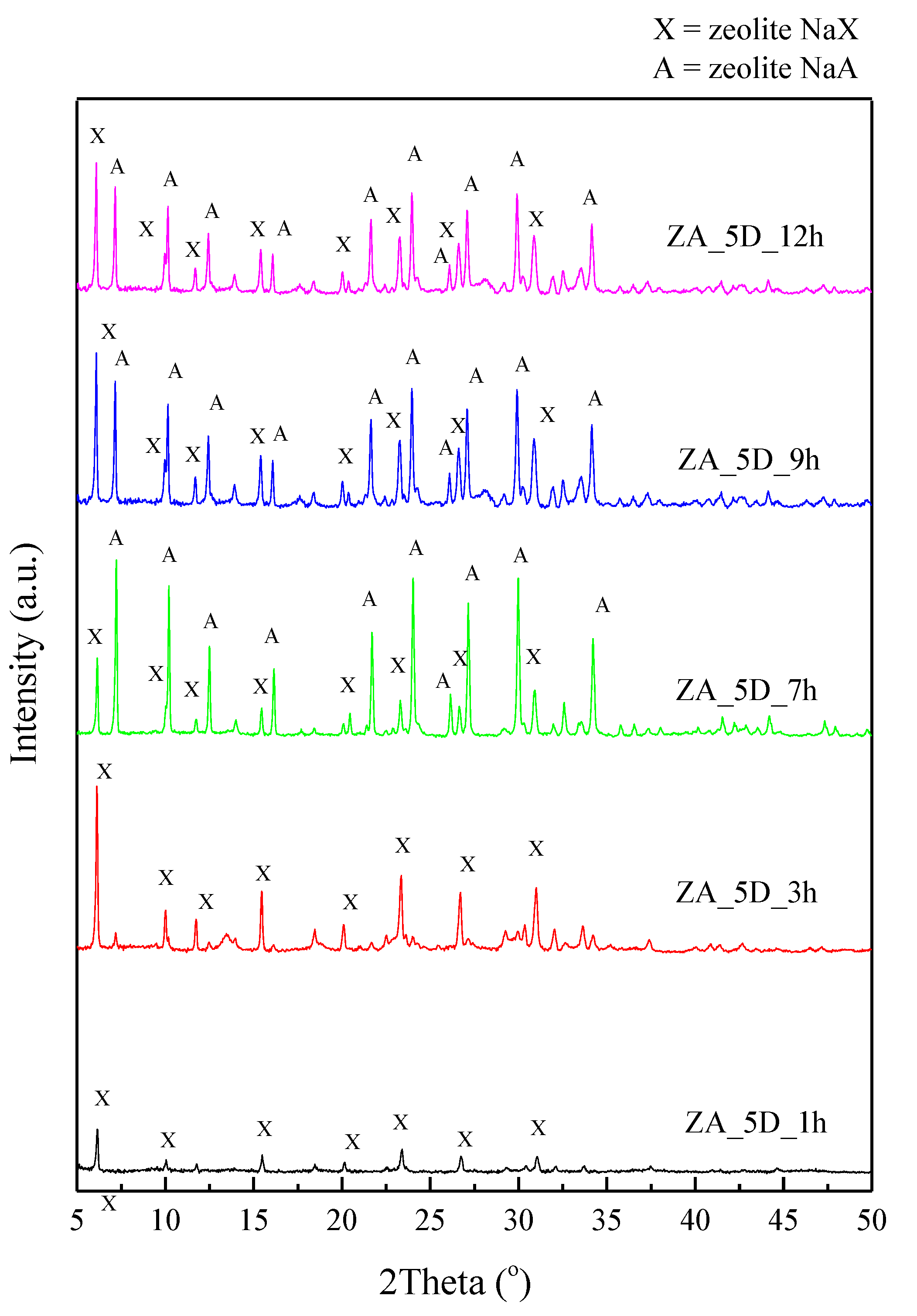
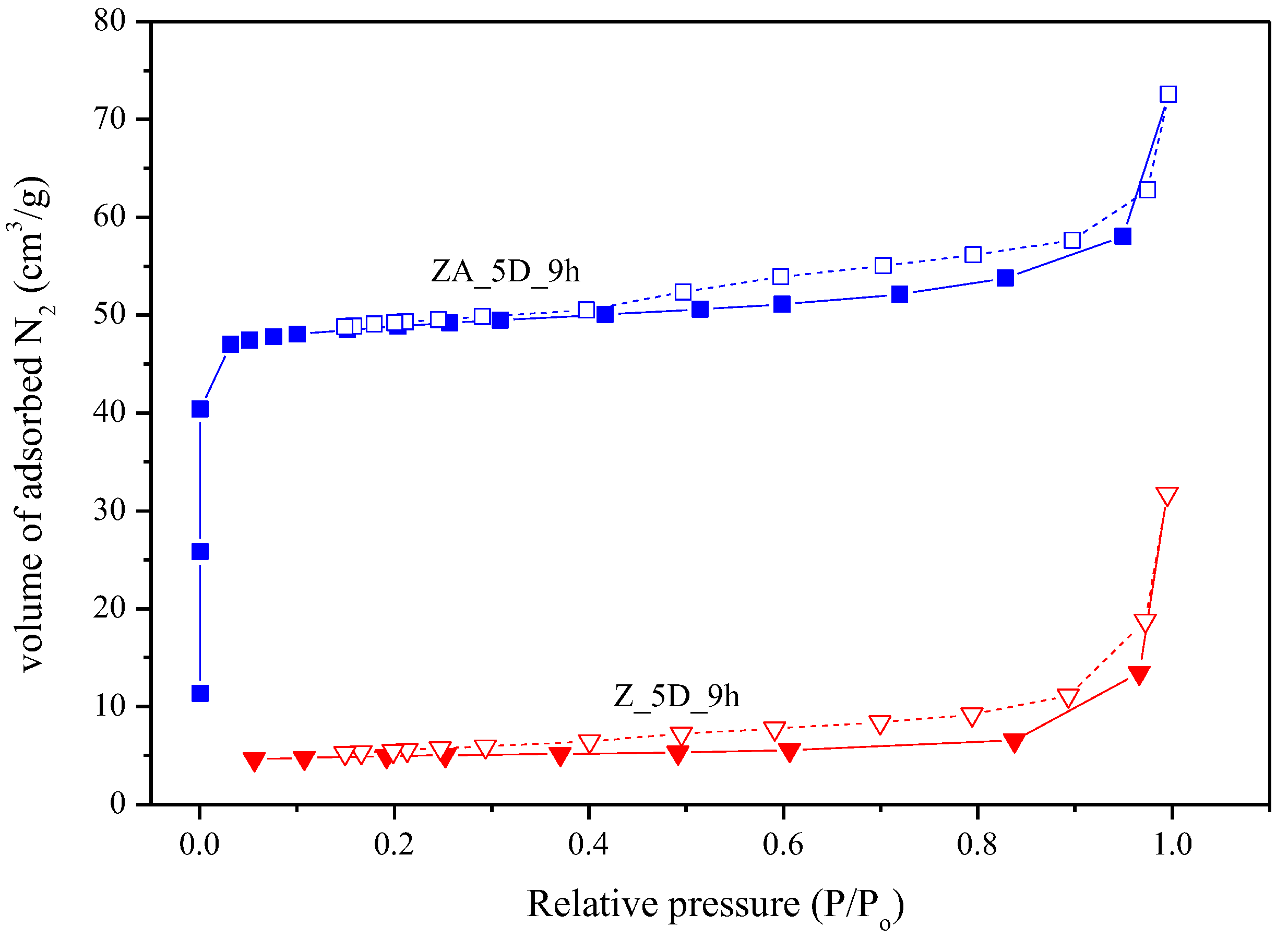
2.1. Effects of Different Synthesis Conditions on Zeolite Properties
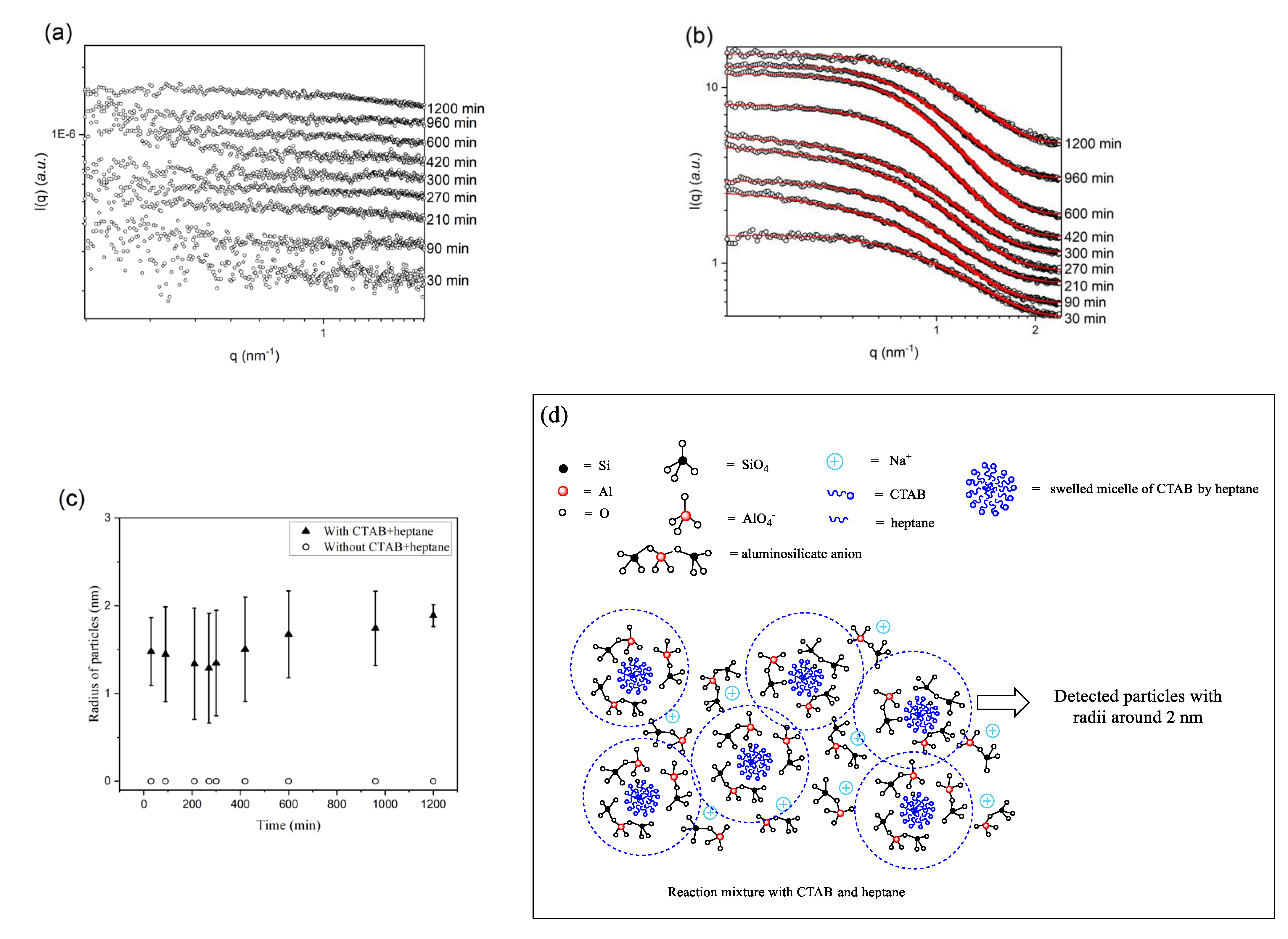
2.2. Effects of CTAB and Heptane on Enhancing Zeolite surface Area and Porosity Investigated by SAXS and WAXS
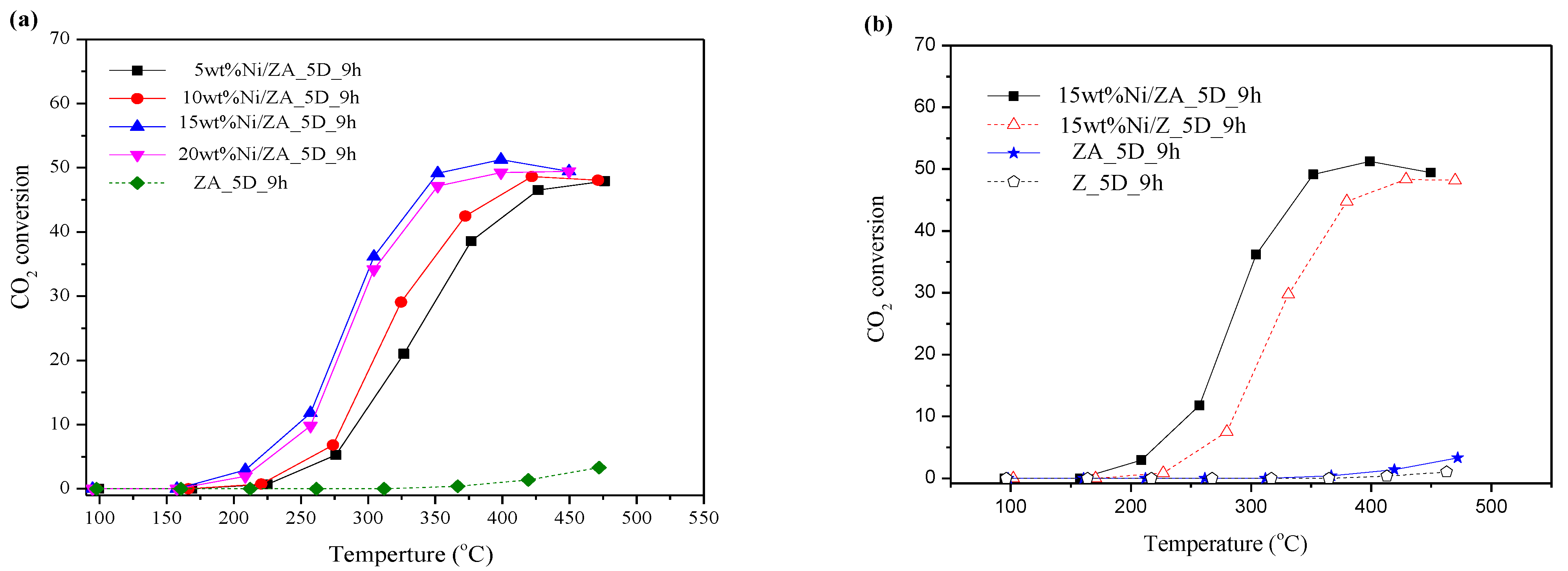
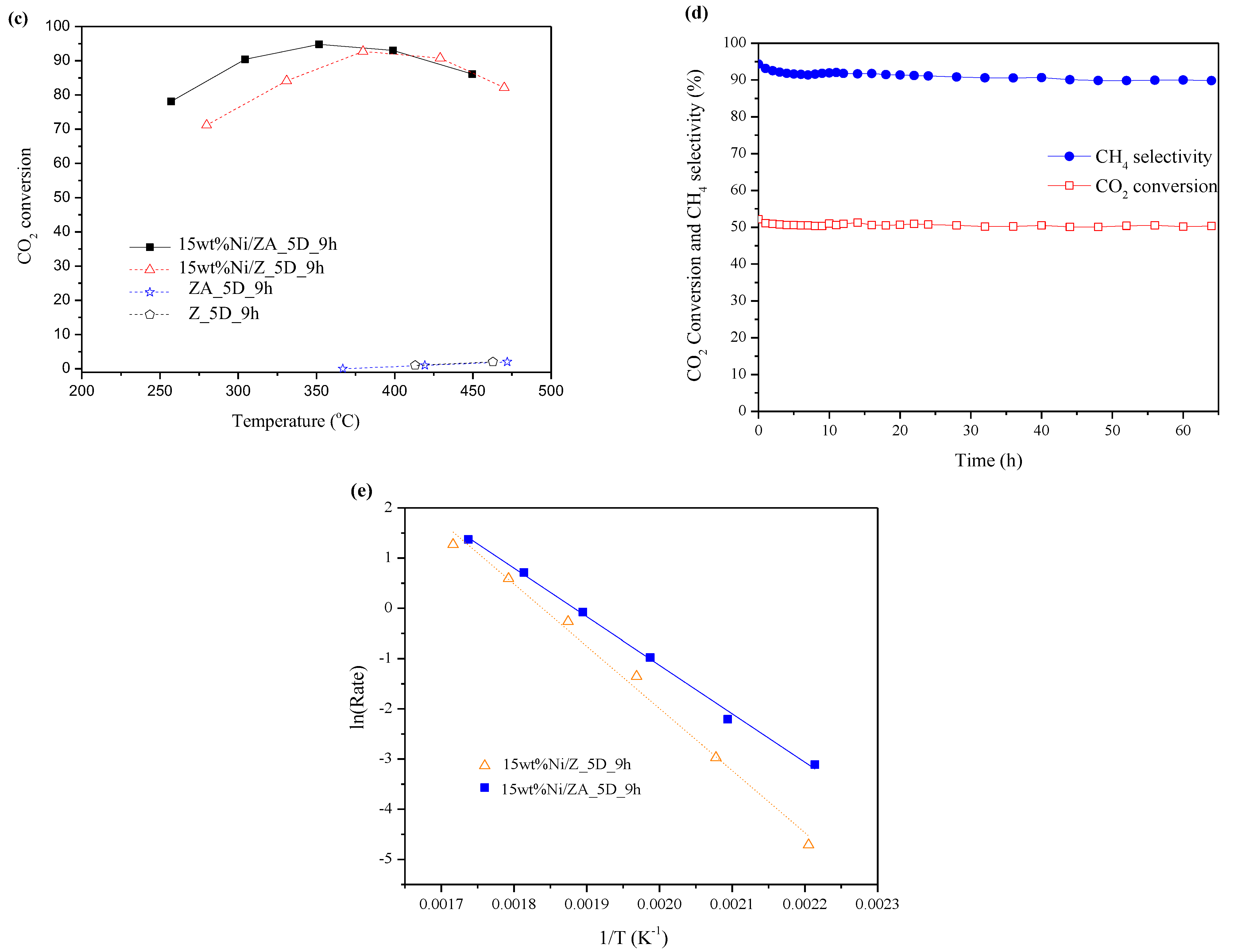
2.3. CO2 Methanation Catalytic Activity
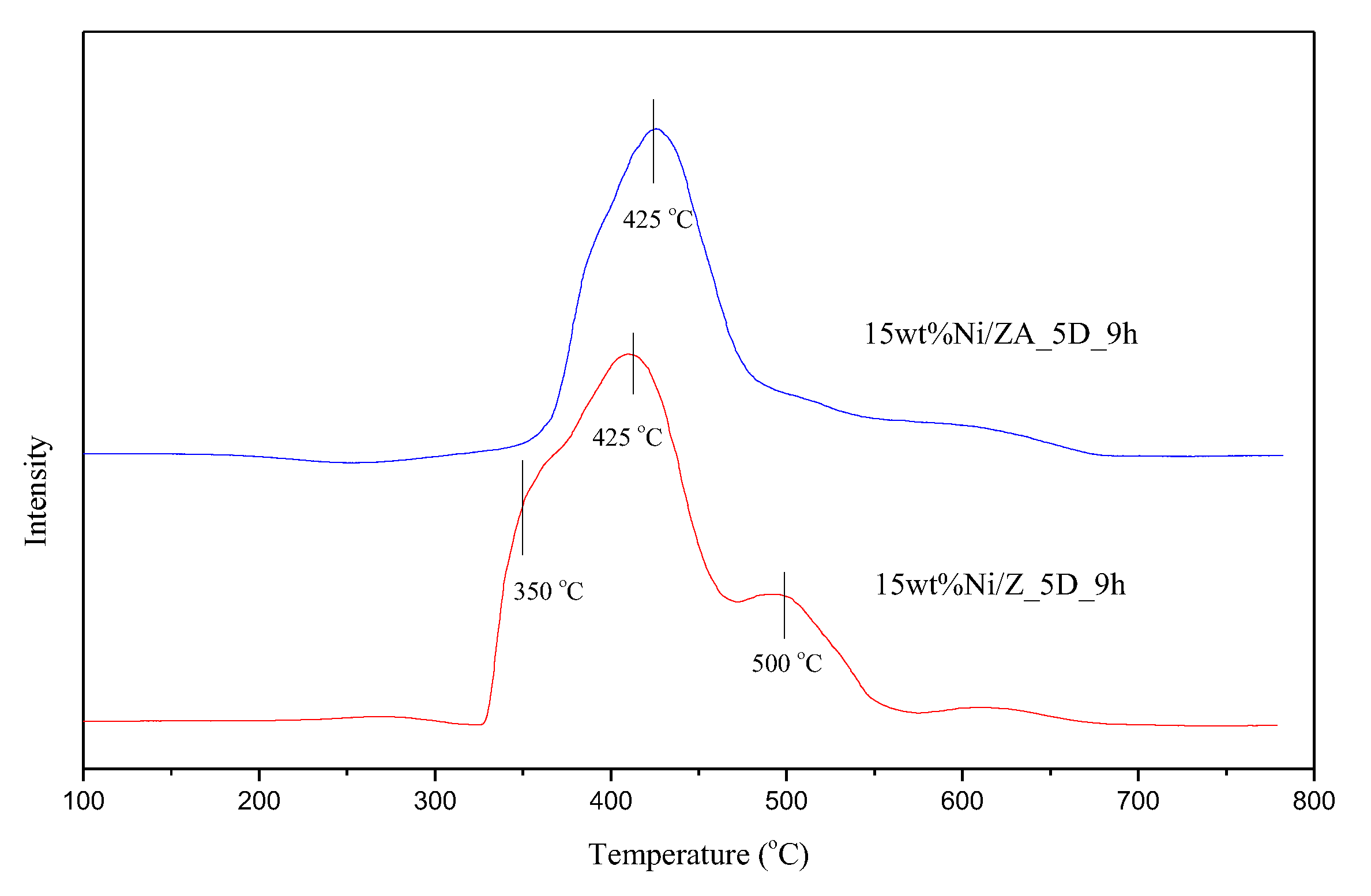
2.4. Catalyst Characterization
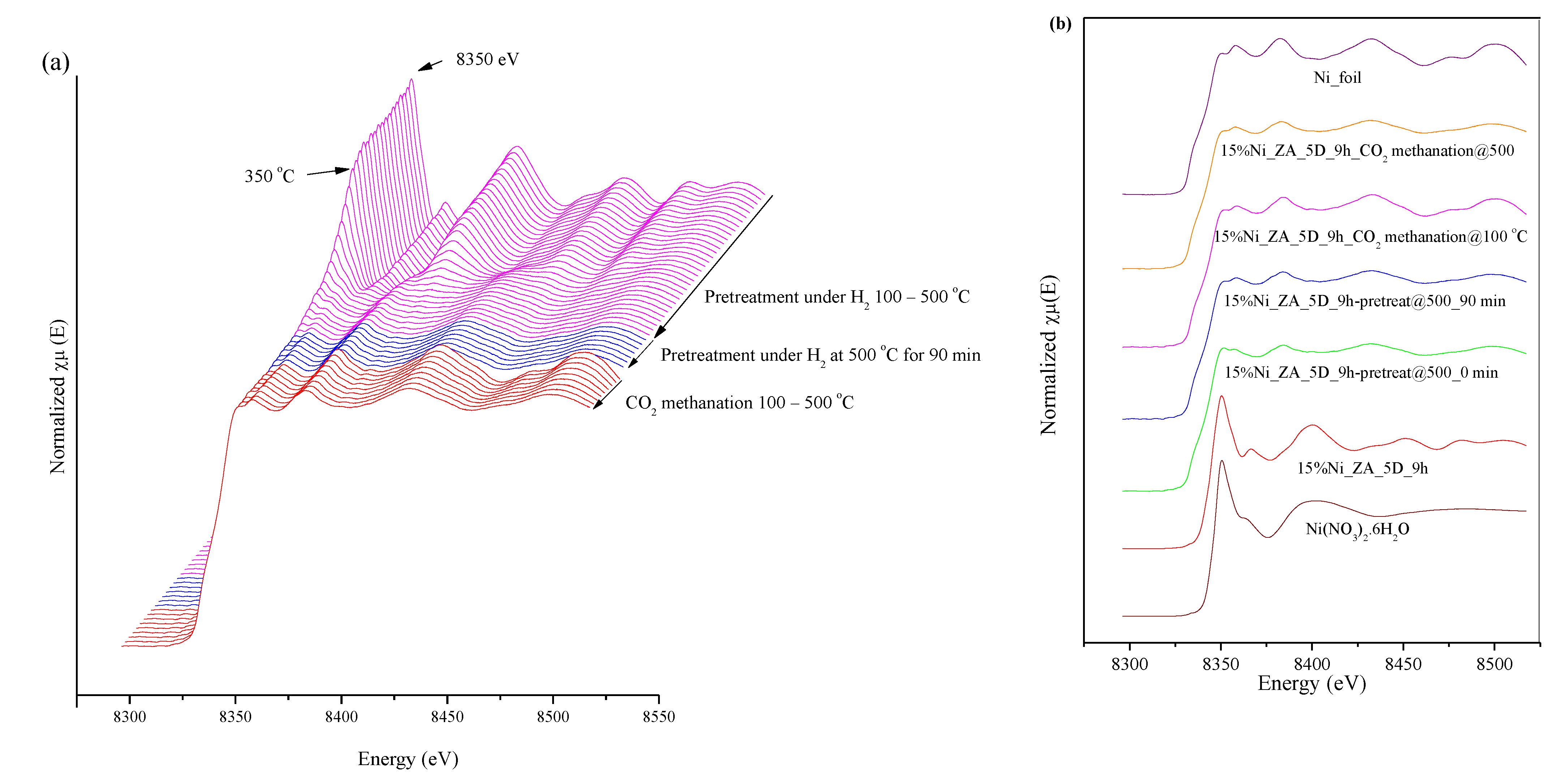
2.5. X-ray Absorption Spectroscopy
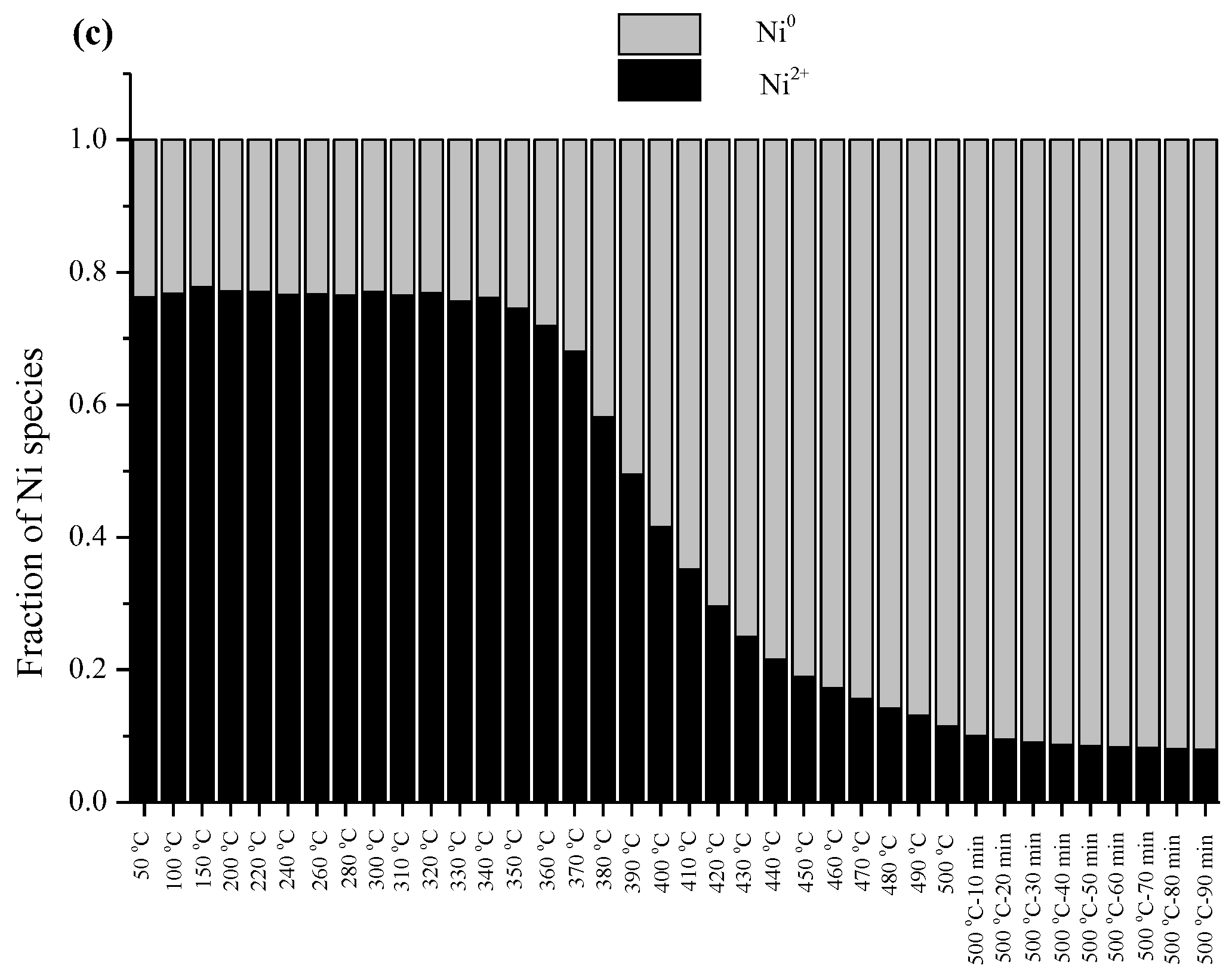
2.5.1. In Situ CO2 Methanation Experiment by Time-Resolved X-ray Absorption Spectroscopy
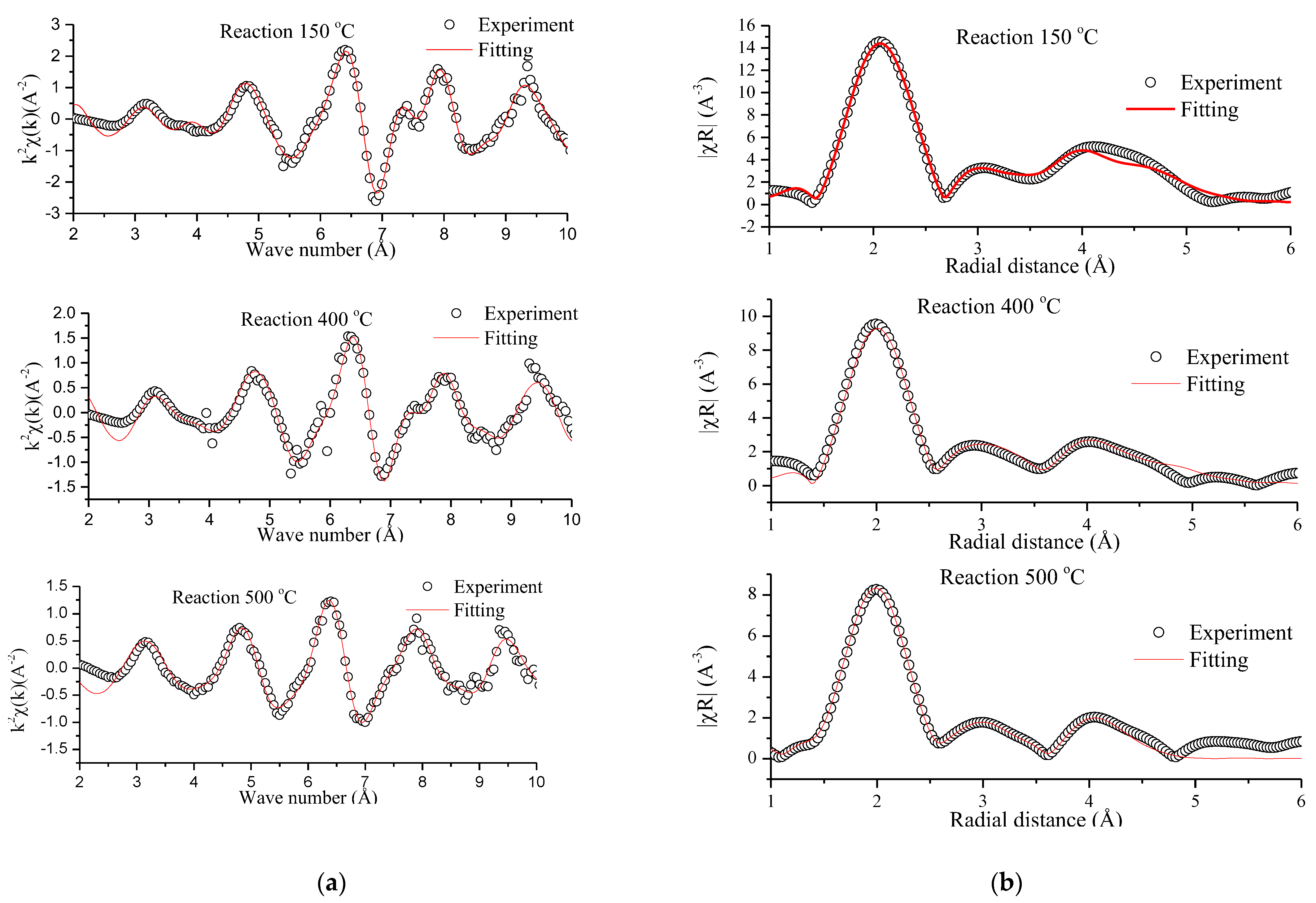
2.5.2. In Situ CO2 Methanation Experiment by X-ray Absorption Spectroscopy
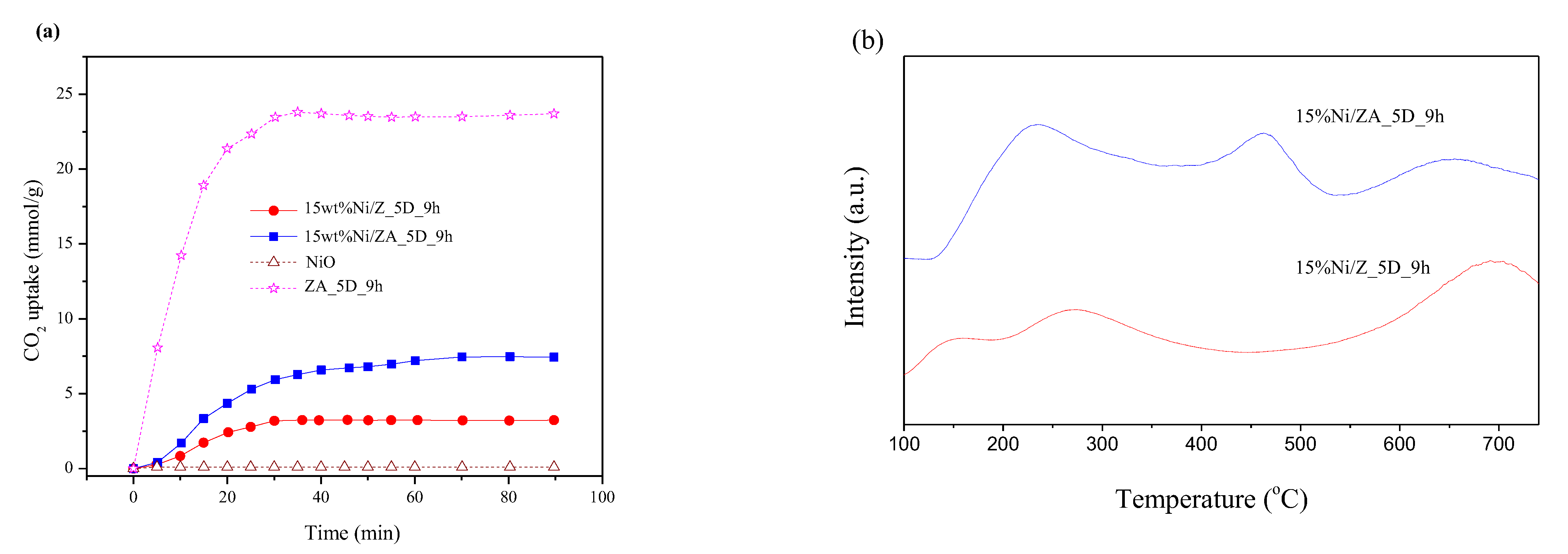
2.6. Effect of Different Surface Properties of Zeolite Support on CO2 Methanation Catalytic Activity
3. Materials and Methods
3.1. Materials and Reagents
3.2. Zeolite Support Synthesis Procedure
3.3. Zeolite-Supported Ni Catalyst Preparation
3.4. Characterization
3.5. CO2 Uptake and CO2-Temperature Program Desorption
3.6. In Situ Small-Angle X-ray Scattering (SAXS) and In Situ Wide-Angle X-ray Scattering (WAXS)
3.7. CO2 Methanation Catalytic Activity Test
3.8. X-ray Absorption Spectroscopy
3.8.1. X-ray Absorption Near-Edge Structure (XANES)
3.8.2. Extended X-ray Absorption Fine Structure (EXAFS)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cui, Q.; Huihui, W.; Xiangyu, J.; Ningbo, G. Effect of carbonization temperature on CO2 adsorption behavior of activated coal char. J. Energy Inst. 2021, 97, 92–99. [Google Scholar]
- Mingming, Z.; Yunxiang, P.; Yuzhu, S.; Ping, L.; Jianguo, Y. Preparation of CaO-Al2O3 sorbent and CO2 capture performance at high temperature. Fuel 2013, 111, 636–642. [Google Scholar]
- Arnost, Z.; Jakub, P.; Jiri, C. MgO-modified mesoporous silicas impregnated by potassium carbonate for carbon dioxide adsorption. Microporous Mesoporous Mater. 2013, 167, 44–50. [Google Scholar]
- Valverde, J.M.; Sanchez-Jimenez, P.E.; Perez-Maqueda, L.A. High and stable CO2 capture capacity of natural limestone at Ca-looping conditions by heat pretreatment and recarbonation synergy. Fuel 2014, 123, 79–85. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yu, P.; Luo, Y. Absorption of CO2 by amino acid-functionalized and traditional dicationic ionic liquids: Properties, Henry’s law constants and mechanisms. Chem. Eng. J. 2013, 214, 355–363. [Google Scholar] [CrossRef]
- Rahbari-Sisakht, M.; Ismail, A.F.; Rana, D.; Matsuura, T. A novel surface modified polyvinylidene fluoride hollow fiber membrane contactor for CO2 absorption. J. Membr. Sci. 2012, 415, 221–228. [Google Scholar] [CrossRef]
- Zain, M.M.; Mohamed, A.R. An overview on conversion technologies to produce value added products from CH4 and CO2 as major biogas constituents. Renew. Sustain. Energy Rev. 2018, 98, 56–63. [Google Scholar] [CrossRef]
- Schwach, P.; Pan, X.; Bao, X. Direct Conversion of Methane to Value-Added Chemicals over Heterogeneous Catalysts: Challenges and Prospects. Chem. Rev. 2017, 117, 8497–8520. [Google Scholar] [CrossRef]
- Zhao, G.; Drewery, M.; Mackie, J.; Oliver, T.; Kennedy, E.M.; Stockenhuber, M. The Catalyzed Conversion of Methane to Value-Added Products. Energy Technol. 2020, 8, 1900665. [Google Scholar] [CrossRef]
- Taifan, W.; Baltrusaitis, J. CH4 conversion to value added products: Potential, limitations and extensions of a single step heterogeneous catalysis. Appl. Catal. B Environ. 2016, 198, 525–547. [Google Scholar] [CrossRef]
- Aziz, M.A.A.; Jalil, A.A.; Triwahyono, S.; Ahmad, A. CO2 methanation over heterogeneous catalysts: Recent progress and future prospects. Green Chem. 2015, 17, 2647–2663. [Google Scholar] [CrossRef]
- Tan, C.H.; Nomanbhay, S.; Shamsuddin, A.H.; Park, Y.-K.; Hernández-Cocoletzi, H.; Show, P.L. Current Developments in Catalytic Methanation of Carbon Dioxide—A Review. Front. Energy Res. 2020, 9, 795423. [Google Scholar] [CrossRef]
- Ashok, J.; Pati, S.; Hongmanorom, P.; Tianxi, Z.; Junmei, C.; Kawi, S. A review of recent catalyst advances in CO2 methanation processes. Catal. Today 2020, 356, 471–489. [Google Scholar] [CrossRef]
- Jiajian, G.; Qing, L.; Fangna, G.; Bin, L.; Ziyi, Z.; Fabing, S. Recent advances in methanation catalysts for the production of synthetic natural gas. RSC Adv. 2015, 5, 22759–22776. [Google Scholar]
- Jianghui, L.; Caiping, M.; Qiong, W.; Yanfei, X.; Guangyuan, M.; Jie, W.; Hongtao, W.; Chenglong, D.; Chenghua, Z.; Mingyue, D. Enhanced low-temperature performance of CO2 methanation over mesoporous Ni/Al2O3-ZrO2 catalysts. Appl. Catal. B Environ. 2019, 243, 262–272. [Google Scholar]
- Ridzuan, N.D.M.; Shaharun, M.S.; Anawar, M.A.; Ud-Din, I. Ni-Based Catalyst for Carbon Dioxide Methanation: A Review on Performance and Progress. Catalysts 2022, 12, 469. [Google Scholar] [CrossRef]
- Li, L.; Zeng, W.; Song, M.; Wu, X.; Li, G.; Hu, C. Research Progress and Reaction Mechanism of CO2 Methanation over Ni-Based Catalysts at Low Temperature: A Review. Catalysts 2022, 12, 244. [Google Scholar] [CrossRef]
- Lv, C.; Xu, L.; Chen, M.; Cui, Y.; Wen, X.; Li, Y.; Wu, C.; Yang, B.; Miao, Z.; Hu, X.; et al. Recent Progresses in Constructing the Highly Efficient Ni Based Catalysts with Advanced Low-Temperature Activity Toward CO2 Methanation. Front. Chem. 2020, 8, 269. [Google Scholar] [CrossRef]
- Guo, X.; Traitangwong, A.; Hu, M.; Zuo, C.; Meeyoo, V.; Peng, Z.; Li, C. Carbon dioxide methanation over Nickel-based catalysts supported on various mesoporous material. Energy Fuels 2018, 32, 3681–3689. [Google Scholar] [CrossRef]
- Du, G.; Lim, S.; Yang, Y.; Wang, C.; Pfefferle, L.; Haller, G.L. Methanation of carbon dioxide on Ni-incorporated MCM-41 catalysts: The influence of catalyst pretreatment and study of steady-state reaction. J. Catal. 2007, 249, 370–379. [Google Scholar] [CrossRef]
- Rahmani, S.; Rezaei, M.; Meshkania, F. Preparation of highly active nickel catalysts supported on mesoporous nanocrystalline γ-Al2O3 for CO2 methanation. J. Ind. Eng. Chem. 2014, 20, 1346–1352. [Google Scholar] [CrossRef]
- Westermann, A.; Azambre, B.; Bacariza, M.C.; Graca, I.; Ribeiro, M.F.; Lopes, J.M. Insights into CO2 methanation mechanism over NiUSY zeolites: An operando IR study. Appl. Catal. B Environ. 2015, 120, 174–175. [Google Scholar] [CrossRef]
- Teh, L.P.; Triwahyono, S.; Jalil, A.A.; Mukti, R.R.; Aziz, M.A.A.; Shishido, T. Mesoporous ZSM5 having both intrinsic acidic and basic sites for cracking and methanation. Chem. Eng. J. 2015, 270, 196–204. [Google Scholar] [CrossRef]
- Graca, I.; González, L.V.; Bacariza, M.C.; Fernandes, A.; Henriques, C.; Lopes, J.M.; Ribeiro, M.F. CO2 hydrogenation into CH4 on NiHNaUSY zeolites. Appl. Catal. B Environ. 2014, 147, 101–110. [Google Scholar] [CrossRef]
- Bacariza, M.C.; Bértolo, R.; Graça, I.; Lopes, J.M.; Henriques, C. The effect of the compensating cation on the catalytic performances of Ni/USY zeolites towards CO2 methanation. J. CO2 Util. 2017, 21, 280–291. [Google Scholar] [CrossRef]
- Xu, L.; Wen, X.; Chen, M.; Lv, C.; Cui, Y.; Wu, X.; Wu, C.; Miao, Z.; Hu, X. Highly dispersed Ni-La catalysts over mesoporous nanosponge MFI zeolite for low-temperature CO2 methanation: Synergistic effect between mesoporous and microporous channels. J. Ind. Eng. Chem. 2021, 100, 159–173. [Google Scholar] [CrossRef]
- Carmen Bacariza, M.; Graça, I.; Lopes, J.M.; Henriques, C. Tuning zeolite properties towards CO2 methanation: An overview. ChemCatChem 2019, 11, 2388–2400. [Google Scholar] [CrossRef]
- Sholeha, N.A.; Mohamad, S.; Bahruji, H.; Prasetyoko, D.; Widiastuti, N.; Fatah, N.A.A.; Jalil, A.A.; Taufiq-Yap, Y.H. Enhanced CO2 methanation at mild temperature on Ni/zeolite from kaolin: Effect of metal–support interface. RSC Adv. 2021, 11, 16376. [Google Scholar] [CrossRef]
- Dreyer, J.A.H.; Li, P.; Zhang, L.; Beh, G.K.; Zhang, R.; Sit, P.H.-L.; Teoh, W.Y. Influence of the oxide support reducibility on the CO2 methanation over Ru-based catalysts. Appl. Catal. B Environ. 2017, 219, 715–726. [Google Scholar] [CrossRef]
- Hu, L.; Urakawa, A. Continuous CO2 capture and reduction in one process: CO2 methanation over unpromoted and promoted Ni/ZrO2. J. CO2 Util. 2018, 25, 323–329. [Google Scholar] [CrossRef] [Green Version]
- Burger, T.; Koschany, F.; Thomys, O.; Köhler, K.; Hinrichsen, O. CO2 methanation over Fe- and Mn-promoted co-precipitated Ni-Al catalysts: Synthesis, characterization and catalysis study. Appl. Catal. A Gen. 2018, 558, 44–54. [Google Scholar] [CrossRef]
- Wierzbicki, D.; Motak, M.; Grzybek, T.; Gálvez, M.E.; Da Costa, P. The influence of lanthanum incorporation method on the performance of nickel-containing hydrotalcite-derived catalysts in CO2 methanation reaction. Catal. Today 2018, 307, 205–211. [Google Scholar] [CrossRef]
- Krachuamram, S.; Danvirutai, C.; Youngme, S.; Chanapattharapol, K.C. Effects of Cetyltrimethylammonium Bromide and Heptane on the Surface Properties and CO2 Adsorption of Zeolite NaX. J. Chin. Chem. Soc. 2017, 64, 658–665. [Google Scholar] [CrossRef]
- Zhu, Y.; Hua, Z.; Zhou, J.; Wang, L.; Zhao, J.; Gong, Y.; Wu, W.; Ruan, M.; Shi, J. Hierarchical Mesoporous Zeolites: Direct Self-Assembly Synthesis in a Conventional Surfactant Solution by Kinetic Control over the Zeolite Seed Formation. Chem. A Eur. J. 2011, 17, 14618–14627. [Google Scholar] [CrossRef] [PubMed]
- Shvets, O.V.; Kasian, N.; Zukal, A.; Pinkas, J.; Cejka, J. The Role of Template Structure and Synergism between Inorganic and Organic Structure Directing Agents in the Synthesis of UTL Zeolite. Chem. Mater. 2010, 22, 3482–3495. [Google Scholar] [CrossRef]
- Xue, T.; Li, S.; Wu, H. Surfactant-promoted synthesis of Hierarchical Zeolite ferrierite nano-sheets. Microporous Mesoporous Mater. 2021, 312, 110748. [Google Scholar] [CrossRef]
- Krachuamram, S.; Chanapattharapol, K.C.; Kamonsutthipaijit, N. Synthesis and characterization of NaX-type zeolites prepared by different silica and alumina sources and their CO2 adsorption properties. Microporous Mesoporous Mater. 2021, 310, 110632. [Google Scholar] [CrossRef]
- Le, T.A.; Kim, J.; Kang, J.K.; Park, E.D. CO and CO2 methanation over M (MMn, Ce, Zr, Mg, K, Zn, or V)-promoted Ni/Al@ Al2O3 catalysts. Catal. Today 2020, 348, 80–88. [Google Scholar] [CrossRef]
- Le, T.A.; Kim, J.; Kang, J.K.; Park, E.D. CO and CO2 methanation over Ni/Al@ Al2O3 core–shell catalyst. Catal. Today 2020, 356, 622–630. [Google Scholar] [CrossRef]
- Lee, Y.H.; Ahn, J.Y.; Nguyen, D.D.; Chang, S.W.; Kim, S.S.; Lee, S.M. Role of oxide support in Ni based catalysts for CO2 methanation. RSC Adv. 2021, 11, 17648. [Google Scholar] [CrossRef]
- Le, M.C.; Van, K.L.; Nguyen, T.H.T.; Nguyen, N.H. The Impact of Ce-Zr Addition on Nickel Dispersion and Catalytic Behavior for CO2 Methanation of Ni/AC Catalyst at Low Temperature. J. Chem. 2017, 2017, 4361056. [Google Scholar] [CrossRef] [Green Version]
- Karam, L.; Bacariza, M.C.; Lopes, J.D.; Henriques, C.; Massiani, P.; Hassan, N.E. Assessing the potential of xNi-yMg-Al2O3 catalysts prepared by EISA-one-pot synthesis towards CO2 methanation: An overall study. Int. J. Hydrogen Energy 2020, 45, 28626–28639. [Google Scholar] [CrossRef]
- Lee, S.M.; Lee, Y.H.; Moon, D.H.; Ahn, J.Y.; Nguyen, D.D.; Chang, S.W.; Kim, S.S. Reaction Mechanism and Catalytic Impact of Ni/CeO2−x Catalyst for Low-Temperature CO2 Methanation. Ind. Eng. Chem. Res. 2019, 58, 8656–8662. [Google Scholar] [CrossRef]
- Breßler, I.; Kohlbrecher, J.; Thünemann, A.F. SASfit: A tool for small-angle scattering data analysis using a library of analytical expressions. J. Appl. Crystallogr. 2015, 48, 1587–1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | SBET (m2/g) | Pore Volume (cm3/g) |
|---|---|---|
| Z_5D_9h | 13.98 | 0.0442 |
| ZA_5D_9h | 166.5 | 0.1090 |
| 15%Ni/Z_5D_9h | 25.66 | 0.0548 |
| 15%Ni/ZA_5D_9h | 14.11 | 0.0495 |
| Reaction Temperature (°C) | Paths | N | R | ||
|---|---|---|---|---|---|
| 150 °C | Ni°–Ni° | 12 | 0.856 | 0.01123 | 2.50 |
| Ni2+–O | 6 | 0.144 | 0.00050 | 2.12 | |
| 400 °C | Ni°–Ni° | 12 | 0.801 | 0.01603 | 2.47 |
| Ni2+–O | 6 | 0.199 | 0.00320 | 2.10 | |
| 500 °C | Ni°–Ni° | 12 | 0.730 | 0.01576 | 2.45 |
| Ni2+–O | 6 | 0.270 | 0.00400 | 2.05 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krachuamram, S.; Kidkhunthod, P.; Poo-arporn, Y.; Kamonsutthipaijit, N.; Chanapattharapol, K.C. On the Optimization of Ni/A and Ni/X Synthesis Procedure toward Active and Selective Catalysts for the Production of CH4 from CO2. Catalysts 2022, 12, 823. https://doi.org/10.3390/catal12080823
Krachuamram S, Kidkhunthod P, Poo-arporn Y, Kamonsutthipaijit N, Chanapattharapol KC. On the Optimization of Ni/A and Ni/X Synthesis Procedure toward Active and Selective Catalysts for the Production of CH4 from CO2. Catalysts. 2022; 12(8):823. https://doi.org/10.3390/catal12080823
Chicago/Turabian StyleKrachuamram, Somkiat, Pinit Kidkhunthod, Yingyot Poo-arporn, Nuntaporn Kamonsutthipaijit, and Kingkaew Chayakul Chanapattharapol. 2022. "On the Optimization of Ni/A and Ni/X Synthesis Procedure toward Active and Selective Catalysts for the Production of CH4 from CO2" Catalysts 12, no. 8: 823. https://doi.org/10.3390/catal12080823
APA StyleKrachuamram, S., Kidkhunthod, P., Poo-arporn, Y., Kamonsutthipaijit, N., & Chanapattharapol, K. C. (2022). On the Optimization of Ni/A and Ni/X Synthesis Procedure toward Active and Selective Catalysts for the Production of CH4 from CO2. Catalysts, 12(8), 823. https://doi.org/10.3390/catal12080823