
Role of N-Doping and O-Groups in Unzipped N-Doped CNT Carbocatalyst for Peroxomonosulfate Activation: Quantitative Structure–Activity Relationship


Abstract
:1. Introduction
2. Results and Discussion
2.1. Structure Characterizations
2.2. Specific Surface Area and Chemical Status
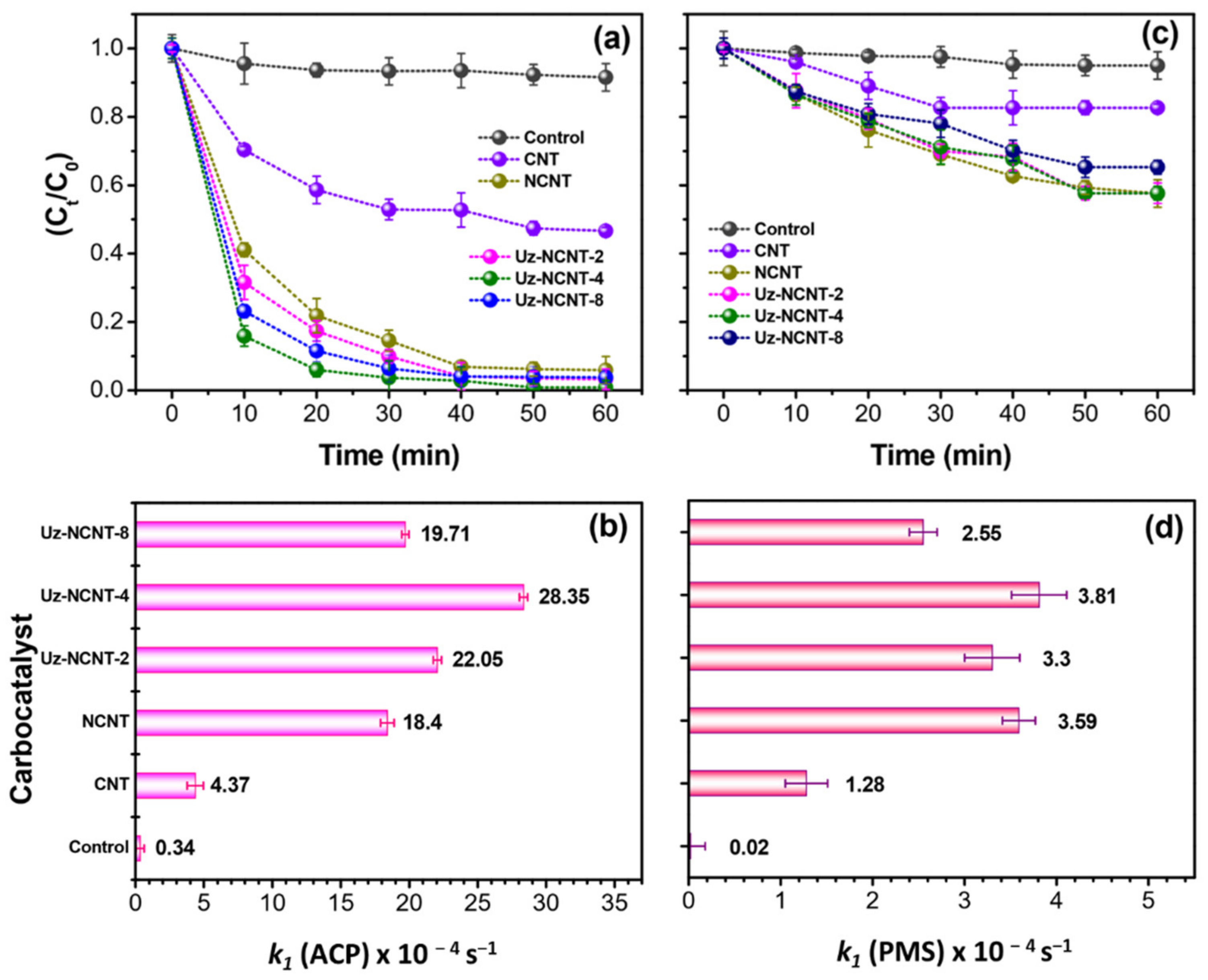
2.3. Catalytic Performance
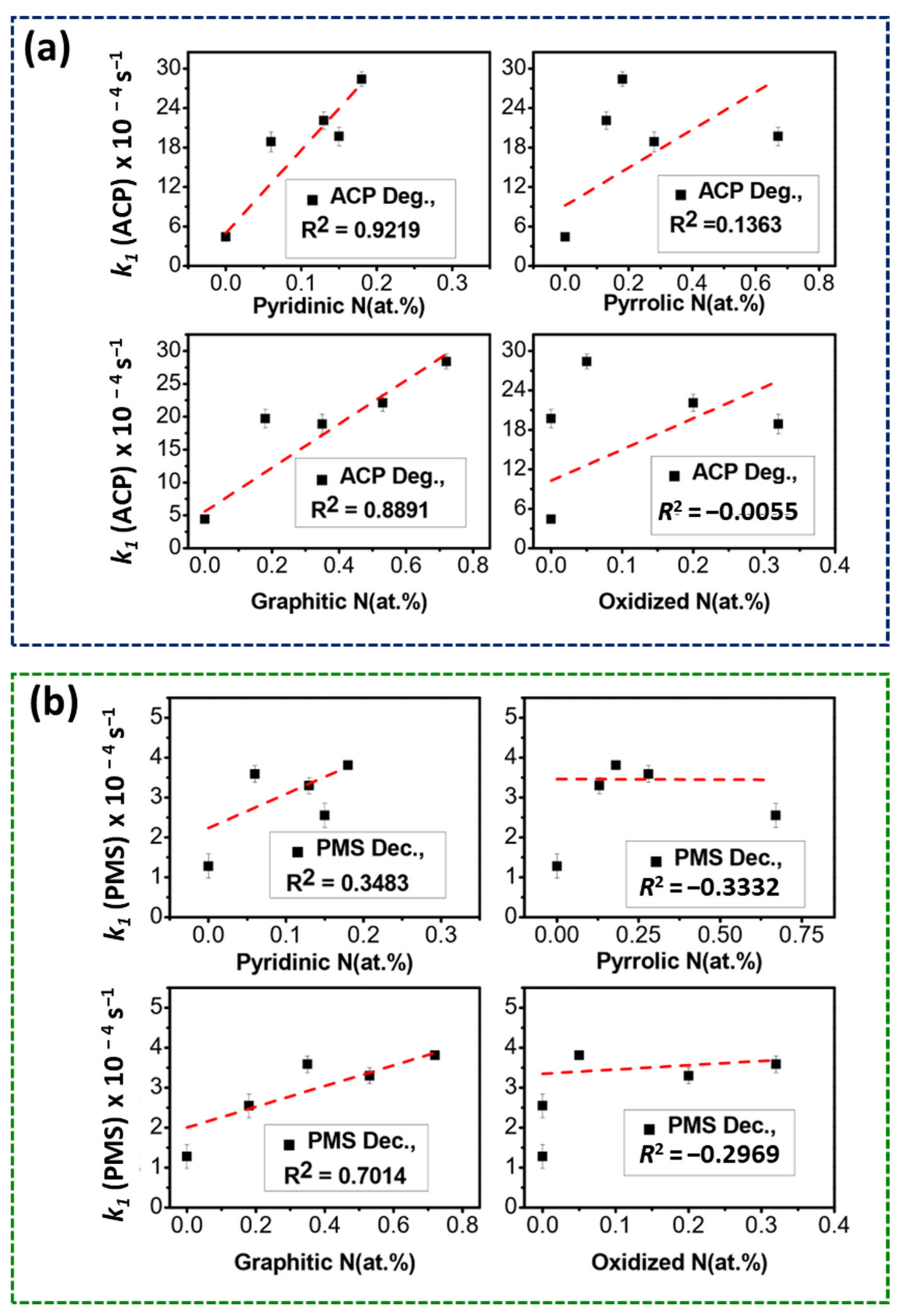
2.3.1. Role of N-Doping on Catalytic Performances
2.3.2. Role of Oxygen Functionalities on Catalytic Performances
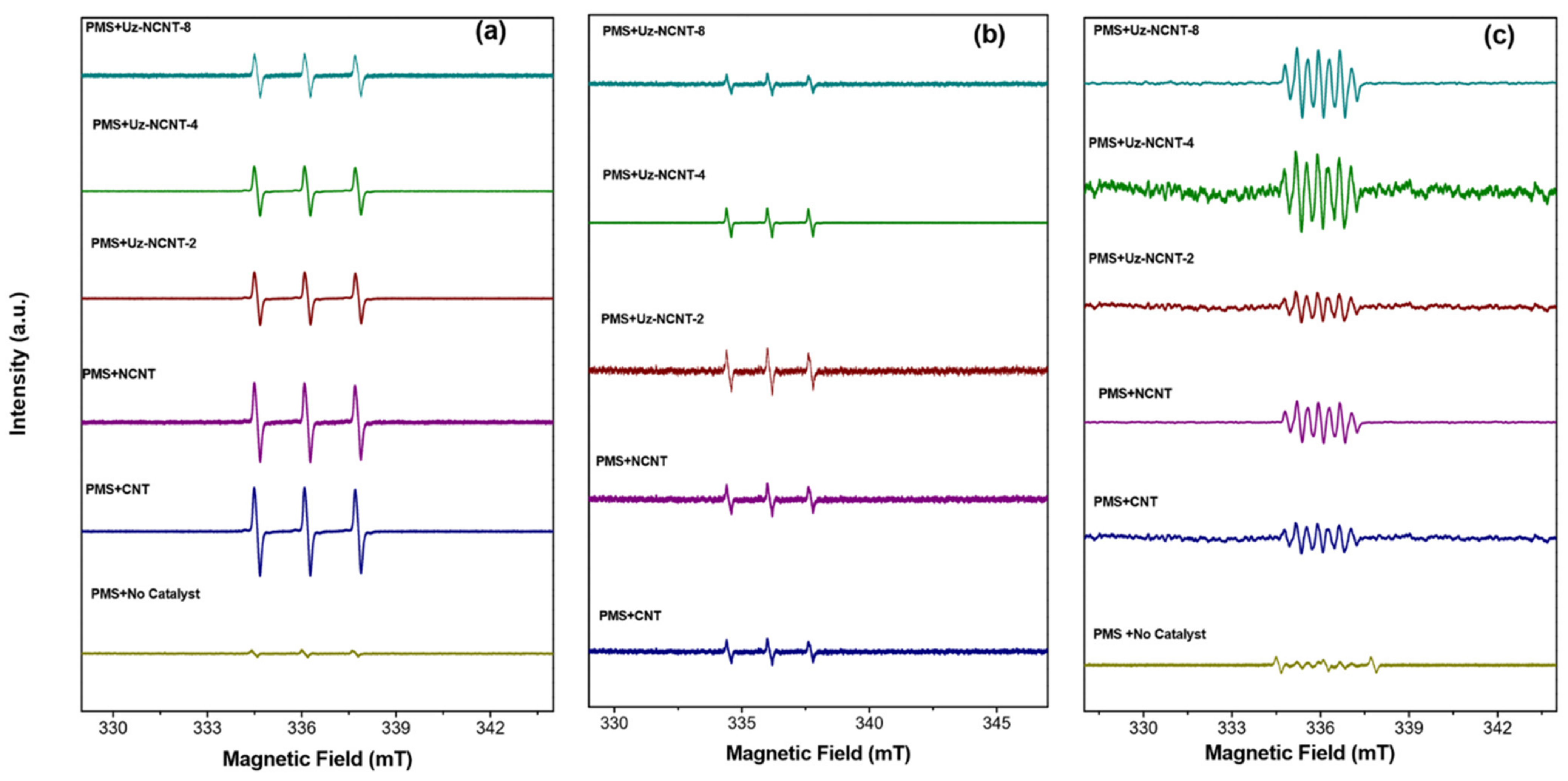
2.4. Identification of Main Reactive Oxidative Species in Carbocatalyst/PMS Systems
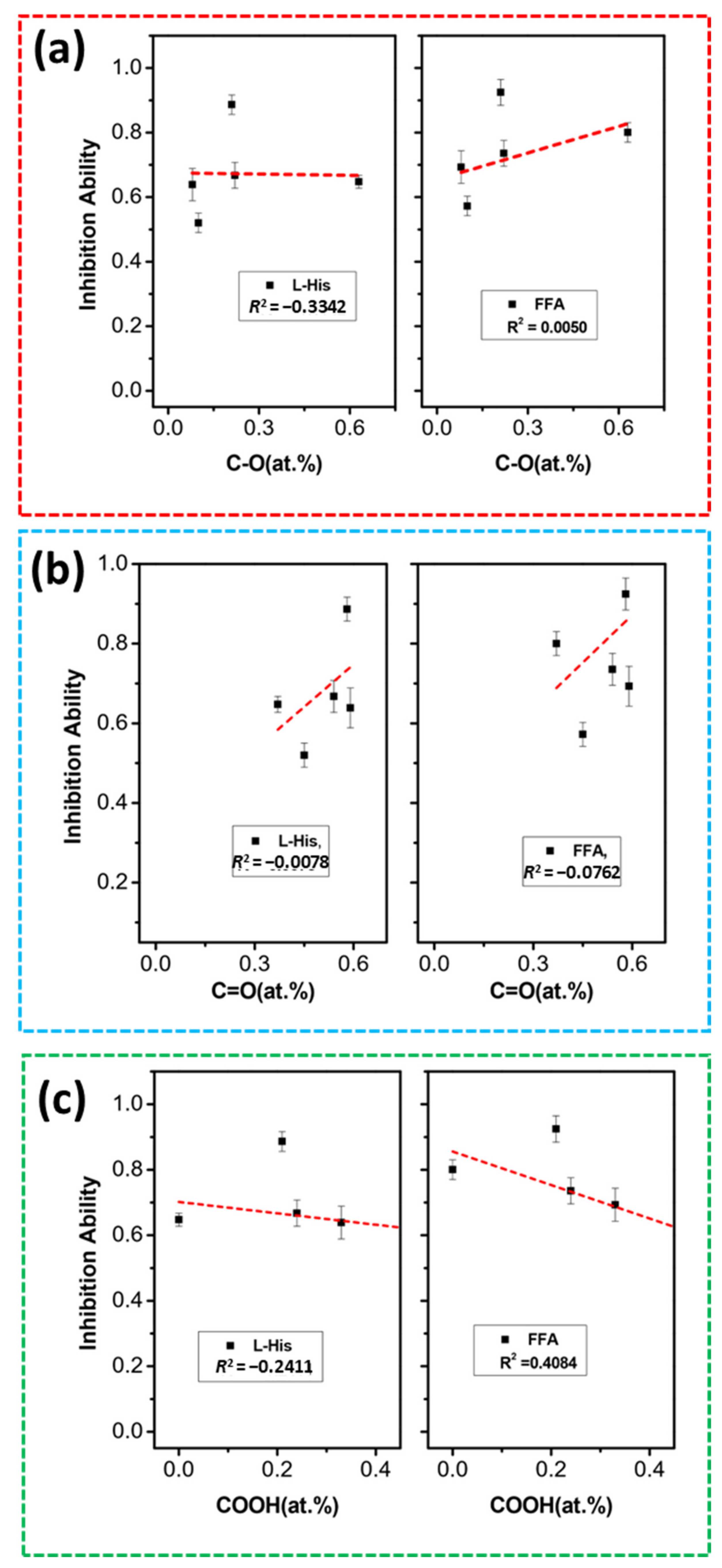
2.5. Classical Quenching Studies
2.5.1. Role of N-Dopants on Radical and Non-Radical Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Scavengers | Molecular Formula | ROS Species | Reaction Rate Constant (M−1 s−1) | References | |
|---|---|---|---|---|---|
| 1 | Ethanol | C2H5OH | SO4• − and •OH | kSO4•− = 9 ×108 kHO• = 1.1 × 106 | [63,71] |
| 2 | tert-Butyl alcohol | C4H10O | •OH | kHO• = 4.5 × 108 | [63] |
| 3 | p-Benzoquinone | C6H4O2 | O2• − | kobs. = 9 ×108 | [64,65] |
| 4 | l-histidine | C6H9N3O2 | 1O2 | kobs. = 3 × 108 | [16,67] |
| 5 | Furfuryl alcohol | C5H6O2 | kobs. = 1.2 × 108 | ||
| 6 | Phenol | C5H5OH | Surface-bound radicals (SO4• − and •OH) | kSO4•− = 8.8 × 109 kHO• = 6.6 × 109 | [68,69] |
| 7 | Potassium iodide | KI | Surface bound complexes | -- | [72,73] |
| 8 | Sodium perchlorate | NaClO4 | Free electrons | -- | [43] |
2.5.2. Role of O-Contents on Radical and Non-Radical Pathway
2.6. Insights into NCNTs/PMS and Unveiling the Active Sites
2.7. Activity Stability Test
3. Materials and Methods
3.1. Materials
3.2. Preparation of NCNT and Uz-NCNTs with Different Degree of Oxidation
3.3. Characterizations of Carbocatalyst
3.4. Evaluation of Catalytic Performance
3.5. Evolution of Reactive Species Quenching Study and EPR Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Oyekunle, D.T.; Zhou, X.; Shahzad, A.; Chen, Z. Review on Carbonaceous Materials as Persulfate Activators: Structure-Performance Relationship, Mechanism and Future Perspectives on Water Treatment. J. Mater. Chem. A 2021, 9, 8012–8050. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, Q.; Ji, G.; Li, A. Degradation of Antibiotic Pollutants by Persulfate Activated with Various Carbon Materials. Chem. Eng. J. 2022, 429, 132387. [Google Scholar] [CrossRef]
- Wu, S.; Yu, L.; Wen, G.; Xie, Z.; Lin, Y. Recent Progress of Carbon-Based Metal-Free Materials in Thermal-Driven Catalysis. J. Energy Chem. 2021, 58, 318–335. [Google Scholar] [CrossRef]
- Huang, W.; Xiao, S.; Zhong, H.; Yan, M.; Yang, X. Activation of Persulfates by Carbonaceous Materials: A Review. Chem. Eng. J. 2021, 418, 129297. [Google Scholar] [CrossRef]
- Chen, Y.; Wei, J.; Duyar, M.S.; Ordomsky, V.V.; Khodakov, A.Y.; Liu, J. Carbon-Based Catalysts for Fischer-Tropsch Synthesis. Chem. Soc. Rev. 2021, 50, 2337–2366. [Google Scholar] [CrossRef]
- Hu, H.; Du, Y.; Duan, X. Functional Materials for Eco-catalysis of Small Molecules. EcoMat 2021, 3, e12121. [Google Scholar] [CrossRef]
- Huang, X.; Wu, S.; Xiao, Z.; Kong, D.; Liang, T.; Li, X.; Luo, B.; Wang, B.; Zhi, L. Predicting the Optimal Chemical Composition of Functionalized Carbon Catalysts towards Oxidative Dehydrogenation of Ethanol to Acetaldehyde. Nano Today 2022, 44, 101508. [Google Scholar] [CrossRef]
- Ding, Y.; Wang, X.; Fu, L.; Peng, X.; Pan, C.; Mao, Q.; Wang, C.; Yan, J. Nonradicals Induced Degradation of Organic Pollutants by Peroxydisulfate (PDS) and Peroxymonosulfate (PMS): Recent Advances and Perspective. Sci. Total Environ. 2021, 765, 142794. [Google Scholar] [CrossRef]
- Duan, X.; Sun, H.; Shao, Z.; Wang, S. Nonradical Reactions in Environmental Remediation Processes: Uncertainty and Challenges. Appl. Catal. B Environ. 2018, 224, 973–982. [Google Scholar] [CrossRef]
- Hu, P.; Su, H.; Chen, Z.; Yu, C.; Li, Q.; Zhou, B.; Alvarez, P.J.J.; Long, M. Selective Degradation of Organic Pollutants Using an Efficient Metal-Free Catalyst Derived from Carbonized Polypyrrole via Peroxymonosulfate Activation. Environ. Sci. Technol. 2017, 51, 11288–11296. [Google Scholar] [CrossRef]
- Zhang, T.; Zhu, H.; Croué, J.P. Production of Sulfate Radical from Peroxymonosulfate Induced by a Magnetically Separable CuFe2O4 Spinel in Water: Efficiency, Stability, and Mechanism. Environ. Sci. Technol. 2013, 47, 2784–2791. [Google Scholar] [CrossRef]
- Anipsitakis, G.P.; Dionysiou, D.D.; Gonzalez, M.A. Cobalt-Mediated Activation of Peroxymonosulfate and Sulfate Radical Attack on Phenolic Compounds. Implications of Chloride Ions. Environ. Sci. Technol. 2006, 40, 1000–1007. [Google Scholar] [CrossRef]
- Anipsitakis, G.P.; Stathatos, E.; Dionysiou, D.D. Heterogeneous Activation of Oxone Using Co3O4. J. Phys. Chem. B 2005, 109, 13052–13055. [Google Scholar] [CrossRef]
- Wang, J.; Wang, S. Activation of Persulfate (PS) and Peroxymonosulfate (PMS) and Application for the Degradation of Emerging Contaminants. Chem. Eng. J. 2018, 334, 1502–1517. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, L.; Dong, Y.; Peng, W.; Fu, Y.; Li, Q.; Fan, Q.; Wang, Y.; Wang, Z. Current Analytical Methods for the Determination of Persulfate in Aqueous Solutions: A Historical Review. Chem. Eng. J. 2021, 416, 129143. [Google Scholar] [CrossRef]
- Chen, X.; Oh, W.D.; Lim, T.T. Graphene- and CNTs-Based Carbocatalysts in Persulfates Activation: Material Design and Catalytic Mechanisms. Chem. Eng. J. 2018, 354, 941–976. [Google Scholar] [CrossRef]
- Cheng, X.; Guo, H.; Zhang, Y.; Wu, X.; Liu, Y. Non-Photochemical Production of Singlet Oxygen via Activation of Persulfate by Carbon Nanotubes. Water Res. 2017, 113, 80–88. [Google Scholar] [CrossRef]
- Liang, P.; Zhang, C.; Duan, X.; Sun, H.; Liu, S.; Tade, M.O.; Wang, S. An Insight into Metal Organic Framework Derived N-Doped Graphene for the Oxidative Degradation of Persistent Contaminants: Formation Mechanism and Generation of Singlet Oxygen from Peroxymonosulfate. Environ. Sci. Nano 2017, 4, 315–324. [Google Scholar] [CrossRef]
- Yun, E.T.; Yoo, H.Y.; Bae, H.; Kim, H.I.; Lee, J. Exploring the Role of Persulfate in the Activation Process: Radical Precursor Versus Electron Acceptor. Environ. Sci. Technol. 2017, 51, 10090–10099. [Google Scholar] [CrossRef]
- Lee, H.; Lee, H.J.; Jeong, J.; Lee, J.; Park, N.B.; Lee, C. Activation of Persulfates by Carbon Nanotubes: Oxidation of Organic Compounds by Nonradical Mechanism. Chem. Eng. J. 2015, 266, 28–33. [Google Scholar] [CrossRef]
- Wang, X.; Qin, Y.; Zhu, L.; Tang, H. Nitrogen-Doped Reduced Graphene Oxide as a Bifunctional Material for Removing Bisphenols: Synergistic Effect between Adsorption and Catalysis. Environ. Sci. Technol. 2015, 49, 6855–6864. [Google Scholar] [CrossRef]
- Duan, X.; Su, C.; Zhou, L.; Sun, H.; Suvorova, A.; Odedairo, T.; Zhu, Z.; Shao, Z.; Wang, S. Surface Controlled Generation of Reactive Radicals from Persulfate by Carbocatalysis on Nanodiamonds. Appl. Catal. B Environ. 2016, 194, 7–15. [Google Scholar] [CrossRef]
- Lee, J.; Von Gunten, U.; Kim, J.H. Persulfate-Based Advanced Oxidation: Critical Assessment of Opportunities and Roadblocks. Environ. Sci. Technol. 2020, 54, 3064–3081. [Google Scholar] [CrossRef]
- Pham, V.L.; Kim, D.G.; Ko, S.O. Catalytic Degradation of Acetaminophen by Fe and N Co-Doped Multi-Walled Carbon Nanotubes. Environ. Res. 2021, 201, 111535. [Google Scholar] [CrossRef]
- Kim, D.G.; Ko, S.O. Advanced Oxidative Degradation of Acetaminophen by Carbon Catalysts: Radical vs Non-Radical Pathways. Environ. Res. 2020, 188, 109767. [Google Scholar]
- Li, J.; Li, M.; Sun, H.; Ao, Z.; Wang, S.; Liu, S. Understanding of the Oxidation Behavior of Benzyl Alcohol by Peroxymonosulfate via Carbon Nanotubes Activation. ACS Catal. 2020, 10, 3516–3525. [Google Scholar] [CrossRef]
- Duan, X.; Sun, H.; Wang, S. Metal-Free Carbocatalysis in Advanced Oxidation Reactions. Acc. Res. 2018, 51, 678–687. [Google Scholar] [CrossRef]
- Duan, X.; Ao, Z.; Zhou, L.; Sun, H.; Wang, G.; Wang, S. Occurrence of Radical and Nonradical Pathways from Carbocatalysts for Aqueous and Nonaqueous Catalytic Oxidation. Appl. Catal. B Environ. 2016, 188, 98–105. [Google Scholar] [CrossRef]
- Ma, W.; Wang, N.; Fan, Y.; Tong, T.; Han, X.; Du, Y. Non-Radical-Dominated Catalytic Degradation of Bisphenol A by ZIF-67 Derived Nitrogen-Doped Carbon Nanotubes Frameworks in the Presence of Peroxymonosulfate. Chem. Eng. J. 2018, 336, 721–731. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, L.; Huang, T.; Li, W.; Wang, Y.; Wang, Z. Decolorization of Azo Dye by Peroxymonosulfate Activated by Carbon Nanotube: Radical versus Non-Radical Mechanism. J. Hazard. Mater. 2016, 320, 571–580. [Google Scholar] [CrossRef]
- Yan, Y.; Miao, J.; Yang, Z.; Xiao, F.X.; Yang, H.B.; Liu, B.; Yang, Y. Carbon Nanotube Catalysts: Recent Advances in Synthesis, Characterization and Applications. Chem. Soc. Rev. 2015, 44, 3295–3346. [Google Scholar] [CrossRef] [PubMed]
- Guan, C.; Jiang, J.; Luo, C.; Pang, S.; Yang, Y.; Wang, Z.; Ma, J.; Yu, J.; Zhao, X. Oxidation of Bromophenols by Carbon Nanotube Activated Peroxymonosulfate (PMS) and Formation of Brominated Products: Comparison to Peroxydisulfate (PDS). Chem. Eng. J. 2018, 337, 40–50. [Google Scholar] [CrossRef]
- Duan, X.; Sun, H.; Wang, Y.; Kang, J.; Wang, S. N-Doping-Induced Nonradical Reaction on Single-Walled Carbon Nanotubes for Catalytic Phenol Oxidation. ACS Catal. 2015, 5, 553–559. [Google Scholar] [CrossRef]
- Ren, W.; Nie, G.; Zhou, P.; Zhang, H.; Duan, X.; Wang, S. The Intrinsic Nature of Persulfate Activation and N-Doping in Carbocatalysis. Environ. Sci. Technol. 2020, 54, 6438–6447. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Kwan, C.K.; Suvorova, A.; Ang, H.M.; Tadé, M.O.; Wang, S. Catalytic Oxidation of Organic Pollutants on Pristine and Surface Nitrogen-Modified Carbon Nanotubes with Sulfate Radicals. Appl. Catal. B Environ. 2014, 154–155, 134–141. [Google Scholar] [CrossRef]
- Duan, X.; Ao, Z.; Sun, H.; Zhou, L.; Wang, G.; Wang, S. Insights into N-Doping in Single-Walled Carbon Nanotubes for Enhanced Activation of Superoxides: A Mechanistic Study. Chem. Commun. 2015, 51, 15249–15252. [Google Scholar] [CrossRef]
- Wang, C.; Kang, J.; Liang, P.; Zhang, H.; Sun, H.; Tadé, M.O.; Wang, S. Ferric Carbide Nanocrystals Encapsulated in Nitrogen-Doped Carbon Nanotubes as an Outstanding Environmental Catalyst. Environ. Sci. Nano 2017, 4, 170–179. [Google Scholar] [CrossRef]
- Yang, Q.; Chen, Y.; Duan, X.; Zhou, S.; Niu, Y.; Sun, H.; Zhi, L.; Wang, S. Unzipping Carbon Nanotubes to Nanoribbons for Revealing the Mechanism of Nonradical Oxidation by Carbocatalysis. Appl. Catal. B Environ. 2020, 276, 119146. [Google Scholar] [CrossRef]
- Xiao, B.; Li, X.; Li, X.; Wang, B.; Langford, C.; Li, R.; Sun, X. Graphene Nanoribbons Derived from the Unzipping of Carbon Nanotubes: Controlled Synthesis and Superior Lithium Storage Performance. J. Phys. Chem. C 2014, 118, 881–890. [Google Scholar] [CrossRef]
- Liu, M.; Song, Y.; He, S.; Tjiu, W.W.; Pan, J.; Xia, Y.Y.; Liu, T. Nitrogen-Doped Graphene Nanoribbons as Efficient Metal-Free Electrocatalysts for Oxygen Reduction. ACS Appl. Mater. Interfaces 2014, 6, 4214–4222. [Google Scholar] [CrossRef]
- Wacławek, S.; Grübel, K.; Černík, M. Simple Spectrophotometric Determination of Monopersulfate. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 149, 928–933. [Google Scholar] [CrossRef]
- Li, H.; Zhang, J.; Gholizadeh, A.B.; Brownless, J.; Fu, Y.; Cai, W.; Han, Y.; Duan, T.; Wang, Y.; Ling, H.; et al. Photoluminescent Semiconducting Graphene Nanoribbons via Longitudinally Unzipping Single-Walled Carbon Nanotubes. ACS Appl. Mater. Interfaces 2021, 13, 52892–52900. [Google Scholar] [CrossRef]
- Chen, X.; Oh, W.D.; Hu, Z.T.; Sun, Y.M.; Webster, R.D.; Li, S.Z.; Lim, T.T. Enhancing Sulfacetamide Degradation by Peroxymonosulfate Activation with N-Doped Graphene Produced through Delicately-Controlled Nitrogen Functionalization via Tweaking Thermal Annealing Processes. Appl. Catal. B Environ. 2018, 225, 243–257. [Google Scholar] [CrossRef]
- Chang, S.S.; Clair, B.; Ruelle, J.; Beauchêne, J.; Di Renzo, F.; Quignard, F.; Zhao, G.J.; Yamamoto, H.; Gril, J. Mesoporosity as a New Parameter for Understanding Tension Stress Generation in Trees. J. Exp. Bot. 2009, 60, 3023–3030. [Google Scholar] [CrossRef]
- Groen, J.C.; Peffer, L.A.A.; Pérez-Ramírez, J. Pore Size Determination in Modified Micro- and Mesoporous Materials. Pitfalls and Limitations in Gas Adsorption Data Analysis. Microporous Mesoporous Mater. 2003, 60, 1–17. [Google Scholar] [CrossRef]
- Duan, X.; Ao, Z.; Sun, H.; Indrawirawan, S.; Wang, Y.; Kang, J.; Liang, F.; Zhu, Z.H.; Wang, S. Nitrogen-Doped Graphene for Generation and Evolution of Reactive Radicals by Metal-Free Catalysis. ACS Appl. Mater. Interfaces 2015, 7, 4169–4178. [Google Scholar] [CrossRef]
- Duan, X.; Indrawirawan, S.; Sun, H.; Wang, S. Effects of Nitrogen-, Boron-, and Phosphorus-Doping or Codoping on Metal-Free Graphene Catalysis. Catal. Today 2015, 249, 184–191. [Google Scholar] [CrossRef]
- Krishnamoorthy, K.; Veerapandian, M.; Yun, K.; Kim, S.J. The Chemical and Structural Analysis of Graphene Oxide with Different Degrees of Oxidation. Carbon 2013, 53, 38–49. [Google Scholar] [CrossRef]
- Dash, S.; Patel, S.; Mishra, B.K. Oxidation by Permanganate: Synthetic and Mechanistic Aspects. Tetrahedron 2009, 65, 707–739. [Google Scholar] [CrossRef]
- Russ, M.T. Dimanganese Heptoxide for the Selective Oxidaion of Organic Substrates. Angew. Chem. Int. Ed. 1987, 26, 1007–1009. [Google Scholar]
- Ma, Y.; Wu, X.; Yu, M.; Li, S.; Liu, J. Turning free-standing three dimensional graphene into electrochemically active by nitrogen doping during chemical vapor deposition process. J. Mater. Sci. Mater. Electron. 2020, 31, 3759–3768. [Google Scholar] [CrossRef]
- Luo, Z.; Lim, S.; Tian, Z.; Shang, J.; Lai, L.; MacDonald, B.; Fu, C.; Shen, Z.; Yu, T.; Lin, J. Pyridinic N Doped Graphene: Synthesis, Electronic Structure, and Electrocatalytic Property. J. Mater. Chem. 2011, 21, 8038–8044. [Google Scholar] [CrossRef]
- Ren, W.; Xiong, L.; Nie, G.; Zhang, H.; Duan, X.; Wang, S. Insights into the Electron-Transfer Regime of Peroxydisulfate Activation on Carbon Nanotubes: The Role of Oxygen Functional Groups. Environ. Sci. Technol. 2020, 54, 1267–1275. [Google Scholar] [CrossRef]
- Frank, B.; Zhang, J.; Blume, R.; Schlögl, R.; Su, D.S. Heteroatoms Increase the Selectivity in Oxidative Dehydrogenation Reactions on Nanocarbons. Angew. Chem. Int. Ed. 2009, 48, 6913–6917. [Google Scholar] [CrossRef]
- Frank, B.; Blume, R.; Rinaldi, A.; Trunschke, A.; Schlögl, R. Oxygen Insertion Catalysis by Sp2 Carbon. Angew. Chem. Int. Ed. 2011, 50, 10226–10230. [Google Scholar] [CrossRef]
- Liu, S.; Peng, W.; Sun, H.; Wang, S. Physical and Chemical Activation of Reduced Graphene Oxide for Enhanced Adsorption and Catalytic Oxidation. Nanoscale 2014, 6, 766–771. [Google Scholar] [CrossRef]
- Gao, Y.; Chen, Z.; Zhu, Y.; Li, T.; Hu, C. New Insights into the Generation of Singlet Oxygen in the Metal-Free Peroxymonosulfate Activation Process: Important Role of Electron-Deficient Carbon Atoms. Environ. Sci. Technol. 2020, 54, 1232–1241. [Google Scholar] [CrossRef]
- Gao, Y.; Zhu, Y.; Chen, Z.; Zeng, Q.; Hu, C. Insights into the Difference in Metal-Free Activation of Peroxymonosulfate and Peroxydisulfate. Chem. Eng. J. 2020, 394, 123936. [Google Scholar] [CrossRef]
- Tian, X.; Gao, P.; Nie, Y.; Yang, C.; Zhou, Z.; Li, Y.; Wang, Y. A Novel Singlet Oxygen Involved Peroxymonosulfate Activation Mechanism for Degradation of Ofloxacin and Phenol in Water. Chem. Commun. 2017, 53, 6589–6592. [Google Scholar] [CrossRef]
- Tang, W.; Zhang, Y.; Guo, H.; Liu, Y. Heterogeneous Activation of Peroxymonosulfate for Bisphenol AF Degradation with BiOI0.5Cl0.5. RSC Adv. 2019, 9, 14060–14071. [Google Scholar] [CrossRef]
- Kim, D.G.; Ko, S.O. Effects of thermal modification of a biochar on persulfate activation and mechanisms of catalytic degradation of a pharmaceutical. Chem. Eng. J. 2020, 399, 125377. [Google Scholar] [CrossRef]
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical Review of Rate Constants for Reactions of Hydrated Electrons, Hydrogen Atoms and Hydroxyl Radicals (⋅OH/⋅O− in Aqueous Solution. J. Phys. Chem. Ref. Data 1988, 17, 513–886. [Google Scholar] [CrossRef]
- Eibenberger, H.; Steenken, S.; O’Neill, P.; Schulte-Frohlinde, D. Pulse Radiolysis and Electron Spin Resonance. J. Phys. Chem. C 1978, 82, 749–750. [Google Scholar] [CrossRef]
- Greenstock, C.L.; Ruddock, G.W. Determination of Superoxide (O2-) Radical Anion Reaction Rates Using Pulse Radiolysis. Int. J. Radiat. Phys. Chem. 1976, 8, 367–369. [Google Scholar] [CrossRef]
- Manring, L.E.; Kramer, M.K.; Foote, C.S. Interception of O2- by Benzoquinone in Cyanoaromatic-Sensitized Photooxygenations. Tetrahedron Lett. 1984, 25, 2523–2526. [Google Scholar] [CrossRef]
- Wilkinson, F.; Brummer, J.G. Rate Constants for the Decay and Reactions of the Lowest Electronically Excited Singlet State of Molecular Oxygen in Solution. J. Phys. Chem. Ref. Data 1981, 10, 809–999. [Google Scholar] [CrossRef]
- Haag, W.R.; Hoigné, J. Singlet Oxygen in Surface Waters. 3. Photochemical Formation and Steady-State Concentrations in Various Types of Waters. Environ. Sci. Technol. 1986, 20, 341–348. [Google Scholar] [CrossRef]
- Lindsey, M.E.; Tarr, M.A. Inhibition of Hydroxyl Radical Reaction with Aromatics by Dissolved Natural Organic Matter. Environ. Sci. Technol. 2000, 34, 444–449. [Google Scholar] [CrossRef]
- Ziajka, J.; Pasiuk-Bronikowska, W. Rate Constants for Atmoshpheric Trace Organics Scavenging SO4- in the Fe-Catalysed Autoxidation of S(IV). Atmos. Environ. 2005, 39, 1431–1438. [Google Scholar] [CrossRef]
- Lin, Y.; Sun, X.; Su, D.S.; Centi, G.; Perathoner, S. Catalysis by Hybrid Sp2/Sp3 Nanodiamonds and Their Role in the Design of Advanced Nanocarbon Materials. Chem. Soc. Rev. 2018, 47, 8438–8473. [Google Scholar] [CrossRef]
- Buxton, G.V.; Elliot, A.J. Rate Constant for Reaction of Hydroxyl Radicals with Bicarbonate Ions. Int. J. Radiat. Appl. Instrum. Part C 1986, 27, 241–243. [Google Scholar] [CrossRef]
- Liang, J.; Xu, X.; Qamar Zaman, W.; Hu, X.; Zhao, L.; Qiu, H.; Cao, X. Different Mechanisms between Biochar and Activated Carbon for the Persulfate Catalytic Degradation of Sulfamethoxazole: Roles of Radicals in Solution or Solid Phase. Chem. Eng. J. 2019, 375, 121908. [Google Scholar] [CrossRef]
- Huang, Z.; Bao, H.; Yao, Y.; Lu, W.; Chen, W. Novel Green Activation Processes and Mechanism of Peroxymonosulfate Based on Supported Cobalt Phthalocyanine Catalyst. Appl. Catal. B Environ. 2014, 154–155, 36–43. [Google Scholar] [CrossRef]
- Duan, X.; Sun, H.; Ao, Z.; Zhou, L.; Wang, G.; Wang, S. Unveiling the Active Sites of Graphene-Catalyzed Peroxymonosulfate Activation. Carbon 2016, 107, 371–378. [Google Scholar] [CrossRef]
- Evans, D.F.; Upton, M.W. Studies on Singlet Oxygen in Aqueous Solution. Part 3. The Decomposition of Peroxy-Acis. J. Chem. Soc. Dalton Trans. 1985, 6, 1151–1153. [Google Scholar] [CrossRef]











| Carbocatalysts | N/at. % | N/at. % | |||
|---|---|---|---|---|---|
| Pyridinic N | Pyrrolic N | Graphitic N | Oxidized N | ||
| CNT | NA | 0.00 | 0.00 | 0.00 | 0.00 |
| NCNT | 0.93 | 0.06 | 0.28 | 0.35 | 0.32 |
| Uz-NCNT-2 | 1.07 | 0.13 | 0.13 | 0.53 | 0.20 |
| Uz-NCNT-4 | 1.04 | 0.18 | 0.18 | 0.72 | 0.05 |
| Uz-NCNT-8 | 0.62 | 0.15 | 0.67 | 0.18 | 0.00 |
| C at. % | O at. % | O/at. % | |||
| C-O | C=O | COOH | |||
| CNT | 99.11 | 0.89 | 0.63 | 0.37 | 0 |
| NCNT | 96.04 | 3.04 | 0.21 | 0.58 | 0.21 |
| Uz-NCNT-2 | 77.43 | 21.5 | 0.22 | 0.54 | 0.24 |
| Uz-NCNT-4 | 74.39 | 24.58 | 0.08 | 0.59 | 0.33 |
| Uz-NCNT-8 | 67.22 | 32.16 | 0.10 | 0.45 | 0.46 |
| Carbocatalyst/PMS Activation Pathway | Reactive Oxidative Species | Governing N-Dopants | Correlation Coefficient (R2) | Governing O-Groups | Correlation Coefficient (R2) |
|---|---|---|---|---|---|
| Radical Species | •OH/SO4• − | Pyridinic N | 0.7359 (Ethanol) 0.7309 (tert-BA) | --- | --- |
| O2• − | Graphitic N | 0.9259 (p-BQ) | -C=O | 0.9827 (p-BQ) | |
| Singlet Oxygen | 1O2 | --- | --- | --- | --- |
| Non-Radical Species | Free flowing electrons | Pyridinic N | 0.8929 (NaClO4) | -C-O | 0.7485 (NaClO4) (Downtrend) |
| Surface-bound radicals | Graphitic N | 0.8572 (KI) | -C=O | 0.9472 (KI) | |
| Carbocatalyst-PMS* activated complex | Pyridinic N | 0.8901 (Phenol) | -C-O | 0.8932 (Phenol) (Downtrend) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Govindan, K.; Kim, D.-G.; Ko, S.-O. Role of N-Doping and O-Groups in Unzipped N-Doped CNT Carbocatalyst for Peroxomonosulfate Activation: Quantitative Structure–Activity Relationship. Catalysts 2022, 12, 845. https://doi.org/10.3390/catal12080845
Govindan K, Kim D-G, Ko S-O. Role of N-Doping and O-Groups in Unzipped N-Doped CNT Carbocatalyst for Peroxomonosulfate Activation: Quantitative Structure–Activity Relationship. Catalysts. 2022; 12(8):845. https://doi.org/10.3390/catal12080845
Chicago/Turabian StyleGovindan, Kadarkarai, Do-Gun Kim, and Seok-Oh Ko. 2022. "Role of N-Doping and O-Groups in Unzipped N-Doped CNT Carbocatalyst for Peroxomonosulfate Activation: Quantitative Structure–Activity Relationship" Catalysts 12, no. 8: 845. https://doi.org/10.3390/catal12080845
APA StyleGovindan, K., Kim, D. -G., & Ko, S. -O. (2022). Role of N-Doping and O-Groups in Unzipped N-Doped CNT Carbocatalyst for Peroxomonosulfate Activation: Quantitative Structure–Activity Relationship. Catalysts, 12(8), 845. https://doi.org/10.3390/catal12080845