

Catalytic Properties of the Spinel-Like CuxMn3−xO4 Copper Manganese Oxides—An Overview



Abstract
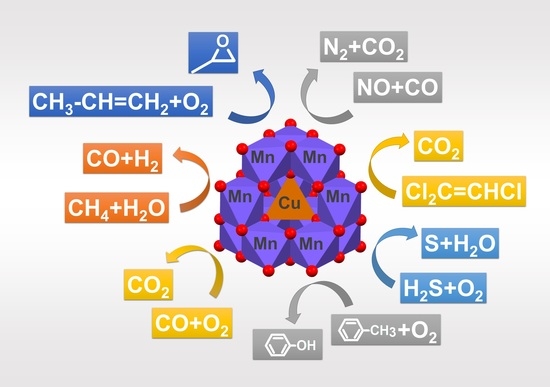
:
1. Introduction
2. Preparation Routes of CuxMn3−xO4 Spinels
- -
- precipitation of copper and manganese hydroxides or carbonates or oxalates directly from aqueous solutions with bases or with special techniques such as spray drying, immersion, or alginate xerogel formation
- -
- hydrothermal and solvothermal methods with the transformation of the precursors containing metal into reactive materials; these methods include hydrolysis or co-precipitation with alkaline materials, such as ammonia that formed from the hydrolysis of urea or hexamethylenetetramine
- -
- solution and solid–phase redox reactions, such as oxidation of low-valence Cu or Mn compounds with permanganate or other oxidants, with subsequent heat treatment or the thermal decomposition of copper permanganate or its complexes that have reducing ligands such as ammonia.
2.1. Ceramic and Related Processes from Solid Precursors
2.2. In Situ Precipitation of Solid Precursors from Solutions
2.3. Thermal Decomposition of the Hydrated and Ammonia-Complexed Copper Permanganate
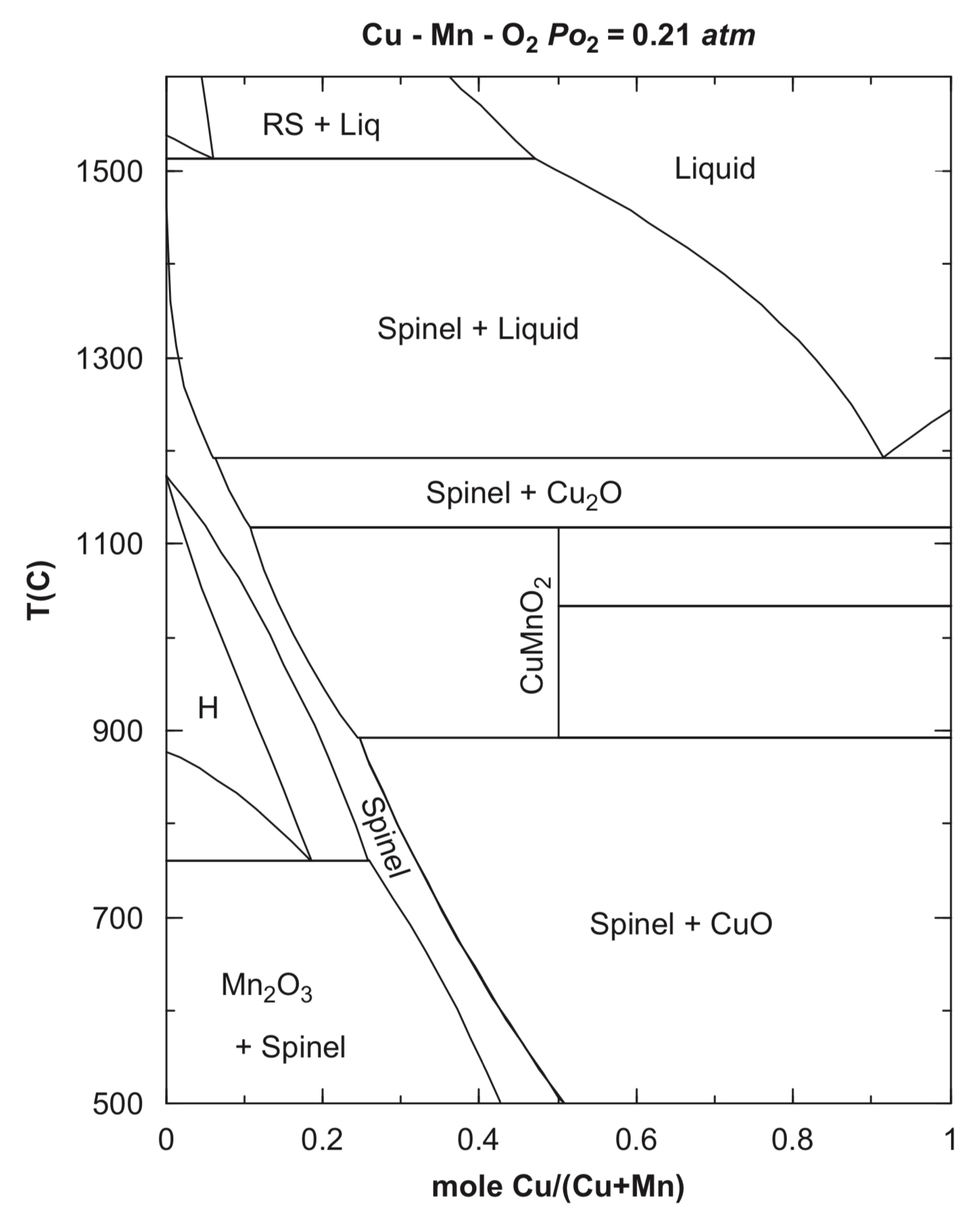
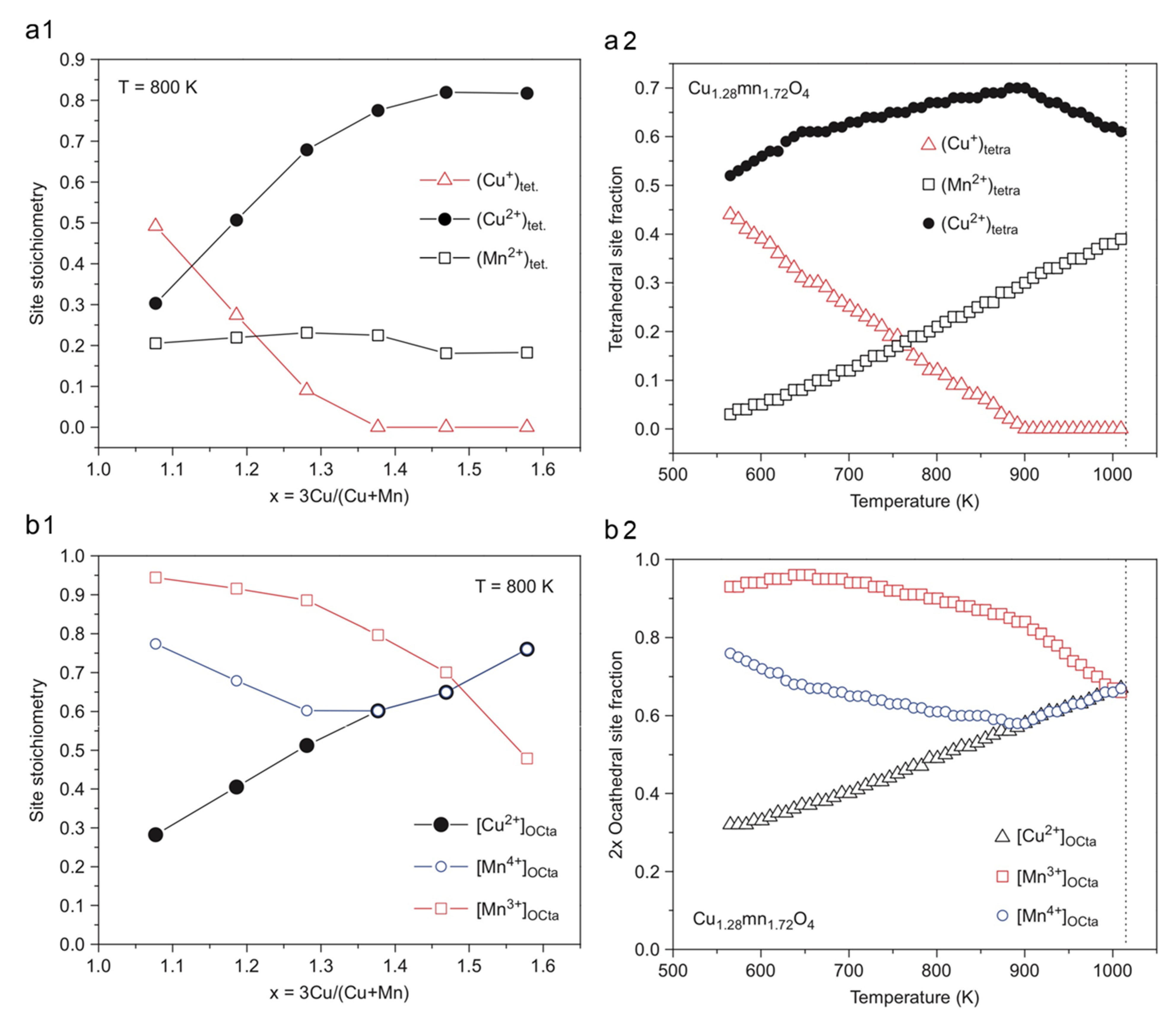
3. Composition and Crystal Structure of CuxMn3−xO4 Spinels
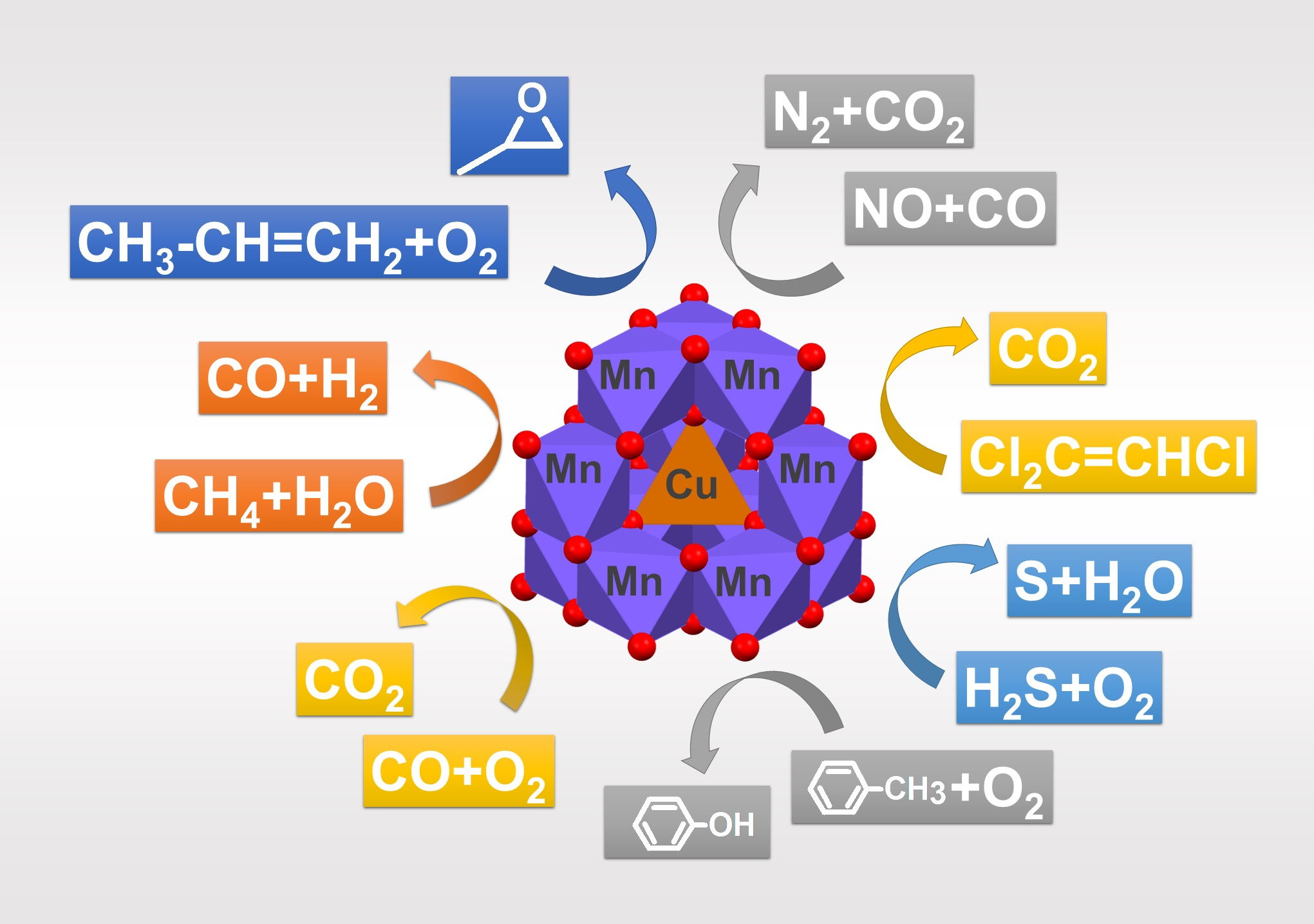
4. Overview of the Catalytic Activity of CuxMn3−xO4 Spinels
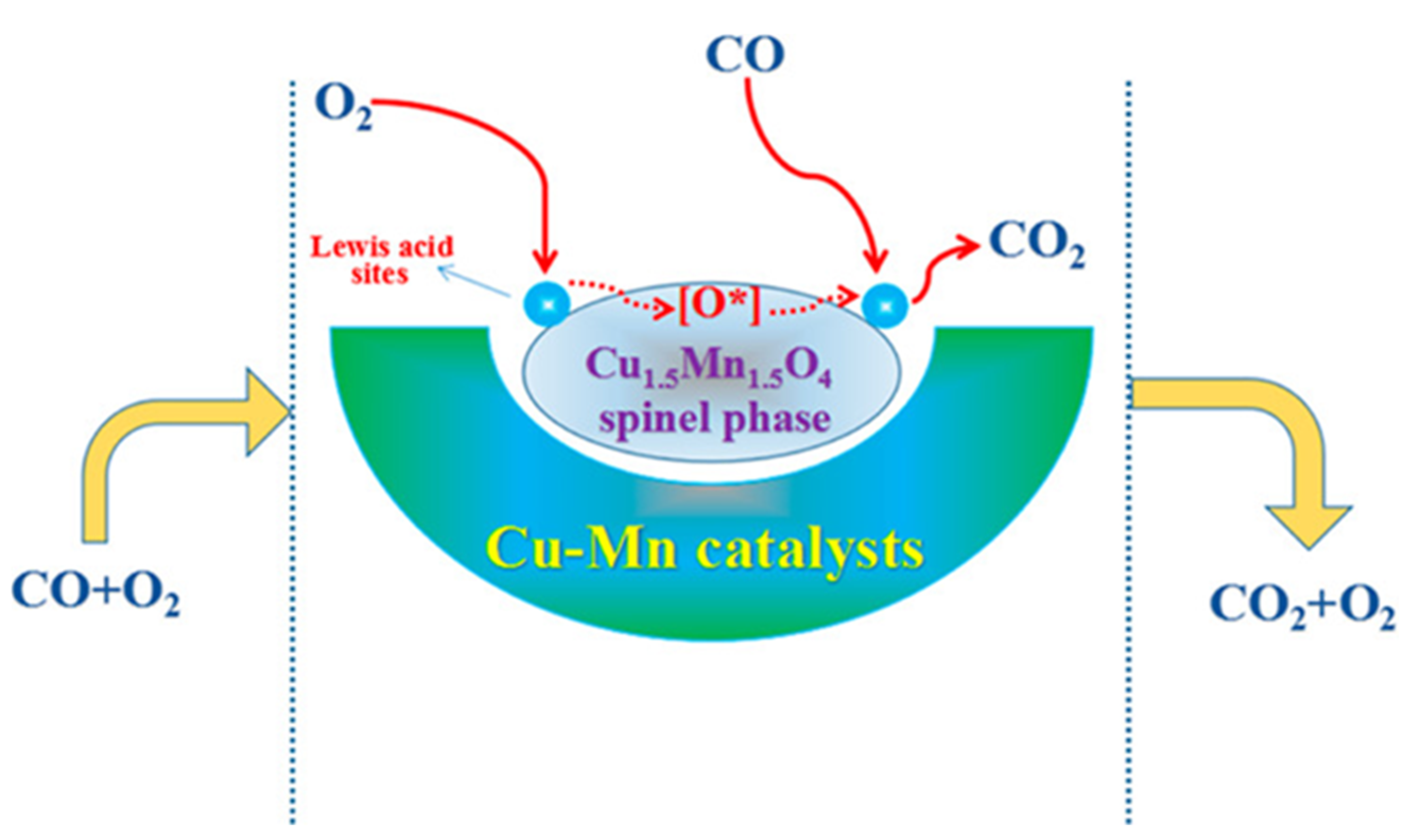
4.1. Removal of Carbon Monoxide from the Air
- (1)
- Molecular oxygen is preferentially adsorbed on the spinel surface and subsequently forms surface-active species containing adsorbed oxygen (O*) such as O2−(ads) and O−(ads).
- (2)
- CO molecules are oxidized by O* species with the formation of gaseous carbon dioxide. The two-step reaction can be formulated as e.g., CO + O−(ads) → CO2 + e− and CO + 2O−(ads) → CO32−(ads).
4.2. Removal of NO from Air by Carbon Monoxide
4.3. Removal of Other Pollutants from the Air and Other Gases
4.4. Removal of Aromatic Hydrocarbons from Air

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CuxMn3−xO4 | Precursors, Preparation Method, and Conditions | Catalyst Efficiency and Reaction Conditions | Refs. | Remarks |
|---|---|---|---|---|
| x = 1.0 | Cu–nitrate, Mn–acetate, Cu:Mn = 1:2, γ–Al2O3, 500 °C for 5 h | 1000 ppm toluene in 7:3 N2:O2, 120,000 h−1 space velocity, T50/T90 = 369/474 °C, resp. | [76] | Supported on Al2O3, SBET = ~101.3 m2 g−1 |
| x = 1.0 | Cu and Mn(II) acetate, co-precipitation at 80 °C with aq. Na2CO3, 500 °C | 0.35% toluene and 9% O2 in Ar, 250 °C | [49] | Mixed phase, SBET = 73 m2 g−1 |
| x = 1.0 | Cu and Mn(II) acetate, hydrothermal treatment in 10 M NaOH at 110 °C for 20 h, 250 °C | 0.35% toluene and 9% O2 in Ar, GHSV 36,000, 210 °C; T10/T50/T95 are 150, 170, 200 °C, resp. | [49] | Nanorods, mixed phase, SBET = 221 m2 g−1 |
| x = 1.0 | Mn and Cu nitrates, co-precipitation with Me4NOH, 400 °C for 5 h | 800 ppm toluene in air, 150 °C for 24 h, GHSV = 27,800 mL g−1 h−1, T10/T50/T90 = 185/195/200 °C, resp. | [26] | SBET = 48 m2 g−1, Vp = 0.30 cm3 g−1 |
| x = 1.2 | Cu–nitrate, Mn–acetate, Cu:Mn = 1:0.5, γ–Al2O3, 500 °C for 5 h | 1000 ppm toluene in 7:3 N2:O2, 120,000 h−1 space velocity, T50/T90 = 330/383 °C, resp. | [76] | Supported on Al2O3, SBET = ~109.2 m2 g−1 |
| x = 1.3 | Cu–nitrate, Mn–acetate, Cu:Mn = 1:1.5, γ–Al2O3, 500 °C for 5 h | 1000 ppm toluene in 7:3 N2:O2, 120,000 h−1 space velocity, T50/T90 = 271–303/293–329 °C, resp., depending on loaded amount on Al2O3 | [76] | Supported on Al2O3, SBET = ~99.0 m2 g−1, TOF = 0.05–0.28 |
| x = 1.4 | Mn and Cu–nitrates, anatase, incipient wetness impregnation, 500 °C for 7 h in air | Atmospheric pressure, 500 ppm toluene in air, 150–300 °C, 5 h, GHSV 5000 h−1 | [18] | Mixed phase, SBET = 34–48 m2 g−1, supported on TiO2 |
| x = 1.4 | Cu:Mn = 1:1, MnCl2, CuCl2, Na2CO3, 450 °C for 5 h | 500 ppm toluene in humid air, GHSV = 50,000 mL g−1 h−1, T50/T90 =265/280 °C, resp., reaction rate = 0.001 mmol g−1 h−1 and 0.005 mmol m−2 h−1 at 200 °C | [13] | SBET = 18 m2 g−1, Pv = 0.02 cm3 g−1, APD = 13 |
| x = 1.5 | Cu–nitrate, Mn–acetate, Cu:Mn = 1:2, γ–Al2O3, 500 °C for 5 h | 1000 ppm toluene in 7:3 N2:O2, 120,000 h−1 space velocity, T50/T90 = 300/343 °C, resp. | [76] | Supported on Al2O3 SBET = ~107.4 m2 g−1 |
| x = 1.5 | ionotropic sodium alginate, metal chlorides, alcoholic dehydration, and calcining the xerogel at 450 °C | Toluene in dry air, 1000 ppm, GHSV = 46,000 mL g−1 h−1, reaction rate = 1.03 mmol g−1 h−1 and 24.52 mmol m−2 h−1 at 200 °C | [38] | cubic, ∼10 nm size, SBET = 42 m2 g−1 |
| x = 1.5 | Mn–acetate, Cu–nitrate, 1.5 M ammonium carbonate, pH = 8, 2 h, calcining at 550 C for 2 h in air | 1000 ppm toluene in air, GHSV 30,000 h−1, T50/T90 = 245/274 °C, resp. | [37] | Mixed phase, undoped, SBET = 784.1 m2 g−1, Vp (meso) = 0.246 and Vp (all) 0.25 cm3 g−1 |
| x = 1.5 | Mn–acetate, Cu–nitrate, La–nitrate, 1.5 M ammonium carbonate, pH = 8, 2 h, calcining at 550 C for 2 h in air | 1000 ppm toluene in air, GHSV 30,000 h−1. T50/T90 for 2, 4, and 6% La content were 218/268, 217/255, and –/280 °C, respectively. | [37] | Mixed phase, 4% La-doping, SBET = 164.2 m2 g−1, Vp (meso) 0.426 and Vp (all) 0.45 cm3 g−1 |
5. Copper Manganese Oxides as Catalysts in Industrial Production Processes
5.1. Copper Manganese Oxide Spinel-Catalyzed Reactions of CO:H2 Mixtures
5.2. Synthesis of Methyl Formate
5.3. Methanol Steam Reforming Process
5.4. Other Industrial Processes Catalyzed by CuxMn3−xO4 Spinels
6. Characterization and Operando Techniques Used in the Studying of CuxMn3−xO4 Spinel Catalysts
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zhang, K.; Ding, H.; Pan, W.; Mu, X.; Qiu, K.; Ma, J.; Zhao, Y.; Song, J.; Zhang, Z. Research Progress of a Composite Metal Oxide Catalyst for VOC Degradation. Environ. Sci. Technol. 2022, 56, 9220–9236. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, L.; Ling, H.; Ge, Z.; Lin, X.; Dai, X.; Chen, H. Critical review of thermochemical energy storage systems based on cobalt, manganese, and copper oxides. Renew. Sustain. Energy Rev. 2022, 158, 112076. [Google Scholar] [CrossRef]
- Tang, Y.; Zhang, H.; Chen, J.; Shen, Y.; Xiong, K.; Zhou, Y.; Li, X.; Hu, Y. Different Active Phases of CuMnOx for Total Oxidation and Partial Oxidation. Cailiao Daobao/Mater. Rep. 2020, 34, 9028–9033. [Google Scholar]
- Yuvaraj, S.; Selvan, R.K.; Lee, Y.S. An overview of AB2O4– and A2BO4–structured negative electrodes for advanced Li–ion batteries. RSC Adv. 2016, 6, 21448–21474. [Google Scholar] [CrossRef]
- Gao, P.-X.; Shimpi, P.; Gao, H.; Liu, C.; Guo, Y.; Cai, W.; Liao, K.-T.; Wrobel, G.; Zhang, Z.; Ren, Z.; et al. Hierarchical assembly of multifunctional oxide–based composite nanostructures for energy and environmental applications. Int. J. Mol. Sci. 2012, 13, 7393–7423. [Google Scholar] [CrossRef] [Green Version]
- Lamb, A.B.; Bray, W.C.; Frazer, J.C.W. The Removal of Carbon Monoxide from Air. J. Ind. Eng. Chem. 1920, 12, 213–221. [Google Scholar] [CrossRef]
- Lu, H.F.; Kong, X.X.; Huang, H.F.; Zhou, Y.; Chen, Y.F. Cu–Mn–Ce Ternary Mixed–Oxide Catalysts for Catalytic Combustion of Toluene. J. Environ. Sci. 2015, 32, 102–107. [Google Scholar] [CrossRef]
- Aguilera, D.A.; Perez, A.; Molina, P.; Moreno, S. Cu–Mn and Co–Mn Catalysts Synthesized from Hydrotalcites and Their Use in the Oxidation of VOCs. Appl. Catal. B Environ. 2011, 104, 144–150. [Google Scholar] [CrossRef]
- Broemme, A.D.D.; Brabers, V.A.M. Preparation and properties of copper– and manganese-containing mixed. Solid State Ion. 1985, 16, 171–178. [Google Scholar] [CrossRef]
- Martin, B.E.; Petric, A. Electrical properties of copper–manganese spinel solutions and their cation valence and cation distribution. J. Phys. Chem. Solids 2007, 68, 2262–2270. [Google Scholar] [CrossRef]
- Múčka, V.; Silber, R. Decomposition of hydrogen peroxide on nickel–silver two–component catalyst. Collect. Czech. Chem. Commun. 1984, 49, 2222–2230. [Google Scholar] [CrossRef]
- Garcıa, E.; Palacios, J.M.; Alonso, L.; Moliner, R. Performance of Mn and Cu Mixed Oxides as Regenerable Sorbents for Hot Coal Gas Desulfurization. Energy Fuels 2000, 14, 1296–1303. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, D.; Li, S.; Zhang, L.; Zheng, G.; Guo, L. Layered copper manganese oxide for the efficient catalytic CO and VOCs oxidation. Chem. Eng. J. 2019, 357, 258–268. [Google Scholar] [CrossRef]
- Kotai, L.; Gacs, I.; Sajo, I.E.; Sharma, P.K.; Banerji, K.K. Beliefs and Facts in Permanganate Chemistry–An Overview on the Synthesis and the Reactivity of Simple and Complex Permanganates. Trends Inorg. Chem. 2009, 11, 25–104. [Google Scholar] [CrossRef]
- Adánez–Rubio, I.; Abad, A.; Gayán, P.; de Diego, L.F.; Adánez, J. CLOU process performance with a Cu–Mn oxygen carrier in the combustion of different types of coal with CO2 capture. Fuel 2018, 212, 605–612. [Google Scholar] [CrossRef] [Green Version]
- Shaheen, W.M.; Selim, M.M. Effect of thermal treatment on physicochemical properties of pure and mixed manganese carbonate and basic copper carbonate. Thermochim. Acta 1998, 322, 117–128. [Google Scholar] [CrossRef]
- Aoki, I. Cation Distribution in CuMn2O4. J. Phys. Soc. Japan 1965, 20, 871. [Google Scholar] [CrossRef]
- Vu, V.H.; Belkouch, J.; Ould-Dris, A.; Taouk, B. Catalytic oxidation of volatile organic compounds on manganese and copper oxides supported on titania. AIChE. J. 2008, 54, 1585–1591. [Google Scholar] [CrossRef]
- Wang, Z.; Zhu, J.; Sun, P.; Zhang, P.; Zeng, Z.; Liang, S.; Zhu, X. Nanostructured Mn–Cu binary oxides for supercapacitor. J. Alloys Compd. 2014, 598, 166–170. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, L.-C.; Chen, M.; Liu, Y.-M.; Cao, Y.; He, H.-Y.; Fan, K.-N. Waste–free Soft Reactive Grinding Synthesis of High–Surface–Area Copper–Manganese Spinel Oxide Catalysts Highly Effective for Methanol Steam Reforming. Catal. Lett. 2008, 121, 144–150. [Google Scholar] [CrossRef]
- Vandenberghe, R.E.; Robbrecht, G.G.; Brabers, V.A.M. On the stability of the cubic spinel structure in the system Cu–Mn–O. Mater. Res. Bull. 1973, 8, 571–580. [Google Scholar] [CrossRef]
- Loginova, M.V.; Stogova, V.A.; Sheftel, I.T. Structural transformations and electrical conductivity of copper–manganese spinel. Izv. Ak. Nauk SSSR Ser. Neorg. Mater. 1971, 1, 120. [Google Scholar]
- Beley, M.; Padel, L.; Bernier, J.C. Etudes et Proprietes des Composes CuxMn3−xO4. Ann. Chim. Fr. 1978, 3, 429–452. [Google Scholar]
- Jarrige, J.; Mexmain, J. Préparation et stabilité des manganites de cuivre. Bull. Soc. Chim. Fr. 1976, 405–409. [Google Scholar]
- Cai, L.N.; Guo, Y.; Lu, A.H.; Branton, P.; Li, W.C. The choice of precipitant and precursor in the co–precipitation synthesis of copper manganese oxide for maximizing carbon monoxide oxidation. J. Mol. Catal. A Chem. 2012, 360, 35–41. [Google Scholar] [CrossRef] [Green Version]
- Ye, Z.; Giraudon, J.-M.; Nuns, N.; Simon, P.; De Geyter, N.; Morent, R.; Lamonier, J.-F. Influence of the preparation method on the activity of copper–manganese oxides for toluene total oxidation. Appl. Catal. B Environ. 2018, 223, 154–166. [Google Scholar] [CrossRef]
- Kótai, L.; Horváth, T.; Szentmihályi, K.; Keszler, Á. Evidenceforquasi–intramolecular acid–base reactions in solutions of transition metal ammine complexes. Transit. Met. Chem. 2000, 25, 293–294. [Google Scholar] [CrossRef]
- Kótai, L.; Gács, I.; Kazinczy, B.; Sajó, I.E. Quasi–intramolecular acid–base reactions in aqueous solutions of metal–complexes of basic ligands I. Generalized theoretical considerations on the deammoniation of [MLm]Xn type ammonia complexes. Transit. Met. Chem. 2003, 28, 292–295. [Google Scholar] [CrossRef]
- Zhao, H.; Fang, K.; Zhou, J.; Lin, M.; Sun, Y. Direct synthesis of methyl formate from syngas on Cu–Mn mixed oxide catalyst. Int. J. Hydrog. Energy 2016, 41, 8819–8828. [Google Scholar] [CrossRef]
- Zhao, H.; Fang, K.; Dong, F.; Lin, M.; Sun, Y.; Tang, Z. Textual properties of Cu–Mn mixed oxides and application for methyl formate synthesis from syngas. J. Ind. Eng. Chem. 2017, 54, 117–125. [Google Scholar] [CrossRef]
- Saravanakumar, B.; Lakshmi, S.M.; Ravi, G.; Ganesh, V.; Sakunthala, A.; Yuvakkumar, R. Electrochemical properties of rice–like copper manganese oxide (CuMn2O4) nanoparticles for pseudocapacitor applications. J. Alloys Compd. 2017, 723, 115–122. [Google Scholar] [CrossRef]
- Liu, T.; Yao, Y.; Wei, L.; Shi, Z.; Han, L.; Yuan, H.; Li, B.; Dong, L.; Wang, F.; Sun, C. Preparation and Evaluation of Copper–Manganese Oxide as a High–Efficiency Catalyst for CO Oxidation and NO Reduction by CO. J. Phys. Chem. C. 2017, 121, 12757–12770. [Google Scholar] [CrossRef]
- Jones, C.; Cole, K.J.; Taylor, S.H.; Crudace, M.J.; Hutchings, G.J. Copper manganese oxide catalysts for ambient temperature carbon monoxide oxidation: Effect of calcination on activity. J. Mol. Catal. A Chem. 2009, 305, 121–124. [Google Scholar] [CrossRef]
- Li, X.; Xu, J.; Zhou, L.; Wang, F.; Gao, J.; Chen, C.; Ning, J.; Ma, H. Liquid–phase oxidation of toluene by molecular oxygen over copper manganese oxides. Catal. Lett. 2006, 110, 149–154. [Google Scholar] [CrossRef]
- Mirzaei, A.A.; Shaterian, H.R.; Habibi, M.; Hutchings, G.J.; Taylor, S.H. Characterisation of copper–manganese oxide catalysts: Effect of precipitate aging upon the structure and morphology of precursors and catalysts. Appl. Catal. A Gen. 2003, 253, 499–508. [Google Scholar] [CrossRef]
- Tabakova, T.; Idakiev, V.; Avgouropoulos, G.; Papavasiliou, J.; Manzoli, M.; Boccuzzi, F.; Ioannides, T. Highly active copper catalyst for low–temperature water–gas shift reaction prepared via a Cu–Mn spinel oxide precursor. Appl. Catal. A Gen. 2013, 451, 184–191. [Google Scholar] [CrossRef]
- Pan, J.; Du, W.; Liu, Y.; Cheng, Y.; Yuan, S. Lanthanum–doped CuMn composite oxide catalysts for catalytic oxidation of toluene. J. Rare Earths 2019, 37, 602–608. [Google Scholar] [CrossRef]
- Behar, S.; Gonzalez, P.; Agulhon, P.; Quignard, F.; Świerczyński, D. New synthesis of nanosized Cu–Mn spinels as efficient oxidation catalysts. Catal. Today 2012, 189, 35–41. [Google Scholar] [CrossRef]
- Krämer, M.; Schmidt, T.; Stöwe, K.; Müller, F.; Natter, H.; Maier, W.F. Structural and catalytic aspects of sol–gel derived copper manganese oxides as low–temperature CO oxidation catalyst. Appl. Catal. A Gen. 2006, 302, 257–263. [Google Scholar] [CrossRef]
- Papavasiliou, J.; Avgouropoulos, G.; Ioannides, T. Steam reforming of methanol over copper–manganese spinel oxide catalysts. Catal. Commun. 2005, 6, 497–501. [Google Scholar] [CrossRef]
- Choi, K.-H.; Lee, D.-H.; Kim, H.-S.; Yoon, Y.-C.; Park, C.-S.; Kim, Y.H. Reaction Characteristics of Precious–Metal–Free Ternary Mn–Cu–M (M = Ce, Co, Cr, and Fe) Oxide Catalysts for Low–Temperature CO Oxidation. Ind. Eng. Chem. Res. 2016, 55, 4443–4450. [Google Scholar] [CrossRef]
- Einaga, H.; Kiya, A.; Yoshioka, S.; Teraoka, Y. Catalytic properties of copper manganese mixed oxides prepared by co–precipitation using tetramethylammonium hydroxide. Catal. Sci. Technol. 2014, 4, 3713–3722. [Google Scholar] [CrossRef]
- Seubsai, A.; Kahn, M.; Zohour, B.; Noon, D.; Charoenpanich, M.; Senka, S. Copper–Manganese Mixed Metal Oxide Catalysts for the Direct Epoxidation of Propylene by Molecular Oxygen. Ind. Eng. Chem. Res. 2015, 54, 2638–2645. [Google Scholar] [CrossRef]
- Bayón, R.; Vicente, G.S.; Maffiotte, C.; Morales, Á. Characterization of copper–manganese–oxide thin films deposited by dip–coating. Sol. Energy Mater. Sol. Cells 2008, 92, 1211–1216. [Google Scholar] [CrossRef]
- Hutchings, G.J.; Mirzaei, A.A.; Joyner, R.W.; Siddiqui, M.R.H.; Taylor, S.H. Effect of preparation conditions on the catalytic performance of copper manganese oxide catalysts for CO oxidation. Appl. Catal. A Gen. 1998, 166, 143–152. [Google Scholar] [CrossRef]
- Zhang, M.; Li, W.; Wu, X.; Zhao, F.; Wang, D.; Zha, X.; Li, S.; Liu, H.; Chen, Y. Low–temperature catalytic oxidation of benzene over nanocrystalline Cu–Mn composite oxides by facile sol–gel synthesis. New J. Chem. 2020, 44, 2442–2451. [Google Scholar] [CrossRef]
- Li, W.B.; Chu, W.B.; Zhuang, M.; Hua, J. Catalytic oxidation of toluene on Mn–containing mixed oxides prepared in reverse microemulsions. Catal. Today 2004, 93–95, 205–209. [Google Scholar] [CrossRef]
- Veerapandian, S.K.P.; Ye, Z.; Giraudon, J.-M.; De Geyter, N.; Morent, R.; Lamonier, J.-F. Plasma-assisted Cu–Mn mixed oxide catalysts for trichloroethylene abatement in moist air. J. Hazard. Mater. 2019, 379, 120781. [Google Scholar] [CrossRef]
- Li, W.B.; Liu, Z.X.; Liu, R.F.; Chen, J.L.; Xu, B.Q. Rod–like CuMnOx transformed from mixed oxide particles by alkaline hydrothermal treatment as a novel catalyst for catalytic combustion of toluene. Phys. Chem. Chem. Phys. 2016, 18, 22794–22798. [Google Scholar] [CrossRef]
- Jarrige, J.; Mexmain, J. Propriétés du Manganite de Cuivre Cu1.5Mn1.5O4. Bull. Soc. Chim. Fr. 1980, 9–10, I-363–I-369. [Google Scholar]
- Kótai, L.; Sajó, I.E.; Gács, I.; Sharma, P.K.; Banerji, K.K. Convenient Routes for the Preparation of Barium Permanganate and other Permanganate Salts. Z. Anorg. Allg. Chem. 2007, 633, 1257–1260. [Google Scholar] [CrossRef]
- Elovich, S.Y.; Roginskii, S.Z.; Shmuk, E.I. Kinetics of the Thermal Decomposition of Solid Permanganates. Izv. AN SSSR Otdel. Khim. Nauk. 1950, 5, 469–474. [Google Scholar]
- Galwey, A.K.; Fakiha, S.A.A.; Abd El-Salaam, K.M. A kinetic and mechanistic study of the thermal decomposition of copper(II) permanganate. Thermochim. Acta 1994, 239, 225–232. [Google Scholar] [CrossRef]
- Ball, M.C.; Dowen, P.; Galwey, A.K. Chemical and physical properties of the solid products of thermal decomposition of copper(II) and nickel(II) permanganates. J. Mater. Chem. 1997, 7, 315–318. [Google Scholar] [CrossRef]
- Yamanaka, S. Anion exchange reactions in layer structured crystals. Stud. Surf. Sci. Catal. 1994, 83, 147–153. [Google Scholar]
- Kótai, L.; Banerji, K.K.; Sajó, I.; Kristóf, J.; Sreedharm, B.; Holly, S.; Keresztury, G.; Rockenbauer, A. An Unprecedented-Type Intramolecular Redox Reaction of Solid [Tetraamminecopper(2+)] Bis (permanganate)([Cu(NH3)4](MnO4)2)–A Low-Temperature Synthesis of Copper Dimanganese Tetraoxide-Type (CuMn2O4) Nanocrystalline Catalyst Precursors. Helv. Chim. Acta 2002, 85, 2316–2327. [Google Scholar] [CrossRef]
- Gorbunov, V.V.; Shmagin, L.F. On the combustion of salts of tetramine copper (II). Fiz. Goreniya Vzriva 1972, 8, 523–526. [Google Scholar]
- Seferiadis, N.; Dubler, E.; Oswald, H.R. Structure of [tetraamminecopper(II)] dipermanganate. Acta Cryst. Sect. C. 1986, 42, 942–945. [Google Scholar] [CrossRef]
- Kótai, L.; Németh, P.; Kocsis, T.; Sajó, I.E.; Pasinszki, T. A new route to synthesize controlled–size MMn2O4–type transition metal (M = Cd, Zn, Cu) nanomanganites. Nano Stud.—Sci. J. 2016, 13, 7–13. [Google Scholar]
- Solt, H.E.; Németh, P.; Mohai, M.; Sajó, I.E.; Klébert, S.; Franguelli, F.P.; Fogaca, L.A.; Pawar, R.P.; Kótai, L. Temperature–Limited Synthesis of Copper Manganites along the Borderline of the Amorphous/Crystalline State and Their Catalytic Activity in CO Oxidation. ACS Omega 2021, 6, 1523–1533. [Google Scholar] [CrossRef] [PubMed]
- Pollert, E. Tetragonal–to–cubic transformation of hausmannite. J. Solid State Chem. 1980, 33, 305–308. [Google Scholar] [CrossRef]
- Miyahara, S. Jahn–Teller distortion in magnetic spinels. J. Phys. Soc. Japan 1962, 17 (Suppl. BI), 181–184. [Google Scholar]
- Radhakrishnan, N.K.; Biswas, A.B. A Neutron Diffraction Study of Cation Distribution in Some Manganites. J. Ind. Chem Soc. 1974, 37, 274–281. [Google Scholar]
- Sinha, A.P.B.; Sanjana, N.R.; Biswas, A.P.B. The crystal structure of copper manganite. J. Phys. Chem. 1958, 62, 191–194. [Google Scholar] [CrossRef]
- Waskowska, A.; Gerward, L.; Olsen, J.S.; Steenstrup, S.; Talik, E. CuMn2O4: Properties and the high–pressure induced Jahn–Teller phase transition. J. Phys. Condens. Matter 2001, 13, 2549–2562. [Google Scholar] [CrossRef]
- Dorris, S.E.; Mason, T.O. Electrical Properties and Cation Valencies in Mn3O4. J. Am. Ceram. Soc. 1988, 71, 379–385. [Google Scholar] [CrossRef]
- Vetter, K.M.; Manecke, G. Der Einstellungsmechanismus des Mn+++/Mn++++–Redoxpotentials an Platin. Z. Phys. Chem. 1950, 195, 270–337. [Google Scholar] [CrossRef]
- Buhl, R. Manganites spinelles purs d’elements de transition preparations et structures cristallographiques. J. Phys. Chem. Solids 1969, 30, 805–812. [Google Scholar] [CrossRef]
- Blasse, G. Ferromagnetism and ferrimagnetism of oxygen spinels containing tetravalent manganese. J. Phys. Chem. Solids 1966, 27, 383–389. [Google Scholar] [CrossRef]
- Miller, A. Determination of the valence state of copper in cubic CuMn2O4 spinel by X–ray absorption edge measurements. J. Phys. Chem. Solids 1967, 29, 633–639. [Google Scholar] [CrossRef]
- Lenglet, M.; D’Huysser, A.; Kasperek, J.; Bonnelle, J.P.; Durr, J. Characterization of the oxidation states of copper and manganese in some manganites by the analysis of the XPS spectrum, X emission, and X absorption thresholds. Mater. Res. Bull. 1985, 20, 745–757. [Google Scholar] [CrossRef]
- Di Castro, V.; Furlani, C.; Gargano, M.; Rossi, M. XPS characterization of the CuO/MnO2 catalyst. Appl. Surf. Sci. 1987, 28, 270–278. [Google Scholar] [CrossRef]
- Radhakrishnan, N.K.; Biwas, A.B. A neutron diffraction study of the spinel oxide CuMn2O4. Phys. Status Solidi (A) Appl. Res. 1977, 44, 45–49. [Google Scholar] [CrossRef]
- Gillot, B.; Buguet, S.; Kester, E.J. Oxidation mechanism and valence states of copper and manganese in tetragonal CuMn2O4. Mater. Chem. 1997, 7, 2513–2517. [Google Scholar] [CrossRef]
- Buciuman, F.C.; Patcas, F.; Hahn, T. Synergy effect between copper and manganese oxides in hopcalite catalysts. Stud. Surf. Sci. Catal. 2001, 138, 315–322. [Google Scholar]
- Wang, H.; Lu, Y.; Han, Y.X.; Lu, C.; Wan, H.; Xu, Z.; Zheng, S. Enhanced catalytic toluene oxidation by the interaction between copper oxide and manganese oxide in Cu–O–Mn/γ–Al2O3 catalysts. Appl. Surf. Sci. 2017, 420, 260–266. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, L.; Shi, L.; Fang, C.; Li, H.; Gao, R.; Huang, L.; Zhang, J. In situ supported MnOx–CeOx on carbon nanotubes for the low–temperature selective catalytic reduction of NO with NH3. Nanoscale 2013, 5, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Takeguchi, T.; Kikuchi, R.; Eguchi, K. Influence of preparation method and additive for Cu–Mn spinel oxide catalyst on water gas shift reaction of reformed fuels. Appl. Catal. A Gen. 2005, 279, 59–66. [Google Scholar] [CrossRef]







| x | Precursors | Synthesis/Annealing Conditions | [Ref.] |
|---|---|---|---|
| 0.98–1.10 | CuO, Mn3O4 | In air, 800–940 °C, 72 h, quenching in H2O | [9] |
| 1.5 | CuO, Mn3O4 | Spray granulation, calcining at 1125 °C for 2 h | [15] |
| 1.1–1.6 | CuO, MnO2 | Grinding, pressing, 940 °C for 24 h, annealing between 500 and 850 °C | [10] |
| 1.15–1.47 | CuO, MnO2 | 5 wt.% graphite as pore modifier, 950 °C for 6 h | [12] |
| 0.98–1.10 | CuO, MnCO3·H2O | In air, 800–940 °C, 72 h, quenching in H2O | [21] |
| 1 | CuCO3·Cu(OH)2, MnCO3 | Heating at 750 and 1000 °C | [16] |
| 1.5 | CuCO3·Cu(OH)2, MnCO3 | 20% excess of solid oxalic or citric acid, grinding at 600 rpm for 2 h, calcining at 300–500 °C in air for 4 h | [20] |
| 1 | Cu(II) and Mn(II) nitrates | Heating in air to decompose into oxides then calcining at 700 °C for 240 h | [17] |
| 1 | Cu(II) acetate tetrahydrate, Mn(II) acetate tetrahydrate | Oxalic acid dihydrate, ball milling at room temperature for 3 h, calcining at 550 °C for 2 h in air | [19] |
| x | Precursors | Synthesis/Annealing Conditions | [Ref.] |
|---|---|---|---|
| 0.30–1.05 | Cu(II) and Mn(II) nitrates | Aq. soln., 25–100% excess of urea, jellifying at 100 °C, combustion at 4–500 °C, calcining at 550 °C for 1 h | [40] |
| 1 | Cu(II) and Mn(II) nitrates | 80 °C, adding of Na2CO3, (2 M), pH = 8.3, calcining in static air at 200–400 °C for 0.5–6 h | [33] |
| 1 | Cu(II) and Mn(II) nitrates | Ethylene-glycol–water–nitric acid mixture, 2 h reflux at 105 °C, calcining at 4–500 °C for 5 h | [39] |
| 1 | Cu(II) and Mn(II) nitrates | Me4NOH, 25 °C, 20 min, drying at 100 °C for 24 h and calcining at 400 °C for 5 h | [26,42] |
| 1–2 | Cu(II) and Mn(II) nitrates | 1−1 M aq. metal salts, 2 M fuel (glycerol, maleic acid, citric acid, ethyl acetoacetate, or nitric acid), 1 M NaCl, jellifying at 150 °C for 12 h, calcining at 400–700 °C for 6 h | [43] |
| 1.1–1.5 | Cu(II)– and Mn(II) nitrates | Spray-drying at 300 °C, firing at 350 °C for 24 h, sintering at 530–940 °C for 48 h, quenching in water | [9] |
| 1.2–1.4 | Cu(II) and Mn(II) nitrates | 80 °C, aq. Na2CO3, pH = 8.9, 10 min, conditioning between 0 and 1440 min at 80 °C in the mother liq., calcining at 500 °C for 17 h | [35] |
| 1.4 | Cu(II) and Mn(II) nitrates | Incipient wetness impregnation, TiO2, 100 °C for 5 h then calcining at 500 °C for 7 h in air | [18] |
| 1.5 | Cu(II) and Mn(II) nitrates | NaOH, 298 K, pH = 11, 30 min, calcining at 300 °C for 2 h | [25] |
| 1.1–1.5 | Cu(II)– and Mn(II) nitrates | NaOH, firing in air at 530–940 °C for 48 h, quenching in H2O | [9,21,22,23] |
| 1.5 | Cu(II) and Mn(II) nitrates | Abs. ethanol, dip-coating, Al foil, room temp., drying at 300 °C for 3 h, calcining at 4–500 °C for 15–90 min | [44] |
| 1.5 | Cu(II) and Mn(II) nitrates | Urea, citric acid, 0.3 M KMnO4, pH = 6–7, 4 h, hydrothermal treatment at 180 °C for 12 h, calcining at 350 °C in air for 4 h | [32] |
| 1.5 | Cu(II) and Mn(II) nitrates | Autoignition, 75% excess of urea, open furnace at 4–500 °C, calcining at 550 °C for 1 h | [36,40] |
| 1.5 | Mn(II) and Cu(II) nitrates | Aq. Na2CO3, 80 °C, pH = 8.3, aging at 80 °C for 6 h, calcining at 700 °C for 7 h | [36,45] |
| 1.5 | Cu(II) and Mn(II) nitrates | 1.2 M Na2CO3, 80 5C, pH = 8.5, calcining at 300–500 °C in air for 4 h | [20] |
| 1.5 | Cu(II) and Mn(II) nitrates | 20% excess of aq. oxalic acid, room temperature, calcining at 300–500 °C for 4 h | [20] |
| 0.5–2 | Cu(II) and Mn(II) nitrates | Aq. citric acid, ethylene glycol, 30 °C for 2 h, jellifying at 70 °C, calcining at 500 °C for 3 h | [46] |
| 1.5 | Mn(II) nitrate, [Cu(NH3)4](II)–hydroxide | Mn nitrate (pH = 3–4) and [Cu(NH3)4](II)–hydroxide (pH = 9–10) solutions were mixed, 30–40 °C, calcining at 550 °C for 4 h | [29,30] |
| 1.8 | Mn(II) nitrate, Cu(II) acetate | Reverse microemulsion, water/triton–X100/n–octanol/cyclohexane, 25% aq. NH4OH. Stirring for 1 h, calcining at 450–1000 °C in oxygen | [47] |
| 1.5 | Mn(II) acetate, Cu(II) nitrate | Aq. Me4NOH, aging for 2 h, calcining in dry air at 400 °C for 5 h | [48] |
| 1.5 | Mn(II) acetate, Cu(II) nitrate | KMnO4, room temp., 24 h, calcining at 300 °C for 2 h | [41] |
| 1.5 | Mn(II) acetate, Cu(II) nitrate | Aq. soln., La nitrate, 1.5 M ammonium carbonate, pH = 8, 2 h aging, calcining at 550 °C for 2 h in air | [37] |
| 1 | Cu(II) and Mn(II) acetates | Aq. Na2CO3, 80 °C, pH = 8–10, 30 min, aging at 80 °C for 12 h, calcining at 500 °C for 20 min, 10 M NaOH, hydrothermal treatment at 110 °C for 20 h, calcining at 250 °C for 1 h | [49] |
| 1.5 | Cu(II) and Mn(II) acetates | 0.2 M aq. Na2CO3, room temp., 4 h, calcining at 500 °C for 6 h | [33,34] |
| 1.5 | Cu(II) and Mn(II) acetates | NaOH, 298 K, pH = 11, 30 min, calcining at 300 °C for 2 h | [25] |
| 1.5 | Cu(II) and Mn(II) chloride | Na2CO3, 3 h aging, drying at 80 °C overnight, calcining at 450 °C for 5 h | [13] |
| 1.5 | Cu(II) and Mn(II) chlorides | Sodium alginate (2 wt.%), room temp. for 12 h, dehydration with successive ethanol−water mixture washing (10–100%), 15 min each, calcining at 450 °C for 8 h | [37] |
| 1.2–1.5 | Cu(II) and Mn(II) chlorides | pH = 4, sodium oxalate, 550–650 °C | [23,50] |
| 1 | Mn(II) sulfate, copper(II) chloride | Solvothermal method, water: methanol = 60:40, ethylene glycol, 1 M NaOH, pH = 10, 120–160 °C foe 12 h | [31] |
| 1.1–1.5 | Cu(II) and Mn(II) sulfates | NaOH, firing in air at 530–940 °C for 48 h, quenching in H2O | [9,21,22,23] |
| Cu/Mn | CuI/CuII | CuII (T–4) (A) | CuII(OC–6)(B) | CuI(T–4) | Preparation Method | Ref. |
|---|---|---|---|---|---|---|
| 0.83 | 0.47 | 25 | 43 | 32 | Dip-coating | [44] |
| 0.88 | 0.47 | 25 | 43 | 32 | Dip-coating | [44] |
| 1.14 | 0 | 33 | 67 | 0 | Dip-coating | [44] |
| 1.08 | 0 | 23 | 77 | 0 | Dip-coating | [44] |
| 1.23 | 0.13 | 41 | 47 | 12 | Dip-coating | [44] |
| 2 | 3.16 | 24 | 76 | Ceramic | [21] | |
| 2 | 1.00 | 50 | 25 | 25 | Precipitation | [73] |
| 2 | 2.00 | 0 | 33 | 66 | Ceramic | [50] |
| 2 | 0.39 | 48 | 24 | 28 | Ceramic | [71] |
| 2 | 0.67 | 0 | 60 | 40 | Ceramic | [74] |
| 1 | 2.00 | 0 | 33 | 66 | Ceramic | [9,21] |
| CuxMn3−xO4 | Precursors, Preparation Method, and Conditions | Catalyst Efficiency and Reaction Conditions | Refs. | Remarks |
|---|---|---|---|---|
| x = 0.5 | Aq. solutions of Cu and Mn nitrates in 1/2 molar ratio were mixed, aq. NaOH solution, 298 K, pH = 11, 300 °C for 2 h | 1% CO, 20% O2 and 79% N2, space velocity was 20,000 mL h−1 g cat−1. The temperature was ramped at a rate of 1 °C min−1 from 0 °C to the final temperature, T50/T100 = 80/120 °C, resp. | [25] | SBET = 46 m2 g−1, Vp = 0.23 cm3 g−1 |
| x = 0.5 | Thermal decomposition of the [Cu2(OH)3MnO4] layered basic salt | It catalyzes CO oxidation at 30 °C | [55] | Mixed phase with CuO |
| x = 1.0 | Mn–nitrate, Cu–nitrate, 0.25 M each, 2 M Na2CO3, pH = 8.3, 400 for 2 | 5000 ppm CO in air, 25 °C, 33,000 h−1, cooling due to 10 °C adiabatic temperature increase | [33] | SBET = 93 m2 g−1, mixed phase |
| x = 1.0 | Metal nitrates (Cu:Mn = 1:4) in ethylene glycol–water–cc. nitric acid mixture, 2 h reflux, drying at 105 °C for 16 h, 450–500 °C | 1 vol.% CO in synthetic air, 100–200 mm particles, 25 °C, flow rate: 30 000 mL h−1 g−1; Experiments with moist air were carried out near low-temperature fuel cell conditions using a different feed gas composition (1 vol.% CO, 4.88 vol.% synthetic air, 20 vol.% CO2 in hydrogen), 60–120 °C, 17–20% CO conversion after 1 h | [39] | SBET = 55–67 m2 g−1 |
| x = 1.0 | Cu–nitrate and Mn–nitrate in 1:2 ratio, 80 °C, 0.25 M Na2CO3, pH = 8.9, aging from 0 to 720 min | 5% CO in He and O2 (1:10 v/v) were fed to the reactor, 20 °C; total GHSV of 33,000 h−1 | [35] | SBET = 26 m2 g−1 |
| x = 1.0 | [Cu(NH3)4](MnO4)2 decomposition in CHCl3 at 61 °C, aq. extraction, 300 °C for 1 h | 0.5% CO and 0.5% O2 in He, (1.37 gCO gcat−1 h−1), T10/T25/T50 = 94/132/163 °C, resp. | [60] | Mixed phase |
| x = 1.0 | [Cu(NH3)4](MnO4)2 decomposition in CHCl3 at 61 °C, 300 °C for 1 h °C | 0.5% CO and 0.5% O2 in He, (1.37 gCO gcat−1 h−1), T10/T25/T50 = 134/159/188 °C, resp. | [60] | – |
| x = 1.0 | [Cu(NH3)4](MnO4)2 decomposition in CCl4 at 77 °C, with aq. extraction (NH4NO3 removal), 200–500 °C for 1–8 h | 0.5% CO and 0.5% O2 in He, (1.37 gCO gcat−1 h−1), without heat treatment T10/T25/T50 = 121/130/161 °C, resp.; 200 °C for 1 and 8 h, T10/T25/T50 = 135/155/173 °C and 127/155/177 °C, resp.; 500 °C for 1 and 8 h, T10/T25/T50 = 111/142/170 and 137/164/190 °C, resp. | [60] | – |
| x = 1.0 | [Cu(NH3)4](MnO4)2 decomposition in CCl4 at 77 °C, 500 °C for 1 h | 0.5% CO and 0.5% O2 in He, (1.37 gCO gcat−1 h−1), T10/T25/T50 = 166/190/>200 °C, resp. | [60] | – |
| x = 1.4 | Cu–nitrate and Mn–nitrate, 1:2 ratio, 80 °C, 0.25 M Na2CO3, pH = 8.9, aging from 0 to 300 and 1440 min | 5% CO in He and O2 (1:10 v/v) were fed to the reactor, 20 °C; total gas hourly space velocity of 33,000 h−1 | [35] | SBET = 23–30 m2 g−1, mixed phase |
| x = 1.4 | Cu:Mn = 1:1, MnCl2, CuCl2, Na2CO3, 3 h aging,, 450 °C for 5 h | 1000 ppm CO, GHSV = 50,000 mL g−1 h−1, T50/T90 = 62/76 °C, resp., reaction rate = 0.36 mmol g−1 h−1 and 1.45 mmol m−2 h−1 at 50 °C; reaction rates were measured at isotherm CO oxidation at 80–180 °C at less than 20% conversion | [13] | SBET = 18 m2 g−1, Vp =0.02 cm3 g−1, APD = 13 |
| x = 1.5 | Mn–acetate, Cu–nitrate, KMnO4, 300 °C for 2 h | 1% CO (20% air in Ar) was fed into the reactor for the CO oxidation reaction, T50/T70/T90 = 59/79/93 °C, resp., the complete transformation was found at 100 °C, Co and Fe doping increases the efficiency | [41] | Mixed phases |
| x = 1.5 | Copper and manganese acetates in 1/2 molar ratio in water, NaOH solution, 298 K, pH 11 300 °C for 2 h | 1% CO, 20% O2 and 79% N2 as feed gas with a space velocity of 20,000 mL h−1 g cat−1. The temperature was ramped at a rate of 1 °C min−1 from 0 °C to the final temperature, T50/T100 = 72/110 °C, resp. | [25] | SBET = 70 m2 g−1, Vp = 0.36 cm3 g−1 |
| x = 1.5 | Hydrothermal method, aq. Cu and Mn nitrates, citric acid, urea, KMnO4 solution, Mn(NO3)2/KMnO4 = 1:2 and urea/citric acid/Mnall/Cu = 1.5:1.5:1:0–0.2; Aq. ammonia (25 wt.%), pH = 6–7, 180 °C for 12 h, 350 °C in air for 4 h | 1.6 vol% CO, 20.8 vol% O2, and 77.6 vol% N2, a space velocity of 30,000 mL·g−1·h−1. It is active at room temperature. 100% CO oxidation was achieved at 65 °C | [32] | Mixed phase, SBET = 112–126 m2 g−1, 5.8–10.1 nm size |
| CuxMn3−xO4 | Precursors, Preparation Method, and Conditions | Catalyst Efficiency and Reaction Conditions | Ref. | Remarks |
|---|---|---|---|---|
| x = 1.5 | Hydrothermal method, aq. Cu and Mn nitrates, citric acid, urea, KMnO4 solution, Mn(NO3)2/KMnO4 = 1:2 and urea/citric acid/Mnall/Cu = 1.5:1.5:1:0–0.2; aq. ammonia (25 wt.%), pH = 6–7, 180 °C for 12 h, 350 °C in air for 4 h. | 5 vol% NO, 10 vol% CO, and 85 vol% He as diluents, GHSV 24,000 mL·g−1·h−1 The 100% NO conversion and 100% N2 selectivity were achieved at 275 °C | [32] | Mixed phase, SBET = 112–126 m2 g−1, 5.8–10.1 nm size |
| CuxMn3−xO4 | Precursors, Preparation Method, and Conditions | Catalyst Efficiency and Reaction Conditions | Ref. | Remarks |
|---|---|---|---|---|
| x = 1.15 | CuO, MnO2, 5% graphite, 950 °C for 6 h | Atmospheric pressure, space velocity of 6000 h−1, 0.5 vol% H2S, 10 vol% H2, 15 vol% H2O(v), 5 vol% CO2, and 15 vol% CO, balance N2, 600 °C | [12] | Mixed oxide phase, porous, Vp = 0.16 cm3 g−1 |
| x = 1.47 | CuO, MnO2, 5% graphite, 950 °C for 6 h | Atmospheric pressure, space velocity of 6000 h−1, 0.5 vol% H2S, 10 vol% H2, 15 vol% H2O(v), 5 vol% CO2, and 15 vol% CO, balance N2, 600 °C | [12] | Mixed oxide phase, porous, Vp = 0.14 cm3 g−1 |
| CuxMn3−xO4 | Precursors, Preparation Method, and Conditions | Catalyst Efficiency and Reaction Conditions | Ref. | Remarks |
|---|---|---|---|---|
| x = 1.5 | Tetramethylammonium hydroxide precipitating agent, aq. copper(II) and manganese(II) salt solution, dry air at 400 °C for 5 h | Decomposition of trichloroethylene in moist air by post–plasma catalysis, 300 °C, GHSV 60,000 mL g−1 h−1, relative humidity (20 °C) 15%, initial TCE concentration 300 ppm, 50% CO2 yield. T50 = 300 °C (244 °C for CO2 formation). | [48] | SiC support, SBET = 30 m2 g−1, Vp = 0.29 cm3 g−1 |
| CuxMn3−xO4 | Precursors, Preparation Method, and Conditions | Catalyst Efficiency and Reaction Conditions | Refs. | Remarks |
|---|---|---|---|---|
| x = 0.5 | Citric acid monohydrate, copper nitrate and 50% manganese nitrate solutions, ethylene glycol, 70 °C, the gel was dried and calcined for 3 h at 500 °C | 100 ppm benzene in synthetic air, GHSV = 60,000 mL g−1 h−1, T10/T50/T90 = 114/164/193 °C, resp. | [46] | Crystallite size 13.2 nm, SBET = 45.00 m2 g−1, Vp = 0.310 cm3 g−1 |
| x = 1.0 | Mn and Cu nitrate, NaHCO3 as precipitating agent, 400–600 °C | 150 ppm benzene concentration, 10% O2 in the gas stream, 2250 ppm ozone, 70 °C | [42] | Mixture with oxide phases |
| x = 1.0 | Citric acid monohydrate, copper nitrate and 50% manganese nitrate solutions, ethylene glycol, 70 °C, the gel was dried and calcined for 3 h at 500 °C | 100 ppm benzene in synthetic air, GHSV = 60,000 mL g−1 h−1, T10/T50/T90 = 96/149/186 °C, resp. | [46] | Crystallite size 11.6 nm, SBET = 68.26 m2 g−1, Vp = 0.314 cm3 g−1 |
| x = 1.5 | Citric acid monohydrate, copper nitrate and 50% manganese nitrate solutions, ethylene glycol, 70 °C, the gel was dried and calcined for 3 h at 500 °C | 100 ppm benzene in synthetic air, GHSV = 60,000 mL g−1 h−1, T10/T50/T90 = 136/188/216 °C, resp. | [46] | Crystallite size 14.7 nm, SBET = 26.53 m2 g−1, Vp = 0.161 cm3 g−1 |
| x = 2 | Citric acid monohydrate, copper nitrate and 50% manganese nitrate solutions, ethylene glycol, 70 °C, the gel was dried and calcined for 3 h at 500 °C | 100 ppm benzene in synthetic air, WHSV = 60,000 mL g−1 h−1, T10/T50/T90 = 149/198/220 °C, resp. | [46] | Crystallite size 18.3 nm, SBET = 25.44 m2 g−1, Vp = 0.177 cm3 g−1 |
| CuxMn3−xO4 | Precursors, Preparation Method, and Conditions | Catalyst Efficiency and Reaction Conditions | Refs. | Remarks |
|---|---|---|---|---|
| 1.5 | Autoignition, 75% excess of urea, manganese nitrate, and copper, heating at 550 °C for 1 h | Space velocity = 4000 h−1, partial pressure of water vapor = 31.1 kPa, 4.492 vol.% CO/Ar + H2O and 15 vol.% CO, 10 vol.% CO2, 63 vol.% H2, 12 vol.% N2 + H2O, CO conversion was 90% at 220 °C | [36] | Multiphase, SBET = 8.0 m2 g−1 |
| 1.5 | aqueous solutions of manganese and copper nitrate, Na2CO3 at 80 °C, pH = 8.3, aging the resulting precipitate at 80 °C for 6 h, 700 °C for 7 h | Space velocity = 4000 h−1, partial pressure of water vapor = 31.1 kPa, 4.492 vol.% CO/Ar + H2O and 15 vol.% CO, 10 vol.% CO2, 63 vol.% H2, 12 vol.% N2 + H2O, 160–240 °C, CO conversion was 80% at 240 °C | [36] | Multiphase, SBET = 5.9 m2 g−1 |
| CuxMn3−xO4 | Precursors, Preparation Method, and Conditions | Catalyst Efficiency and Reaction Conditions | Refs. | Remarks |
|---|---|---|---|---|
| x = 1.5 | Mn(II)–nitrate:Cu(NH3)42+ (Cu:Mn = 1:1), 35 °C, 550 °C for 4 h | MeOH–DMF as liquid phase, H2/CO = 2:1, 160 °C, 30 min | [30] | Mixed phase, SBET = 15 m2 g−1, Vp = 0.03 cm3 g−1 size is 38.4 nm |
| 1.5 | Mn(II)–nitrate:Cu(NH3)42+ (Cu:Mn = 1:1), 35 °C, 550 °C for 4 h | CaO–ZrO2 as co-catalyst, 1.5:3.5 (v/v) methanol–DMF as a solvent, 2:1 H2:CO molar ratio, room temperature up to 4.0 MPa, heating up to 160 °C in 1 h, stirring for 8 h; 12.7–16.6% CO conversion, STY = 2.17–2.63 g L−1 h−1, methyl formate selectivity was 81.3–83.6% | [29] | Mixed phase, SBET = 10.5–18.4 m2 g−1 and Vp = 0.02–0.04 cm3 g−1, nanosized sample |
| CuxMn3−xO4 | Precursors, Preparation Method, and Conditions | Catalyst Efficiency and Reaction Conditions | Refs. | Remarks |
|---|---|---|---|---|
| x = 0.3 | Combustion method, 50% excess of urea, Cu, and Mn–nitrate (4–500 °C), heat treatment at 550 °C for 1 h | Atmospheric pressure, 240–280 °C, W/F = 0.257 g s−1 cm−3, 5% MeOH, 7.5% H2O and He; 61/97% MeOH conversion, 1.7/5.4% CO and 96.3/94.6% H2 selectivity, at 240 and 280 °C, resp. | [40] | SBET = 4.1 m2 g−1 |
| x = 0.6 | Combustion method, 50% excess of urea, Cu, and Mn–nitrate (4–500 °C), heat treatment at 550 °C for 1 h | Atmospheric pressure, 240–280 °C, W/F = 0.257 g s−1 cm−3, 5% MeOH, 7.5% H2O and He; 83.6 1/100% MeOH conversion, 2.1/5.7% CO and 96.5/94.3% H2 selectivity, at 240 and 280 °C, resp. | [40] | SBET = 7.5 m2 g−1 |
| x = 0.75 | combustion method, 50% excess of urea, Cu, and Mn–nitrate (4–500 °C), heat treatment at 550 °C for 1 h | Atmospheric pressure, 240–280 °C, W/F = 0.257 g s−1 cm−3, 5% MeOH, 7.5% H2O and He; 91.6/100% MeOH conversion, 1.7/6.4% CO and 97.5/93.6% H2 selectivity, at 240 and 280 °C, resp. | [40] | SBET = 6.0 m2 g−1 |
| x = 0.9 | Combustion method, 50 % excess of urea, Cu, and Mn–nitrate (4–500 °C), heat treatment at 550 °C for 1 h | Atmospheric pressure, 240–280 °C, W/F = 0.257 g s−1 cm−3, 5% MeOH, 7.5% H2O and He; 93.1/100% MeOH conversion, 2.1/7.5% CO and 92.5/97.9% H2 selectivity, at 240 and 280 °C, resp. | [40] | SBET = 4.8–8.6 m2 g−1 |
| x = 1.05 | Combustion method, 50% excess of urea, Cu, and Mn–nitrate (4–500 °C), heat treatment at 550 °C for 1 h | Atmospheric pressure, 240–280 °C, W/F = 0.257 g s−1 cm−3, 5% MeOH, 7.5% H2O and He; 93.8/100% MeOH conversion, 2.1/9.1% CO and 91.9/97.9% H2 selectivity, at 240 and 280 °C, resp. | [40] | SBET = 6.2 m2 g−1 |
| x = 1.5 | 1.2 M aqueous solution of Na2CO3, Cu, and Mn nitrates (each 0.5 M), pH at 8.5, 300 °C in air for 4 h | Atmospheric pressure, premixed water, and methanol with an H2O/MeOH molar ratio of 1.3; 51.5/65.7% MeOH conversion, 1.2/1.1 CO, and 99.6/99.1. H2 selectivity at 240 and 260 °C, 246 and 157 mmol gcat−1 h−1 production rate, resp. | [20] | SBET = 55.2 m2 g−1, Vp = 0.14 cm3 g−1, dp = 8.5 |
| x = 1.5 | 1.2 M aqueous solution of Na2CO3, Cu and Mn nitrates (each 0.5 M), pH = 8.5, 500 °C in air for 4 h | Atmospheric pressure, premixed water, and methanol with an H2O/MeOH molar ratio of 1.3; 45.5/62% MeOH conversion, 1.7/1.5 CO, and 99.4/99.1 H2 selectivity at 240 and 260 °C, with 139 and 189 mmol gcat−1 h−1 production rate, resp. | [20] | SBET = 14.9 m2 g−1, Vp = 0.07 cm3 g−1, dp = 18.5 nm |
| x = 1.5 | Aq. copper and manganese nitrate solution, 20% excess of oxalic acid at room temperature, 300 °C for 4 h | Atmospheric pressure, premixed water, and methanol with an H2O/MeOH molar ratio of 1.3; 43/60% MeOH conversion, 1.5/1.4% CO and 99.1–99.1% H2 selectivity at 240 and 260 °C, with 131 and 182 mmol gcat−1 h−1 production rate, resp. | [20] | SBET = 9.6 m2 g−1, Vp = 0.06 cm3 g−1, dp = 18.2 nm |
| x = 1.5 | Aq. copper and manganese nitrate solution, 20% excess of oxalic acid at room temperature, 500 C for 4 h | Atmospheric pressure, premixed water, and methanol with an H2O/MeOH molar ratio of 1.3; 35/52.5% MeOH conversion, 1.5/1.5% CO and 99.1/99.4% H2 selectivity at 240 and 260 °C, with 106 and 160 mmol gcat−1 h−1 production rate, resp. | [20] | SBET = 4.7 m2 g−1, Vp = 0.05 cm3 g−1, dp = 35.6 nm |
| x = 1.5 | A 1:2 molar ratio amount of Cu2CO3(OH)2 and MnCO3, 20% stoichiometric excess of solid oxalic acid dihydrate, grinding under ambient conditions in a planetary mill at a speed of 600 rpm for 2 h, 300 °C in air for 4 h | Atmospheric pressure, premixed water, and methanol with an H2O/MeOH molar ratio of 1.3; 74/88.5% MeOH conversion, 0.8/0.8% CO and 99–4/99.4 H2 selectivity at 240 and 260 °C, with 226 and 170 mmol gcat−1 h−1 production rate, resp. | [20] | SBET = 96.1 m2 g−1, Vp = 0.35 cm3 g−1, dp = 12.5 nm |
| x = 1.5 | A 1:2 molar ratio amount of Cu2CO3(OH)2 and MnCO3, 20% stoichiometric excess of solid oxalic acid dihydrate, grinding under ambient conditions in a planetary mill at a speed of 600 rpm for 2 h, 500 °C in air for 4 h | Atmospheric pressure, premixed water, and methanol with an H2O/MeOH molar ratio of 1.3; 65/80.5% MeOH conversion, 1.3/1.5 CO, and 99.1/99.4% H2 selectivity at 240 and 260 °C, with 199 and 246 mmol gcat−1 h−1 production rate, resp. | [20] | SBET = 24.6 m2 g−1, Vp = 0.18 cm3 g−1, dp = 26.5 nm |
| x = 1.5 | A 1:2 molar ratio amount of Cu2CO3(OH)2 and MnCO3 premixed with 20% stoichiometric excess of solid oxalic acid, grinding under ambient conditions in a planetary mill at a speed of 600 rpm for 2 h with higher intensity (more balls), calcination at 300 °C in air for 4 h. | Atmospheric pressure, premixed water, and methanol with an H2O/MeOH molar ratio of 1.3; 79.9/92.9% MeOH conversion, 0.7/0.7% CO and 99.5/99.5% H2 selectivity at 240 and 260 °C, with 244 and 284 mmol gcat−1 h−1 production rate, resp. | [20] | SBET = 118.1 m2 g−1, Vp = 0.39 cm3 g−1, dp = 11.1 nm |
| x = 1.5 | A 1:2 molar ratio of Cu2CO3(OH)2 and MnCO3 premixed with 20% stoichiometric excess of solid citric acid, grinding under ambient conditions in a planetary mill (600 rpm) for 2 h, 500 °C in air for 4 h | Atmospheric pressure, premixed water, and methanol with an H2O/MeOH molar ratio of 1.3; 57/69% MeOH conversion, 1.5/1.6% CO and 99.4/99.6% H2 selectivity at 240 and 260 °C, with 173 and 209 mmol gcat−1 h−1 production rate, resp. | [20] | SBET = 60.6 m2 g−1, Vp = 0.30 cm3 g−1, dp = 17.5 nm |
| x | Preparation Method, Precursors, and Conditions | Reaction, Catalyst Efficiency, Catalytic Reaction Conditions | Ref. | Remark |
|---|---|---|---|---|
| x = 1 | Precipitation, Cu– and Mn–acetate, Na2CO3, calcination at 700 °C | Toluene conversion into benzyl alcohol, benzaldehyde, and benzoic acid, ~21.6% conversion at 190 °C and 1.0 MPa O2 pressure, 2 h | [34] | Size is 37 nm |
| x = 1–2 | 1 M copper nitrate and 1 M manganese nitrate 2 M fuel (glycerol), 1 M NaCl, 150 °C for 12 h, combustion at 400–700 °C for 6 h, self–propagating reaction | Propylene conversion into propylene oxide, 2:2:5 propylene:oxygen:helium volume ratio, 250–300 °C, 1 atm, GHSV = 20 000 h−1, 30–37 % selectivity at 1–3–1.5 % conversion | [43] | Multiphase, SBET = 6.2 m2 g−1, Pd 39.4 nm |
| x = 1.5 | Spray granulation of CuO and Mn3O4 in a spouted bed system, calcining for 2 h at 1125 °C | Chemical Looping with Oxygen Uncoupling in coal combustion with CO2 capture, 50 h | [15] | Mixed phase, SBET = 1.0, porosity is 12.1–18.7% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kótai, L.; Petruševski, V.M.; Bereczki, L.; Béres, K.A. Catalytic Properties of the Spinel-Like CuxMn3−xO4 Copper Manganese Oxides—An Overview. Catalysts 2023, 13, 129. https://doi.org/10.3390/catal13010129
Kótai L, Petruševski VM, Bereczki L, Béres KA. Catalytic Properties of the Spinel-Like CuxMn3−xO4 Copper Manganese Oxides—An Overview. Catalysts. 2023; 13(1):129. https://doi.org/10.3390/catal13010129
Chicago/Turabian StyleKótai, László, Vladimir M. Petruševski, Laura Bereczki, and Kende Attila Béres. 2023. "Catalytic Properties of the Spinel-Like CuxMn3−xO4 Copper Manganese Oxides—An Overview" Catalysts 13, no. 1: 129. https://doi.org/10.3390/catal13010129
APA StyleKótai, L., Petruševski, V. M., Bereczki, L., & Béres, K. A. (2023). Catalytic Properties of the Spinel-Like CuxMn3−xO4 Copper Manganese Oxides—An Overview. Catalysts, 13(1), 129. https://doi.org/10.3390/catal13010129