
Synthesis of 3,4-Dihydropyridin-2-ones via Domino Reaction under Phase Transfer Catalysis Conditions


Abstract
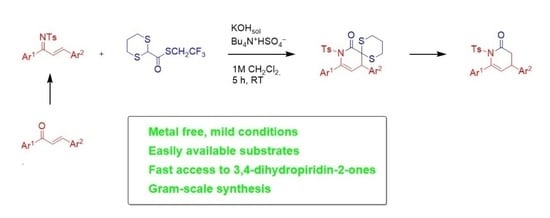
:
1. Introduction
2. Results
3. Conclusions
4. Materials and Methods
4.1. Synthesis of S-2,2,2-Trifluoroethyl 1,3-dithiane-2-carbothioate (1b)
4.2. General Procedure for N-Tosylimine 3 Synthesis
4.3. General Procedure for the Synthesis of 3,4-Dihydropyridin-2-ones 4a–j
4.4. General Procedure for the Synthesis of 3,4-Dihydropyridin-2-ones 5a–b
4.5. Synthesis of 4,6-Diphenyl-3,4-dihydropyridin-2-one (6a)
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Tripathi, S.K.; Biswal, B.K. Piperlongumine, a potent anticancer phytotherapeutic: Perspectives on contemporary status and future possibilities as an anticancer agent. Pharmacol. Res. 2020, 156, 104772–104789. [Google Scholar] [CrossRef] [PubMed]
- Lodin-Friedman, A.; Carmeli, S. Metabolites from Microcystis aeruginosa Bloom Material Collected at a Water Reservoir near Kibbutz Hafetz Haim, Israel. J. Nat. Prod. 2013, 76, 1196–1200. [Google Scholar] [CrossRef] [PubMed]
- Turdi, H.; Chao, H.; Hangeland, J.J.; Ahmad, S.; Meng, W.; Brigance, R.; Zhao, G.; Wang, W.; Moore, F.; Ye, X.Y.; et al. Screening Hit to Clinical Candidate: Discovery of BMS-963272, a Potent, Selective MGAT2 Inhibitor for the Treatment of Metabolic Disorders. J. Med. Chem. 2021, 64, 14773–14792. [Google Scholar] [CrossRef] [PubMed]
- Kempson, J.; Hou, X.; Sun, J.-H.; Wong, M.; Pawluczyk, J.; Li, J.; Krishnananthan, S.; Simmons, E.M.; Hsiao, Y.; Li, Y.-X.; et al. Synthesis Optimization, Scale-Up, and Catalyst Screening Efforts toward the MGAT2 Clinical Candidate, BMS-963272. Org. Process Res. Dev. 2022, 26, 1327–1335. [Google Scholar] [CrossRef]
- Ruebsam, F.; Murphy, D.E.; Tran, C.V.; Li, L.-S.; Zhao, J.; Dragovich, P.S.; McGuire, H.M.; Xiang, Q.; Sun, Z.; Ayida, B.K.; et al. Discovery of tricyclic 5,6-dihydro-1H-pyridin-2-ones as novel, potent, and orally bioavailable inhibitors of HCV NS5B polymerase. Bioorg. Med. Chem. Lett. 2009, 19, 6404–6412. [Google Scholar] [CrossRef]
- Xiao, Y.; Wang, J.; Xia, W.; Shu, S.; Jao, S.; Zhou, Y. γ-Carbon Activation through N-Heterocyclic Carbene/Bronsted Acids Cooperative Catalysis: A Highly Enantioselective Route to δ-Lactams. Org. Lett. 2015, 17, 3850–3853. [Google Scholar] [CrossRef]
- Gabriel, P.; Almehmadi, A.; Wong, Z.R.; Dixon, D.J. A General Iridium-Catalyzed Reductive Dienamine Synthesis of Catharanthine via the Elusive Dehydrosecodine. J. Am. Chem. Soc. 2021, 143, 10828–10835. [Google Scholar] [CrossRef]
- Tanaka, N.; Yoshino, Y.; Nakano, F.; Kurimoto, S.-I.; Kawazoe, K.; Tsuji, D.; Itoh, K.; Li, S.-L.; Sun, H.-D.; Takaishi, Y.; et al. Lanicepines A and B, Sesquiterpenes with Amino Acid-Derived Substituents from the Flowering Aerial Parts of Saussurea laniceps. J. Nat. Prod. 2022, 85, 1180–1185. [Google Scholar] [CrossRef]
- Yan, L.; Wang, H.; Xiong, F.; Tao, Y.; Wu, Y.; Chen, F. Chloramphenicol base chemistry. Part 11: Chloramphenicol base-derived thiourea-catalyzed enantioselective Michael addition of malononitrile to α,β-unsaturated ketones. Tetrahedron Asymmetry 2017, 28, 921–929. [Google Scholar] [CrossRef]
- Huang, X.; Zhu, J.; Broadbent, S. The first asymmetric synthesis of a 4-aryl-substituted 5-carboxy-3,4-dihydropyridin-2-one derivative. Tetrahedron Lett. 2010, 51, 1554–1557. [Google Scholar] [CrossRef]
- Huang, X.; Broadbent, S.; Dvorak, C.; Zhao, S.-H. Pilot-Plant Preparation of 3,4-Dihydropyridin-2-one Derivatives, the Core Structures of P2X7 Receptor Antagonists. Org. Process Res. Dev. 2010, 14, 612–616. [Google Scholar] [CrossRef]
- Lopez-Tapia, F.; Walker, K.A.M.; Brotherton-Pleiss, C.; Caroon, J.; Nitzan, D.; Lowrie, L.; Gleason, S.; Zhao, S.-H.; Berger, J.; Cockayne, D.; et al. Novel Series of Dihydropyridinone P2X7 Receptor Antagonists. J. Med. Chem. 2015, 58, 8413–8426. [Google Scholar] [CrossRef] [PubMed]
- Nantermet, P.G.; Barrow, J.C.; Selnick, H.G.; Homnick, C.F.; Freidinger, R.M.; Chang, S.L.; O’Malley, S.S.; Reiss, D.R.; Broten, T.P.; Ransom, R.W.; et al. Selective α1a adrenergic receptor antagonists based on 4-aryl-3,4-dihydropyridine-2-ones. Bioorg. Med. Chem. Lett. 2000, 10, 1625–1628. [Google Scholar] [CrossRef] [PubMed]
- Goodman, K.B.; Cui, H.; Dowdell, S.E.; Gaitanopoulos, D.E.; Ivy, R.L.; Sehon, C.A.; Stavenger, R.A.; Wang, G.Z.; Viet, A.Q.; Xu, W.; et al. Development of Dihydropyridone Indazole Amides as Selective Rho-Kinase Inhibitors. J. Med. Chem. 2007, 50, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-Q.; Liu, B.-K.; Wu, Q.; Lin, X.-F. Diastereoselective enzymatic synthesis of highly substituted 3,4-dihydropyridin-2-ones via domino Knoevenagel condensation–Michael addition–intramolecular cyclization. Tetrahedron 2011, 67, 9736–9740. [Google Scholar] [CrossRef]
- Cheng, J.; Huang, Z.; Chi, Y.R. NHC Organocatalytic Formal LUMO Activation of α,β-Unsaturated Esters for Reaction with Enamides. Angew. Chem. Int. Ed. 2013, 52, 8592–8596. [Google Scholar] [CrossRef]
- Xia, W.; Yao, H.; Liu, D.; Zhao, L.; Zhou, Y.; Liu, H. Enantioselective N-Heterocyclic Carbene-Catalyzed [3+3] Annulation of α,β-Unsaturated Esters with Methyl Ketoimine. Adv. Synth. Catal. 2016, 358, 1864–1869. [Google Scholar] [CrossRef]
- Yeh, P.-P.; Daniels, D.S.B.; Fallan, C.; Gould, E.; Simal, C.; Taylor, J.E.; Slawin, A.M.Z.; Smith, A.D. Exploring the scope of the isothiourea-mediated synthesis of dihydropyridinones. Org. Biomol. Chem. 2015, 13, 2177–2191. [Google Scholar] [CrossRef] [PubMed]
- Pranabes, B.; Koyel, P.; Sanjay, P.; Asish, R.D. Nano crystalline ZnO catalyzed one pot multicomponent reaction for an easy access of fully decorated 4H-pyran scaffolds and its rearrangement to 2-pyridone nucleus in aqueous media. Tetrahedron Lett. 2012, 53, 4687–4691. [Google Scholar]
- Kumar, J.B.S.; Kumari, N.; Luthra, P.M. One-Pot Synthesis of 3,4-Dihydropyridin-2-one via Michael Addition of in situ–Generated Enaminones. Synth. Commun. 2013, 43, 3010–3019. [Google Scholar] [CrossRef]
- Zhiqiang, L.; Lu, T.; Qi, W.; Xianfu, L. Imidazole-catalyzed Three-component Cascade Reaction for the Facile Synthesis of Highly Substituted 3,4-Dihydropyridin-2-one Derivatives. Chin. J. Chem. 2012, 30, 2343–2348. [Google Scholar]
- Yang, L.; Zheng, Q.-Y.; Wang, D.-X.; Huang, Z.-T.; Wang, M.-X. Reversal of Nucleophilicity of Enamides in Water: Control of Cyclization Pathways by Reaction Media for the Orthogonal Synthesis of Dihydropyridinone and Pyrrolidinone Clausena Alkaloids. Org. Lett. 2008, 10, 2461–2464. [Google Scholar] [CrossRef]
- Meng, T.; Liu, L.; Jia, H.; Ren, L.; Feng, C.; Wang, X.; Zhao, W. One-pot synthesis of 3,4-dihydropyridin-2-ones via tandem reaction of Blaise reaction intermediate and acrylic ester. Appl. Organomet. Chem. 2016, 30, 47–50. [Google Scholar] [CrossRef]
- Luo, T.; Xu, H.; Liu, Y. Aqueous Synthesis of 3,4-Dihydropyridinones from Acryloyl Chloride and Enaminones by Domino Amidation and Intramolecular Michael Addition. ChemSelect 2019, 4, 10621–10623. [Google Scholar] [CrossRef]
- Fuentes, N.; Kong, W.; Fernández-Sánchez, L.; Merino, E.; Nevado, C. Cyclization Cascades via N-Amidyl Radicals toward Highly Functionalized Heterocyclic Scaffolds. J. Am. Chem. Soc. 2015, 137, 964–973. [Google Scholar] [CrossRef]
- Li, L.-L.; Ding, D.; Song, J.; Han, Z.-Y.; Gong, L.-Z. Catalytic Generation of C1 Ammonium Enolates from Halides and CO for Asymmetric Cascade Reactions. Angew. Chem. Int. Ed. 2019, 58, 7647–7651. [Google Scholar] [CrossRef] [PubMed]
- Foschi, F.; Albanese, D.; Pecnikaj, I.; Tagliabue, A.; Penso, M. Regioselective O-Sulfonylation of N,N-Bis(2-hydroxyalkyl)tosylamides as a Synthetic Key Step to Enantiopure Morpholines. Org. Lett. 2017, 19, 70–73. [Google Scholar] [CrossRef]
- Albanese, D.; Landini, D.; Penso, M.; Tagliabue, A.; Carlini, E. Concise Synthesis of C-2-Symmetrical 2,6-Disubstituted Morpholines by N → O Boc Migration under SL-PTC Conditions. Org. Process Res. Dev. 2010, 14, 705–711. [Google Scholar] [CrossRef]
- Albanese, D.; Landini, D.; Penso, M. Novel concise synthesis of 2-substituted 3,4-dihydro-2H-1,4-benzoxazines by ring opening of glycidols under solid-liquid phase transfer catalysis conditions. Chem. Comm. 1999, 20, 2095–2096. [Google Scholar] [CrossRef]
- Destro, D.; Bottinelli, C.; Ferrari, L.; Albanese, D.C.M.; Bencivenni, G.; Gillick-Healy, M.W.; Kelly, B.G.; Adamo, M.F.A. Enantioselective Synthesis of 3,4-Dihydropyran-2-ones via Phase-Transfer-Catalyzed Addition−Cyclization of Acetylacetone to Cinnamic Thioesters. J. Org. Chem. 2020, 85, 5183–5192. [Google Scholar] [CrossRef]
- Gaggero, N.; Albanese, D.C.M.; Nava, D. A new approach to 4,6-dihydropyran-2-ones by Domino Michael addition-cyclization reaction under PTC conditions. Tetrahedron 2014, 70, 8744–8749. [Google Scholar] [CrossRef]
- Yus, M.; Nàjera, C.; Foubelo, F. The role of 1,3-dithianes in natural product synthesis. Tetrahedron 2003, 59, 6147–6212. [Google Scholar] [CrossRef]
- Xie, L.; Bors, D.A.; Streitwieser, A. Carbon acidity. 82. Equilibrium Cesium Ion Pair Acidities of Some Substituted 1,3-Dithianes. J. Org. Chem. 1992, 57, 4986–4990. [Google Scholar] [CrossRef]
- Massolo, E.; Benaglia, M.; Genoni, A.; Annunziata, R.; Celentano, G.; Gaggero, N. Stereoselective reaction of 2-carboxythioesters-1,3-dithiane with nitroalkenes: An organocatalytic strategy for the asymmetric addition of a glyoxylate anion equivalent. Org. Biomol. Chem. 2015, 13, 5591–5596. [Google Scholar] [CrossRef]
- Massolo, E.; Brenna, D.; Cozzi, F.; Raimondi, L.; Gaggero, N.; Benaglia, M. 2-Carboxythioesters-1,3-dithiane: A Functionalized Masked Carbonyl Nucleophile for the Organocatalytic Enantioselective Michael Addition to Enones. Synlett 2016, 27, 2716–2720. [Google Scholar]
- Albanese, D. Phase Transfer Catalysis. In Kirk-Othmer Encyclopedia of Chemical Technology; John Wiley & Sons: New York, NY, USA, 2020. [Google Scholar]
- Albanese, D. Phase Transfer Catalysis (PTC): A powerful tool for the organic synthesis. Mini-Rev. Org. Chem. 2006, 3, 195–217. [Google Scholar] [CrossRef]
- Back, T.G.; Baron, D.L.; Yang, K. Desulfurization with Nickel and Cobalt Boride: Scope, Selectivity, Stereochemistry, and Deuterium-Labeling Studies. J. Org. Chem. 1993, 58, 2407–2413. [Google Scholar] [CrossRef]
- Zaman, S.S.; Sarmah, P.; Barua, N.C.; Sharma, R.P. Nickel Boride Desulfurization of α,β-Unsaturated Ethylene Dithioketals. Chem. Ind. 1989, 21, 806–807. [Google Scholar]
- Liu, B.; Wang, W.; Huang, R.; Yan, J.; Wu, J.; Xue, W.; Yang, S.; Jin, Z.; Chi, Y.R. Direct Activation of β-sp3-Carbons of Saturated Carboxylic Esters as Electrophilic Carbons via Oxidative Carbene Catalysis. Org. Lett. 2018, 20, 260–263. [Google Scholar] [CrossRef]
- Zhang, S.; Bacheley, L.; Young, C.M.; Stark, D.G.; O’Riordan, T.; Slawin, A.M.Z.; Smith, A.D. Isothiourea-Catalyzed Functionalization of Pyrrolyl- and Indolylacetic Acid: Enantioselective Synthesis of Dihydropyridine ones and One-pot Synthesis of Pyridinones. Asian J. Org. Chem. 2020, 9, 1562–1566. [Google Scholar] [CrossRef]
- Espinosa, M.; Blay, G.; Cardona, L.; Pedro, J.R. Asymmetric Conjugate Addition of Malonate Esters to α,β-Unsaturated N-SulfonylImines: An Expeditious Route to Chiral δ-Aminoesters and Piperidones. Chem. Eur. J. 2013, 19, 14861–14866. [Google Scholar] [CrossRef] [PubMed]
- Stokes, S.; Mead, K.T. Synthesis of Quinolines from N-Tosyl-1-azadienes. Synth. Commun. 2013, 43, 2627–2633. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
| Entry | Thioester | 3a (eq.) | Cat. (0.1 eq.) | Base (eq.) | Solvent | t (h) | 4a (Yield %) b |
| 1 | 1a | 1.05 | TBAHSO4 | KOH (1.1) | DCM | 22 | 51 |
| 2 | 1a | 1.05 | TEBA | KOH (1.1) | DCM | 22 | 50 |
| 3 | 1a | 1.25 | TBAHSO4 | KOH (1.1) | DCM | 22 | 61 |
| 4 | 1a | 1.25 | TBAHSO4 | KOH (1.5) | DCM | 22 | 62 |
| 5 | 1a | 1.25 | TBAHSO4 | K2CO3 (1.5) | DCM | 22 | 38 |
| 6 | 1a | 1.25 | – | KOH (1.1) | DCM | 22 | 13 |
| 7 c | 1a | 1.25 | TBAHSO4 | – | DCM | 22 | 20 |
| 8 | 1b | 1.25 | TBAHSO4 | KOH (1.5) | DCM | 5 | 85 |
| 9 d | 1b | 1.25 | TBAHSO4 | KOH (1.5) | DCM | 5 | 88 |
| 10 e | 1b | 1.25 | TBAHSO4 | KOH (1.5) | toluene | 5 | 38 |
| 11 e | 1b | 1.25 | TBAHSO4 | KOH (1.5) | 2MeTHF | 5 | 44 |
| 12 e | 1b | 1.25 | TBAHSO4 | KOH (1.5) | CH3CN | 5 | 54 |
| 13 e | 1b | 1.25 | TBAHSO4 | KOH (1.5) | 1,2,3-trimethylbenzene | 5 | 47 |
| 14 | 1b | 1.15 | TBAHSO4 | KOH (1.5) | DCM | 5 | 77 |
| 15 | 1b | 1.15 | TBAHSO4 | – | DCM | 5 | 0 |
| 16 e | 1b | 1.15 | – | KOH (1.5) | DCM | 5 | 33 |
 | |||
| R1 | R2 | Yield (%) | |
| 3a,4a | Ph | Ph | 85 |
| 3b,4b | 4-C6H4-Cl | Ph | 62 |
| 3c,4c | 4-C6H4-Br | Ph | 64 |
| 3d,4d | 4-C6H4-OMe | 4-C6H4-OMe | 60 b |
| 3e,4e | Ph | 4-C6H4-OMe | 59 |
| 3f,4f | Ph | 4-C6H4-Me | 70 |
| 3g,4g | Ph | 4-C6H4-NO2 | – |
| 3h,4h | Ph | 2-C6H4-Br | 50 |
| 3i,4i | Ph | 3-methylthienyl | 72 |
| 3j,4j | Ph | 2-methylfuryl | 76 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Albanese, D.C.M.; Gaggero, N.; Prenga, K. Synthesis of 3,4-Dihydropyridin-2-ones via Domino Reaction under Phase Transfer Catalysis Conditions. Catalysts 2023, 13, 170. https://doi.org/10.3390/catal13010170
Albanese DCM, Gaggero N, Prenga K. Synthesis of 3,4-Dihydropyridin-2-ones via Domino Reaction under Phase Transfer Catalysis Conditions. Catalysts. 2023; 13(1):170. https://doi.org/10.3390/catal13010170
Chicago/Turabian StyleAlbanese, Domenico C. M., Nicoletta Gaggero, and Kamila Prenga. 2023. "Synthesis of 3,4-Dihydropyridin-2-ones via Domino Reaction under Phase Transfer Catalysis Conditions" Catalysts 13, no. 1: 170. https://doi.org/10.3390/catal13010170
APA StyleAlbanese, D. C. M., Gaggero, N., & Prenga, K. (2023). Synthesis of 3,4-Dihydropyridin-2-ones via Domino Reaction under Phase Transfer Catalysis Conditions. Catalysts, 13(1), 170. https://doi.org/10.3390/catal13010170