
Catalysis for CO2 Hydrogenation—What We Have Learned/Should Learn from the Hydrogenation of Syngas to Methanol

 ,
, 

Abstract
:1. Introduction
2. Current Status of Syngas to Methanol
2.1. Cu-Based Catalysts
2.2. Precious Metal Catalysts
2.3. Metal Oxide Catalysts
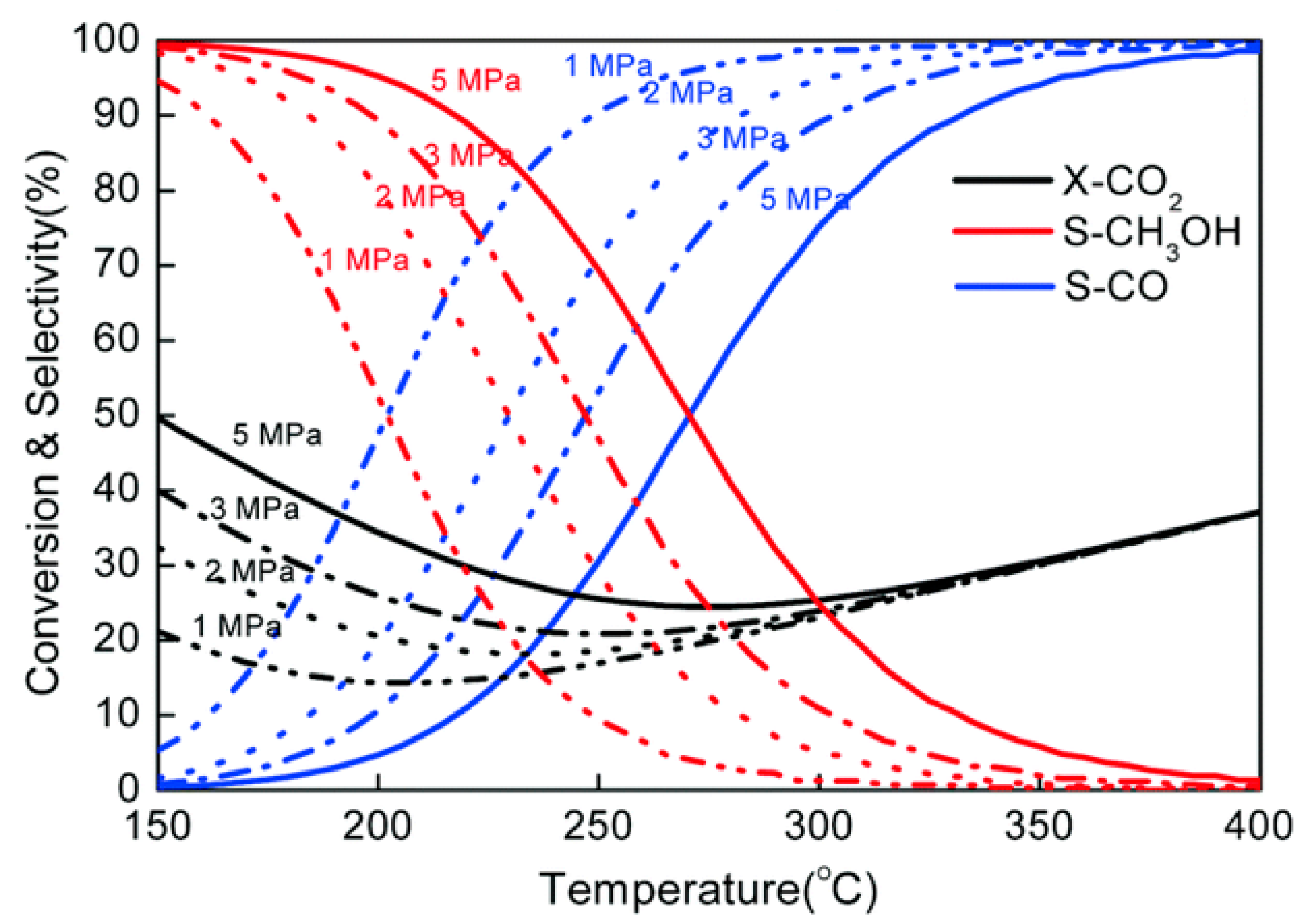
3. Methanol Synthesis from CO2
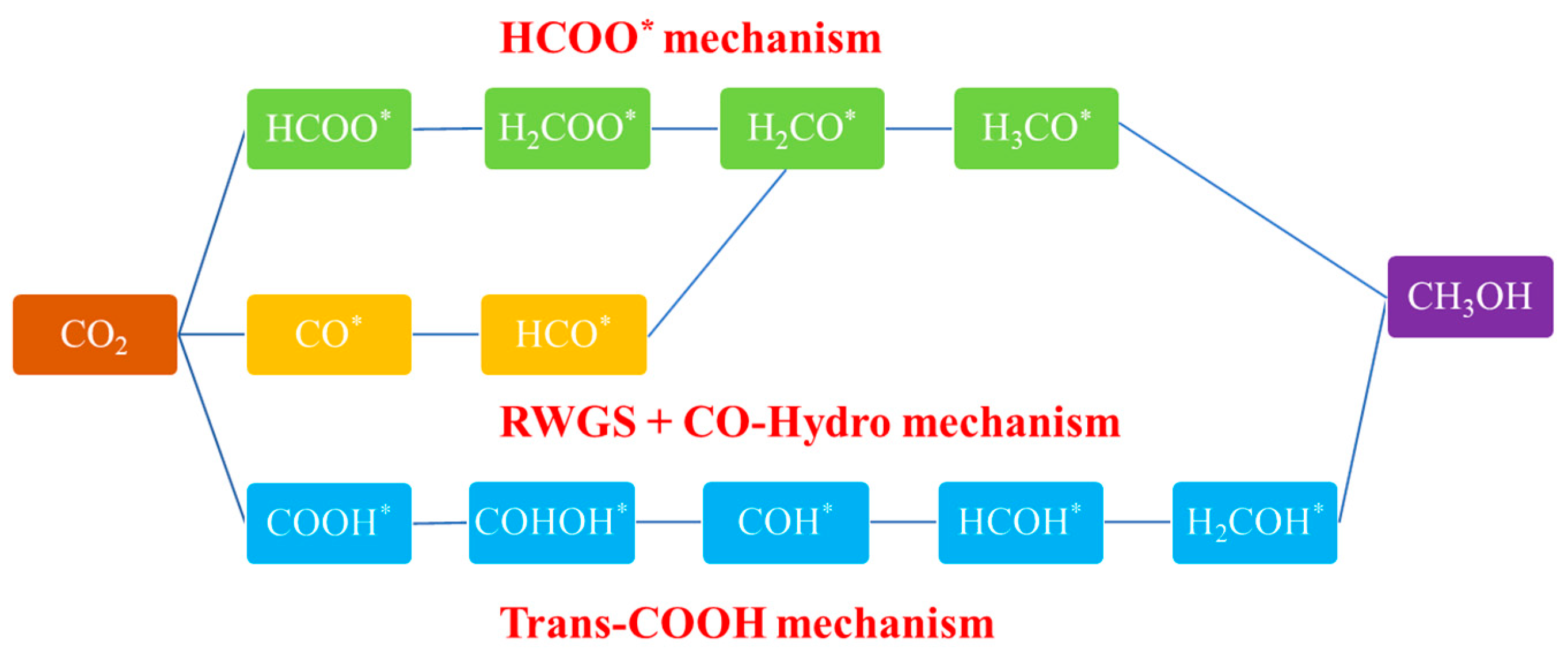
3.1. Reaction Pathways of CO2 Hydrogenation to Methanol
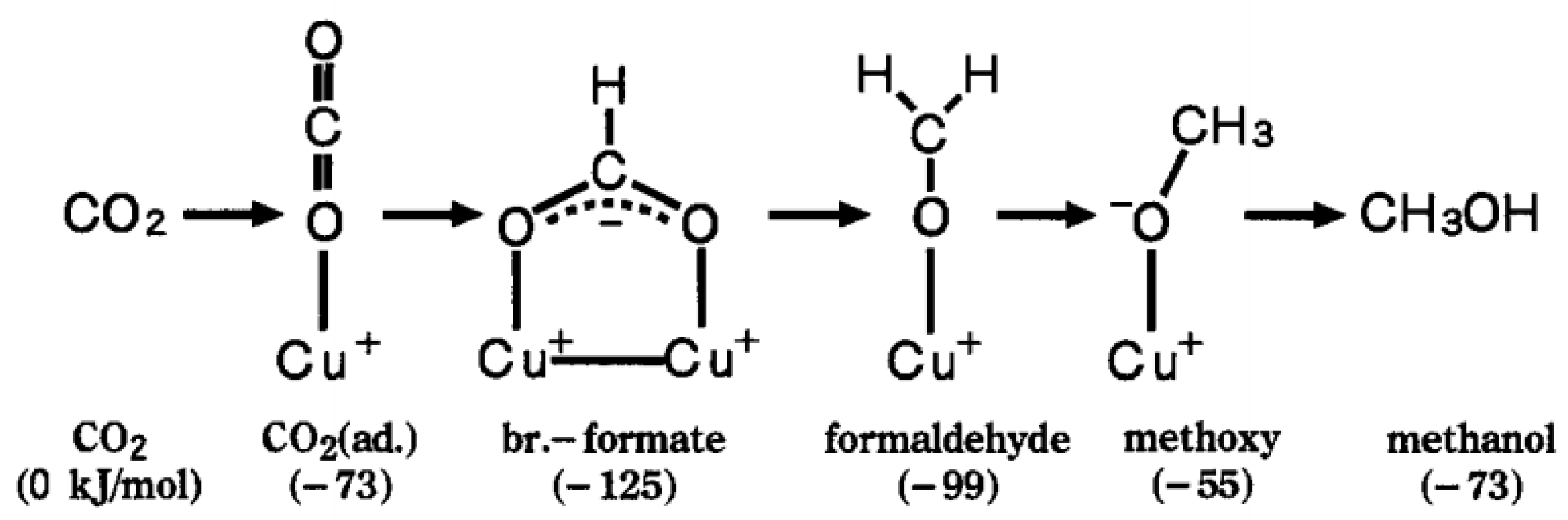
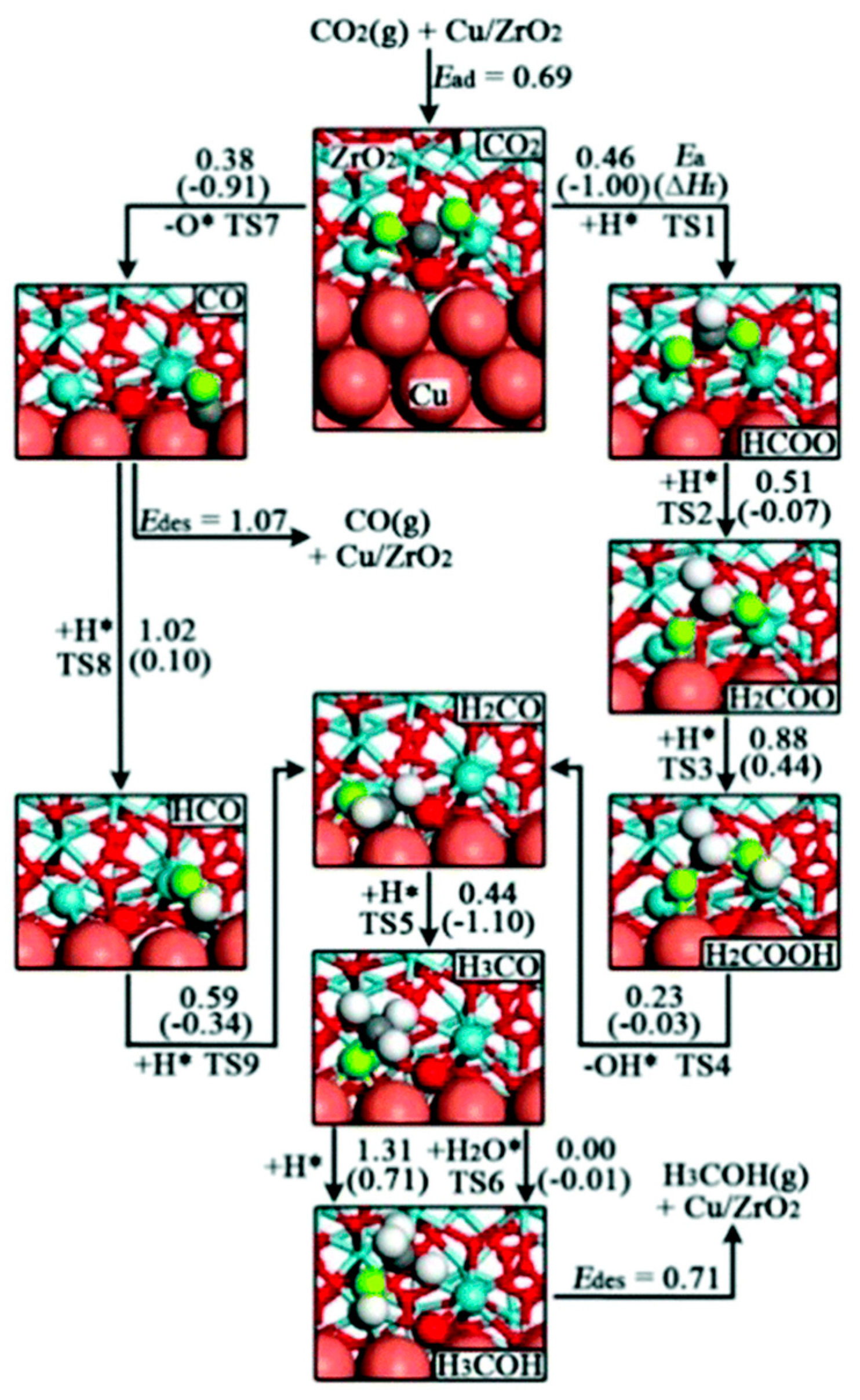
3.1.1. Formate (HCOO*) Pathway
3.1.2. RWGS + CO Hydrogenation Pathway

3.1.3. Trans-COOH Pathway
3.2. Insights into the Active Phase of Cu-Based CO2 Hydrogenation to Methanol Catalysts
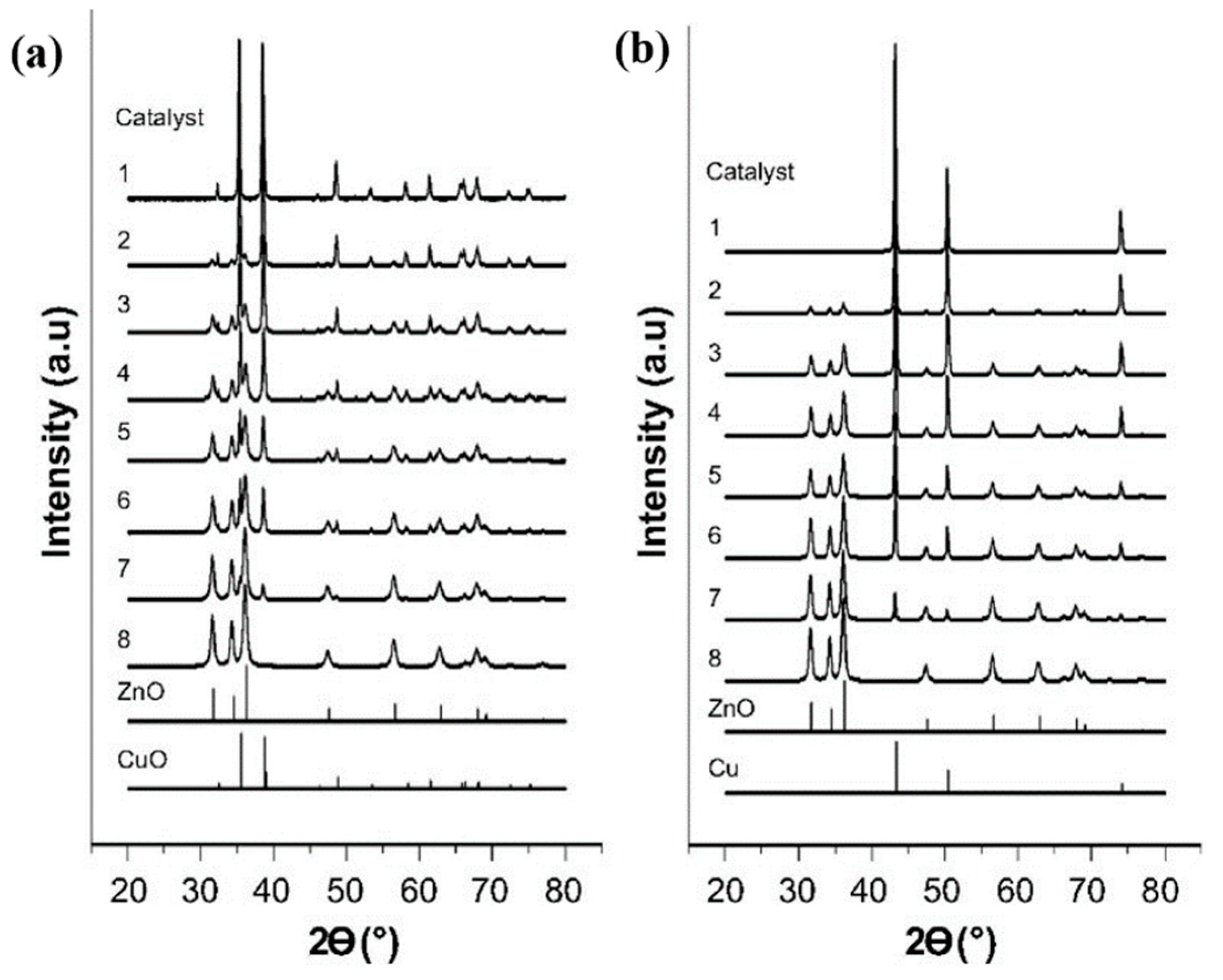
3.2.1. Chemical State of Cu Species
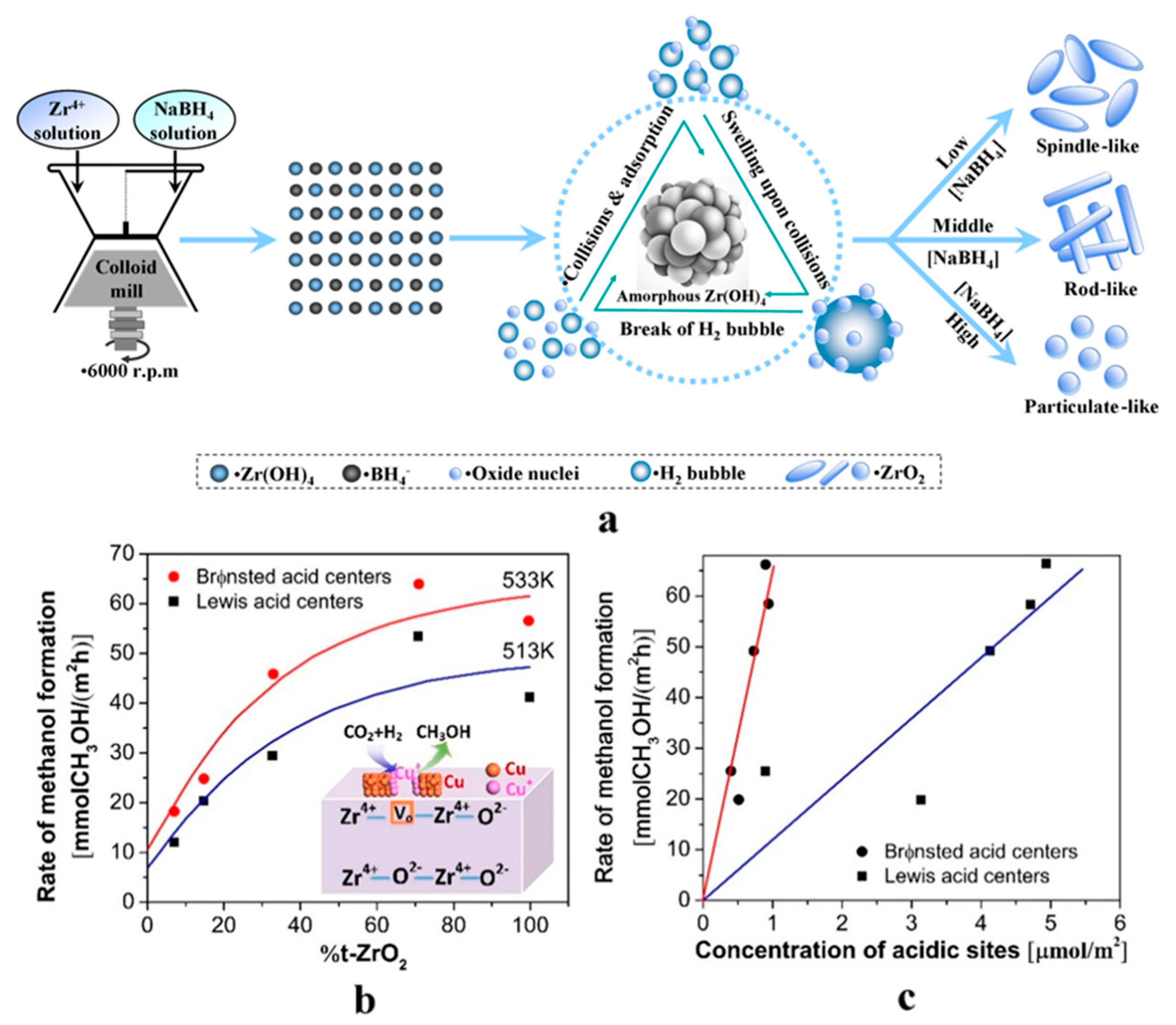
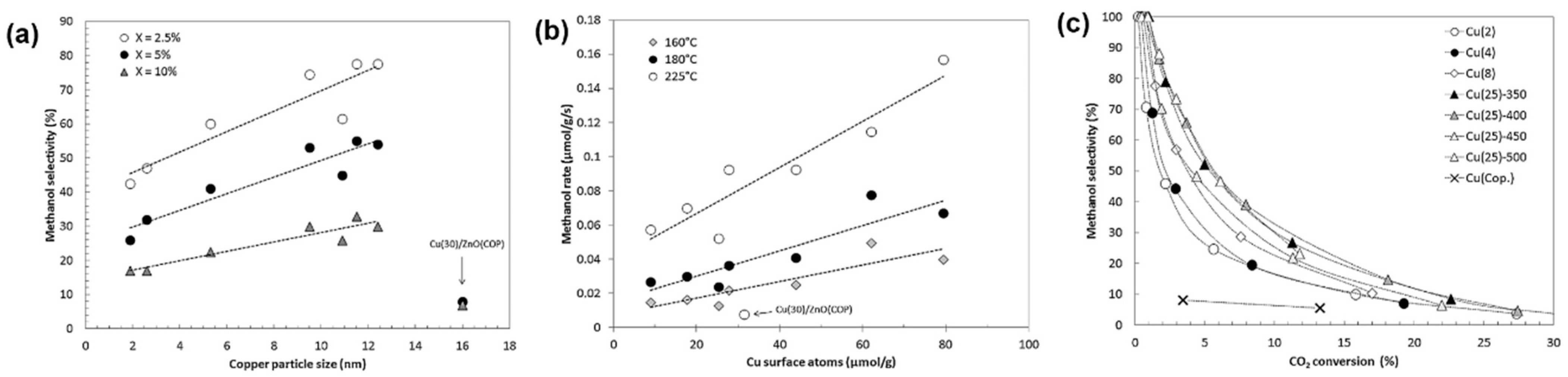
3.2.2. Morphology of Metal Nanoparticles
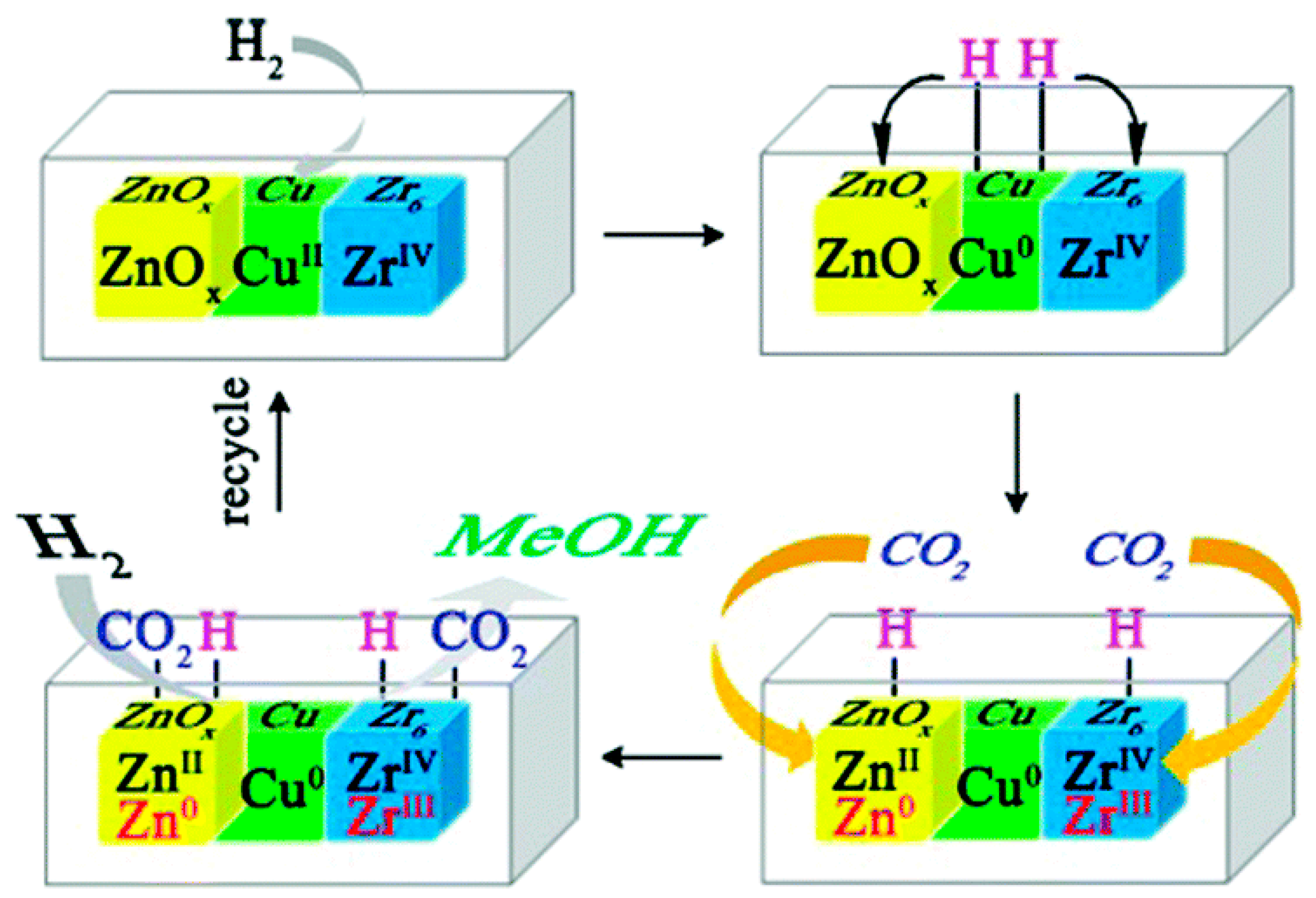
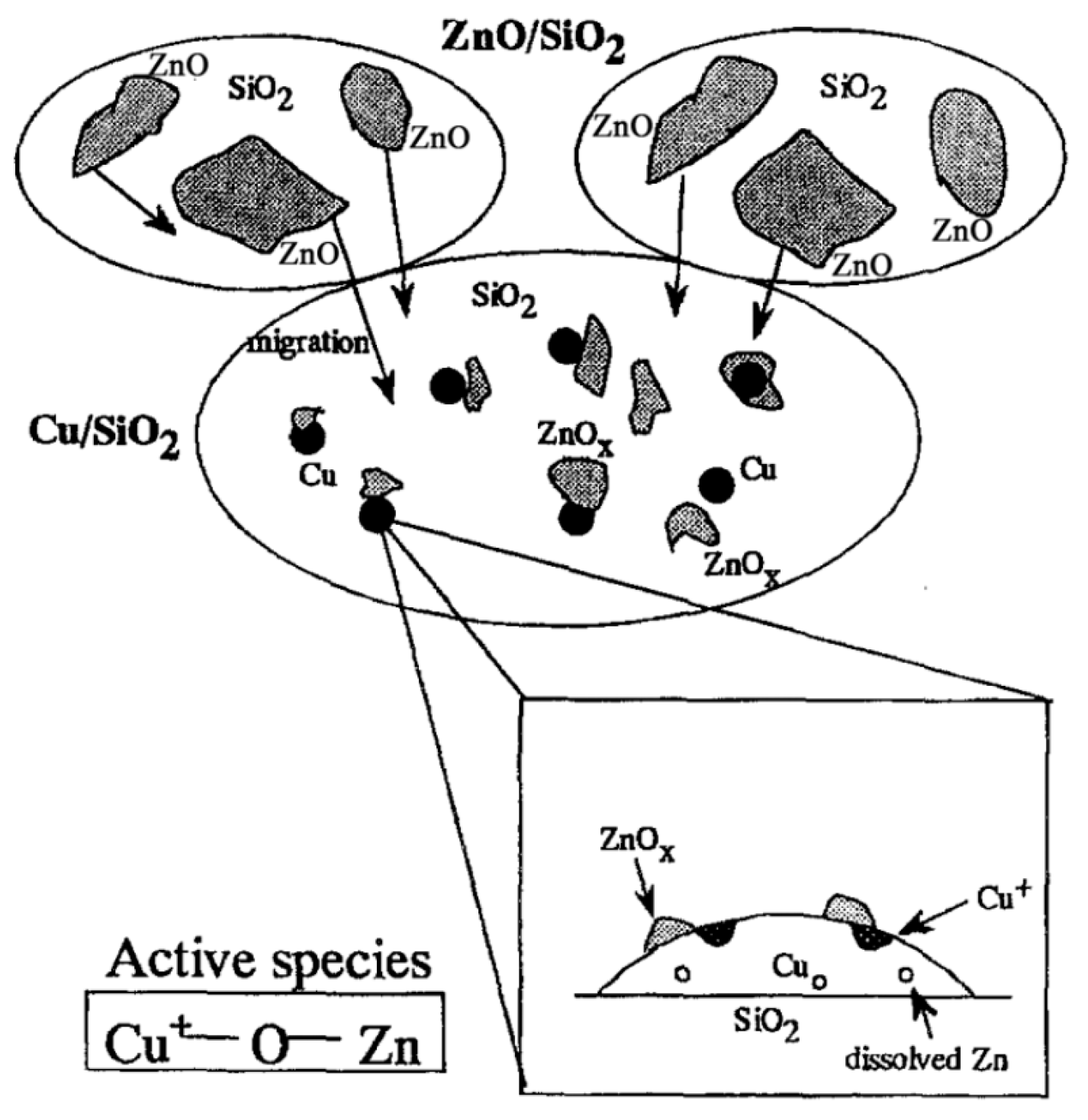
3.2.3. Metal–Support Interactions
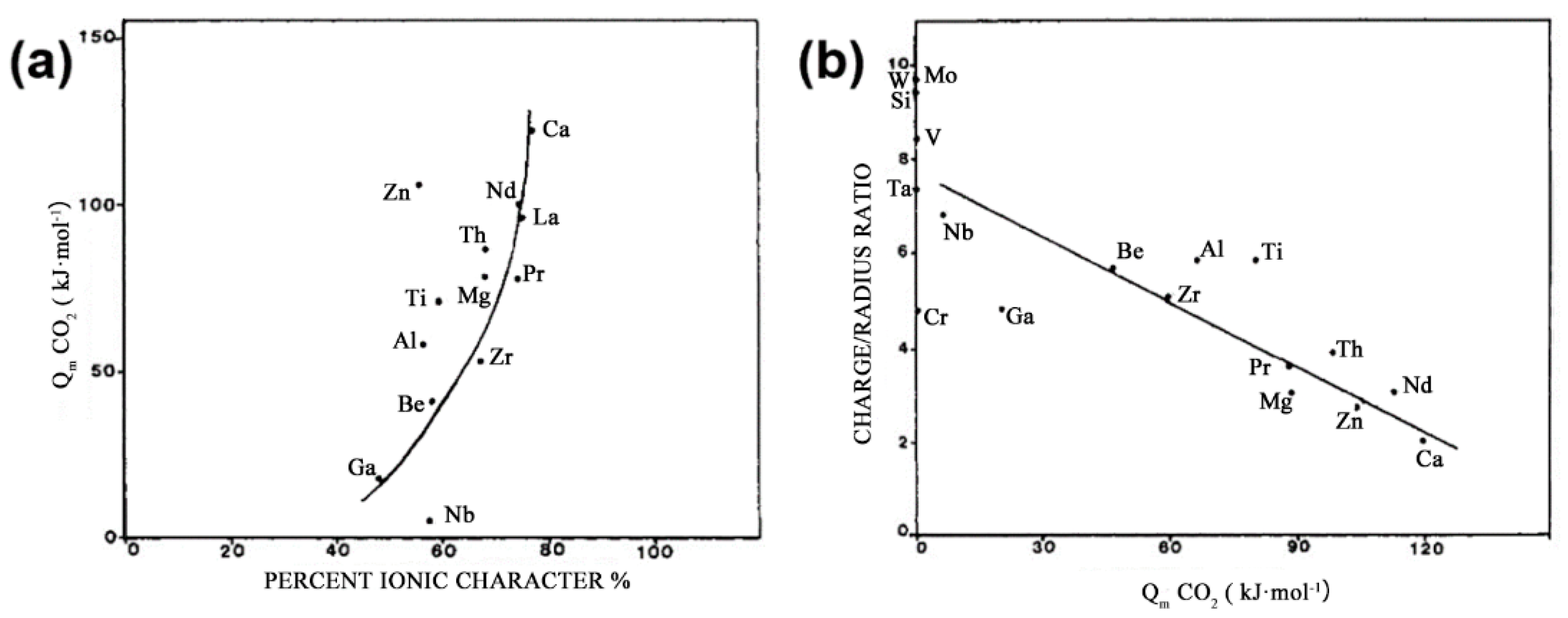
3.2.4. Active Sites of CO2 Activation
4. Conclusions and Perspective
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Xue, B.L.Y.; Wang, C. Progress on carbon capture, storge and utilization technology and coal seam CO2 storage. Chem. World 2020, 61, 294–297. [Google Scholar]
- Jiang, K.; Ashworth, P.; Zhang, S.; Liang, X.; Sun, Y.; Angus, D. China’s carbon capture, utilization and storage (CCUS) policy: A critical review. Renew. Sustain. Energy Rev. 2020, 119, 109601. [Google Scholar] [CrossRef]
- Li, Q.W.Z.; Lou, S. Research progress in methanol production from carbon dioxide hydrogenation. Mod. Chem. Ind. 2019, 39, 19–23. [Google Scholar]
- McFarlan, A. Techno-economic assessment of pathways for electricity generation in northern remote communities in Canada using methanol and dimethyl ether to replace diesel. Renew. Sustain. Energy Rev. 2018, 90, 863–876. [Google Scholar] [CrossRef]
- Goeppert, A.; Czaun, M.; Jones, J.P.; Prakash, G.K.S.; Olah, G.A. recycling of carbon dioxide to methanol and derived products—Closing the loop. Chem. Soc. Rev. 2014, 43, 7995–8048. [Google Scholar] [CrossRef]
- Olah, G.A.; Prakash, G.K.S.; Goeppert, A. Anthropogenic Chemical Carbon Cycle for a Sustainable Future. J. Am. Chem. Soc. 2011, 133, 12881–12898. [Google Scholar] [CrossRef]
- Zhong, J.; Yang, X.; Liang, Z.; Huang, B.; Zhang, Y. State of the art and perspectives in heterogeneous catalysis of CO2 hydrogenation to methanol. Chem. Soc. Rev. 2020, 49, 1385–1413. [Google Scholar] [CrossRef]
- Shen, W.J.; Jun, K.W.; Choi, H.S.; Lee, K.W. Thermodynamic investigation of methanol and dimethyl ether synthesis from CO2 hydrogenation. Korean J. Chem. Eng. 2000, 17, 210–216. [Google Scholar] [CrossRef]
- Ipatieff, V.N.; Monroe, G.S. Synthesis of methanol from carbon dioxide and hydrogen over Copper-Alumina catalysts. mechanism of reaction. J. Am. Chem. Soc. 2002, 67, 2168–2171. [Google Scholar] [CrossRef]
- Zhang, Z.L.F.; Kebin, C. New development of methanol technology. Liaoning Chem. Ind. 2001, 30, 4. [Google Scholar]
- Ling, Y.; Luo, J.; Ran, Y.; Liu, Z.; Li, W.-X.; Yang, F. Atomic-Scale Visualization of Heterolytic H2 Dissociation and COx Hydrogenation on ZnO under Ambient Conditions. J. Am. Chem. Soc. 2023, 145, 22697–22707. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lin, S.; Li, M.; Zhu, C.; Yang, H.; Dong, P.; Lu, M.; Wang, W.; Cao, J.; Liu, Q.; et al. Boosting CO2 hydrogenation of Fe-based monolithic catalysts via 3D printing technology-induced heat/mass-transfer enhancements. Appl. Catal. B Environ. 2024, 340, 123211. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Z.; Luo, P.; Cui, N.; Wang, K.; Huang, W. Size-dependent and sensitivity of copper particle in ternary CuZnAl catalyst for syngas to ethanol. Appl. Catal. B Environ. 2023, 336, 122949. [Google Scholar] [CrossRef]
- Wei, Z.Z.; Bai, B.; Bai, H.; Duan, Y.L.; Yang, M.X.; Cao, H.J.; Zuo, Z.J.; Zuo, J.P.; Wang, Q.; Huang, W. Mechanistic exploration of syngas conversion at the interface of graphene/Cu(111): Identifying the effect of promoted electron transfer on the product selectivity. Catal. Sci. Technol. 2023. [Google Scholar] [CrossRef]
- Luk, H.T.; Novak, G.; Safonova, O.V.; Siol, S.; Stewart, J.A.; Curulla Ferré, D.; Mondelli, C.; Pérez-Ramírez, J. CO2-Promoted Catalytic Process Forming Higher Alcohols with Tunable Nature at Record Productivity. ChemCatChem 2020, 12, 2732–2744. [Google Scholar] [CrossRef]
- Kunkes, E.L.; Studt, F.; Abild-Pedersen, F.; Schlögl, R.; Behrens, M. Hydrogenation of CO2 to methanol and CO on Cu/ZnO/Al2O3: Is there a common intermediate or not? J. Catal. 2015, 328, 43–48. [Google Scholar] [CrossRef]
- Zhu, J.; Su, Y.; Chai, J.; Muravev, V.; Hensen, E.J.M. Mechanism and Nature of Active Sites for Methanol Synthesis from CO/CO2 on Cu/CeO2. ACS Catal. 2020, 10, 11532–11544. [Google Scholar] [CrossRef]
- Nielsen, N.D.; Jensen, A.D.; Christensen, J.M. The roles of CO and CO2 in high pressure methanol synthesis over Cu-based catalysts. J. Catal. 2021, 393, 324–334. [Google Scholar] [CrossRef]
- Bansode, A.; Tidona, B.; Rohr, P.R.V.; Urakawa, A. Impact of K and Ba promoters on CO2 hydrogenation over Cu/Al2O3 catalysts at high pressure. Catal. Sci. Technol. 2013, 3, 767–778. [Google Scholar] [CrossRef]
- Ladera, R.; Pérez-Alonso, F.J.; González-Carballo, J.M.; Ojeda, M.; Rojas, S.; Fierro, J.L.G. Catalytic valorization of CO2 via methanol synthesis with Ga-promoted Cu–ZnO–ZrO2 catalysts. Appl. Catal. B Environ. 2013, 142, 241–248. [Google Scholar] [CrossRef]
- Toyir, J.; Piscina, P.R.R.D.L.; Fierro, J.L.G.; Homs, N.S. Highly effective conversion of CO2 to methanol over supported and promoted copper-based catalysts: Influence of support and promoter. Appl. Catal. B Environ. 2001, 29, 207–215. [Google Scholar] [CrossRef]
- Słoczyński, J.; Grabowski, R.; Olszewski, P.; Kozłowska, A.; Stoch, J.; Lachowska, M.; Skrzypek, J. Effect of metal oxide additives on the activity and stability of Cu/ZnO/ZrO2 catalysts in the synthesis of methanol from CO2 and H2. Appl. Catal. A Gen. 2006, 310, 127–137. [Google Scholar] [CrossRef]
- Li, M.J.; Zeng, Z.; Liao, F.; Hong, X.; Tsang, S.C.E. Enhanced CO2 hydrogenation to methanol over CuZn nanoalloy in Ga modified Cu/ZnO catalysts. J. Catal. 2016, 343, 157–167. [Google Scholar] [CrossRef]
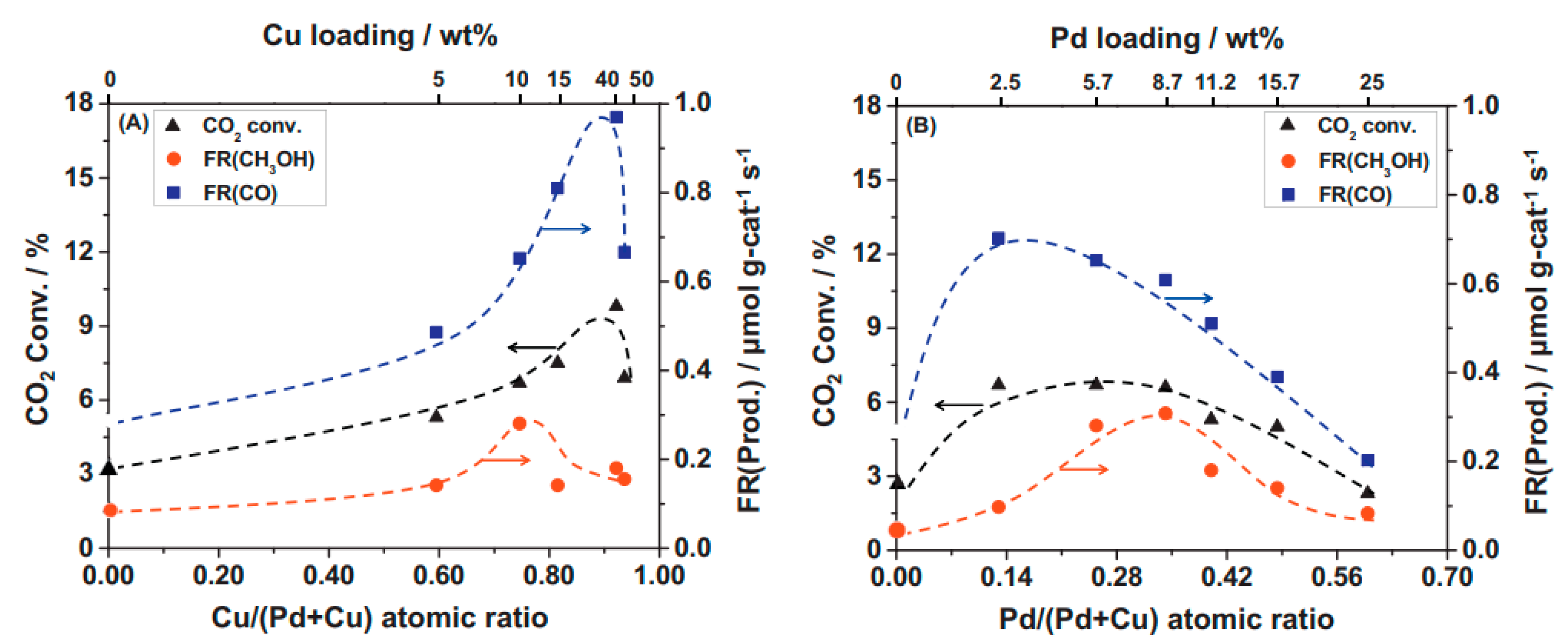
- Jiang, X.; Koizumi, N.; Guo, X.; Song, C. Bimetallic Pd-Cu Catalysts for Selective CO2 Hydrogenation to Methanol. Appl. Catal. B Environ. 2015, 170–171, 173–185. [Google Scholar] [CrossRef]
- Liang, X.L.; Xie, J.R.; Liu, Z.M. A Novel Pd-decorated Carbon Nanotubes-promoted Pd-ZnO Catalyst for CO2 Hydrogenation to Methanol. Catal. Lett. 2015, 145, 1138–1147. [Google Scholar] [CrossRef]
- Kong, H.; Li, H.Y.; Zhang, L.H.B. Pd-Decorated CNT-Promoted Pd-Ga2O3 Catalyst for Hydrogenation of CO2 to Methanol. Catal. Lett. 2011, 141, 886–894. [Google Scholar] [CrossRef]
- Qu, J.; Xu, F.; Zhou, X. Shape Effect of Pd-Promoted Ga2O3 Nanocatalysts for Methanol Synthesis by CO2 Hydrogenation. J. Phys. Chem. C. Nanomater. Interfaces 2014, 118, 24452–24466. [Google Scholar] [CrossRef]
- Collins, S.E.; Chiavassa, D.L.; Bonivardi, A.L.; Baltanás, M.A. Hydrogen Spillover in Ga2O3–Pd/SiO2 Catalysts for Methanol Synthesis from CO2/H2. Catal. Lett. 2005, 103, 83–88. [Google Scholar] [CrossRef]
- Song, Y.; Liu, X.; Xiao, L.; Wu, W.; Zhang, J. Pd-Promoter/MCM-41: A Highly Effective Bifunctional Catalyst for Conversion of Carbon Dioxide. Catal. Lett. 2015, 145, 1272–1280. [Google Scholar] [CrossRef]
- Martin, O.; Martín, D.A.J.; Mondelli, C.; Mitchell, S.; Segawa, T.F.; Hauert, R.; Drouilly, C.; Curulla-Ferré, D.D.; Pérez-Ramírez, P.J. Indium Oxide as a Superior Catalyst for Methanol Synthesis by CO2 Hydrogenation. Angew. Chem. Int. Ed. 2016, 55, 6261–6265. [Google Scholar] [CrossRef]
- Chen, T.-Y.; Chen, C.; Ding, T.-B.; Huang, X.; Shen, H.; Cao, L.; Zhu, X.; Xu, M.; Gao, J.; Han, J.; et al. Unraveling Highly Tunable Selectivity in CO2 Hydrogenation over Bimetallic In-Zr Oxide Catalysts. ACS Catal. 2019, 9, 8785–8797. [Google Scholar] [CrossRef]
- Sun, K.; Fan, Z.; Ye, J.; Yan, J.; Ge, Q.; Li, Y.; He, W.; Yang, W.; Liu, C. Hydrogenation of CO2 to methanol over In2O3 catalyst. J. CO2 Util. 2015, 12, 1–6. [Google Scholar] [CrossRef]
- Wang, J.; Li, G.; Li, Z.; Tang, C.; Feng, Z.; An, H.; Liu, H.; Liu, T.; Li, C. A highly selective and stable ZnO-ZrO2 solid solution catalyst for CO2 hydrogenation to methanol. Sci. Adv. 2017, 3, e1701290. [Google Scholar] [CrossRef] [PubMed]
- Lunkenbein, T.; Schumann, J.; Behrens, M.; Schloegl, R.; Willinger, M.G. Formation of a ZnO Overlayer in Industrial Cu/ZnO/Al2O3 Catalysts Induced by Strong Metal-Support Interactions. Angew. Chem.-Int. Ed. 2015, 54, 4544–4548. [Google Scholar] [CrossRef]
- Cao, W.; Kang, J.; Fan, G.; Yang, L.; Li, F. Fabrication of Porous ZrO2 Nanostructures with Controlled Crystalline Phases and Structures via a Facile and Cost-Effective Hydrothermal Approach. Ind. Eng. Chem. Res. 2015, 54, 12795–12804. [Google Scholar] [CrossRef]
- Samson, K.; Sliwa, M.; Socha, R.P. Influence of ZrO2 Structure and Copper Electronic State on Activity of Cu/ZrO2 Catalysts in Methanol Synthesis from CO2. ACS Catal. 2014, 4, 3730–3741. [Google Scholar] [CrossRef]
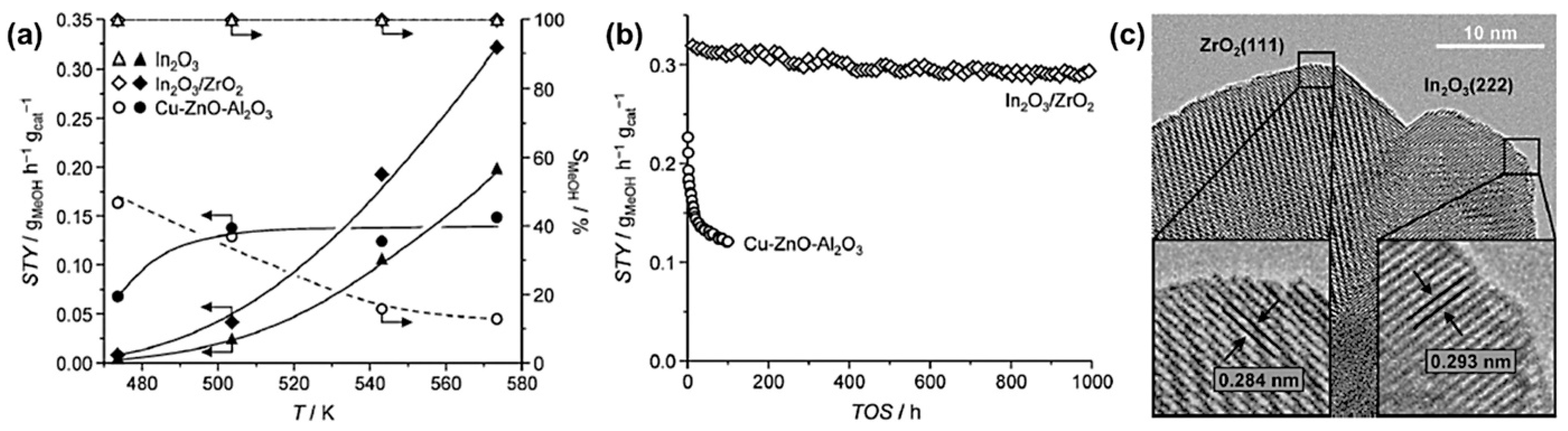
- Li, K.; Chen, J.G. CO2 Hydrogenation to Methanol over ZrO2-Containing Catalysts: Insights into ZrO2 Induced Synergy. ACS Catal. 2019, 9, 7840–7861. [Google Scholar] [CrossRef]
- Graciani, J.; Mudiyanselage, K.; Xu, F.; Baber, A.E.; Evans, J.; Senanayake, S.D.; Stacchiola, D.J.; Liu, P.; Hrbek, J.; Sanz, J.F.; et al. Highly active copper-ceria and copper-ceria-titania catalysts for methanol synthesis from CO2. Science 2014, 345, 546–550. [Google Scholar] [CrossRef]
- Tan, Q.; Shi, Z.; Wu, D. CO2 Hydrogenation to Methanol over a Highly Active Cu-Ni/CeO2—Nanotube Catalyst. Ind. Eng. Chem. Res. 2018, 57, 10148–10158. [Google Scholar] [CrossRef]
- Yang, H.; Gao, P.; Zhang, C.; Zhong, L.; Li, X.; Wang, S.; Wang, H.; Wei, W.; Sun, Y. Core–shell structured Cu@m-SiO2 and Cu/ZnO@m-SiO2 catalysts for methanol synthesis from CO2 hydrogenation. Catal. Commun. 2016, 84, 56–60. [Google Scholar] [CrossRef]
- Yu, J.; Yang, M.; Zhang, J.; Ge, Q.; Zimina, A.; Pruessmann, T.; Zheng, L.; Grunwaldt, J.-D.; Sun, J. Stabilizing Cu+ in Cu/SiO2 Catalysts with a Shattuckite-Like Structure Boosts CO2 Hydrogenation into Methanol. ACS Catal. 2020, 10, 14694–14706. [Google Scholar] [CrossRef]
- Liu, C.; Guo, X.; Guo, Q.; Mao, D.; Yu, J.; Lu, G. Methanol synthesis from CO2 hydrogenation over copper catalysts supported on MgO-modified TiO2. J. Mol. Catal. A Chem. 2016, 425, 86–93. [Google Scholar] [CrossRef]
- Chang, K.; Wang, T.; Chen, J.G. Hydrogenation of CO2 to methanol over CuCeTiOx catalysts. Appl. Catal. B Environ. Int. J. Devoted Catal. Sci. Its Appl. 2017, 206, 704–711. [Google Scholar] [CrossRef]
- Wang, G.; Chen, L.; Sun, Y.; Wu, J.; Fu, M.; Ye, D. Carbon dioxide hydrogenation to methanol over Cu/ZrO2/CNTs: Effect of carbon surface chemistry. RSC Adv. 2015, 5, 45320–45330. [Google Scholar] [CrossRef]
- Sun, Y.; Chen, L.; Bao, Y.; Wang, G.; Zhang, Y.; Fu, M.; Wu, J.; Ye, D. Roles of nitrogen species on nitrogen-doped CNTs supported Cu-ZrO2 system for carbon dioxide hydrogenation to methanol. Catal. Today 2018, 307, 212–223. [Google Scholar] [CrossRef]
- An, B.; Zhang, J.; Cheng, K.; Ji, P.; Wang, C.; Lin, W. Confinement of Ultrasmall Cu/ZnOx Nanoparticles in Metal-Organic Frameworks for Selective Methanol Synthesis from Catalytic Hydrogenation of CO2. J. Am. Chem. Soc. 2017, 139, 3834. [Google Scholar] [CrossRef]
- Jiang, X.; Nie, X.; Guo, X.; Song, C.; Chen, J.G. Recent Advances in Carbon Dioxide Hydrogenation to Methanol via Heterogeneous Catalysis. Chem. Rev. 2020, 120, 7984–8034. [Google Scholar] [CrossRef]
- Ren, H.; Xu, C.H.; Zhao, H.Y.; Wang, Y.X.; Liu, J.; Liu, J.Y. Methanol synthesis from CO2 hydrogenation over Cu/γ-Al2O3 catalysts modified by ZnO, ZrO2 and MgO. J. Ind. Eng. Chem. 2015, 28, 261–267. [Google Scholar] [CrossRef]
- Li, S.; Guo, L.; Ishihara, T. Hydrogenation of CO2 to methanol over Cu/AlCeO catalyst. Catal. Today 2019, 339, 352–361. [Google Scholar] [CrossRef]
- Chen, K.; Fang, H.; Wu, S.; Liu, X.; Zheng, J.; Zhou, S.; Duan, X.; Zhuang, Y.; Tsang, S.C.E.; Yuan, Y. CO2 hydrogenation to methanol over Cu catalysts supported on La-modified SBA-15: The crucial role of Cu-LaOx interfaces. Appl. Catal. B Environ. Int. J. Devoted Catal. Sci. Its Appl. 2019, 251, 119–129. [Google Scholar] [CrossRef]
- Bell, F.A.T. In-SituInfrared Study of Methanol Synthesis from H2/CO2 over Cu/SiO2and Cu/ZrO2/SiO2. J. Catal. 1997, 172, 222–237. [Google Scholar]
- Arena, F.; Italiano, G.; Barbera, K.; Bordiga, S.; Bonura, G.; Spadaro, L.; Frusteri, F. Solid-state interactions, adsorption sites and functionality of Cu-ZnO/ZrO2 catalysts in the CO2 hydrogenation to CH3OH. Appl. Catal. A Gen. 2008, 350, 16–23. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Liu, P.; Graciani, J.; Senanayake, S.D.; Grinter, D.; Stacchiola, D.; Hrbek, J.; Fernandez Sanz, J. Inverse Oxide/Metal Catalysts in Fundamental Studies and Practical Applications: A Perspective of Recent Developments. J. Phys. Chem. Lett. 2016, 7, 2627–2639. [Google Scholar] [CrossRef]
- Wu, C.; Lin, L.; Liu, J.; Zhang, J.; Ma, D. Inverse ZrO2/Cu as a highly efficient methanol synthesis catalyst from CO2 hydrogenation. Nat. Commun. 2020, 11, 5767. [Google Scholar] [CrossRef]
- Tisseraud, C.; Comminges, C.; Pronier, S.; Pouilloux, Y.; Valant, A.L. The Cu–ZnO synergy in methanol synthesis Part 3: Impact of the composition of a selective Cu@ZnOx core–shell catalyst on methanol rate explained by experimental studies and a concentric spheres model. J. Catal. 2016, 343, 106–114. [Google Scholar] [CrossRef]
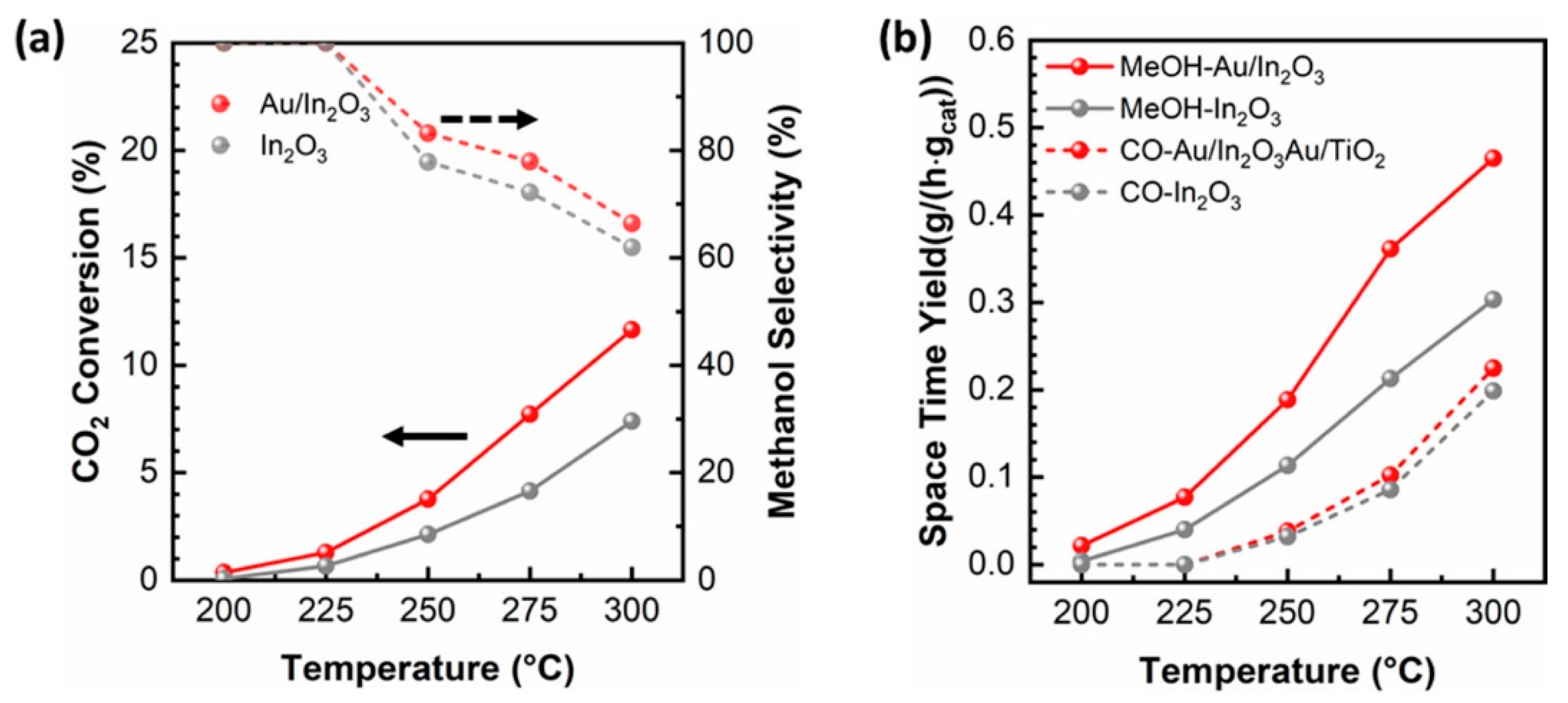
- Rui, N.; Zhang, F.; Sun, K.; Liu, Z.; Liu, C.J. Hydrogenation of CO2 to Methanol on a Auδ+–In2O3-x Catalyst. ACS Catal. 2020, 10, 11307–11317. [Google Scholar] [CrossRef]
- Koizumi, N.; Jiang, X.; Kugai, J.; Song, C. Effects of mesoporous silica supports and alkaline promoters on activity of Pd catalysts in CO2 hydrogenation for methanol synthesis. Catal. Today 2012, 194, 16–24. [Google Scholar] [CrossRef]
- Choi, E.J.; Lee, Y.H.; Lee, D.W.; Moon, D.J.; Lee, K.Y. Hydrogenation of CO2 to methanol over Pd–Cu/CeO2 catalysts. Mol. Catal. 2017, 434, 146–153. [Google Scholar] [CrossRef]
- Vu, T.T.N.; Desgagnés, A.; Iliuta, M.C. Efficient approaches to overcome challenges in material development for conventional and intensified CO2 catalytic hydrogenation to CO, methanol, and DME. Appl. Catal. A Gen. 2021, 617, 118119. [Google Scholar] [CrossRef]
- Liu, P.N.; Rskov, J.K. Ligand and ensemble effects in adsorption on alloy surfaces. Phys. Chem. Chem. Phys. 2001, 3, 3814–3818. [Google Scholar] [CrossRef]
- Liu, L.; Yao, H.; Jiang, Z.; Fang, T. Theoretical study of methanol synthesis from CO2 hydrogenation on PdCu3(111) surface. Appl. Surf. Sci. A J. Devoted Prop. Interfaces Relat. Synth. Behav. Mater. 2018, 451, 333–345. [Google Scholar] [CrossRef]
- Liu, L.; Fan, F.; Jiang, Z.; Gao, X.; Wei, J.; Fang, T. Mechanistic Study of Pd-Cu Bimetallic Catalysts for Methanol Synthesis from CO2 Hydrogenation. J. Phys. Chem. C 2017, 121, 26287–26299. [Google Scholar] [CrossRef]
- Qin, B.; Li, S. First principles investigation of dissociative adsorption of H2 during CO2 hydrogenation over cubic and hexagonal In2O3 catalysts. Phys. Chem. Chem. Phys. 2020, 22, 3390–3399. [Google Scholar] [CrossRef]
- Zhang, M.; Dou, M.; Yu, Y. Theoretical study of the promotional effect of ZrO2 on In2O3 catalyzed methanol synthesis from CO2 hydrogenation. Appl. Surf. Sci. 2018, 433, 780–789. [Google Scholar] [CrossRef]
- Ye, J.; Liu, C.; Ge, Q. DFT Study of CO2 Adsorption and Hydrogenation on the In2O3 Surface. Am. Chem. Soc. 2012, 116, 7817–7825. [Google Scholar] [CrossRef]
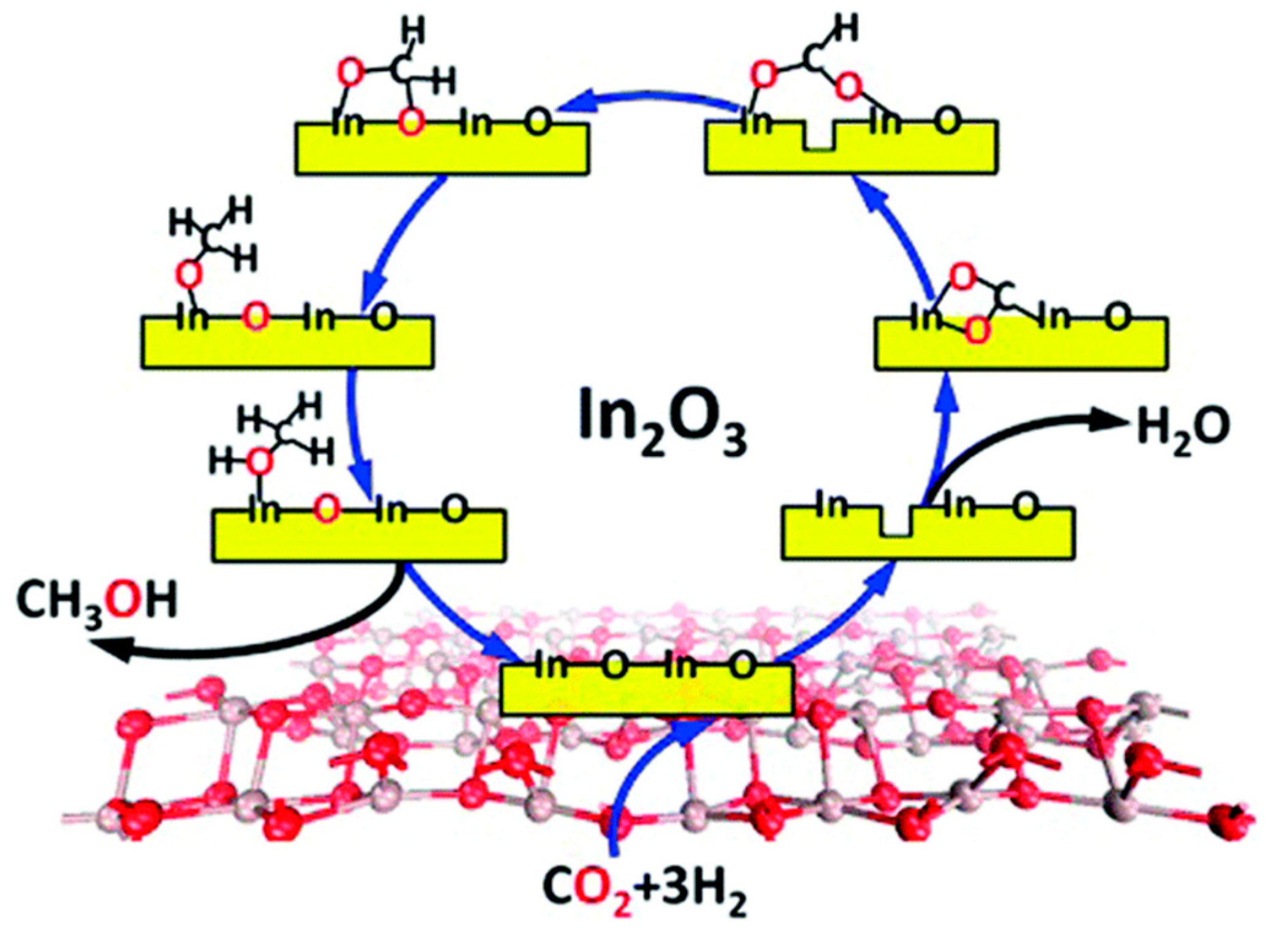
- Ye, J.; Liu, C.J.; Mei, D.; Ge, Q. Active Oxygen Vacancy Site for Methanol Synthesis from CO2 Hydrogenation on In2O3(110): A DFT Study. ACS Catal. 2013, 3, 1296–1306. [Google Scholar] [CrossRef]
- Martin, O.; Pérez-Ramírez, J. New and revisited insights into the promotion of methanol synthesis catalysts by CO2. Catal. Sci. Technol. 2013, 3, 3343–3352. [Google Scholar] [CrossRef]
- Dou, M.; Zhang, M.; Chen, Y.; Yu, Y. Theoretical study of methanol synthesis from CO2 and CO hydrogenation on the surface of ZrO2 supported In2O3 catalyst. Surf. Sci. 2018, 672, 7–12. [Google Scholar] [CrossRef]
- Shanshan, D.; Peng, G.; Ziyu, L.; Xinqing, C.; Chengguang, Y.; Hui, W.; Liangshu, Z.; Shenggang, L.; Yuhan, S. Role of zirconium in direct CO2 hydrogenation to lower olefins on oxide/zeolite bifunctional catalysts. J. Catal. 2018, 364, 382–393. [Google Scholar]
- Sharafutdinov, I.; Elkjær, C.F.; de Carvalho, H.W.P.; Gardini, D.; Chiarello, G.L.; Damsgaard, C.D.; Wagner, J.B.; Grunwaldt, J.D.; Dahl, S.; Chorkendorff, I. Intermetallic compounds of Ni and Ga as catalysts for the synthesis of methanol. J. Catal. 2014, 320, 77–88. [Google Scholar] [CrossRef]
- Sha, F.; Han, Z.; Tang, S.; Wang, J.; Li, C. Hydrogenation of Carbon Dioxide to Methanol over Non-Cu-based Heterogeneous Catalysts. ChemSusChem 2020, 13, 6160–6181. [Google Scholar] [CrossRef] [PubMed]
- Kakumoto, T.; Watanabe, T. A theoretical study for methanol synthesis by CO2 hydrogenation. Catal. Today 1997, 36, 39–44. [Google Scholar] [CrossRef]
- Kim, Y.; Trung, T.S.B.; Yang, S.; Kim, S.; Lee, H. Mechanism of the Surface Hydrogen Induced Conversion of CO2 to Methanol at Cu(111) Step Sites. ACS Catal. 2016, 6, 1037–1044. [Google Scholar] [CrossRef]
- Yang, Y.; Evans, J.; Rodriguez, J.A.; White, M.G.; Liu, P. Fundamental studies of methanol synthesis from CO2 hydrogenation on Cu(111), Cu clusters, and Cu/ZnO(000). Phys. Chem. Chem. Phys. 2010, 12, 9909–9917. [Google Scholar] [CrossRef] [PubMed]
- Larmier, K.; Liao, W.C.; Tada, S.; Lam, E.; Verel, D.R.; Bansode, A.; Urakawa, A.; Comas-Vives, A.; Copéret, P.C. CO2-to-Methanol Hydrogenation on Zirconia-Supported Copper Nanoparticles: Reaction Intermediates and the Role of the Metal–Support Interface. Angew. Chem. 2017, 56, 2318–2323. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Li, S.; Bu, X.; Dang, S.; Liu, Z.; Wang, H.; Zhong, L.; Qiu, M.; Yang, C.; Cai, J. Direct conversion of CO2 into liquid fuels with high selectivity over a bifunctional catalyst. Nat. Chem. 2017, 9, 1019–1024. [Google Scholar] [CrossRef]
- Grabow, L.C.; Mavrikakis, M. Mechanism of Methanol Synthesis on Cu through CO2 and COHydrogenation. ACS Catal. 2011, 1, 365–384. [Google Scholar] [CrossRef]
- Nie, X.; Jiang, X.; Wang, H.; Luo, W.; Janik, M.J.; Chen, Y.; Song, C. Mechanistic Understanding of Alloy Effect and Water Promotion for Pd-Cu Bimetallic Catalysts in CO2 Hydrogenation to Methanol. ACS Catal. 2018, 8, 4873–4892. [Google Scholar] [CrossRef]
- Zhang, M.; Dou, M.; Yu, Y. DFT study of CO2 conversion on InZr3(110) surface. Phys. Chem. Chem. Phys. PCCP 2017, 19, 28917–28927. [Google Scholar] [CrossRef]
- Yang, Y.; White, M.G.; Liu, P. Theoretical Study of Methanol Synthesis from CO2 Hydrogenation on Metal-Doped Cu(111) Surfaces. J. Phys. Chem. C 2012, 116, 248–256. [Google Scholar] [CrossRef]
- Liu, L.; Fan, F.; Bai, M.; Xue, F.; Ma, X.; Jiang, Z.; Fang, T. Mechanistic study of methanol synthesis from CO2 hydrogenation on Rh-doped Cu(111) surfaces. Mol. Catal. 2019, 466, 26–36. [Google Scholar] [CrossRef]
- Kattel, S.; Yan, B.; Yang, Y.; Chen, J.G.; Liu, P. Optimizing Binding Energies of Key Intermediates for CO2 Hydrogenation to Methanol over Oxide-Supported Copper. J. Am. Chem. Soc. 2016, 138, 12440. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.L.; Hong, Q.J.; Liu, Z.P. CO2 fixation into methanol at Cu/ZrO2 interface from first principles kinetic Monte Carlo. J. Catal. 2009, 263, 114–122. [Google Scholar] [CrossRef]
- Yang, Y.; Mei, D.; Peden, C.H.F.; Campbell, C.T.; Mims, C.A. Surface-Bound Intermediates in Low-Temperature Methanol Synthesis on Copper: Participants and Spectators. ACS Catal. 2015, 5, 7328–7337. [Google Scholar] [CrossRef]
- Zhao, Y.-F.; Yang, Y.; Mims, C.; Peden, C.H.F.; Li, J.; Mei, D. Insight into methanol synthesis from CO2 hydrogenation on Cu(1 1 1): Complex reaction network and the effects of H2O. J. Catal. 2011, 281, 199–211. [Google Scholar] [CrossRef]
- Tang, Q.; Shen, Z.; Russell, C.K.; Fan, M. Thermodynamic and Kinetic Study on Carbon Dioxide Hydrogenation to Methanol over a Ga3Ni5(111) Surface: The Effects of Step Edge. J. Phys. Chem. C. Nanomater. Interfaces 2018, 122, 315–330. [Google Scholar] [CrossRef]
- Tang, Q.; Shen, Z.; Huang, L.; He, T.; Fan, M. Synthesis of methanol from CO2 hydrogenation promoted by dissociative adsorption of hydrogen on a Ga3Ni5(221) surface. Phys. Chem. Chem. Phys. PCCP 2017, 19, 18539–18555. [Google Scholar] [CrossRef]
- Chinchen, G.C.; Waugh, K.C.; Whan, D.A. The activity and state of the copper surface in methanol synthesis catalysts. Appl. Catal. 1986, 25, 101–107. [Google Scholar] [CrossRef]
- Liu, X.M.; Lu, G.Q.; Yan, Z.F.; Beltramini, J. Recent advances in catalysts for methanol synthesis via hydrogenation of CO and CO2. Ind. Eng. Chem. Res. 2003, 42, 6518–6530. [Google Scholar] [CrossRef]
- Pan, W.X.; Cao, R.; Roberts, D.L.; Griffin, G.L. Methanol synthesis activity of Cu/ZnO catalysts. Cheminform 1989, 2, 440–446. [Google Scholar] [CrossRef]
- Chorkendorff, P.B.R.K. Synthesis of methanol from a mixture of H2 and CO2 on Cu(100). Surf. Sci. 1994, 318, 267–280. [Google Scholar]
- Askgaard, T.S.; Norskov, J.K.; Ovesen, C.V.; Stoltze, P. A Kinetic Model of Methanol Synthesis. J. Catal. 1995, 156, 229–242. [Google Scholar] [CrossRef]
- Karelovic, A.; Bargibant, A.; Fernández, C.; Ruiz, P. Effect of the structural and morphological properties of Cu/ZnO catalysts prepared by citrate method on their activity toward methanol synthesis from CO2 and H2 under mild reaction conditions. Catal. Today 2012, 197, 109–118. [Google Scholar] [CrossRef]
- Clausen, B.S.; Steffensen, G.; Fabius, B.; Villadsen, J.; Feidenhans’L, R.; TopsøE, H. In situ cell for combined XRD and on-line catalysis tests: Studies of Cu-based water gas shift and methanol catalysts. J. Catal. 1991, 132, 524–535. [Google Scholar] [CrossRef]
- Liu, J.; Shi, J.; He, D.; Zhang, Q.; Wu, X.; Liang, Y.; Zhu, Q. Surface active structure of ultra-fine Cu/ZrO2 catalysts used for the CO2+H2 to methanol reaction. Appl. Catal. A Gen. 2001, 218, 113–119. [Google Scholar] [CrossRef]
- Herman, R.G.; Klier, K.; Simmons, G.W.; Finn, B.P.; Bulko, J.B.; Kobylinski, T.P. Catalytic synthesis of methanol from CO/H2I. Phase composition, electronic properties, and activities of the Cu/ZnO/M2O3 catalysts. J. Catal. 1979, 56, 407–429. [Google Scholar] [CrossRef]
- Szanyi, J.; Goodman, D.W. Methanol synthesis on a Cu(100) catalyst. Catal. Lett. 1991, 10, 383–390. [Google Scholar] [CrossRef]
- Jedrecy, N.; Gallini, S.; Sauvage-Simkin, M.; Pinchaux, R. Copper growth on the O-terminated ZnO (0001) surface: Structure and morphology. Phys. Rev. B 2001, 64, 085424. [Google Scholar] [CrossRef]
- Arena, F.; Di Chio, R.; Filiciotto, L.; Trunfio, G.; Espro, C.; Palella, A.; Patti, A.; Spadaro, L. Probing the functionality of nanostructured MnCeOx catalysts in the carbon monoxide oxidation Part II. Reaction mechanism and kinetic modelling. Appl. Catal. B Environ. 2017, 218, 803–809. [Google Scholar] [CrossRef]
- Li, L.J.G.F. Influence of copper content on structural features and performance of pre-reduced LaMn1-xCuxO3 (0 ≤ x < 1) catalysts for methanol synthesis from CO2/H2. J. Rare Earths 2010, 25, 747–751. [Google Scholar]
- Nakamura, J.; Uchijima, T.; Kanai, Y.; Fujitani, T. The role of ZnO in Cu/ZnO methanol synthesis catalysts. Catal. Today 1996, 28, 223–230. [Google Scholar] [CrossRef]
- Arena, F.; Italiano, G.; Barbera, K.; Bonura, G.; Spadaro, L.; Frusteri, F. Basic evidences for methanol-synthesis catalyst design. Catal. Today 2009, 143, 80–85. [Google Scholar] [CrossRef]
- Kasatkin, I.; Kurr, P.; Kniep, B.; Trunschke, A.; Schlögl, R. Role of Lattice Strain and Defects in Copper Particles on the Activity of Cu/ZnO/Al2O3 Catalysts for Methanol Synthesis. Angew. Chem. Int. Ed. 2007, 46, 7324–7327. [Google Scholar] [CrossRef] [PubMed]
- Behrens, M. Meso- and nano-structuring of industrial Cu/ZnO/(Al2O3) catalysts. J. Catal. 2009, 267, 24–29. [Google Scholar] [CrossRef]
- Mehta, S.; Simmons, G.W.; Klier, K.; Herman, R.G. Catalytic synthesis of methanol from ja:math: II. Electron microscopy (TEM, STEM, microdiffraction, and energy dispersive analysis) of the ja:math and Cu/ZnO/Cr2O3 catalysts. J. Catal. 1979, 57, 339–360. [Google Scholar] [CrossRef]
- Natesakhawat, S.; Lekse, J.W.; Baltrus, J.P.; Ohodnicki, P.R.; Howard, B.H.; Deng, X.; Matranga, C. Active Sites and Structure–Activity Relationships of Copper-Based Catalysts for Carbon Dioxide Hydrogenation to Methanol. Acs Catal. 2012, 2, 1667–1676. [Google Scholar] [CrossRef]
- Oguchi, H.; Kanai, H.; Utani, K.; Matsumura, Y.; Imamura, S. Cu2O as active species in the steam reforming of methanol by CuO/ZrO2 catalysts. Appl. Catal. A Gen. 2005, 293, 64–70. [Google Scholar] [CrossRef]
- Liao, F.; Huang, Y.; Ge, J.; Zheng, W.; Tedsree, K.; Collier, P.; Hong, X.; Tsang, S.C. Morphology-Dependent Interactions of ZnO with Cu Nanoparticles at the Materials’ Interface in Selective Hydrogenation of CO2 to CH3OH. Angew. Chem. Int. Ed. 2011, 50, 2162–2165. [Google Scholar] [CrossRef]
- Melián-Cabrera, I.; Granados, M.L.; Fierro, J.L.G. Pd-Modified Cu–Zn Catalysts for Methanol Synthesis from CO2/H2 Mixtures: Catalytic Structures and Performance. J. Catal. 2002, 210, 285–294. [Google Scholar] [CrossRef]
- Zhang, Q.X.Z.; Qian, Z.H. Synthesis of methanol from CO2/H2 at low pressure on CuO-ZnO and CuO-ZnO-ZrO2 catalysts. J. Catal. 1989, 10, 22–28. [Google Scholar]
- Kanai, Y.; Watanabe, T.; Fujitani, T.; Uchijima, T.; Nakamura, J. The synergy between Cu and ZnO in methanol synthesis catalysts. Catal. Lett. 1996, 38, 157–163. [Google Scholar] [CrossRef]
- Grunwaldt, J.D.; Molenbroek, A.M.; Topse, N.Y.; Topse, H.; Clausen, B.S. In Situ Investigations of Structural Changes in Cu/ZnO Catalysts. J. Catal. 2000, 194, 452–460. [Google Scholar] [CrossRef]
- Behrens, M.; Studt, F.; Kasatkin, I.; Kuehl, S.; Haevecker, M.; Abild-Pedersen, F.; Zander, S.; Girgsdies, F.; Kurr, P.; Kniep, B.-L.; et al. The Active Site of Methanol Synthesis over Cu/ZnO/Al2O3 Industrial Catalysts. Science 2012, 336, 893–897. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, M.; Strunk, J.; Hinrichsen, O.; Muhler, M.; Fink, K.; Meyer, B.; Wöll, C. Active Sites on Oxide Surfaces: ZnO-Catalyzed Synthesis of Methanol from CO and H2. Angew. Chem. Int. Ed. 2005, 44, 2790–2794. [Google Scholar] [CrossRef] [PubMed]
- Le Valant, A.; Comminges, C.; Tisseraud, C.; Canaff, C.; Pinard, L.; Pouilloux, Y. The Cu-ZnO synergy in methanol synthesis from CO2, Part 1: Origin of active site explained by experimental studies and a sphere contact quantification model on Cu plus ZnO mechanical mixtures. J. Catal. 2015, 324, 41–49. [Google Scholar] [CrossRef]
- Kattel, S.; Ramírez, P.J.; Chen, J.G.; Rodriguez, J.A.; Liu, P. Active sites for CO2 hydrogenation to methanol on Cu/ZnO catalysts. Science 2017, 355, 1296–1299. [Google Scholar] [CrossRef] [PubMed]
- Habraken, F.H.P.M.; Mesters, C.M.A.M.; Bootsma, G.A. The adsorption and incorporation of oxygen on Cu (100) and its reaction with carbon monoxide; comparison with Cu (111) and Cu (110). Surf. Sci. 1980, 97, 264–282. [Google Scholar] [CrossRef]
- Norton, P.R.; Tapping, R.L. Photoelectron spectroscopic studies of the adsorption of CO and CO2 on nickel, platinum and copper. Chem. Phys. Lett. 1976, 38, 207–212. [Google Scholar] [CrossRef]
- Chinchen, G.C.; Spencer, M.S.; Waugh, K.C.; Whan, D.A. Promotion of methanol synthesis and the water-gas shift reactions by adsorbed oxygen on supported copper catalysts. J. Chem. Soc. Faraday Trans. Phys. Chem. Condens. Phases 1987, 83, 2193–2212. [Google Scholar] [CrossRef]
- Wachs, I.E.; Madix, R.J. Selective oxidation of CH3OH to H2CO on a copper(110) catalyst. J. Catal. 1978, 53, 208–227. [Google Scholar] [CrossRef]
- Hadden, R.A.; Vandervell, H.D.; Waugh, K.C.; Webb, G. The adsorption and decomposition of carbon dioxide on polycrystalline copper. Catal. Lett. 1988, 1, 27–33. [Google Scholar] [CrossRef]
- Sakakini, B.H.; Tabatabaei, J.; Watson, M.J.; Waugh, K.C. Structural changes of the Cu surface of a Cu/ZnO/Al2O3 catalyst, resulting from oxidation and reduction, probed by CO infrared spectroscopy. J. Mol. Catal. A Chem. 2000, 162, 297–306. [Google Scholar] [CrossRef]
- Copperthwaite, R.G.; Davies, P.R.; Morris, M.A.; Roberts, M.W.; Ryder, R.A. The reactive chemisorption of carbon dioxide at magnesium and copper surfaces at low temperature. Catal. Lett. 1988, 1, 11–19. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Liu, P.; Stacchiola, D.; Senanayake, S.D.; White, M.G.; Chen, J.G. Hydrogenation of CO2 to Methanol: Importance of Metal-Oxide and Metal-Carbide Interfaces in the Activation of CO2. Acs Catal. 2015, 5, 6696–6706. [Google Scholar] [CrossRef]
- Burch, R.; Golunski, S.E.; Spencer, M.S. The role of copper and zinc oxide in methanol synthesis catalysts. J. Chem. Soc. Faraday Trans. 1990, 86, 2683–2691. [Google Scholar] [CrossRef]
- Auroux, A.; Gervasini, A. Microcalorimetric Study of the Acidity and Basicity of Metal Oxide Surfaces. Cheminform 1990, 21, 4. [Google Scholar] [CrossRef]
- Bianchi, D.; Chafik, T.; Khalfallah, M.; Teichner, S.J. Intermediate species on zirconia supported methanol aerogel catalysts: IV. Adsorption of carbon dioxide. Appl. Catal. A Gen. 1994, 112, 219–235. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | Temperature (°C) | H2: CO2 Ratio | Pressure (MPa) | CO2 Conversion (%) | MeOH Selectivity (%) | Yield (gMeOH·kgcat−1·h−1) |
|---|---|---|---|---|---|---|
| Cu/γ-Al2O3 [19] | 200 | 3.8 | 10 | N/A | 45 | N/A |
| Cu-K/γ-Al2O3 [19] | 200 | 3.8 | 10 | N/A | 5 | N/A |
| Cu-Ba/γ-Al2O3 [19] | 200 | 3.8 | 10 | N/A | 63 | N/A |
| Ga-Cu-Zn/ZrO2 [20] | 250 | 3 | 4 | 18 | 69 | 512 |
| Cu-Zn/SiO2 [21] | 270 | 3 | 2 | 2 | 47.2 | 64 |
| Cu-Zn-Ga/SiO2 [21] | 270 | 3 | 2 | 2 | 99.8 | 128 |
| Cu-Zn-Ga/H-SiO2 [21] | 270 | 3 | 2 | 5.6 | 99.5 | 352 |
| Cu/ZnO/ZrO4Ga4O3 [22] | 250 | 3 | 8 | N/A | 75 | 324 |
| Cu/ZnGa4O4 [23] | 240 | 2.8 | 4.5 | 26 | 48 | N/A |
| Cu/SiO2 [24] | 250 | 3 | 4.1 | 2.8 | 15 | N/A |
| Pd/SiO2 [24] | 250 | 3 | 4.1 | 3.0 | 23 | N/A |
| Pd-Zn/CNT [25] | 270 | 3 | 5 | 19.6 | 35.5 | 343 |
| Pd-Ga/CNT [26] | 250 | 3 | 5 | 16.5 | 52.5 | 512 |
| Pd/Ga2O3 [27] | 250 | 3 | 5 | 17.3 | 51.6 | 175.6 |
| Ga4O3-Pd/SiO4 [28] | 250 | 3 | 3 | 1.34 | 58.9 | 283.4 |
| Pd-CaO/MCM-41 [29] | 250 | 3 | 3 | 12.1 | 65.2 | N/A |
| In2O3/ZrO2 [30] | 300 | 4 | 5 | 5.2 | 99.8 | 295 |
| In2.5/ZrO2 [31] | 300 | 4 | 5 | 5.7 | 46.5 | 160 |
| In2O3 [32] | 270 | 3 | 4 | 1.1 | 54.9 | 25 |
| In2O3 [32] | 330 | 3 | 4 | 7.1 | 40.0 | 118 |
| ZnO-ZrO2 [33] | 320 | 4 | 5 | 10 | 91.0 | 730 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Z.; Guo, D.; Dong, S.; Wu, J.; Zhu, M.; Han, Y.-F.; Liu, Z.-W. Catalysis for CO2 Hydrogenation—What We Have Learned/Should Learn from the Hydrogenation of Syngas to Methanol. Catalysts 2023, 13, 1452. https://doi.org/10.3390/catal13111452
Yang Z, Guo D, Dong S, Wu J, Zhu M, Han Y-F, Liu Z-W. Catalysis for CO2 Hydrogenation—What We Have Learned/Should Learn from the Hydrogenation of Syngas to Methanol. Catalysts. 2023; 13(11):1452. https://doi.org/10.3390/catal13111452
Chicago/Turabian StyleYang, Zixu, Derun Guo, Shengbin Dong, Jiayi Wu, Minghui Zhu, Yi-Fan Han, and Zhong-Wen Liu. 2023. "Catalysis for CO2 Hydrogenation—What We Have Learned/Should Learn from the Hydrogenation of Syngas to Methanol" Catalysts 13, no. 11: 1452. https://doi.org/10.3390/catal13111452
APA StyleYang, Z., Guo, D., Dong, S., Wu, J., Zhu, M., Han, Y. -F., & Liu, Z. -W. (2023). Catalysis for CO2 Hydrogenation—What We Have Learned/Should Learn from the Hydrogenation of Syngas to Methanol. Catalysts, 13(11), 1452. https://doi.org/10.3390/catal13111452