

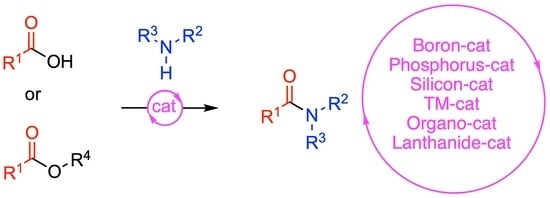
Direct Catalytic Amidations from Carboxylic Acid and Ester Derivatives: A Review




Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
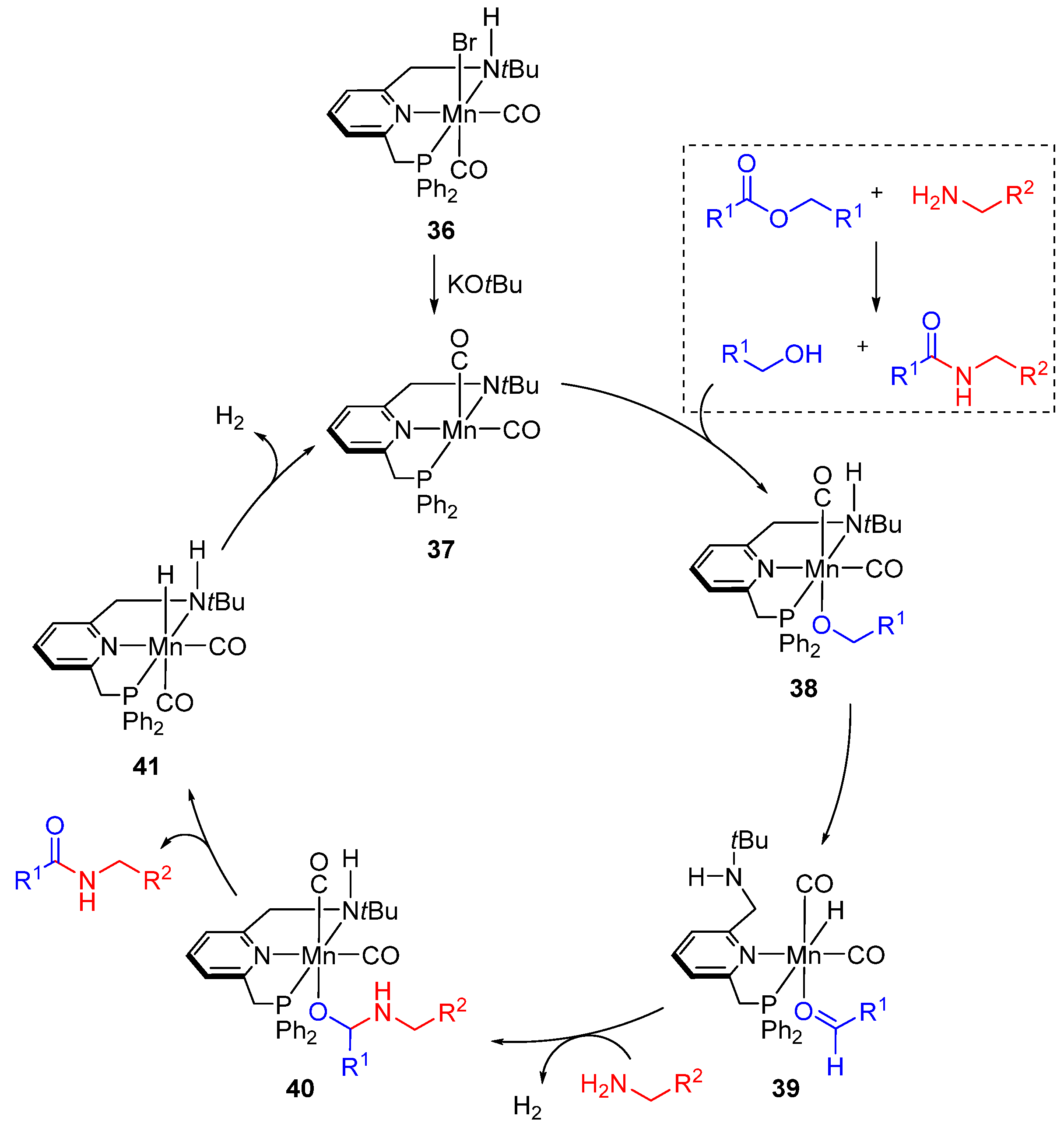{kind=link}
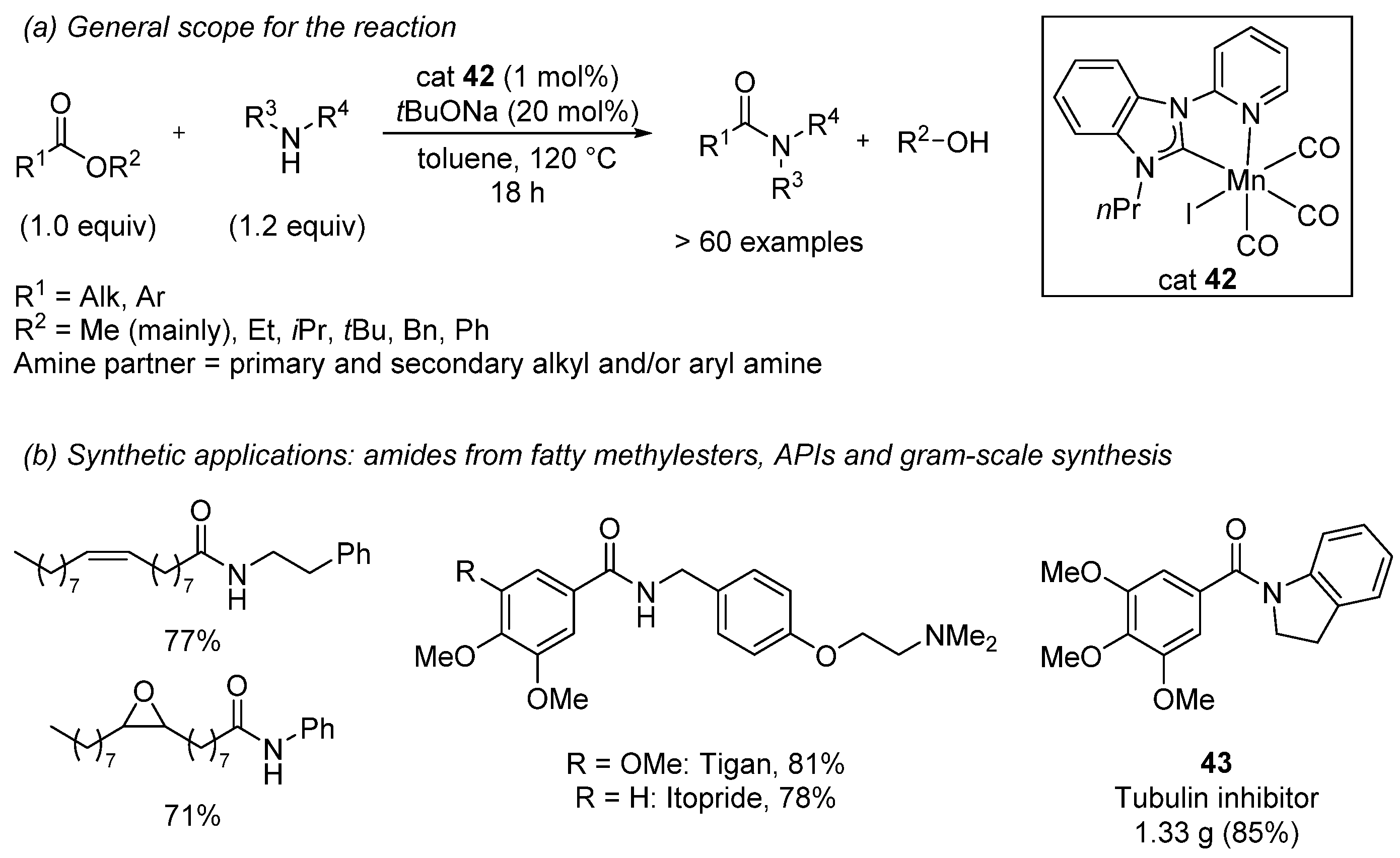{kind=link}
{kind=link}
{kind=link}
{kind=link}
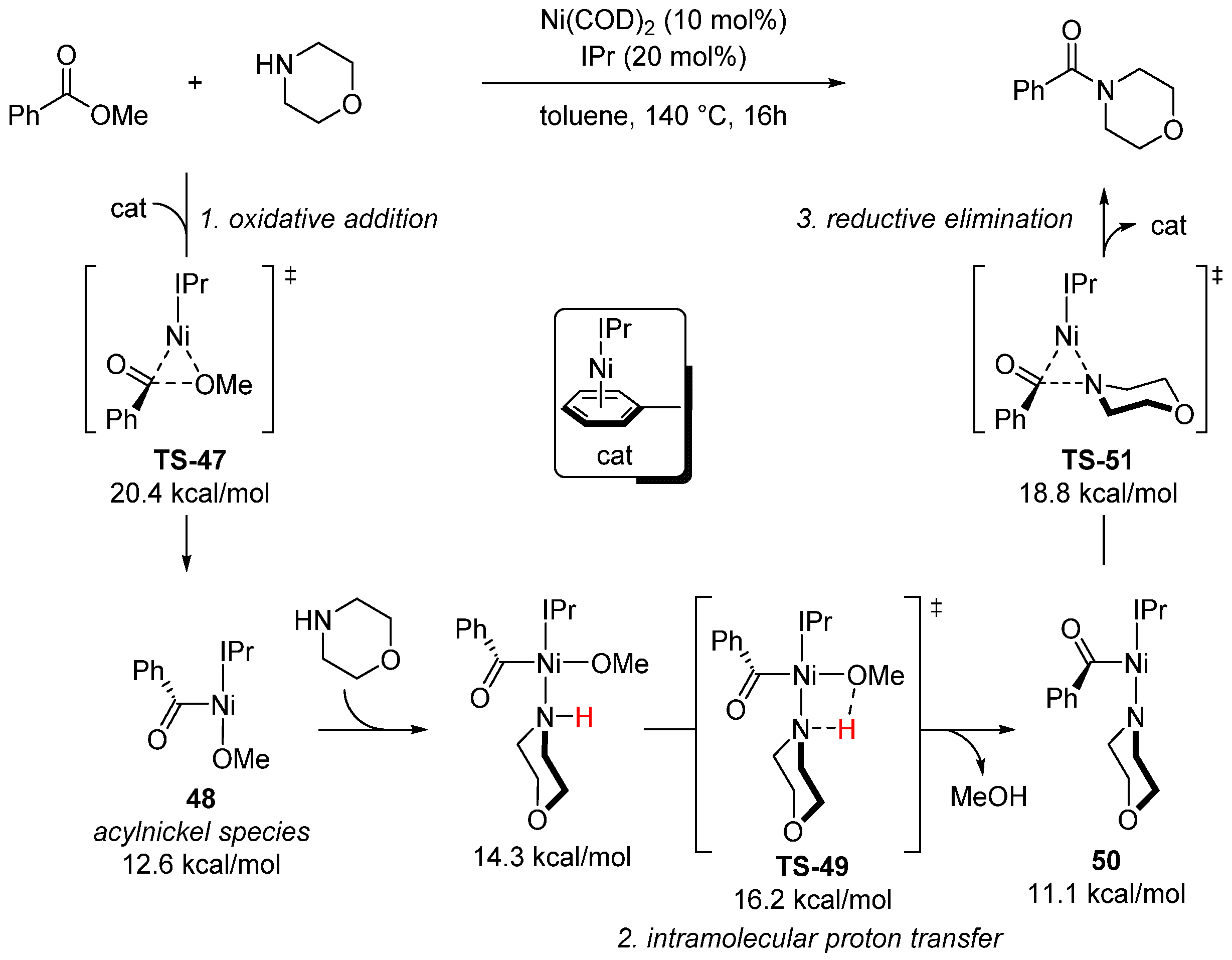{kind=link}
{kind=link}
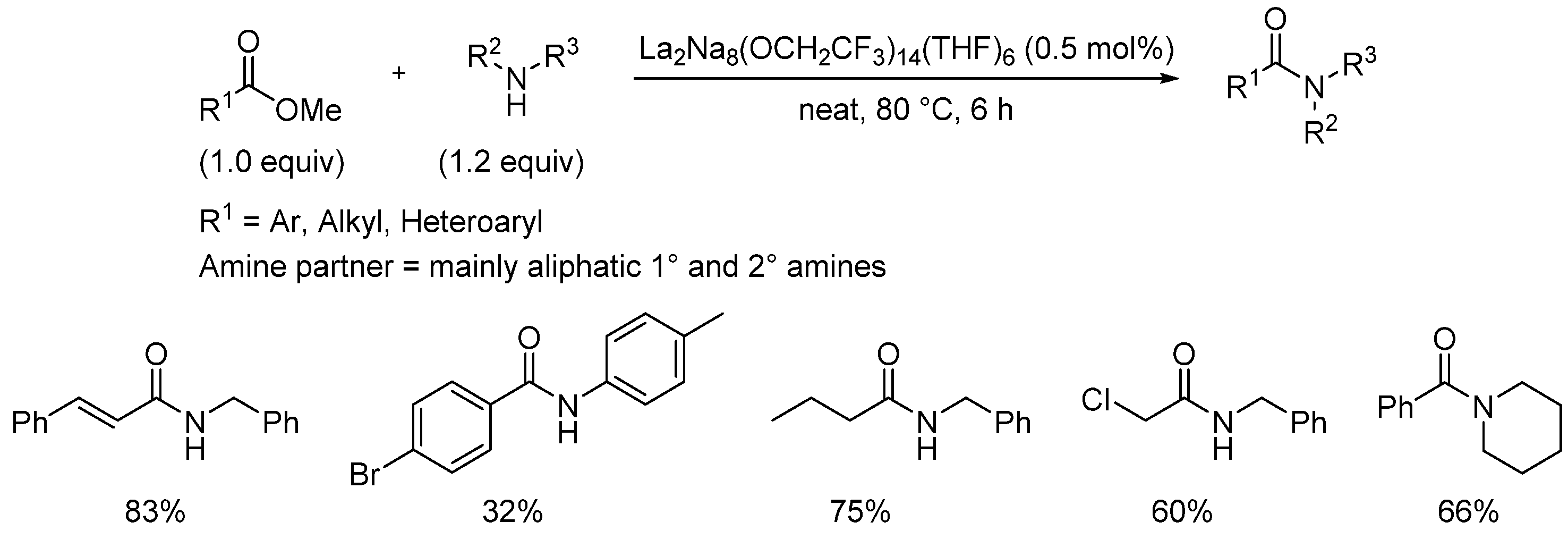{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
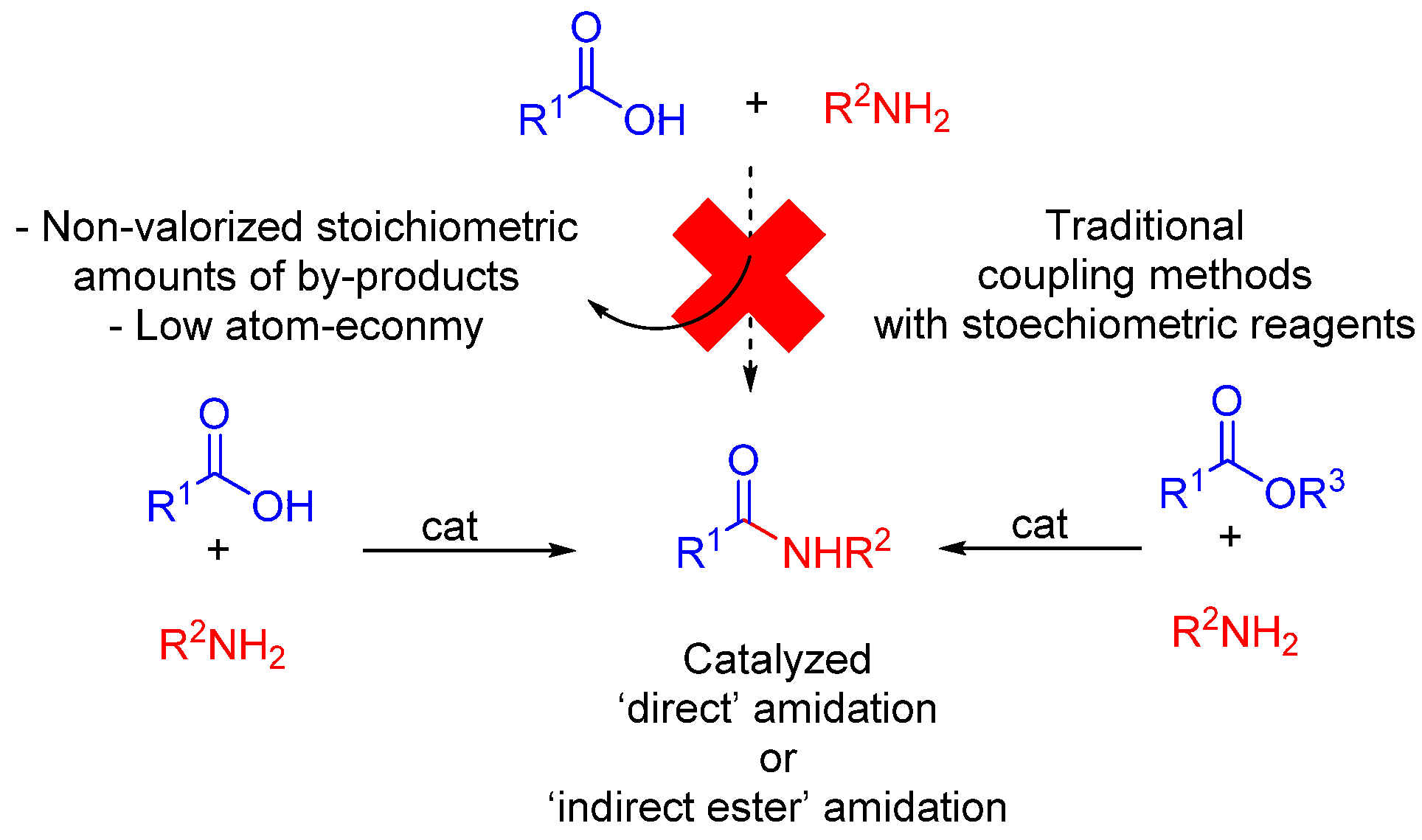
1. Introduction
2. Direct Amidations of Carboxylic Acids with Amines
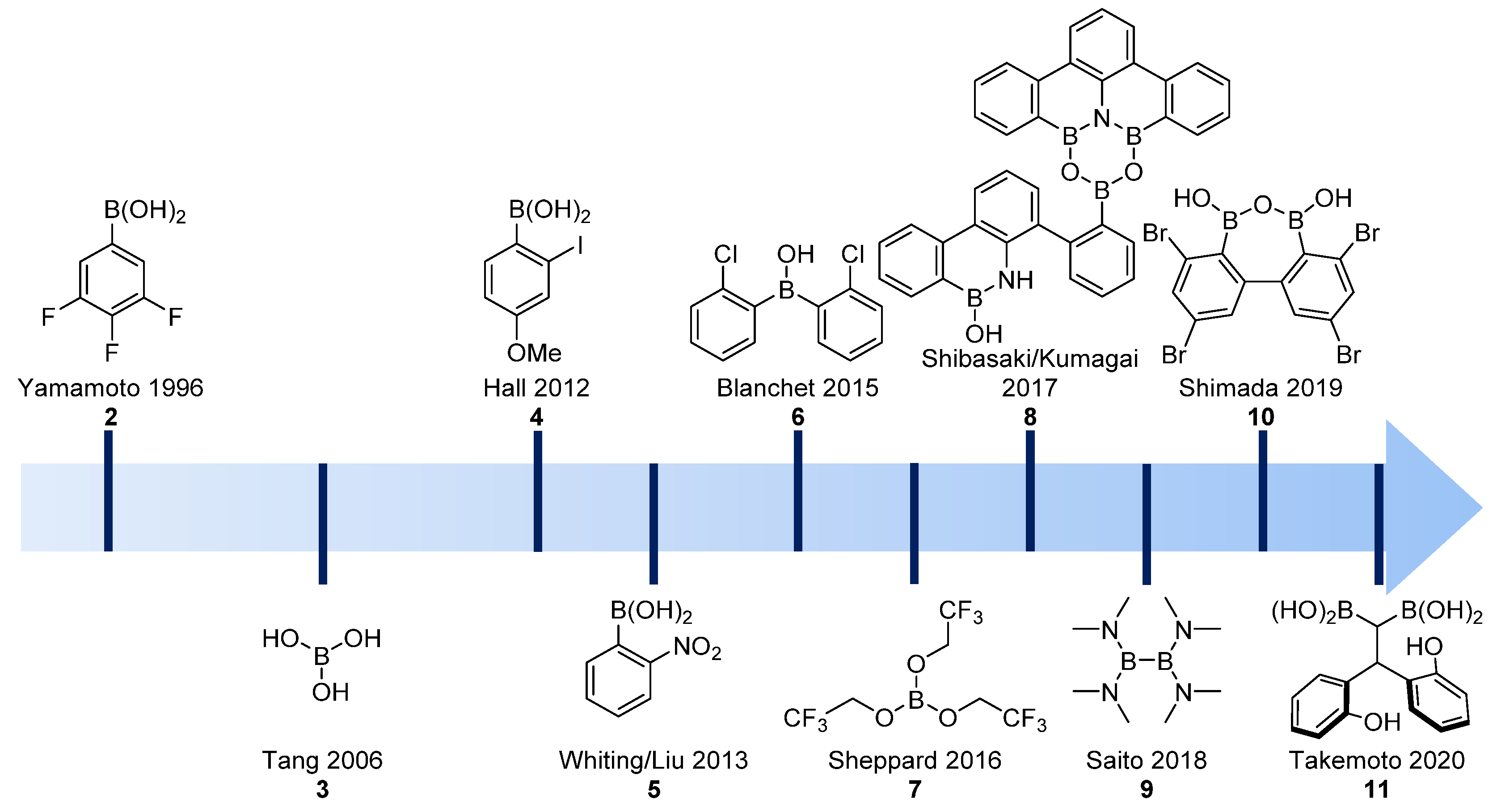
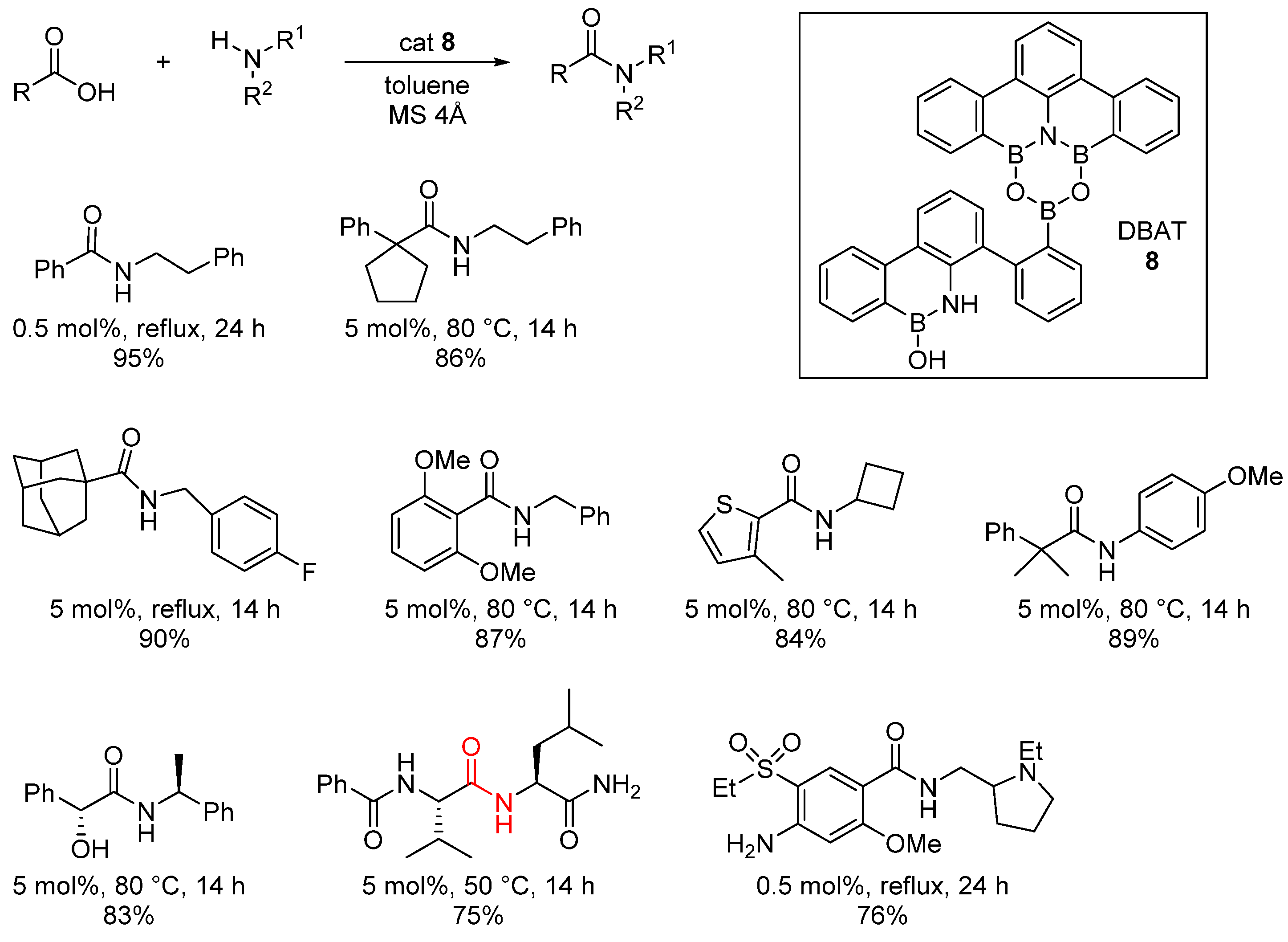
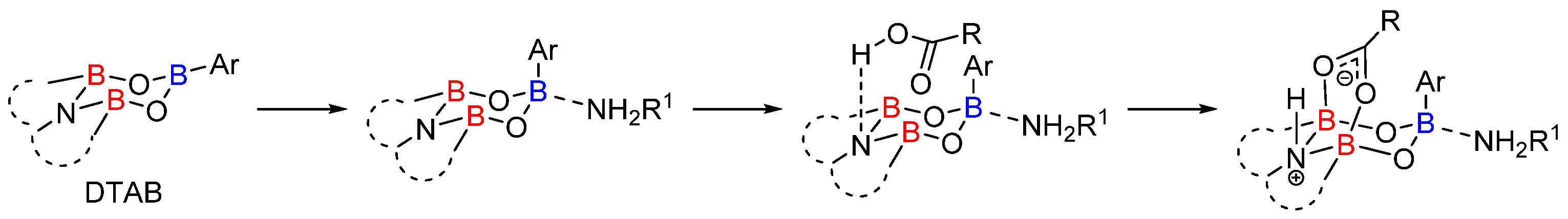
2.1. Boron-Derived Catalysts
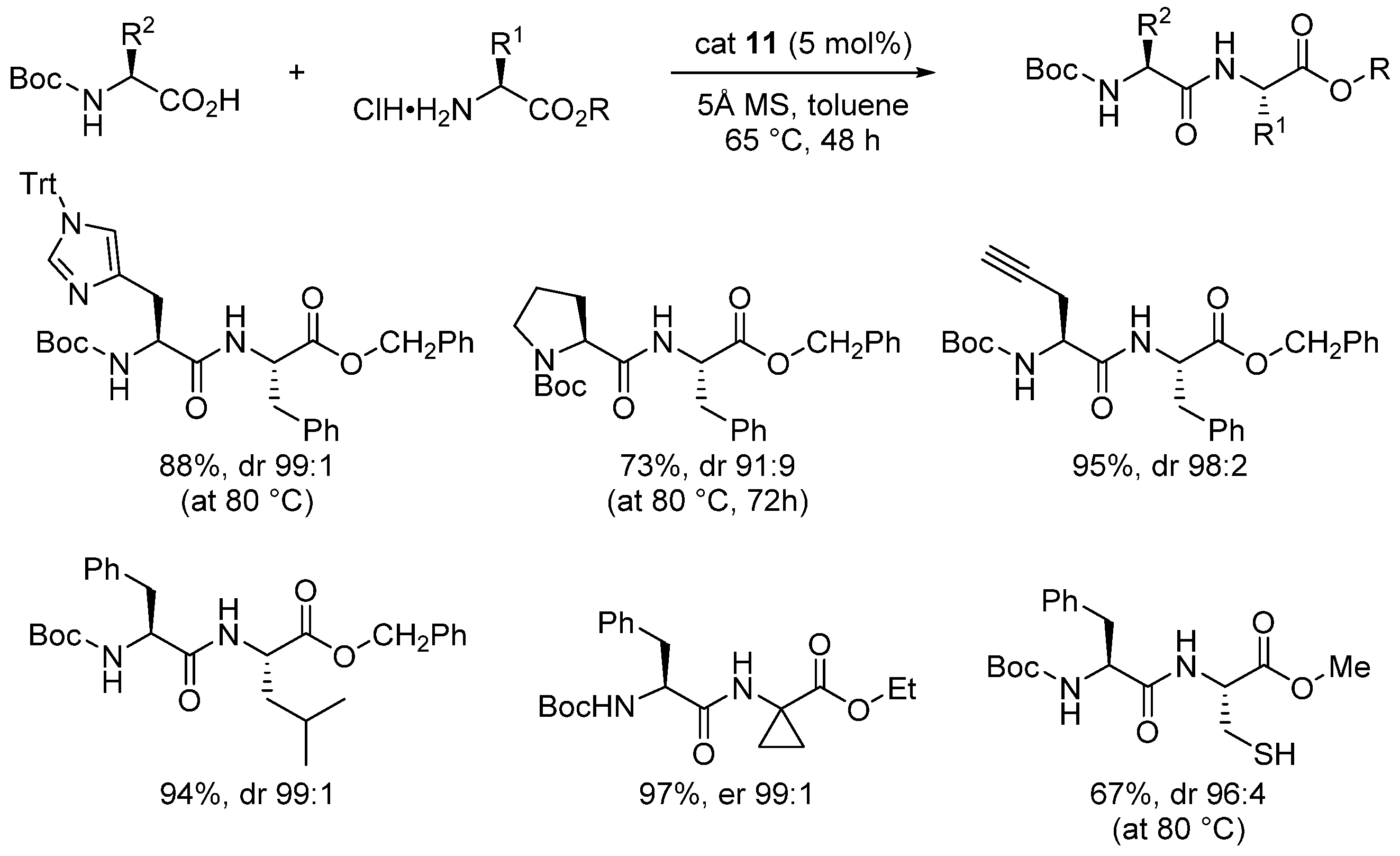
- The formation of the bicyclic intermediate 13 is highly dependent of the quantity and nature of molecular sieves; a large quantity of 5 Å MS is required and less than 5% conversion was observed with 4 Å MS (see Scheme 3);
- Boroxine 15 could be considered as a poorly reactive ‘resting state’ intermediate. As determined by DFT, the use of o-substituted aromatic boronic acids destabilizes the formation of the corresponding boroxine 15 derivatives, thus explaining the efficiency and success of such ortho-substituted catalysts (see Figure 1);
- Finally, it should be noted that for borinic acid 6, the actual catalyst can be determined to be ortho-chloro boronic acid (obtained by protodeborylation of borinic acid 6).
2.2. Phosporus- and Silicon-Derived Catalysts
2.3. Organo-Catalyzed Reactions
2.4. Metal-Catalyzed Reactions
2.5. Miscellaneous
3. Direct Amidations of Carboxylic Esters with Amines
3.1. Transition Metal-Derived Catalysts
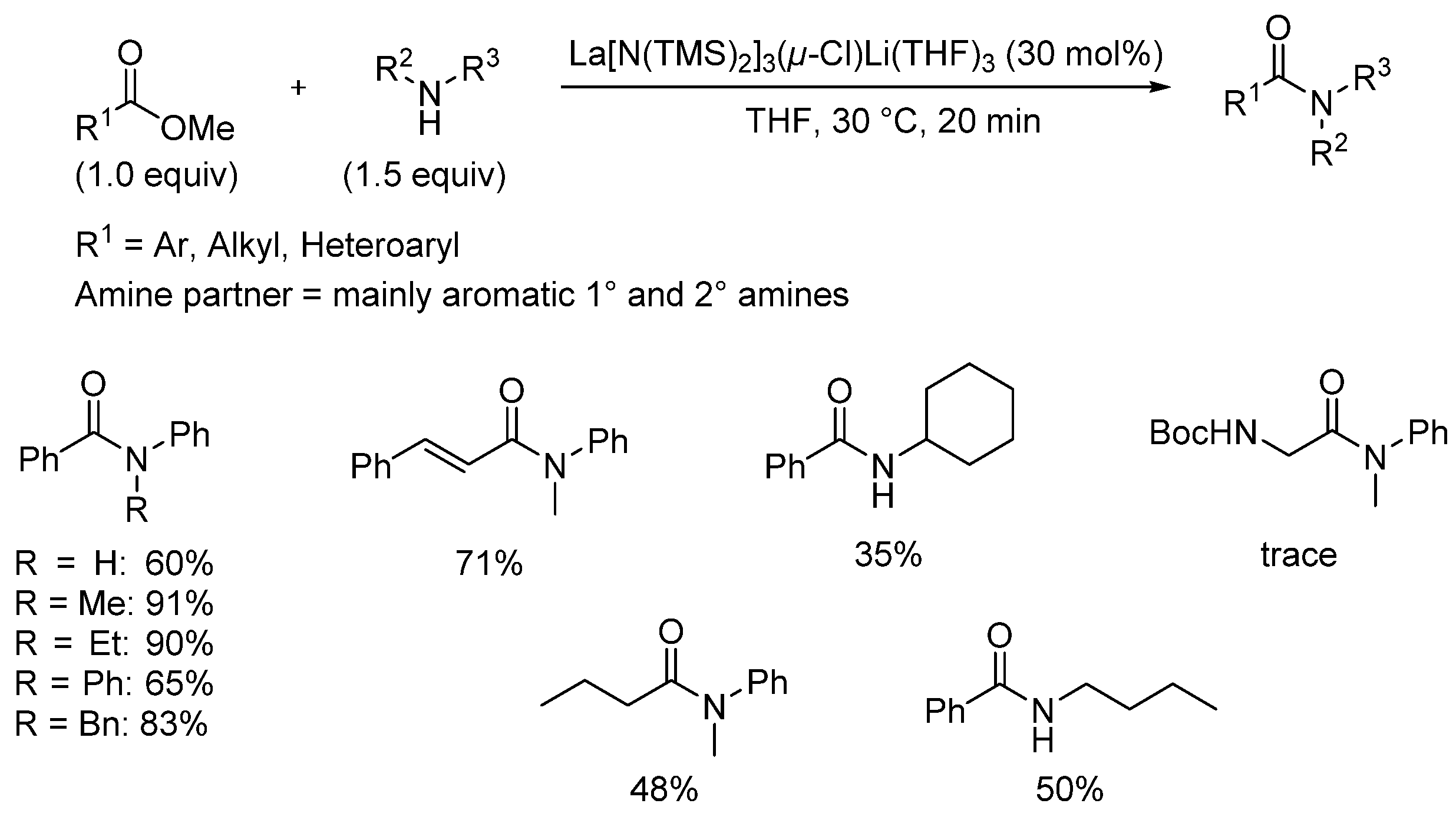
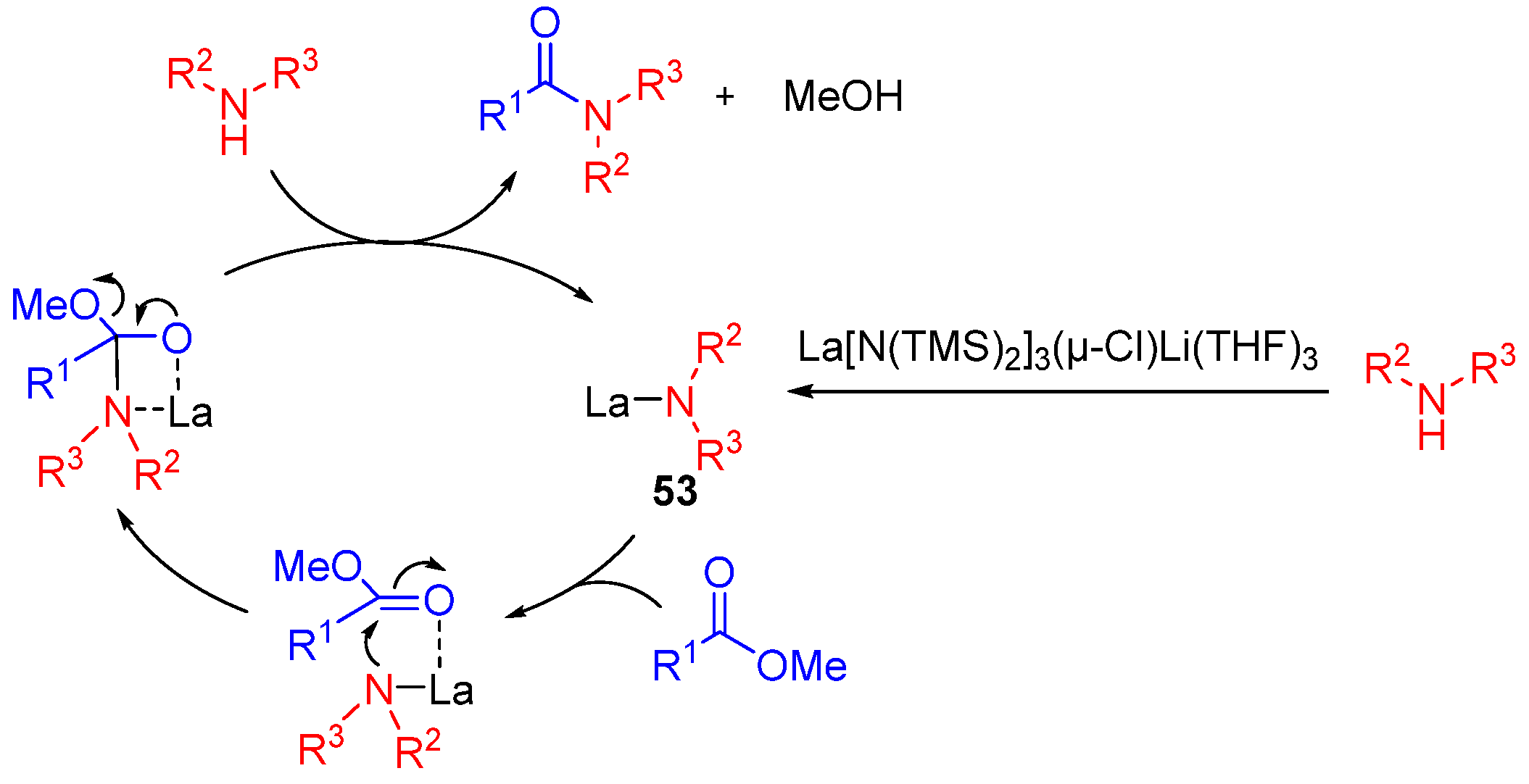
3.2. Lanthanide-Derived Catalysts
3.3. Miscellaneous
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Greenberg, A.; Breneman, C.M.; Liebman, J.F. The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials Science. Wiley-Interscience: New York, NY, USA, 2000. [Google Scholar]
- Rajput, P.; Sharma, A. Synthesis and biological importance of amide analogues. J. Pharmacol. Med. Chem. 2018, 2, 22. [Google Scholar]
- Carey, J.S.; Laffan, D.; Thomsonc, C.; Williams, M.T. Analysis of the reactions used for the preparation of drug candidate molecules. Org. Biomol. Chem. 2006, 4, 2337. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.G.; Boström, J. Analysis of past and present synthetic methodologies on medicinal chemistry: Where have all the new reactions gone? J. Med. Chem. 2016, 59, 4443. [Google Scholar] [CrossRef] [PubMed]
- De Figueiredo, R.M.; Suppo, J.-S.; Campagne, J.-M. Nonclassical routes for amide bond formation. Chem. Rev. 2016, 116, 12029. [Google Scholar] [CrossRef] [PubMed]
- Ojeda-Porras, A.; Gamba-Sánchez, D. Recent developments in amide synthesis using nonactivated starting materials. J. Org. Chem. 2016, 81, 11548. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, H.; Tinnis, F.; Selander, N.; Adolfsson, H. Catalytic amide formation from non-activated carboxylic acids and amines. Chem. Soc. Rev. 2014, 43, 2714. [Google Scholar] [CrossRef]
- Sabatini, M.T.; Boulton, L.T.; Sneddon, H.F.; Sheppard, T.D. A green chemistry perspective on catalytic amide bond formation. Nat. Catal. 2019, 2, 10. [Google Scholar] [CrossRef]
- Wang, X. Challenges and outlook for catalytic direct amidation reactions. Nat. Catal. 2019, 2, 98. [Google Scholar] [CrossRef]
- Todorovic, M.; Perrin, D.M. Recent developments in catalytic amide bond formation. Pept. Sci. 2020, 112, e24210. [Google Scholar] [CrossRef]
- Massolo, E.; Pirola, M.; Benaglia, M. Amide bond formation strategies: Latest advances on a dateless transformation. Eur. J. Org. Chem. 2020, 2020, 4641–4645. [Google Scholar] [CrossRef]
- Santos, A.S.; Silva, A.M.S.; Marques, M.M.B. Sustainable amidation reactions—Recent advances. Eur. J. Org. Chem. 2020, 2020, 2501. [Google Scholar] [CrossRef]
- Tosi, E.; de Figueiredo, R.M.; Campagne, J.-M. Enantioselective catalytic C−H amidations: An highlight. Catalysts 2021, 11, 471. [Google Scholar] [CrossRef]
- Cossy, J.; Pale-Grosdemange, C. A convenient synthesis of amides from carboxylic acids and primary amines. Tetrahedron Lett. 1989, 30, 2771. [Google Scholar] [CrossRef]
- Arnold, K.; Davies, B.; Giles, R.; Grosjean, C.; Smith, G.; Whiting, A. To catalyze or not to catalyze? Insight into direct amide bond formation from amines and carboxylic acids under thermal and catalyzed conditions. Adv. Synth. Catal. 2006, 348, 813. [Google Scholar] [CrossRef]
- Charville, H.; Jackson, D.A.; Hodges, G.; Whiting, A.; Wilson, M.R. The uncatalyzed direct amide formation reaction—Mechanism studies and the key role of carboxylic acid H-bonding. Eur. J. Org. Chem. 2011, 2011, 5981. [Google Scholar] [CrossRef]
- Unpublished result from this laboratory.
- Perreux, L.; Loupy, A.; Volatron, F. Solvent-free preparation of amides from acids and primary amines under microwave irradiation. Tetrahedron 2002, 58, 2155. [Google Scholar] [CrossRef]
- Zarecki, A.P.; Kolanowski, J.L.; Markiewicz, W.T. Microwave-assisted catalytic method for a green synthesis of amides directly from amines and carboxylic acids. Molecules 2020, 25, 1761. [Google Scholar] [CrossRef]
- Magano, J. Large-scale amidations in process chemistry: Practical considerations for reagent selection and reaction execution. Org. Process Res. Dev. 2022, 26, 1562. [Google Scholar] [CrossRef]
- El Dine, T.M.; Erb, W.; Berhault, Y.; Rouden, J.; Blanchet, J. Catalytic chemical amide synthesis at room temperature: One more step toward peptide synthesis. J. Org. Chem. 2015, 80, 4532. [Google Scholar] [CrossRef]
- Hall, D.G. Boronic acid catalysis. Chem. Soc. Rev. 2019, 48, 3475. [Google Scholar] [CrossRef]
- Wang, K.; Lu, Y.; Ishihara, K. The ortho-substituent on 2,4-bis(trifluoromethyl)phenylboronic acid catalyzed dehydrative condensation between carboxylic acids and amines. Chem. Commun. 2018, 54, 5410. [Google Scholar] [CrossRef] [PubMed]
- Al-Zoubi, R.M.; Al-Jammal, W.K.; McDonald, R. Regioselective synthesis of ortho-iodobiphenylboronic acid derivatives: A superior catalyst for carboxylic acid activation. New J. Chem. 2020, 44, 3612. [Google Scholar] [CrossRef]
- Ishihara, K.; Lu, Y. Boronic acid–DMAPO cooperative catalysis for dehydrative condensation between carboxylic acids and amines. Chem. Sci. 2016, 7, 1276. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wang, K.; Ishihara, K. Design of boronic acid-base complexes as reusable homogeneous catalysts in dehydrative condensations between carboxylic acids and amines. Asian J. Org. Chem. 2017, 6, 1191. [Google Scholar] [CrossRef]
- Khaldoun, K.; Safer, A.; Saidi-Besbes, S.; Carboni, B.; Le Guével, R.; Carreaux, F. An efficient solvent-free microwave-assisted synthesis of cinnamamides by amidation reaction using phenylboronic acid/Lewis base Co-catalytic system. Synthesis 2019, 51, 3891. [Google Scholar] [CrossRef]
- Arkhipenko, S.; Sabatini, M.T.; Batsanov, A.S.; Karaluka, V.; Sheppard, T.D.; Rzepa, H.S.; Whiting, A. Mechanistic insights into boron-catalysed direct amidation reactions. Chem. Sci. 2018, 9, 1058. [Google Scholar] [CrossRef]
- Noda, H.; Furutachi, M.; Asada, Y.; Shibasaki, M.; Kumagai, N. Unique physicochemical and catalytic properties dictated by the B3NO2 ring system. Nat. Chem. 2017, 9, 571. [Google Scholar] [CrossRef]
- Liu, Z.; Noda, H.; Shibasaki, M.; Kumagai, N. Catalytic oligopeptide synthesis. Org. Lett. 2018, 20, 612. [Google Scholar] [CrossRef]
- Opie, C.R.; Noda, H.; Shibasaki, M.; Kumagai, N. All non-carbon B3NO2 exotic heterocycles: Synthesis, dynamics, and catalysis. Chem. Eur. J. 2019, 25, 4648. [Google Scholar] [CrossRef]
- Noda, H.; Asada, Y.; Shibasaki, M.; Kumagai, N. Neighboring protonation unveils Lewis acidity in the B3NO2 heterocycle. J. Am. Chem. Soc. 2019, 141, 1546. [Google Scholar] [CrossRef]
- Sawant, D.N.; Bagal, D.B.; Ogawa, S.; Selvam, K.; Saito, S. Diboron-catalyzed dehydrative amidation of aromatic carboxylic acids with amines. Org. Lett. 2018, 20, 4397. [Google Scholar] [CrossRef] [PubMed]
- Shimada, N.; Hirata, M.; Koshizuka, M.; Ohse, N.; Kaito, R.; Makino, K. Diboronic acid anhydrides as effective catalysts for the hydroxy-directed dehydrative amidation of carboxylic acids. Org. Lett. 2019, 21, 4303. [Google Scholar] [CrossRef]
- Koshizuka, M.; Makino, K.; Shimada, N. Diboronic acid anhydride-catalyzed direct peptide bond formation enabled by hydroxy-directed dehydrative condensation. Org. Lett. 2020, 22, 8658. [Google Scholar] [CrossRef] [PubMed]
- Shimada, N.; Takahashi, N.; Ohse, N.; Koshizuka, M.; Makino, K. Synthesis of Weinreb amides using diboronic acid anhydride-catalyzed dehydrative amidation of carboxylic acids. Chem. Commun. 2020, 56, 13145. [Google Scholar] [CrossRef]
- Shimada, N.; Ohse, N.; Takahashi, N.; Urata, S.; Koshizuka, M.; Makino, K. Direct synthesis of N-protected serine- and threonine-derived Weinreb amides via diboronic acid anhydride-catalyzed dehydrative amidation: Application to the concise synthesis of Garner’s aldehyde. Synlett 2021, 32, 1024. [Google Scholar] [CrossRef]
- Michigami, K.; Sakaguchi, T.; Takemoto, Y. Catalytic dehydrative peptide synthesis with gem-diboronic acids. ACS Catal. 2020, 10, 683. [Google Scholar] [CrossRef]
- Yohda, M.; Yamamoto, Y. Enantioselective addition of arylboronic acids to methyl 2-formylbenzoates by using a ruthenium/Me-BIPAM catalyst for synthesis of chiral 3-aryl-isobenzofuranones. Org. Biomol. Chem. 2015, 13, 10874. [Google Scholar] [CrossRef] [PubMed]
- Sabatini, M.T.; Boulton, L.T.; Sheppard, T.D. Borate esters: Simple catalysts for the sustainable synthesis of complex amides. Sci. Adv. 2017, 3, e1701028. [Google Scholar] [CrossRef]
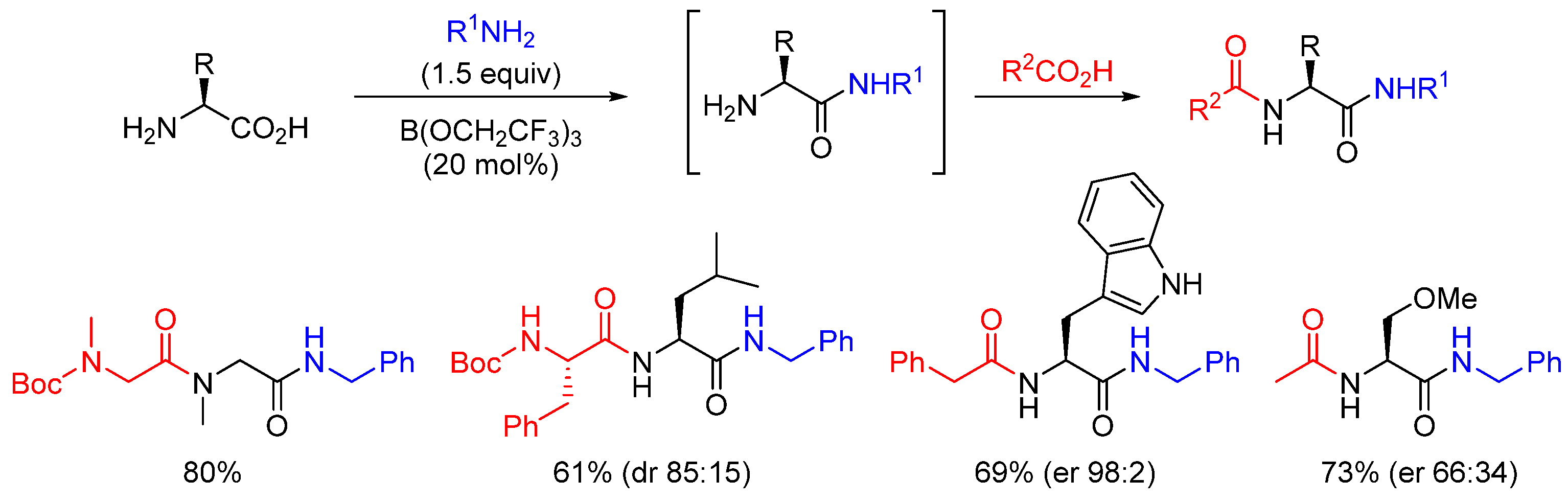
- Sabatini, M.T.; Karaluka, V.; Lanigan, R.M.; Boulton, L.T.; Badland, M.; Sheppard, T.D. Protecting-group-free amidation of amino acids using Lewis acid catalysts. Chem. Eur. J. 2018, 24, 7033. [Google Scholar] [CrossRef]
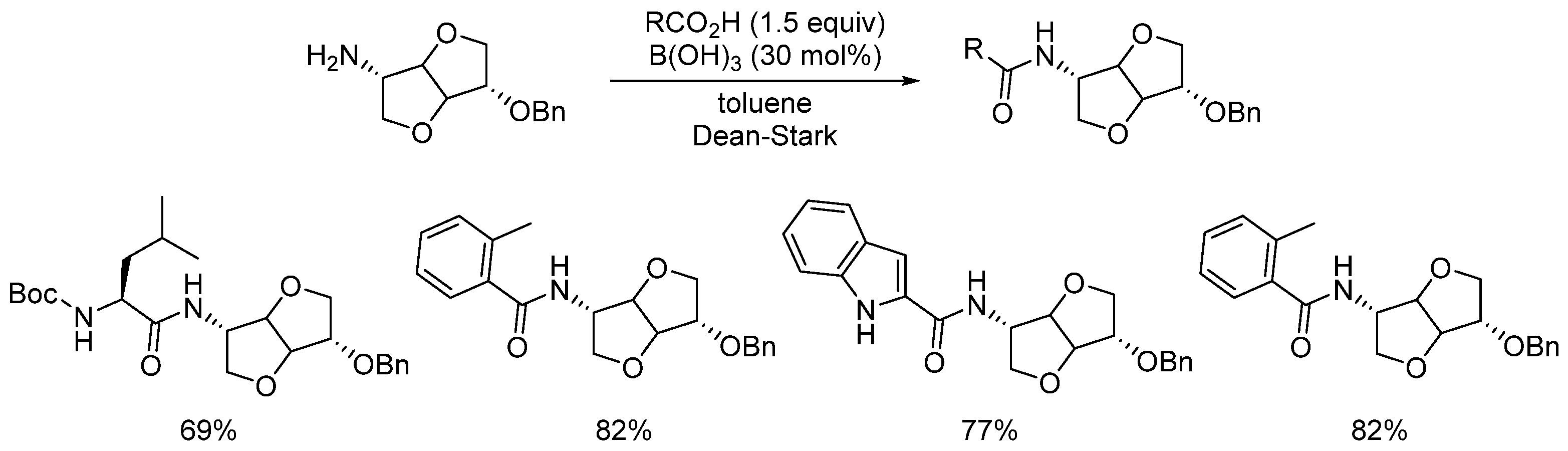
- Coomber, C.E.; Laserna, V.; Martin, L.T.; Smith, P.D.; Hailes, H.C.; Porter, M.J.; Sheppard, T.D. Catalytic direct amidations in tert-butyl acetate using B(OCH2CF3)3. Org. Biomol. Chem. 2019, 17, 6465. [Google Scholar] [CrossRef]
- Mylavarapu, R.K.; GCM, K.; Kolla, N.; Veeramalla, R.; Koilkonda, P.; Bhattacharya, A.; Bandichhor, R. Boric acid catalyzed amidation in the synthesis of active pharmaceutical ingredients. Org. Proc. Res. Dev. 2007, 11, 1065. [Google Scholar] [CrossRef]
- Shinde, G.B.; Niphade, N.C.; Deshmukh, S.P.; Toche, R.B.; Mathad, V.T. Industrial application of the Forster reaction: Novel one-pot synthesis of cinacalcet hydrochloride, a calcimimetic agent. Org. Proc. Res. Dev. 2011, 15, 455. [Google Scholar] [CrossRef]
- Tang, P.W. Boric acid catalyzed formation from carboxylic acids and amines: N-Benzyl-4-phenylbutyramide. Org. Synth. 2012, 89, 432. [Google Scholar]
- Janvier, M.; Moebs-Sanchez, S.; Popowycz, F. Bio-based amides from renewable isosorbide by a direct and atom-economic boric acid amidation methodology. Eur. J. Org. Chem. 2016, 2016, 2308–2318. [Google Scholar] [CrossRef]
- Du, Y.; Barber, T.; Lim, S.E.; Rzepa, H.S.; Baxendale, H.S.; Whiting, A. A solid-supported arylboronic acid catalyst for direct amidation. Chem. Commun. 2019, 55, 2916. [Google Scholar] [CrossRef]
- Baraniak, M.K.; Lalancette, R.A.; Jäkle, F. Electron-deficient borinic acid polymers: Synthesis, supramolecular assembly, and examination as catalysts in amide bond formation. Chem. Eur. J. 2019, 25, 13799. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.H.; Wong, L.T.L. Silicon tetrachloride as a coupling reagent for amide formation. J. Org. Chem. 1969, 34, 2766. [Google Scholar] [CrossRef]
- Chan, T.H.; Wong, L.T.L. Evaluation of acyloxysilane as acylating agent for peptide synthesis. J. Org. Chem. 1971, 36, 850. [Google Scholar] [CrossRef]
- Davies, J.J.; Braddock, D.C.; Lickiss, P.D. Silicon compounds as stoichiometric coupling reagents for direct amidation. Org. Biomol. Chem. 2021, 19, 6746. [Google Scholar] [CrossRef]
- For the recent use of stoichiometric amount of silicon reagent in the presence of a co-catalytic amount of another silicon reagent in peptide synthesis, see: Muramatsu, W.; Manthena, C.; Nakashima, E.; Yamamoto, H. Peptide bond-forming reaction via amino acid silyl esters: New catalytic reactivity of an aminosilane. ACS Catal. 2020, 10, 9594. [Google Scholar] [CrossRef]
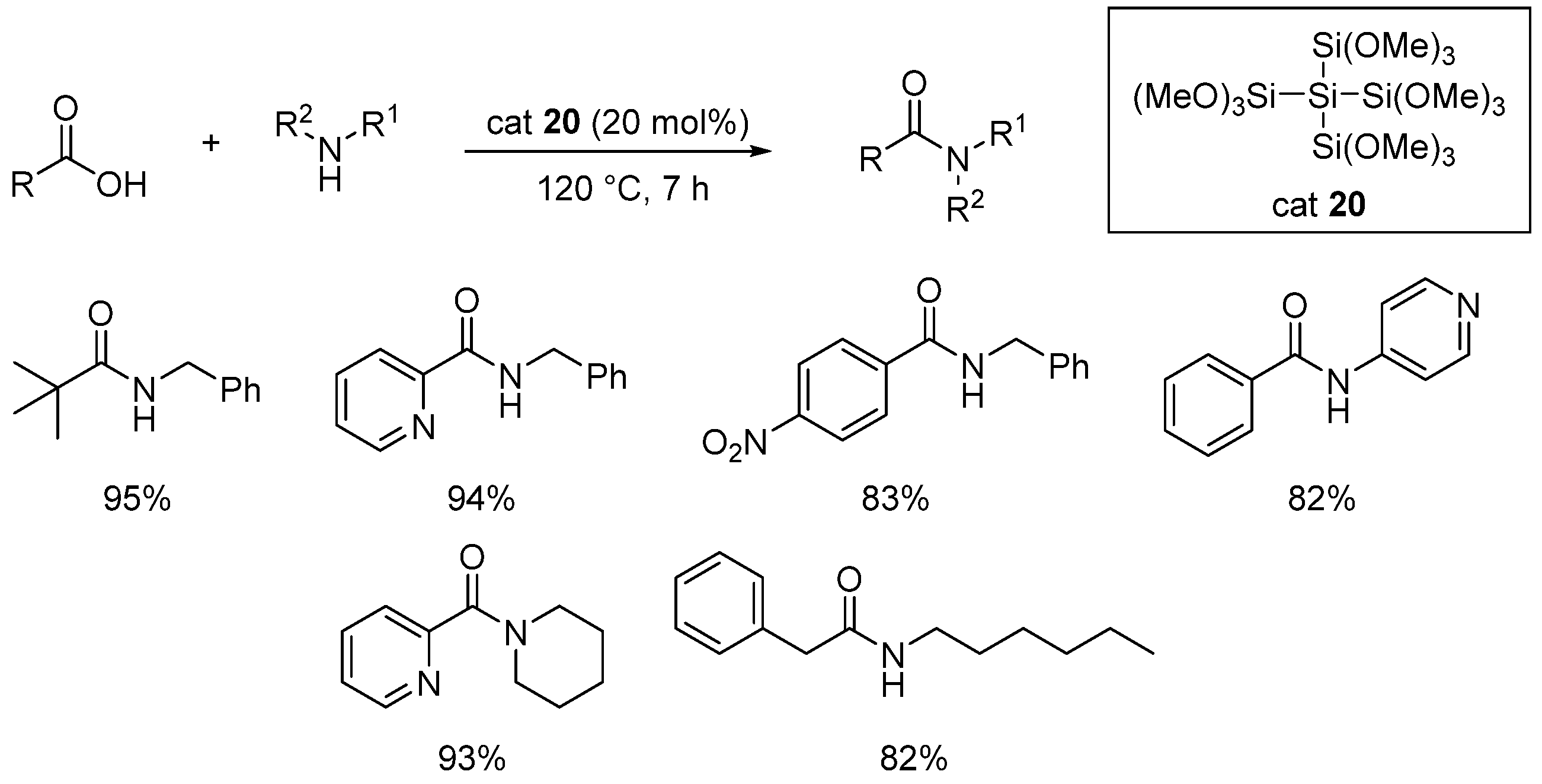
- Lainer, T.; Czerny, F.; Haas, M. Solvent-free amide bond formation using a variety of methoxysilanes as coupling agent. Org. Biomol. Chem. 2022, 20, 3717. [Google Scholar] [CrossRef] [PubMed]
- Ojeda-Porras, A.; Hernández-Santana, A.; Gamba-Sánchez, D. Direct amidation of carboxylic acids with amines under microwave irradiation using silica gel as a solid support. Green Chem. 2015, 17, 3157. [Google Scholar] [CrossRef]
- Rimola, A.; Fabbiani, M.; Sodupe, M.; Ugliengo, P.; Martra, G. How does silica catalyze the amide bond formation under dry conditions? Role of specific surface silanol pairs. ACS Catal. 2018, 8, 4558. [Google Scholar] [CrossRef]
- Lenstra, D.C.; Rutjes, F.P.J.T.; Mecinović, J. Triphenylphosphine-catalysed amide bond formation between carboxylic acids and amines. Chem. Commun. 2014, 50, 5763. [Google Scholar] [CrossRef]
- Hamstra, D.F.J.; Lenstra, D.C.; Koenders, T.J.; Rutjes, F.P.J.T.; Mecinović, J. Poly(methylhydrosiloxane) as a green reducing agent in organophosphorus-catalysed amide bond formation. Org. Biomol. Chem. 2017, 15, 6426. [Google Scholar] [CrossRef]
- Ren, J.-W.; Tong, M.-N.; Zhao, Y.-F.; Ni, F. Synthesis of dipeptide, amide, and ester without racemization by oxalyl chloride and catalytic triphenylphosphine oxide. Org. Lett. 2021, 23, 7497. [Google Scholar] [CrossRef]
- Movahed, F.S.; Sawant, D.N.; Bagal, D.B.; Saito, S. Tris(o-phenylenedioxy)cyclotriphosphazene as a promoter for the formation of amide bonds between aromatic acids and amines. Synthesis 2020, 52, 3253. [Google Scholar]
- Liebeskind, L.S.; Gangireddy, P.; Lindale, M.G. Benzoisothiazolone organo/copper-cocatalyzed redox dehydrative construction of amides and peptides from carboxylic acids using (EtO)3P as the reductant and O2 in air as the terminal oxidant. J. Am. Chem. Soc. 2016, 138, 6715. [Google Scholar] [CrossRef]
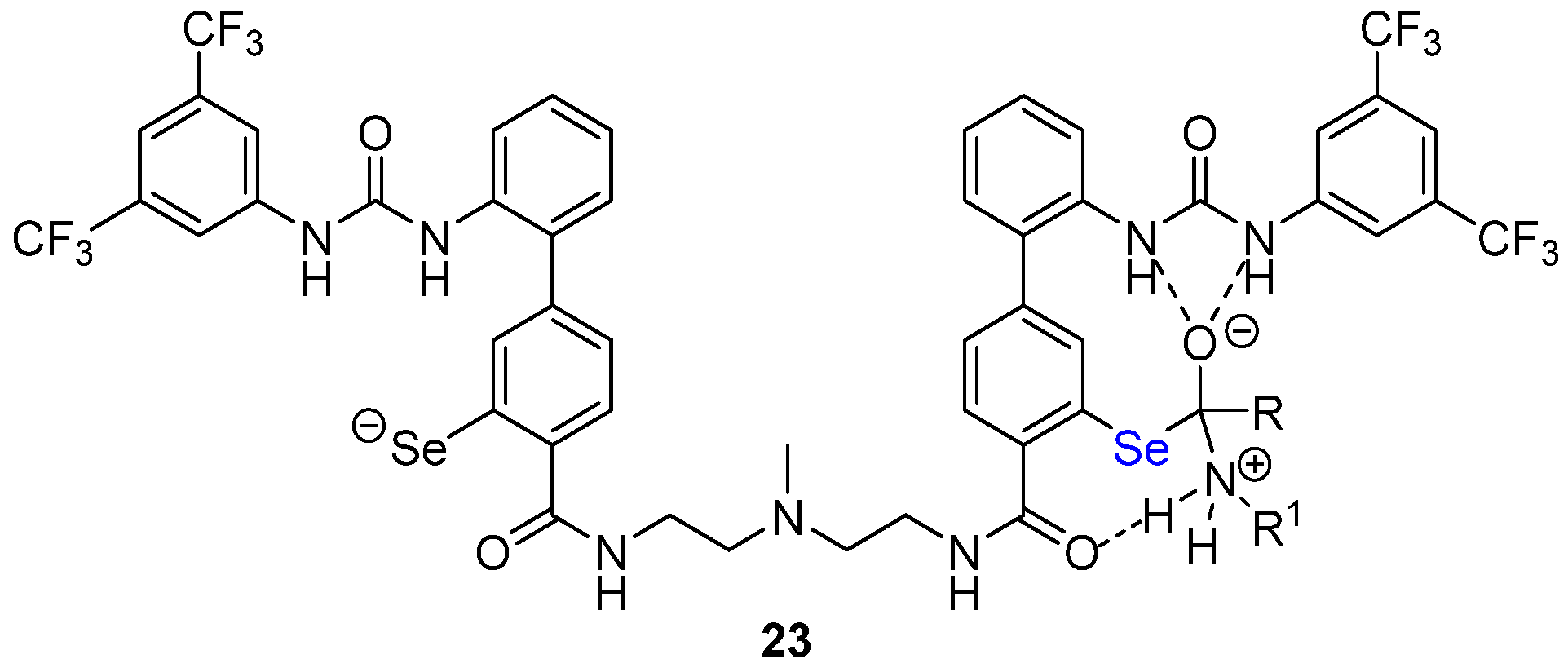
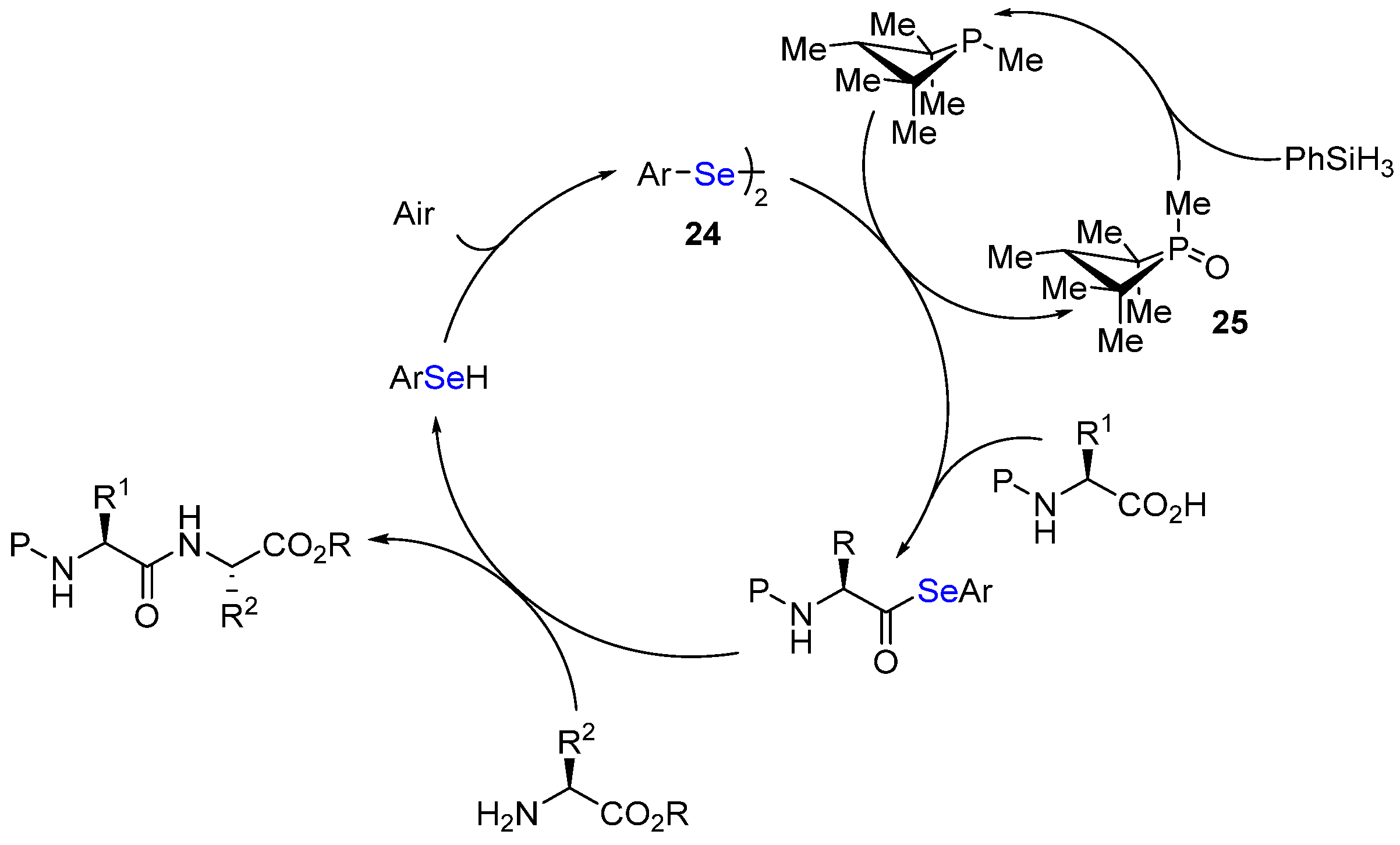
- Akondi, S.M.; Gangireddy, P.; Pickel, T.C.; Liebeskind, L.S. Aerobic, diselenide-catalyzed redox dehydration: Amides and peptides. Org. Lett. 2018, 20, 538. [Google Scholar] [CrossRef]
- Satishkumar, S.; Panigrahi, N.R.; Arora, P.S. Rational design of an organocatalyst for peptide bond formation. J. Am. Chem. Soc. 2019, 141, 15977. [Google Scholar]
- Panigrahi, N.R.; Arora, P.S. Two-component redox organocatalyst for peptide bond formation. J. Am. Chem. Soc. 2022, 144, 3637. [Google Scholar]
- Nguyen, T.V.; Lyons, D.J.M. A novel aromatic carbocation-based coupling reagent for esterification and amidation reactions. Chem. Commun. 2015, 51, 3131. [Google Scholar] [CrossRef] [PubMed]
- Huy, P.H.; Mbouhom, C. Formamide catalyzed activation of carboxylic acids—Versatile and cost-efficient amidation and esterification. Chem. Sci. 2019, 10, 7399. [Google Scholar] [CrossRef] [PubMed]
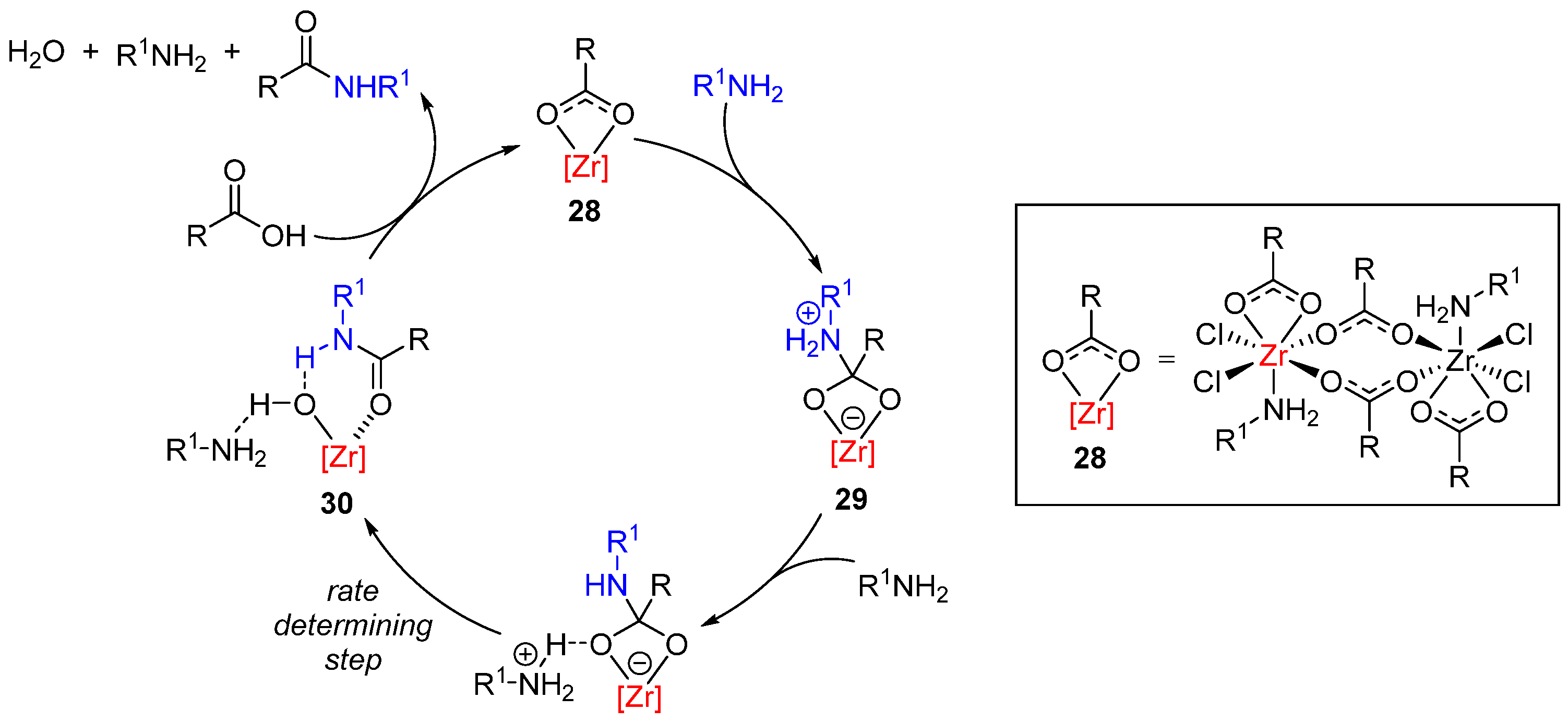
- Lundberg, H.; Tinnis, F.; Adolfsson, H. Direct amide coupling of non-activated carboxylic acids and amines catalysed by zirconium(IV) chloride. Chem. Eur. J. 2012, 18, 3822. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.L.; Chhatwal, A.R.; Williams, J.M.J. Direct amide formation from unactivated carboxylic acids and amines. Chem. Commun. 2012, 48, 666. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, H.; Tinnis, F.; Zhang, J.; Algarra, A.G.; Himo, F.; Adolfsson, H. Mechanistic elucidation of zirconium-catalyzed direct amidation. J. Am. Chem. Soc. 2017, 139, 2286. [Google Scholar] [CrossRef]
- Li, N.; Wang, L.; Zhang, L.; Zhao, W.; Qiao, J.; Xu, X.; Liang, Z. Air-stable Bis(pentamethylcyclopentadienyl) zirconium perfluorooctanesulfonate as an efficient and recyclable catalyst for the synthesis of N-substituted amides. ChemCatChem 2018, 10, 3532. [Google Scholar] [CrossRef]
- Lundberg, H.; Tinnis, F.; Adolfsson, H. Zirconium catalyzed amide formation without water scavenging. Appl. Organometal. Chem. 2019, 33, e5062. [Google Scholar] [CrossRef]
- Lundberg, H.; Adolfsson, H. Hafnium-catalyzed direct amide formation at room temperature. ACS Catal. 2015, 5, 3271. [Google Scholar] [CrossRef]
- De Azambuja, F.; Parac-Vogt, T.N. Water-tolerant and atom economical amide bond formation by metal-substituted polyoxometalate catalysts. ACS Catal. 2019, 9, 10245. [Google Scholar] [CrossRef]
- De Azambuja, F.; Loosen, A.; Conic, D.; van den Besselaar, M.; Harvey, J.N.; Parac-Vogt, T.N. En route to a heterogeneous catalytic direct peptide bond formation by Zr-based metal-organic framework catalysts. ACS Catal. 2021, 11, 7647. [Google Scholar] [CrossRef]
- Wang, A.; Xie, Y.; Wang, J.; Shi, D.; Yu, H. Atom-economic amide synthesis by using an iron-substituted polyoxometalate catalyst. Chem. Commun. 2022, 58, 1127. [Google Scholar] [CrossRef] [PubMed]
- Abánades Lázaro, I.; Forgan, R.S.; Cirujano, F.G. MOF nanoparticles as heterogeneous catalysts for direct amide bond formations. Dalton Trans. 2022, 51, 8368. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.A.; Siddiki, S.M.A.H.; Onodera, W.; Kon, K.; Shimizu, K.-i. Amidation of carboxylic acids with amines by Nb2O5 as a reusable Lewis acid catalyst. ChemCatChem 2015, 7, 3555. [Google Scholar] [CrossRef]
- Shinde, V.N.; Kumar, A. KPF6-mediated rsterification and amidation of carboxylic acids. J. Org. Chem. 2022, 87, 2651. [Google Scholar]
- Großmann, L.M.; Beier, V.; Duttenhofer, L.; Lennartz, L.; Opatz, T. An iodide-mediated anodic amide coupling. Chem. Eur. J. 2022, 28, e202201768. [Google Scholar] [CrossRef]
- Nagahara, S.; Okada, Y.; Kitano, Y.; Chiba, K. Biphasic electrochemical peptide synthesis. Chem. Sci. 2021, 12, 12911. [Google Scholar] [CrossRef]
- Yang, B.; Niu, K.; Haag, F.; Cao, N.; Zhang, J.; Zhang, H.; Li, Q.; Allegretti, F.; Björk, J.; Barth, J.V.; et al. Abiotic formation of an amide bond via surface-supported direct carboxyl-amine coupling. Angew. Chem. Int. Ed. 2022, 61, e202113590. [Google Scholar]
- Basha, A.; Lipton, M.F.; Weinreb, S.M. A mild, general method for conversion of esters to amides. Tetrahedron Lett. 1977, 48, 4171. [Google Scholar] [CrossRef]
- Lipton, M.F.; Basha, A.; Weinreb, S.M. Conversion of esters to amides with dimethylaluminium amides: N,N-dimethylcyclohexanecarboxamide. Org. Synth. 1979, 59, 49. [Google Scholar]
- Ishihara, K.; Kuroki, Y.; Hanaki, N.; Ohara, S.; Yamamoto, H. Antimony-templated macrolactamization of tetraamino esters. Facile synthesis of macrocyclic spermine alkaloids, (±)-buchnerine, (±)-verbacine, (±)-verbaskine, and (±)-verbascenine. J. Am. Chem. Soc. 1996, 118, 1569. [Google Scholar] [CrossRef]
- Wang, W.-B.; Roskamp, E.J. Tin(II) amides: New reagents for the conversion of esters to amides. J. Org. Chem. 1992, 57, 6101. [Google Scholar] [CrossRef]
- De M. Muñoz, J.; Alcázar, J.; de la Hoz, A.; Díaz-Ortiz, Á.; de Diego, S.-A.A. Preparation of amides mediated by isopropylmagnesium chloride under continuous flow conditions. Green Chem. 2012, 14, 1335. [Google Scholar] [CrossRef]
- Kim, B.R.; Lee, H.-G.; Kang, S.-B.; Sung, G.H.; Kim, J.-J.; Park, J.K.; Lee, S.-G.; Yoon, Y.-J. tert-Butoxide-assisted amidation of esters under green conditions. Synthesis 2012, 1, 42. [Google Scholar]
- Nad, P.; Gupta, K.; Sen, A.; Mukherjee, A. Mechanistic understanding of KOtBu-mediated direct amidation of esters with anilines: An experimental study and computational approach. Chem. Asian J. 2022, 17, e202200800. [Google Scholar] [CrossRef]
- Li, G.; Szostak, M. Highly selective transition-metal-free transamidation of amides and amidation of esters at room temperature. Nat. Commun. 2018, 9, 4165. [Google Scholar] [CrossRef]
- Li, G.; Ji, C.-L.; Hong, X.; Szostak, M. Highly chemoselective, transition-metal-free transamidation of unactivated amides and direct amidation of alkyl esters by N−C/O−C cleavage. J. Am. Chem. Soc. 2019, 141, 11161. [Google Scholar] [CrossRef]
- Rzhevskiy, S.A.; Ageshina, A.A.; Chesnokov, G.; Gribanov, P.S.; Topchiy, M.A.; Nechaev, M.S.; Asachenko, A.F. Solvent- and transition metal-free amide synthesis from phenyl esters and aryl amines. RSC Adv. 2019, 9, 1536. [Google Scholar] [CrossRef]
- Zhang, R.; Yao, W.-Z.; Qian, L.; Sang, W.; Yuan, Y.; Du, M.-C.; Cheng, H.; Chen, C.; Qin, X. A practical and sustainable protocol for direct amidation of unactivated esters under transition-metal-free and solvent-free conditions. Green Chem. 2021, 23, 3972. [Google Scholar] [CrossRef]
- Ranu, B.C.; Dutta, P. A simple and convenient procedure for the conversion of esters to secondary amides. Synth. Commun. 2003, 33, 297. [Google Scholar] [CrossRef]
- Han, C.; Lee, J.P.; Lobkovsky, E.; Porco, J.A., Jr. Catalytic ester-amide exchange using group (IV) metal alkoxide-activator complexes. J. Am. Chem. Soc. 2005, 127, 10039. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.T.; Lenstra, D.C.; Mecinović, J. Chemoselective calcium-catalysed direct amidation of carboxylic esters. RSC Adv. 2015, 5, 77658. [Google Scholar] [CrossRef]
- Arora, R.; Paul, S.; Gupta, R. A mild and efficient procedure for the conversion of aromatic carboxylic esters to secondary amides. Can. J. Chem. 2005, 83, 1137. [Google Scholar] [CrossRef]
- Ohshima, T.; Hayashi, Y.; Agura, K.; Fujii, Y.; Yoshiyama, A.; Mashima, K. Sodium methoxide: A simple but highly efficient catalyst for the direct amidation of esters. Chem. Commun. 2012, 48, 5434. [Google Scholar] [CrossRef] [PubMed]
- Price, K.E.; Larrivée-Aboussafy, C.; Lillie, B.M.; McLaughlin, R.W.; Mustakis, J.; Hettenbach, K.W.; Hawkins, J.M.; Vaidyanathan, R. Mild and efficient DBU-catalyzed amidation of cyanoacetates. Org. Lett. 2009, 11, 2003. [Google Scholar] [CrossRef]
- Yang, X.; Birman, V.B. Acyl transfer catalysis with 1,2,4-triazole anion. Org. Lett. 2009, 11, 1499. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Muthaiah, S.; Hong, S.H. Tandem synthesis of amides and secondary amines from esters with primary amines under solvent-free conditions. Adv. Synth. Catal. 2014, 356, 2653. [Google Scholar] [CrossRef]
- Vandavasi, J.K.; Hsiao, C.-T.; Hu, W.-P.; Boominathan, S.S.K.; Wang, J.-J. Silver(I)-catalyzed tandem approach to β-oxo amides. Eur. J. Org. Chem. 2015, 2015, 3171. [Google Scholar] [CrossRef]
- Morimoto, H.; Fujiwara, R.; Shimizu, Y.; Morisaki, K.; Ohshima, T. Lanthanum(III) triflate catalyzed direct amidation of esters. Org. Lett. 2014, 16, 2018. [Google Scholar] [CrossRef]
- Furukawa, S.; Fukuyama, T.; Matsui, A.; Kuratsu, M.; Nakaya, R.; Ineyama, T.; Ueda, H.; Ryu, I. Coupling-reagent-free synthesis of dipeptides and tripeptides using amino acid ionic liquids. Chem. Eur. J. 2015, 21, 11980. [Google Scholar] [CrossRef]
- Hie, L.; Fine Nathel, N.F.; Hong, X.; Yang, Y.-F.; Houk, K.N.; Garg, N.K. Nickel-catalyzed activation of acyl C−O bonds of methyl esters. Angew. Chem. Int. Ed. 2016, 55, 2810. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Siddiki, S.M.A.H.; Kon, K.; Shimizu, K.-i. A heterogeneous Niobium(V) oxide catalyst for the direct amidation of esters. ChemCatChem 2015, 7, 2705. [Google Scholar] [CrossRef]
- Gnanaprakasam, B.; Milstein, D. Synthesis of amides from esters and amines with liberation of H2 under neutral conditions. J. Am. Chem. Soc. 2011, 133, 1682. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Wang, Y.; Du, G.-F.; Dai, B.; He, L. N-Heterocyclic carbene-catalysed amidation of vinyl esters with aromatic amines. Tetrahedron 2015, 71, 3472. [Google Scholar] [CrossRef]
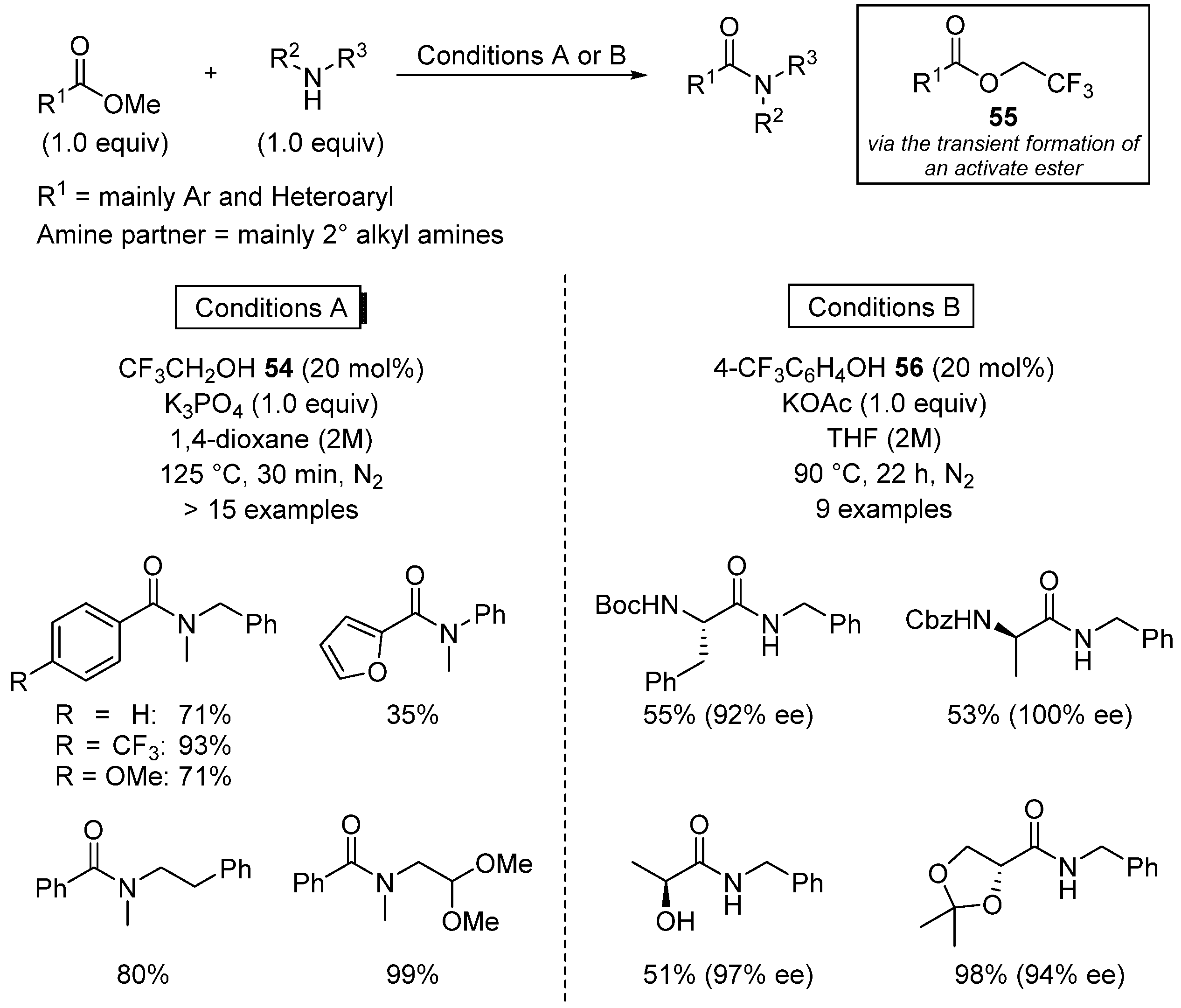
- Caldwell, N.; Jamieson, C.; Simpson, I.; Watson, A.J.B. Catalytic amidation of unactivated ester derivatives mediated by trifluoroethanol. Chem. Commun. 2015, 51, 9495. [Google Scholar] [CrossRef] [PubMed]
- Lenstra, D.C.; Nguyen, D.T.; Mecinović, J. Zirconium-catalyzed direct amide bond formation between carboxylic esters and amines. Tetrahedron 2015, 71, 5547. [Google Scholar] [CrossRef]
- Caldwell, N.; Jamieson, C.; Simpson, I.; Tuttle, T. Organobase-catalyzed amidation of esters with amino alcohols. Org. Lett. 2013, 15, 2506. [Google Scholar] [CrossRef]
- Sabot, C.; Kumar, K.A.; Meunier, S.; Mioskowski, C. A convenient aminolysis of esters catalyzed by 1,5,7-triazabicyclo [4.4.0]dec-5-ene (TBD) under solvent-free conditions. Tetrahedron Lett. 2007, 48, 3863. [Google Scholar] [CrossRef]
- Movassaghi, M.; Schmidt, M.A. N-Heterocyclic Carbene-catalyzed amidation of unactivated esters with amino alcohols. Org. Lett. 2005, 7, 2453. [Google Scholar] [CrossRef]
- Allen, C.L.; Williams, J.M. Metal-catalysed approaches to amide bond formation. Chem. Soc. Rev. 2011, 40, 3405. [Google Scholar] [CrossRef]
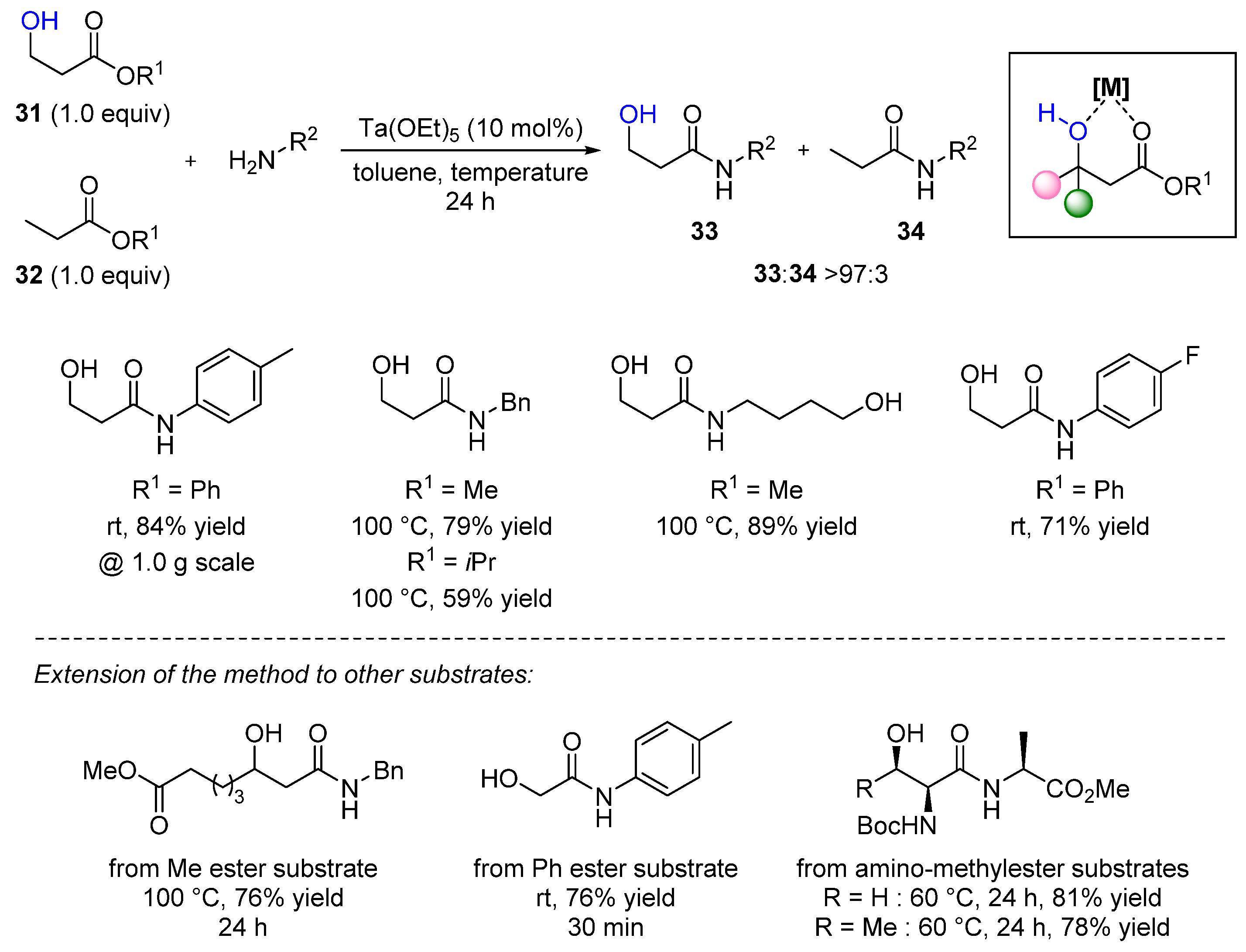
- Tsuji, H.; Yamamoto, H. Hydroxy-directed amidation of carboxylic acid esters using a tantalum alkoxide catalyst. J. Am. Chem. Soc. 2016, 138, 14218. [Google Scholar] [CrossRef] [PubMed]
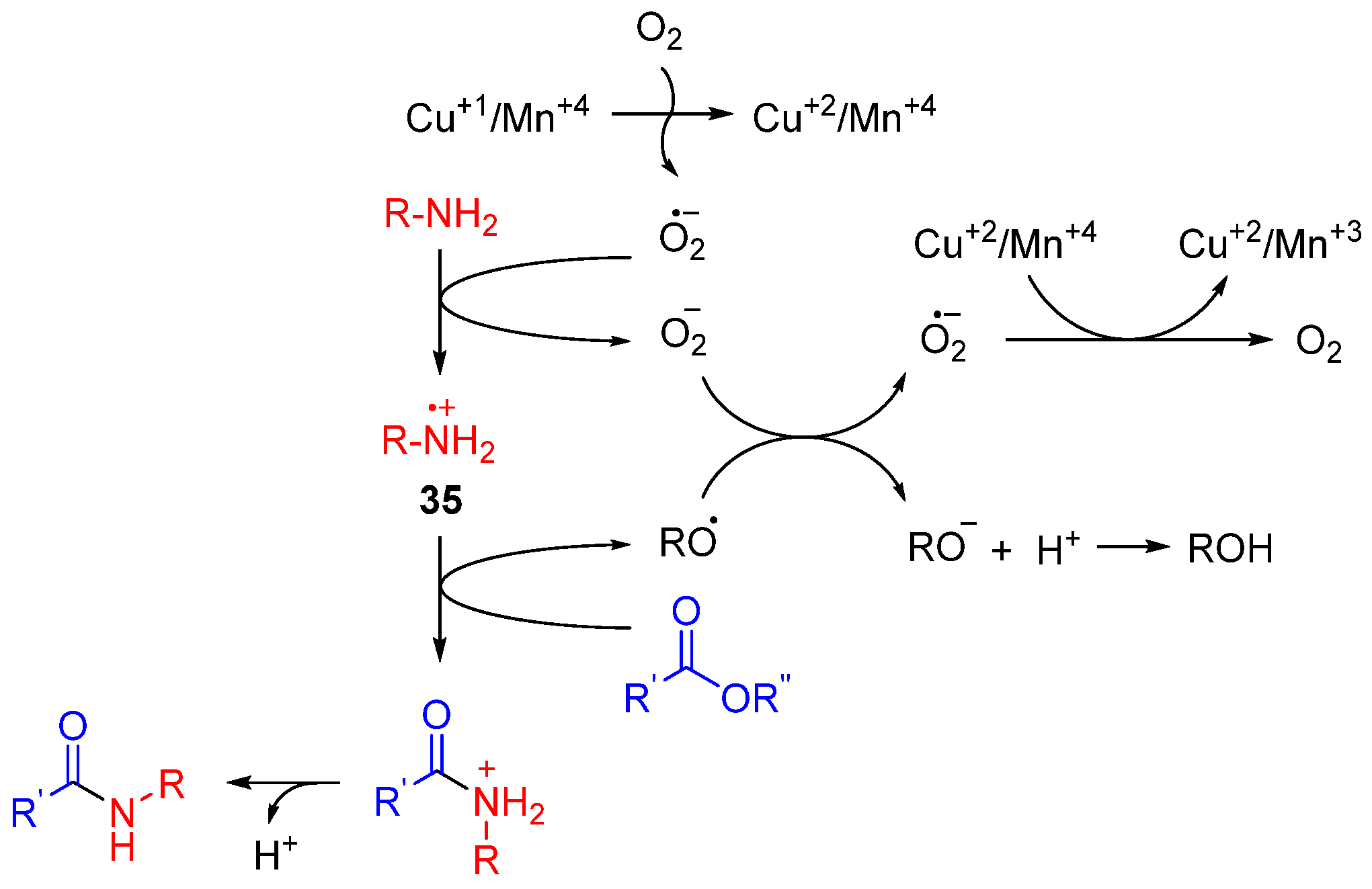
- Sultan, S.; Kumar, M.; Devari, S.; Mukherjee, D.; Ali Shah, B. Copper-manganese spinel oxide catalyzed synthesis of amides and azobenzenes via aminyl radical cations. ChemCatChem 2016, 8, 703. [Google Scholar] [CrossRef]
- Kumar, A.; Espinosa-Jalapa, N.A.; Leitus, G.; Diskin-Posner, Y.; Avram, L.; Milstein, D. Direct synthesis of amides by dehydrogenative coupling of amines with either alcohols or esters: Manganese pincer complex as catalyst. Angew. Chem. Int. Ed. 2017, 56, 14992. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Wang, X.; Tao, S.; Bu, Q.; Wei, D.; Liu, N. Manganese catalyzed direct amidation of esters with amines. J. Org. Chem. 2021, 86, 2339. [Google Scholar] [CrossRef]
- Ben Halima, T.; Vandavasi, J.K.; Shkoor, M.; Newman, S.G. A cross-coupling approach to amide bond formation from esters. ACS Catal. 2017, 7, 2176. [Google Scholar] [CrossRef]
- Shi, S.; Szostak, M. Pd–PEPPSI: A general Pd–NHC precatalyst for Buchwald–Hartwig cross-coupling of esters and amides (transamidation) under the same reaction conditions. Chem. Commun. 2017, 53, 10584. [Google Scholar] [CrossRef]
- Dardir, A.H.; Melvin, P.R.; Davis, R.M.; Hazari, N.; Mohadjer Beromi, M. Rapidly activating Pd-precatalyst for Suzuki-Miyaura and Buchwald-Hartwig couplings of aryl esters. J. Org. Chem. 2018, 83, 469. [Google Scholar] [CrossRef]
- Zhou, T.; Li, G.; Nolan, S.P.; Szostak, M. [Pd(NHC)(acac)Cl]: Well-defined, air-stable, and readily available precatalysts for Suzuki and Buchwald-Hartwig cross-coupling (transamidation) of amides and esters by N–C/O–C activation. Org. Lett. 2019, 21, 3304. [Google Scholar] [CrossRef]
- Muto, K.; Yamaguchi, J.; Musaev, D.G.; Itami, K. Decarbonylative organoboron cross-coupling of esters by nickel catalysis. Nat. Commun. 2015, 6, 7508. [Google Scholar] [CrossRef]
- Hong, X.; Liang, Y.; Houk, K.N. Mechanisms and origins of switchable chemoselectivity of Ni-catalyzed C(aryl)-O and C(acyl)-O activation of aryl esters with phosphine ligands. J. Am. Chem. Soc. 2014, 136, 2017. [Google Scholar] [CrossRef]
- Ben Halima, T.; Zhang, W.; Yalaoui, I.; Hong, X.; Yang, Y.-F.; Houk, K.N.; Newman, S.G. Palladium-catalyzed Suzuki–Miyaura coupling of aryl esters. J. Am. Chem. Soc. 2017, 139, 1311. [Google Scholar] [CrossRef] [PubMed]
- Pu, X.; Hu, J.; Zhao, Y.; Shi, Z. Nickel-catalyzed decarbonylative borylation and silylation of esters. ACS Catal. 2016, 6, 6692. [Google Scholar] [CrossRef]
- Lu, Q.; Yu, H.; Fu, Y. Mechanistic study of chemoselectivity in Ni-catalyzed coupling reactions between azoles and aryl carboxylates. J. Am. Chem. Soc. 2014, 136, 8252. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, S.-L.; Fu, Y.; Guo, Q.-X.; Liu, L. Mechanism of Ni-catalyzed selective C−O bond activation in cross-coupling of aryl esters. J. Am. Chem. Soc. 2009, 131, 8815. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Muto, K.; Yamaguchi, J.; Zhao, C.; Itami, K.; Musaev, D.G. Key mechanistic features of Ni-catalyzed C–H/C–O biaryl coupling of azoles and naphthalen-2-yl pivalates. J. Am. Chem. Soc. 2014, 136, 14834. [Google Scholar] [CrossRef]
- Ben Halima, T.; Masson-Makdissi, J.; Newman, S.G. Nickel-catalyzed amide bond formation from methyl esters. Angew. Chem. Int. Ed. 2018, 57, 12925. [Google Scholar] [CrossRef]
- Zheng, Y.-L.; Newman, S.G. Methyl esters as cross-coupling electrophiles: Direct synthesis of amide bonds. ACS Catal. 2019, 9, 4426. [Google Scholar] [CrossRef]
- Hie, L.; Fine Nathel, N.; Shah, T.K.; Baker, E.L.; Hong, X.; Yang, Y.-F.; Liu, P.; Houk, K.N.; Garg, N.K. Conversion of amides to esters by the nickel-catalysed activation of amide C–N bonds. Nature 2015, 524, 79. [Google Scholar] [CrossRef]
- Chu, C.-q.; Dang, L. Esterification of aryl and alkyl amides enabled by tailor-made and proposed nickel catalyst: Insights from theoretical investigation. J. Org. Chem. 2018, 83, 5009. [Google Scholar] [CrossRef]
- Ji, C.-L.; Xie, P.-P.; Hong, X. Computational study of mechanism and thermodynamics of Ni/IPr-catalyzed amidation of esters. Molecules 2018, 23, 2681. [Google Scholar] [CrossRef]
- Mkhonazi, B.D.; Shandu, M.; Tshinavhe, R.; Simelane, S.B.; Moshapo, P.T. Solvent-free iron(III) chloride-catalyzed direct amidation of esters. Molecules 2020, 25, 1040. [Google Scholar] [CrossRef] [PubMed]
- Rashed, M.N.; Masuda, K.; Ichitsuka, T.; Koumura, N.; Sato, K.; Kobayashi, S. Zirconium oxide-catalyzed direct amidation of unactivated esters under continuous-flow conditions. Adv. Synth. Catal. 2021, 363, 2529. [Google Scholar] [CrossRef]
- Li, Z.; Wang, C.; Wang, Y.; Yuan, D.; Yao, Y. Heterobimetallic lanthanide–sodium alkoxides catalyze the amidation of esters. Asian J. Org. Chem. 2018, 7, 810. [Google Scholar] [CrossRef]
- Li, Z.; Guo, C.; Chen, J.; Yao, Y.; Luo, Y. Facile amidation of esters with aromatic amines promoted by lanthanide tris (amide) complexes. Appl. Organometal. Chem. 2020, 34, e5517. [Google Scholar] [CrossRef]
- McPherson, C.G.; Caldwell, N.; Jamieson, C.; Simpson, I.; Watson, A.J.B. Amidation of unactivated ester derivatives mediated by trifluoroethanol. Org. Biomol. Chem. 2017, 15, 3507. [Google Scholar] [CrossRef] [PubMed]
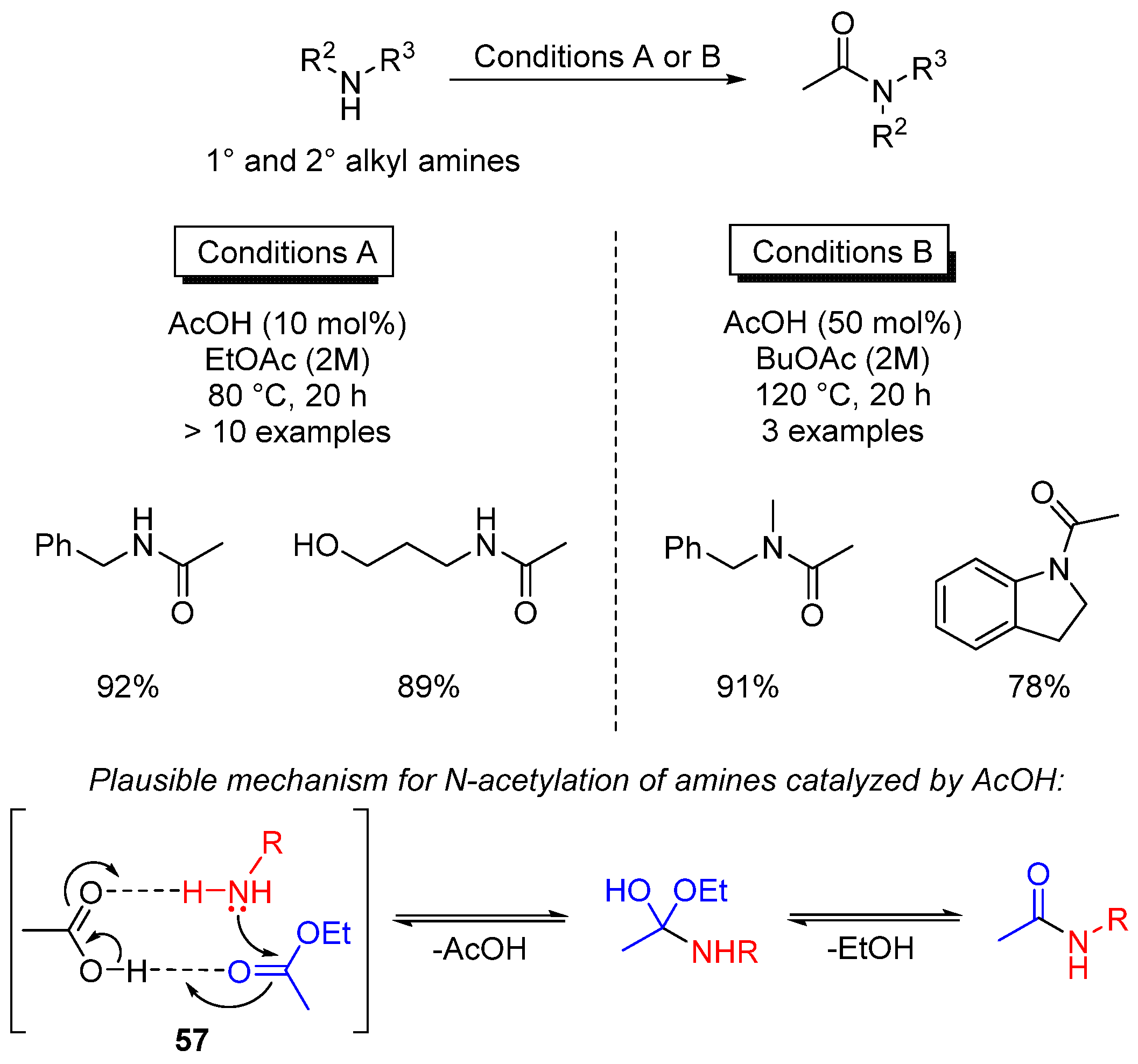
- Sanz Sharley, D.D.; Williams, J.M.J. Acetic acid as a catalyst for the N-acylation of amines using esters as the acyl source. Chem. Commun. 2017, 53, 2020. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, E.; Yamamoto, H. Biomimetic peptide catalytic bond-forming utilizing a mild Brønsted acid. Chem. Eur. J. 2022, 28, e202103989. [Google Scholar] [CrossRef]
- Abedelnour, E.; Ognier, S.; Zhang, M.; Schio, L.; Venier, O.; Cossy, J.; Tatoulian, M. Plasma flow chemistry for direct N-acylation of amines by esters. Chem. Commun. 2022, 58, 7281. [Google Scholar] [CrossRef]
- Nicholson, W.I.; Barreteau, F.; Leitch, J.A.; Payne, R.; Priestley, I.; Godineau, E.; Battilocchio, C.; Browne, D.L. Direct amidation of esters by ball milling. Angew. Chem. Int. Ed. 2021, 60, 21868. [Google Scholar] [CrossRef]
- Lee, H.-J.; Choi, E.-S.; Maruoka, K. Development of a catalytic ester activation protocol for the efficient formation of amide bonds using an Ar−I/HF⋅pyridine/mCPBA system. Asian J. Org. Chem. 2022, 11, e202200483. [Google Scholar] [CrossRef]














































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taussat, A.; de Figueiredo, R.M.; Campagne, J.-M. Direct Catalytic Amidations from Carboxylic Acid and Ester Derivatives: A Review. Catalysts 2023, 13, 366. https://doi.org/10.3390/catal13020366
Taussat A, de Figueiredo RM, Campagne J-M. Direct Catalytic Amidations from Carboxylic Acid and Ester Derivatives: A Review. Catalysts. 2023; 13(2):366. https://doi.org/10.3390/catal13020366
Chicago/Turabian StyleTaussat, Armand, Renata Marcia de Figueiredo, and Jean-Marc Campagne. 2023. "Direct Catalytic Amidations from Carboxylic Acid and Ester Derivatives: A Review" Catalysts 13, no. 2: 366. https://doi.org/10.3390/catal13020366
APA StyleTaussat, A., de Figueiredo, R. M., & Campagne, J. -M. (2023). Direct Catalytic Amidations from Carboxylic Acid and Ester Derivatives: A Review. Catalysts, 13(2), 366. https://doi.org/10.3390/catal13020366