

Heteroleptic Copper Complexes as Catalysts for the CuAAC Reaction: Counter-Ion Influence in Catalyst Efficiency



Abstract
:
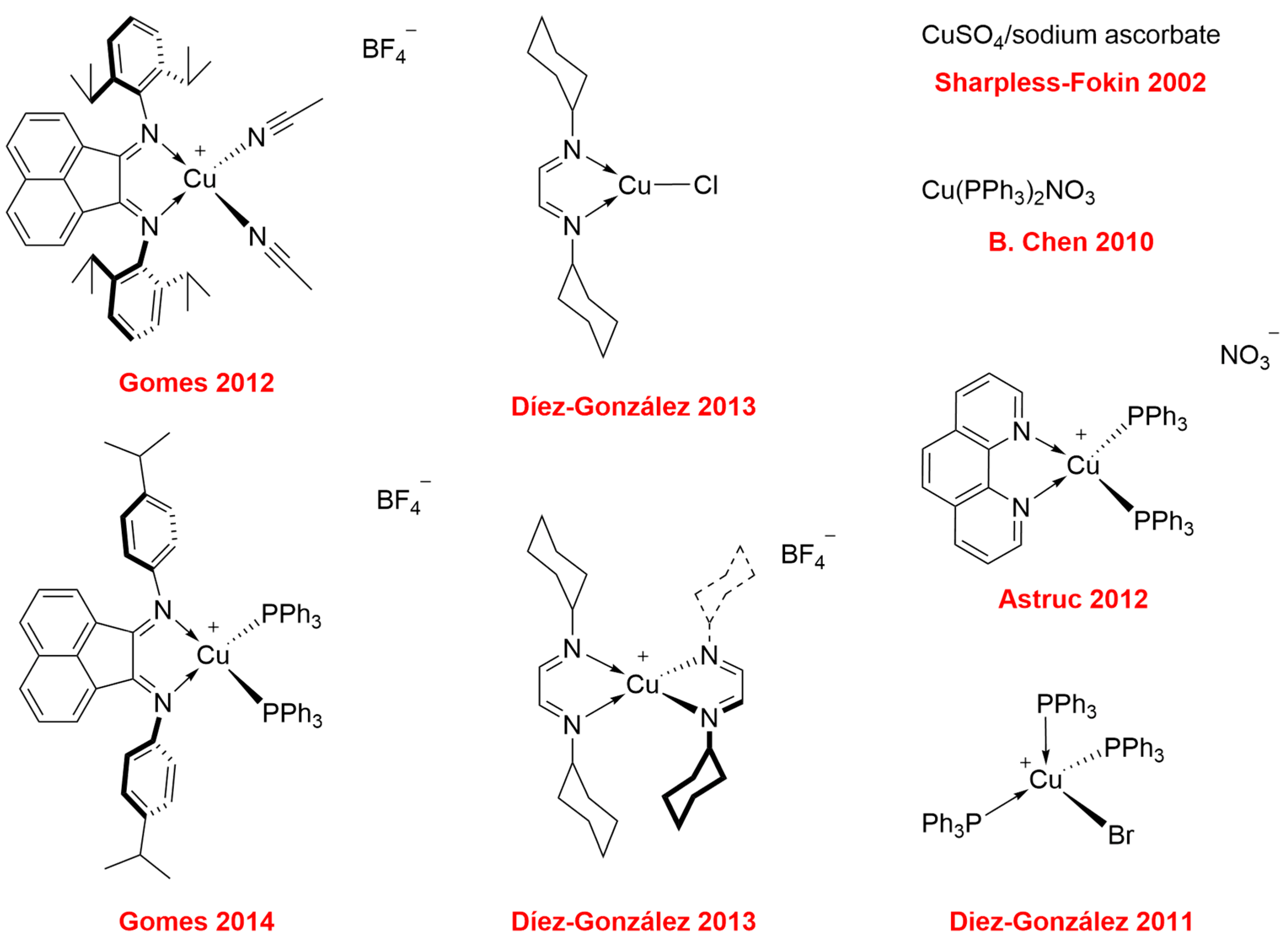
1. Introduction
2. Results
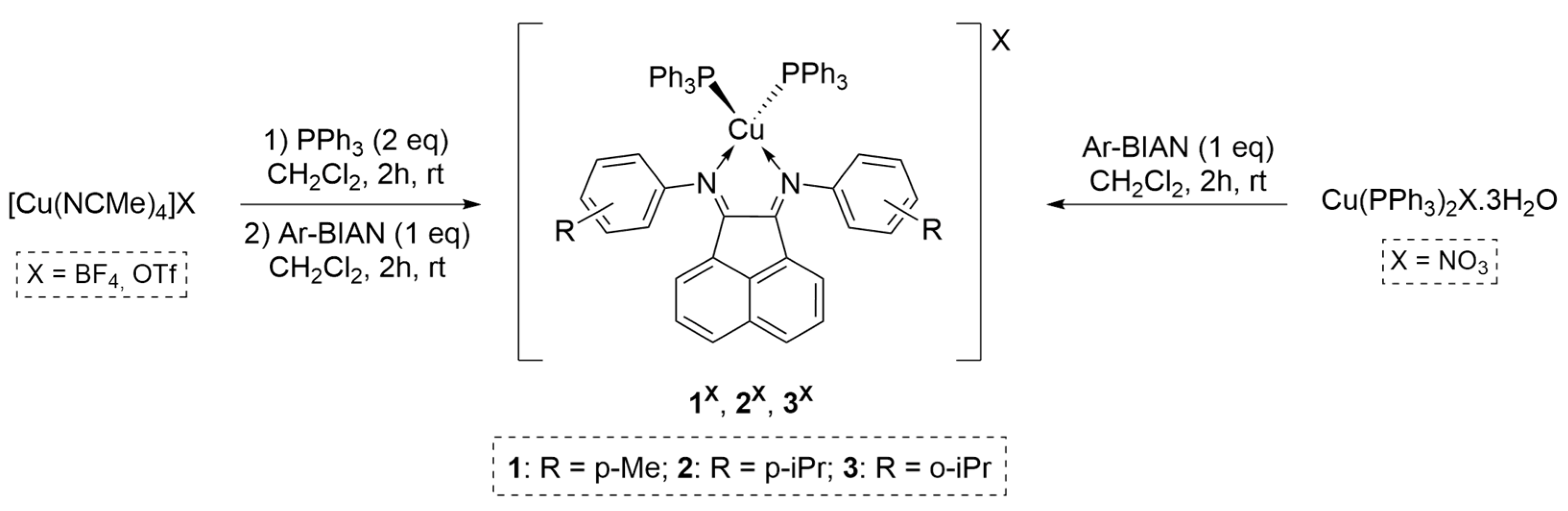
2.1. Synthesis and Characterization
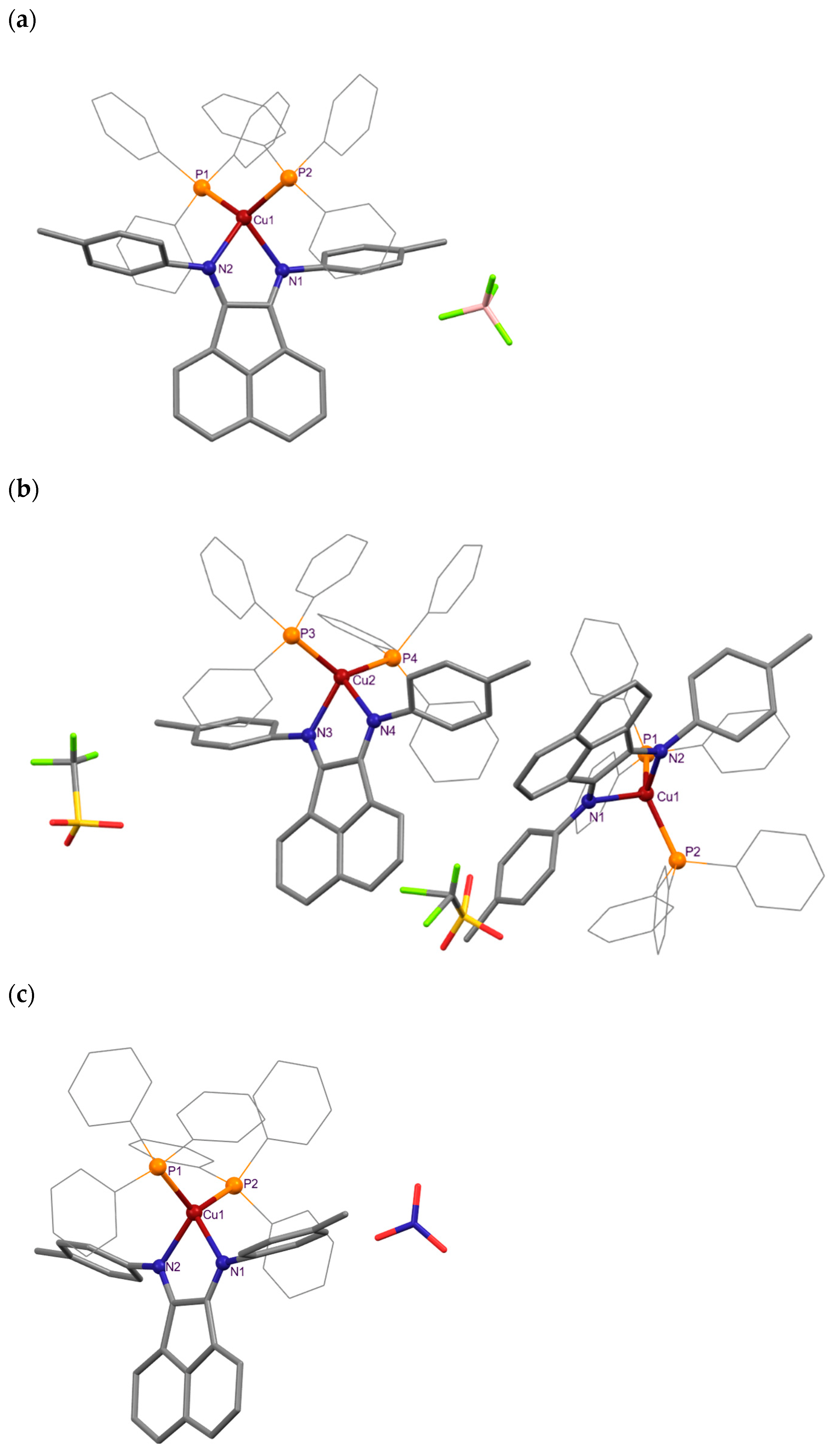
2.2. X-ray Diffraction Studies
2.3. Catalytic Studies
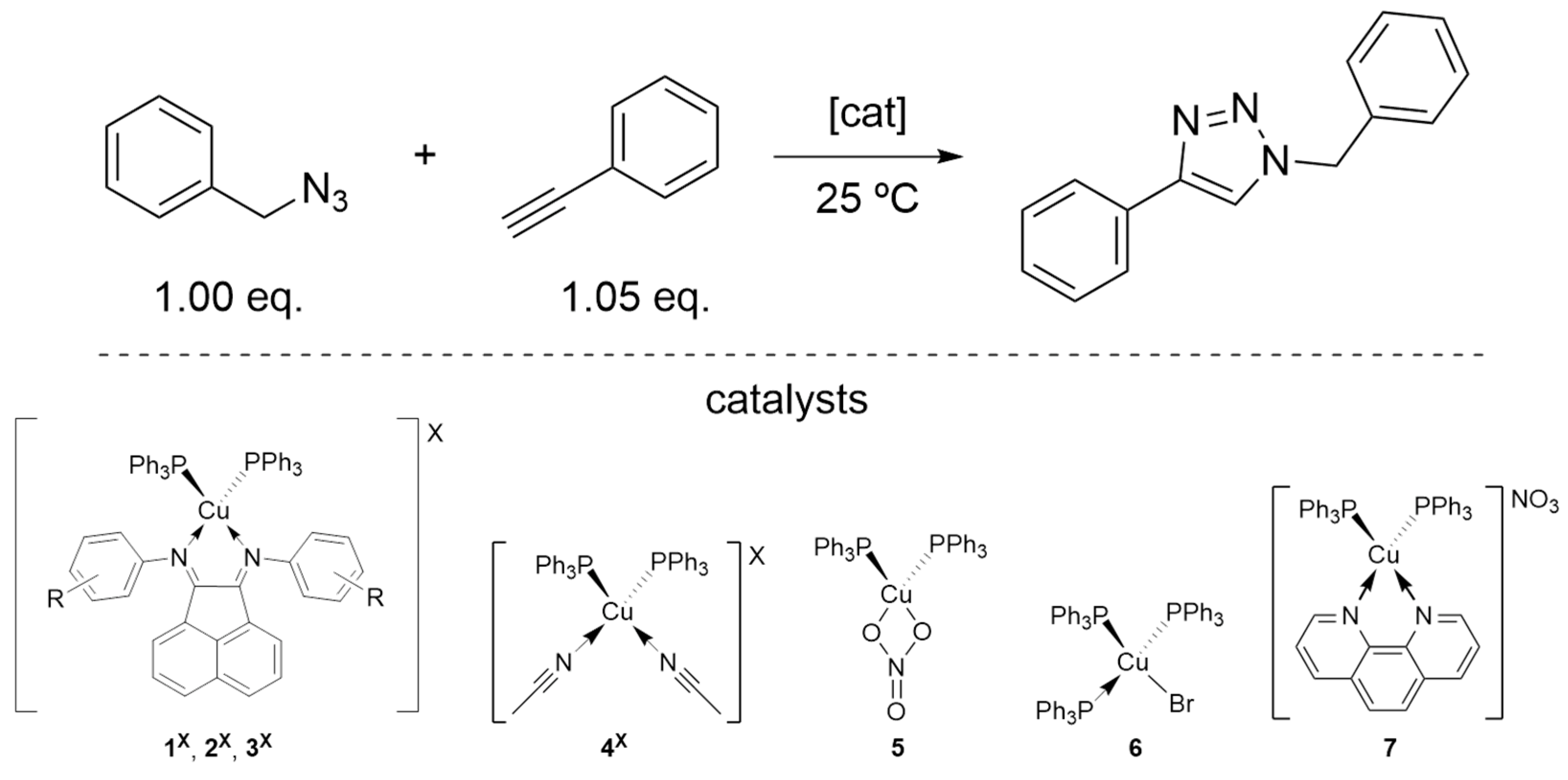
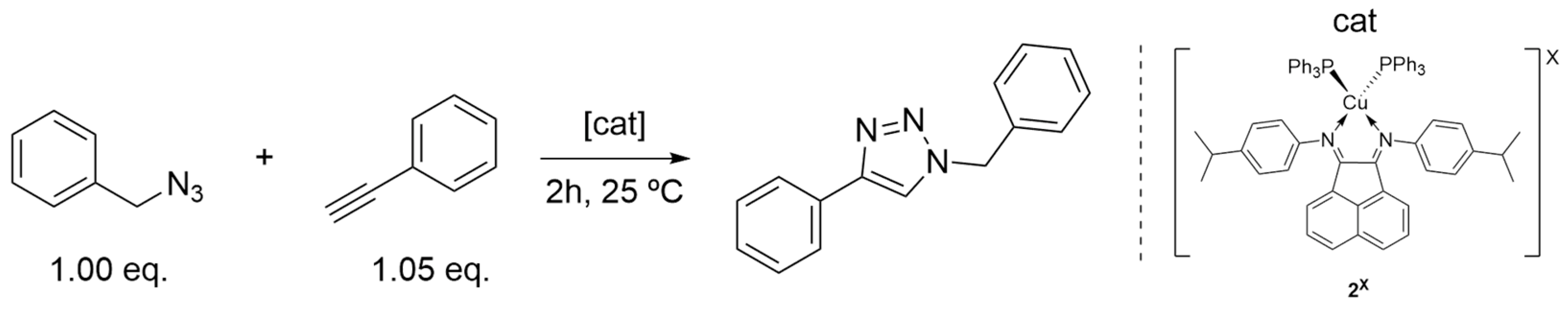
2.3.1. Catalysts’ Screening
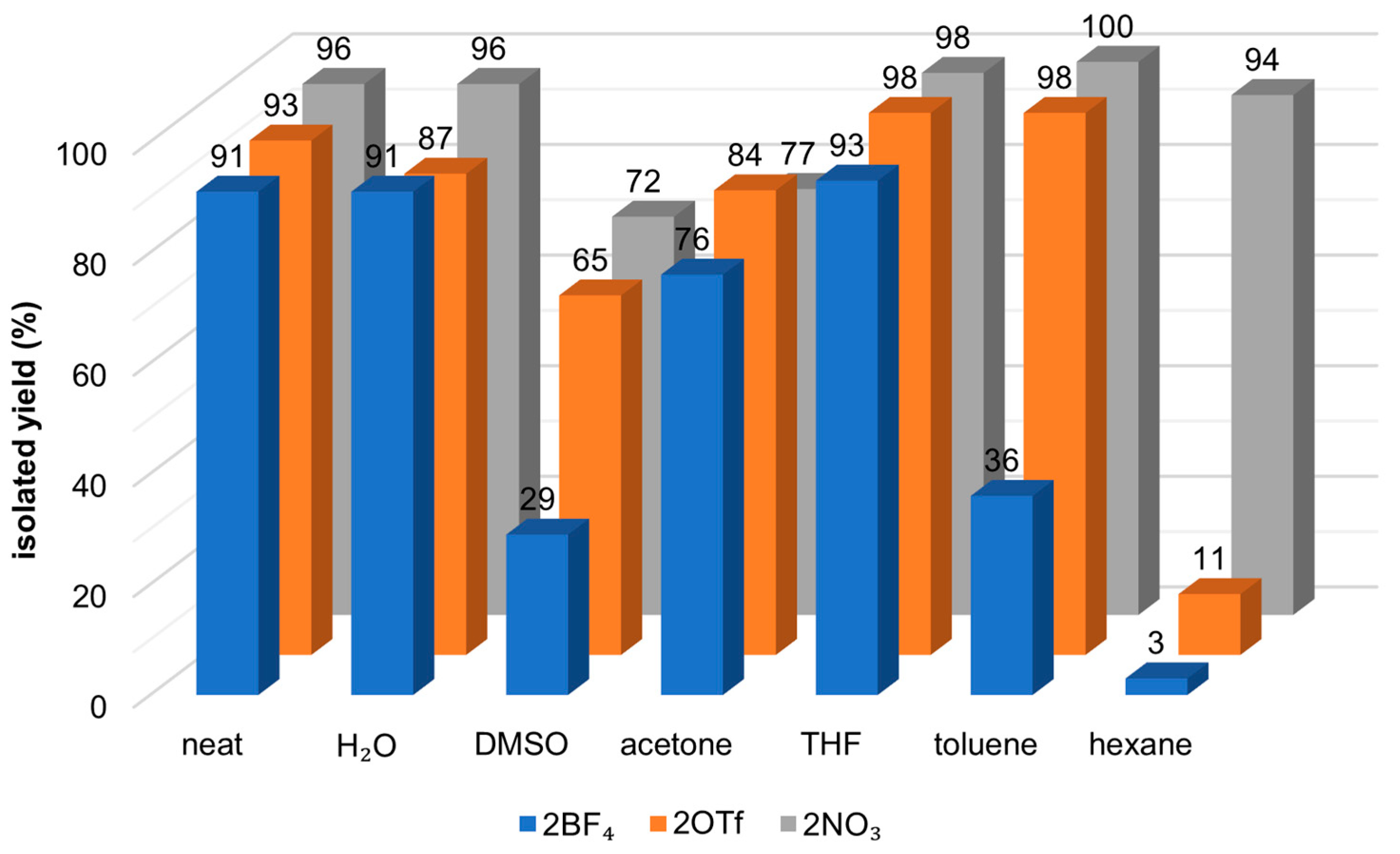
2.3.2. Solvents’ Screening
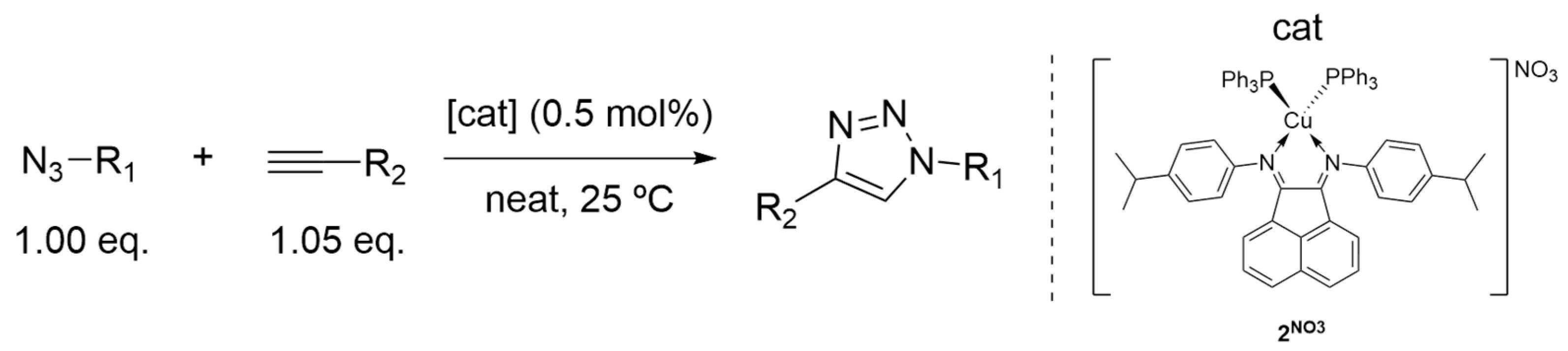
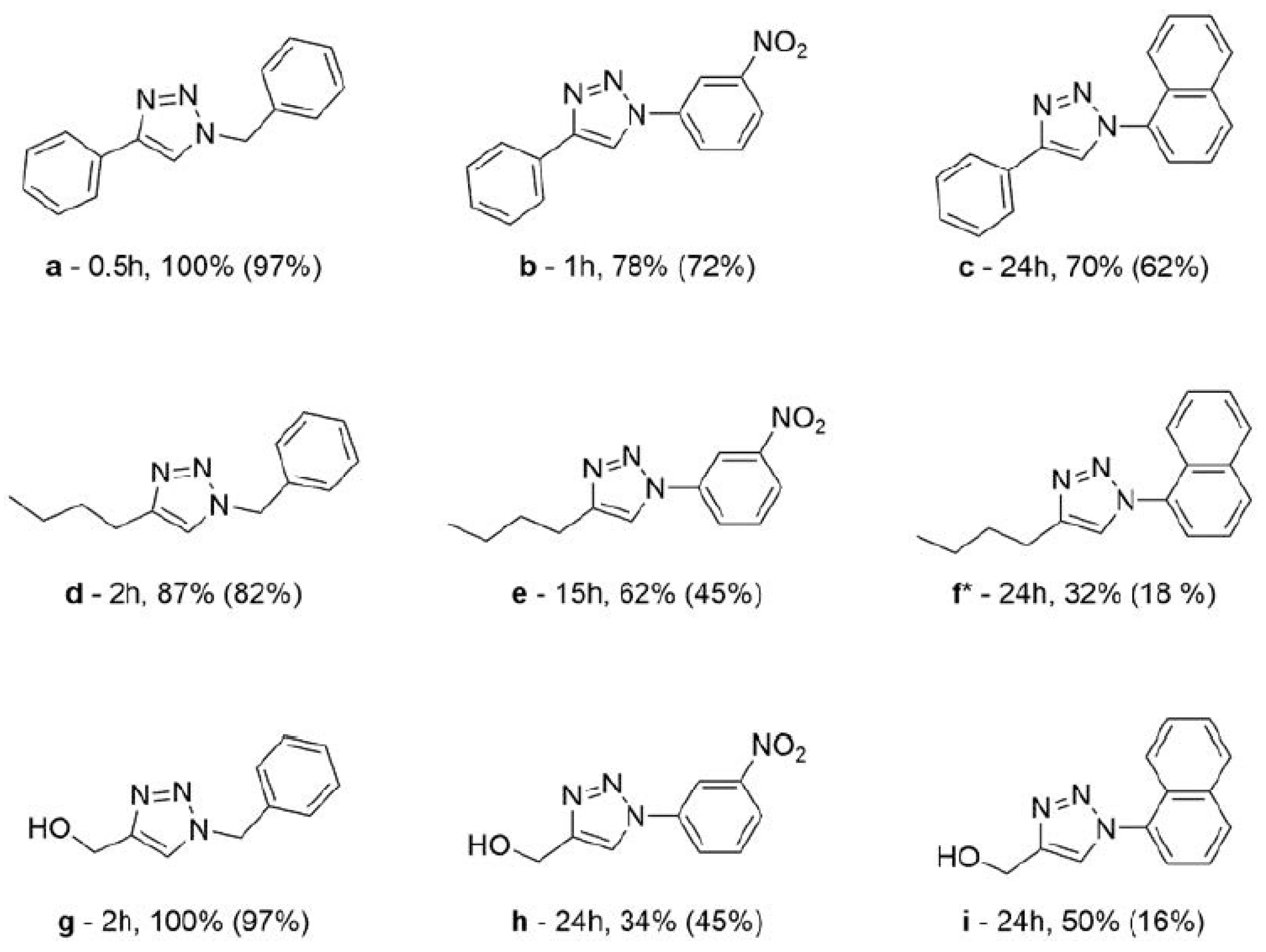
2.3.3. Catalysis Scope
3. Materials and Methods
3.1. General Considerations
3.2. Synthetic Procedures
3.2.1. Syntheses of Complexes 1BF4, 1OTf, 2OTf and 3OTf
3.2.2. Syntheses of Complexes 1NO3, 2NO3 and 3NO3
3.2.3. Synthesis of Azides
3.3. X-ray Diffraction
3.4. General Procedures for the CuAAC Reaction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Meldal, M.; Diness, F. Recent Fascinating Aspects of the CuAAC Click Reaction. Trends Chem. 2020, 2, 569–584. [Google Scholar] [CrossRef]
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase: [1,2,3]-Triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise huisgen cycloaddition process: Copper(I)-catalyzed regioselective ‘ligation’ of azides and terminal alkynes. Angew. Chemie Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- The Royal Swedish Academy of Sciences. The Nobel Prize in Chemistry 2022; The Royal Swedish Academy of Sciences: Stockholm, Sweden, 2022. [Google Scholar]
- Meldal, M.; Tomøe, C.W. Cu-catalyzed azide-Alkyne cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [Google Scholar] [CrossRef]
- Wang, C.; Ikhlef, D.; Kahlal, S.; Saillard, J.Y.; Astruc, D. Metal-catalyzed azide-alkyne ‘click’ reactions: Mechanistic overview and recent trends. Coord. Chem. Rev. 2016, 316, 1–20. [Google Scholar] [CrossRef]
- Rodionov, V.O.; Presolski, S.I.; Díaz, D.D.; Fokin, V.V.; Finn, M.G. Ligand-accelerated Cu-catalyzed azide-alkyne cycloaddition: A mechanistic report. J. Am. Chem. Soc. 2007, 129, 12705–12712. [Google Scholar] [CrossRef]
- Liang, L.; Astruc, D. The copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC) ‘click’ reaction and its applications. An overview. Coord. Chem. Rev. 2011, 255, 2933–2945. [Google Scholar] [CrossRef]
- Hein, J.E.; Fokin, V.V. Copper-catalyzed azide-alkyne cycloaddition (CuAAC) and beyond: New reactivity of copper(i) acetylides. Chem. Soc. Rev. 2010, 39, 1302–1315. [Google Scholar] [CrossRef]
- Fokin, V.V.; Matyjaszewski, K. Organic Chemistry-Breakthroughs, Perspectives; Wiley: Hoboken, NJ, USA, 2012; pp. 247–277. [Google Scholar]
- Moorman, R.M.; Collier, M.B.; Frohock, B.H.; Womble, M.D.; Chalker, J.M. Halide inhibition of the copper-catalysed azide-alkyne cycloaddition. Org. Biomol. Chem. 2014, 13, 1974–1978. [Google Scholar] [CrossRef]
- Chan, T.R.; Hilgraf, R.; Sharpless, K.B.; Fokin, V.V. Polytriazoles as copper(I)-stabilizing ligands in catalysis. Org. Lett. 2004, 6, 2853–2855. [Google Scholar] [CrossRef]
- Díez-González, S.; Nolan, S.P. [(NHC)2Cu]X complexes as efficient catalysts for azide-alkyne click chemistry at low catalyst loadings. Angew. Chem. Int. Ed. 2008, 47, 8881–8884. [Google Scholar] [CrossRef]
- Barta, J.M.; Díez-González, S. Well-Defined Diimine Copper(I) Complexes as Catalysts in Click Azide-Alkyne Cycloaddition Reactions. Molecules 2013, 18, 8919–8928. [Google Scholar] [CrossRef]
- Lal, S.; Díez-González, S. [CuBr(PPh3)3] for azide-alkyne cycloaddition reactions under strict click conditions. J. Org. Chem. 2011, 76, 2367–2373. [Google Scholar] [CrossRef]
- Lazreg, F.; Slawin, A.M.Z.; Cazin, C.S.J. Heteroleptic bis(N-heterocyclic carbene)copper(I) complexes: Highly efficient systems for the [3+2] cycloaddition of azides and alkynes. Organometallics 2012, 31, 7969–7975. [Google Scholar] [CrossRef]
- Sau, S.C.; Roy, S.R.; Sen, T.K.; Mullangi, D.; Mandal, S.K. An abnormal N-heterocyclic carbene-copper(I) complex in click chemistry. Adv. Synth. Catal. 2013, 355, 2982–2991. [Google Scholar] [CrossRef]
- Wang, D.; Li, N.; Zhao, M.; Shi, W.; Ma, C.; Chen, B. Solvent-free synthesis of 1,4-disubstituted 1,2,3-triazoles using a low amount of Cu(PPh3)2NO3 complex. Green Chem. 2010, 12, 2120–2123. [Google Scholar] [CrossRef]
- Li, L.; Lopes, P.S.; Rosa, V.; Figueira, C.A.; Lemos, M.A.N.D.A.; Duarte, M.T.; Avilés, T.; Gomes, P.T. Synthesis and structural characterisation of (aryl-BIAN)copper(i) complexes and their application as catalysts for the cycloaddition of azides and alkynes. Dalton Trans. 2012, 41, 5144–5154. [Google Scholar] [CrossRef]
- Fliedel, C.; Rosa, V.; Santos, C.I.M.; Gonzalez, P.J.; Almeida, R.M.; Gomes, C.S.B.; Gomes, P.T.; Lemos, M.A.N.D.A.; Aullón, G.; Welter, R.; et al. Copper(ii) complexes of bis(aryl-imino)acenaphthene ligands: Synthesis, structure, DFT studies and evaluation in reverse ATRP of styrene. Dalton Trans. 2014, 43, 13041–13054. [Google Scholar] [CrossRef]
- Li, L.; Lopes, P.S.; Figueira, C.A.; Gomes, C.S.B.; Duarte, M.T.; Rosa, V.; Fliedel, C.; Avilés, T.; Gomes, P.T. Cationic and Neutral (Ar-BIAN)Copper(I) Complexes Containing Phosphane and Arsane Ancillary Ligands: Synthesis, Molecular Structure and Catalytic Behaviour in Cycloaddition Reactions of Azides and Alkynes. Eur. J. Inorg. Chem. 2013, 2013, 1404–1417. [Google Scholar] [CrossRef]
- van Asselt, R.; Elsevier, C.J.; Smeets, W.J.J.; Spek, A.L.; Benedix, R. Synthesis and characterization of rigid bidentate nitrogen ligands and some examples of coordination to divalent palladium. X-ray crystal structures of bis (p-tolylimino) acenaphthene and methylchloro [bis(o,o′-diisopropylphenyl-imino) acenaphthene] palla. Recl. Trav. Chim. Pays-Bas 1994, 113, 88–98. [Google Scholar] [CrossRef]
- Rosa, V.; Santos, C.I.M.; Welter, R.; Aullón, G.; Lodeiro, C.; Avilés, T. Comparison of the structure and stability of new α-diimine complexes of copper(I) and silver(I): Density functional theory versus experimental. Inorg. Chem. 2010, 49, 8699–8708. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge structural database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- McCormick, T.; Jia, W.-L.; Wang, S. Phosphorescent Cu(I) complexes of 2-(2′-pyridylbenzimidazolyl) benzene: Impact of phosphine ancillary ligands on electronic and photophysical properties of the Cu(I) complexes. Inorg. Chem. 2005, 45, 147–155. [Google Scholar] [CrossRef]
- Saravanabharathi, D.; Monika; Venugopalan, P.; Samuelson, A.G. Solid state structural and solution studies on the formation of a flexible cavity for anions by copper(I) and 1,2-bis(diphenylphosphino)ethane. Polyhedron 2002, 21, 2433–2443. [Google Scholar] [CrossRef]
- Wang, D.; Zhao, M.; Liu, X.; Chen, Y.; Li, N.; Chen, B. Quick and highly efficient copper-catalyzed cycloaddition of organic azides with terminal alkynes. Org. Biomol. Chem. 2011, 10, 229–231. [Google Scholar] [CrossRef]
- Ferretti, F.; Rota, L.; Ragaini, F. Unexpected coordination behavior of ruthenium to a polymeric α-diimine containing the poly[bis(arylimino)acenaphthene] fragment. Inorg. Chim. Acta 2021, 518, 120257. [Google Scholar] [CrossRef]
- Angelic, R.J. Inorganic Syntheses. In Reagents for Transition Metal Complex and Organometallic Syntheses; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1990; Volume 28, pp. 68–70. [Google Scholar] [CrossRef]
- Knight, D.A.; Keller, S.W. Solvent dependant ligand displacement and anion coordination in selected copper(I) coordination compounds. J. Chem. Crystallogr. 2006, 36, 531–542. [Google Scholar] [CrossRef]
- Gujadhur, R.; Venkataraman, D.; Kintigh, J.T. Formation of aryl-nitrogen bonds using a soluble copper(I) catalyst. Tetrahedron Lett. 2001, 42, 4791–4793. [Google Scholar] [CrossRef]
- Gysling, H.J.; Kubas, G.J. Coordination Complexes of Copper(I) Nitrate. Inorg. Synth. 1979, 19, 92–97. [Google Scholar] [CrossRef]
- Villarreal, W.; Colina-Vegas, L.; Visbal, G.; Corona, O.; Corrêa, R.S.; Ellena, J.; Cominetti, M.R.; Batista, A.A.; Navarro, M. Copper(I)−Phosphine Polypyridyl Complexes: Synthesis, Characterization, DNA/HSA Binding Study, and Antiproliferative Activity. Inorg. Chem. 2017, 56, 3781–3793. [Google Scholar] [CrossRef]
- Zhao, J.; Guo, J.; Huang, M.; You, Y.; Wu, Z. Design, synthesis and biological evaluation of new steroidal β -triazoly enones as potent antiproliferative agents. Steroids 2019, 150, 108431. [Google Scholar] [CrossRef]
- Campbell-Verduyn, L.; Elsinga, P.H.; Mirfeizi, L.; Dierckx, R.A.; Feringa, B.L. Copper-free ‘click’: 1,3-dipolar cycloaddition of azides and arynes. Org. Biomol. Chem. 2008, 6, 3461–3463. [Google Scholar] [CrossRef]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef]
- Burla, M.C.; Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; Cuocci, C.; Giacovazzo, C.; Mallamo, M.; Mazzone, A.; Polidori, G. Crystal structure determination and refinement via SIR2014. J. Appl. Crystallogr. 2015, 48, 306–309. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Beltrán, Á.; Gata, I.; Maya, C.; Avó, J.; Lima, J.C.; Laia, C.A.T.; Peloso, R.; Outis, M.; Nicasio, M.C. Dinuclear Cu(I) Halides with Terphenyl Phosphines: Synthesis, Photophysical Studies, and Catalytic Applications in CuAAC Reactions. Inorg. Chem. 2020, 59, 10894–10906. [Google Scholar] [CrossRef]
- Wang, S.; Jia, K.; Cheng, J.; Chen, Y.; Yuan, Y. Dual roles of substituted thiourea as reductant and ligand in CuAAC reaction. Tetrahedron Lett. 2017, 58, 3717–3721. [Google Scholar] [CrossRef]
- Jardim, G.A.M.; Cruz, E.H.G.; Rodrigues, B.L.; Ramos, D.F.; Oliveira, R.N.; Silva, P.E.A. On the Search for Potential Antimycobacterial Drugs: Synthesis of Naphthoquinoidal, Phenazinic and 1,2,3-Triazolic Compounds and Evaluation Against Mycobacterium tuberculosis. J. Braz. Chem. Soc. 2015, 26, 1013–1027. [Google Scholar] [CrossRef]
- Gannarapu, M.R.; Vasamsetti, S.B.; Punna, N.; Kotamraju, S.; Banda, N. Synthesis of novel 1-substituted triazole linked 1,2-benzothiazine 1,1-dioxido propenone derivatives as potent anti-inflammatory agents and inhibitors of monocyte-to-macrophage differentiation. MedChemComm 2015, 6, 1494–1500. [Google Scholar] [CrossRef]
- Dutta, S.; Gupta, S.J.; Sen, A.K. Silver trifluoromethanesulfonate and metallic copper mediated syntheses of 1,2,3-triazole-O- and triazolyl glycoconjugates: Consecutive glycosylation and cyclization under one-pot condition. Tetrahedron Lett. 2016, 57, 3086–3090. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| L3 | 1 BF4 | 1OTf | 1NO3 | 2NO3 | 3NO3 | ||
|---|---|---|---|---|---|---|---|
| Molecule 1 | Molecule 2 | ||||||
| Cu1–N1 | 2.121(3) | 2.161(4) | 2.109(4) | 2.089(3) | 2.122(3) | 2.165(5) | |
| Cu1–N2 | 2.100(3) | 2.091(4) | 2.125(4) | 2.092(3) | 2.104(3) | 2.157(5) | |
| Cu1–P1 | 2.2571(10) | 2.2580(14) | 2.2522(14) | 2.2551(11) | 2.2606(11) | 2.2800(19) | |
| Cu1–P2 | 2.2656(10) | 2.2709(14) | 2.2573(14) | 2.2608(11) | 2.2763(10) | 2.2788(19) | |
| C1–N1 | 1.277(3) | 1.272(5) | 1.282(6) | 1.271(6) | 1.273(5) | 1.280(4) | 1.274(9) |
| C11–N2 | 1.275(3) | 1.281(5) | 1.286(6) | 1.266(6) | 1.288(5) | 1.278(4) | 1.263(8) |
| N1–Cu1–N2 | 78.84(11) | 78.63(15) | 78.76(15) | 79.44(12) | 78.90(11) | 78.79(18) | |
| P1–Cu1–P2 | 122.10(4) | 118.37(5) | 130.45(6) | 120.46(4) | 124.34(4) | 123.21(7) | |
| ω a | 104.73(8) | 103.36(11) | 97.96(10) | 97.64(7) | 80.97(9) | 105.99(14) | |
| φ1 b | 111.65(7) | 70.45(11) | 58.41(16) | 60.56(16) | 77.02(12) | 61.30(13) | 72.4(2) |
| φ2 b | 65.41(6) | 108.28(9) | 104.59(17) | 62.25(17) | 71.63(14) | 109.70(12) | 72.2(2) |
| χ c | 47.57(9) | 43.21(13) | 49.3(2) | 115.5(2) | 141.63(16) | 51.86(16) | 142.3(3) |
| Entry a | Cat. | Cat. Loading (mol%) | Time (h) | Conversion (%) b | η (%) c |
|---|---|---|---|---|---|
| 1 | 1BF4 | 0.5 | 1 | 98 | 89 |
| 2 | 1Otf | 0.5 | 1 | 99 | 94 |
| 3 | 1NO3 | 0.1 | 0.5 | 98 | 92 |
| 4 | 2BF4 | 0.5 | 1 | 99 | 91 |
| 5 | 2Otf | 0.3 | 0.5 | 99 | 93 |
| 6 | 2NO3 | 0.1 | 0.5 | 97 | 96 |
| 7 | 3BF4 | 0.5 | 1 | 98 | 89 |
| 8 | 3Otf | 0.5 | 1 | 100 | 92 |
| 9 | 3NO3 | 0.1 | 0.5 | 98 | 92 |
| 10 | 4BF4 | 0.5 | 0.5 | 2 | / |
| 11 | 4Otf | 0.5 | 0.5 | 2 | / |
| 12 | 5 | 0.5 | 0.5 | 8 | / |
| 13 | 6 | 0.5 | 0.5 | 97 | 94 |
| 14 | 7 | 0.5 | 0.5 | 100 | 87 |
| 15 | / | / | 0.5 | 0 | 0 |
| L3 | 1BF4 | 1OTf | |
|---|---|---|---|
| Formula | C30H28N2 | C63H52BCl2Cu2F4N2P2 | C63H50CuF3N2O3P2S |
| M | 416.54 | 1120.25 | 1097.59 |
| λ (Å) | 0.71073 | 0.71073 | 0.71073 |
| T (K) | 110(2) | 110(2) | 296(2) |
| Crystal system | Triclinic | Orthorhombic | Triclinic |
| Space group | P-1 | Pbca | P-1 |
| a (Å) | 8.9160(7) | 17.4954(15) | 15.2564(10) |
| b (Å) | 11.0941(10) | 23.7716(18) | 16.0253(11) |
| c (Å) | 12.5663(10) | 26.188(2) | 23.0757(15) |
| α (°) | 94.783(3) | 90 | 90.355(3) |
| β (°) | 103.724(3) | 90 | 93.137(3) |
| γ (°) | 107.064(3) | 90 | 98.235(3) |
| V (Å3) | 1138.58(17) | 10,891.2(15) | 5574.6(6) |
| Z | 2 | 8 | 4 |
| ρcalc (g·cm−3) | 1.215 | 1.366 | 1.308 |
| µ (mm−1) | 0.071 | 0.614 | 0.544 |
| Crystal size | 0.40 × 0.20 × 0.20 | 0.40 × 0.20 × 0.20 | 0.30 × 0.20 × 0.20 |
| Crystal color | Yellow | Red | Red |
| Crystal description | Prism | Block | Prism |
| θmax (°) | 25.680 | 25.740 | 25.350 |
| Total data | 25,850 | 112,589 | 189,596 |
| Unique data | 4323 | 10,365 | 20,386 |
| Rint | 0.1400 | 0.1721 | 0.1505 |
| R [I > 2σ(I)] | 0.0609 | 0.0581 | 0.0788 |
| Rw | 0.1266 | 0.1464 | 0.2088 |
| Goodness of fit | 1.002 | 1.048 | 1.047 |
| ρmin ρmax | −0.371 0.230 | −1.240 0.888 | −0.698 0.954 |
| 1NO3 | 2NO3 | 3NO3 | |
|---|---|---|---|
| Formula | C63H51Cl3CuN3O3P2 | C67H59Cl3CuN3O3P2 | C69H61Cl9CuN3O3P2 |
| M | 1129.89 | 1186.00 | 1097.59 |
| λ (Å) | 0.71073 | 0.71073 | 1424.73 |
| T (K) | 296(2) | 110(2) | 296(2) |
| Crystal system | Monoclinic | Orthorhombic | Tetragonal |
| Space group | P21/c | Pbca | P41 |
| a (Å) | 16.7053(12) | 17.979(3) | 13.2253(4) |
| b (Å) | 14.7608(11) | 24.990(4) | 13.2253(4) |
| c (Å) | 22.8229(17) | 27.167(4) | 40.3250(19) |
| α (°) | 90 | 90 | 90 |
| β (°) | 95.361(2) | 90 | 90 |
| γ (°) | 90 | 90 | 90 |
| V (Å3) | 5603.1(7) | 12,206(3) | 7053.2(5) |
| Z | 4 | 8 | 4 |
| ρcalc (g·cm−3) | 1.339 | 1.291 | 1.342 |
| µ (mm−1) | 0.639 | 0.590 | 0.742 |
| Crystal size | 0.40 × 0.26 × 0.14 | 0.30 × 0.24 × 0.20 | 0.26 × 0.20 × 0.16 |
| Crystal color | Red | Red | Red |
| Crystal description | Prism | Prism | Prism |
| θmax (°) | 26.815 | 25.772 | 25.749 |
| Total data | 104,765 | 216,199 | 21,487 |
| Unique data | 11,970 | 11,645 | 10,328 |
| Rint | 0.1603 | 0.1057 | 0.0503 |
| R [I > 2σ(I)] | 0.0728 | 0.0638 | 0.0611 |
| Rw | 0.1987 | 0.1818 | 0.1616 |
| Goodness of fit | 1.093 | 1.056 | 1.047 |
| ρmin ρmax | −0.732 1.527 | −0.615 0.777 | −0.652 0.579 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viana, M.S.; Gomes, C.S.B.; Rosa, V. Heteroleptic Copper Complexes as Catalysts for the CuAAC Reaction: Counter-Ion Influence in Catalyst Efficiency. Catalysts 2023, 13, 386. https://doi.org/10.3390/catal13020386
Viana MS, Gomes CSB, Rosa V. Heteroleptic Copper Complexes as Catalysts for the CuAAC Reaction: Counter-Ion Influence in Catalyst Efficiency. Catalysts. 2023; 13(2):386. https://doi.org/10.3390/catal13020386
Chicago/Turabian StyleViana, Maria S., Clara S. B. Gomes, and Vitor Rosa. 2023. "Heteroleptic Copper Complexes as Catalysts for the CuAAC Reaction: Counter-Ion Influence in Catalyst Efficiency" Catalysts 13, no. 2: 386. https://doi.org/10.3390/catal13020386
APA StyleViana, M. S., Gomes, C. S. B., & Rosa, V. (2023). Heteroleptic Copper Complexes as Catalysts for the CuAAC Reaction: Counter-Ion Influence in Catalyst Efficiency. Catalysts, 13(2), 386. https://doi.org/10.3390/catal13020386