
Impact of Geometric and Electronic Factors on Selective Hydro-Deoxygenation of Guaiacol by Surface-Rich Metal/Silica Catalysts


Abstract
:1. Introduction
2. Results and Discussion
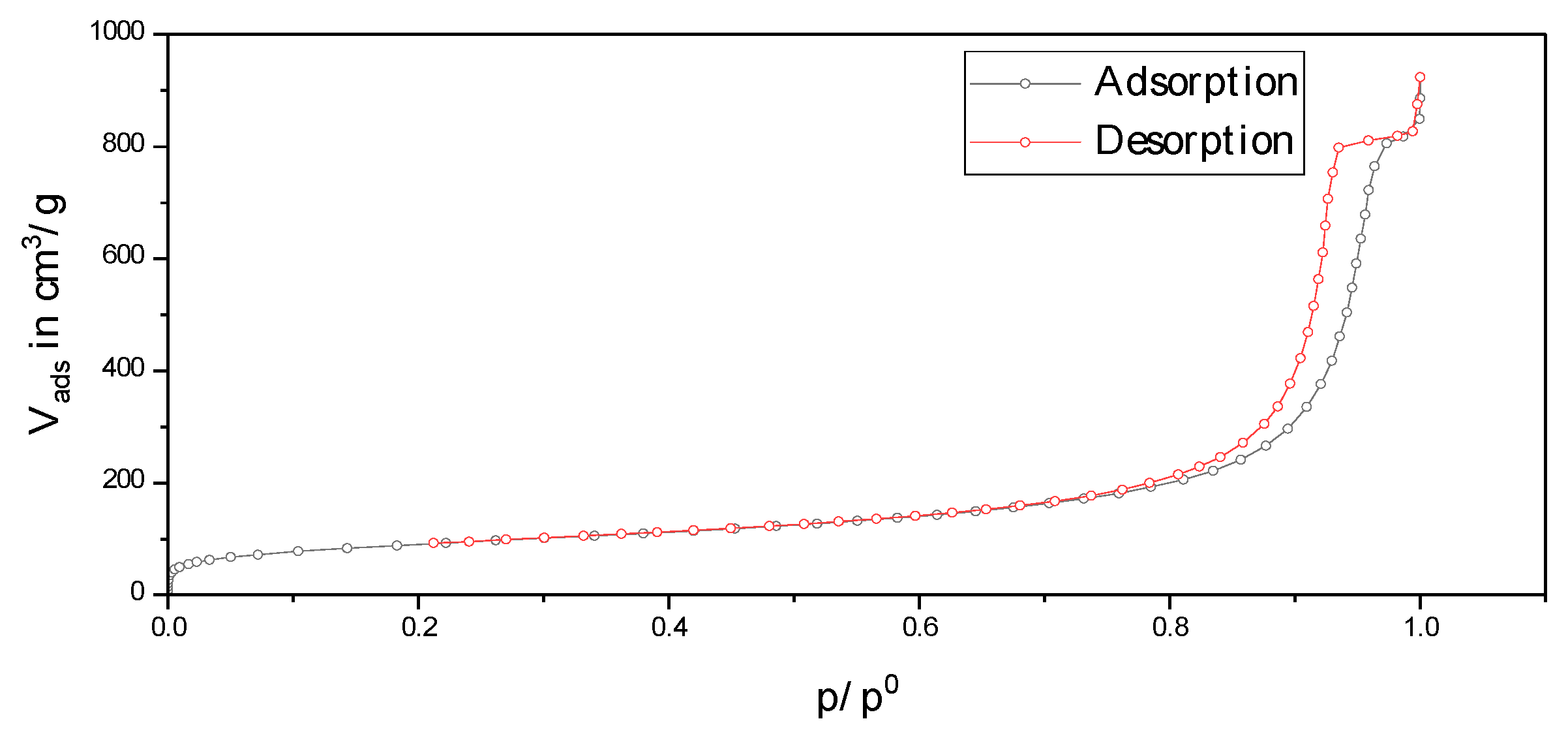
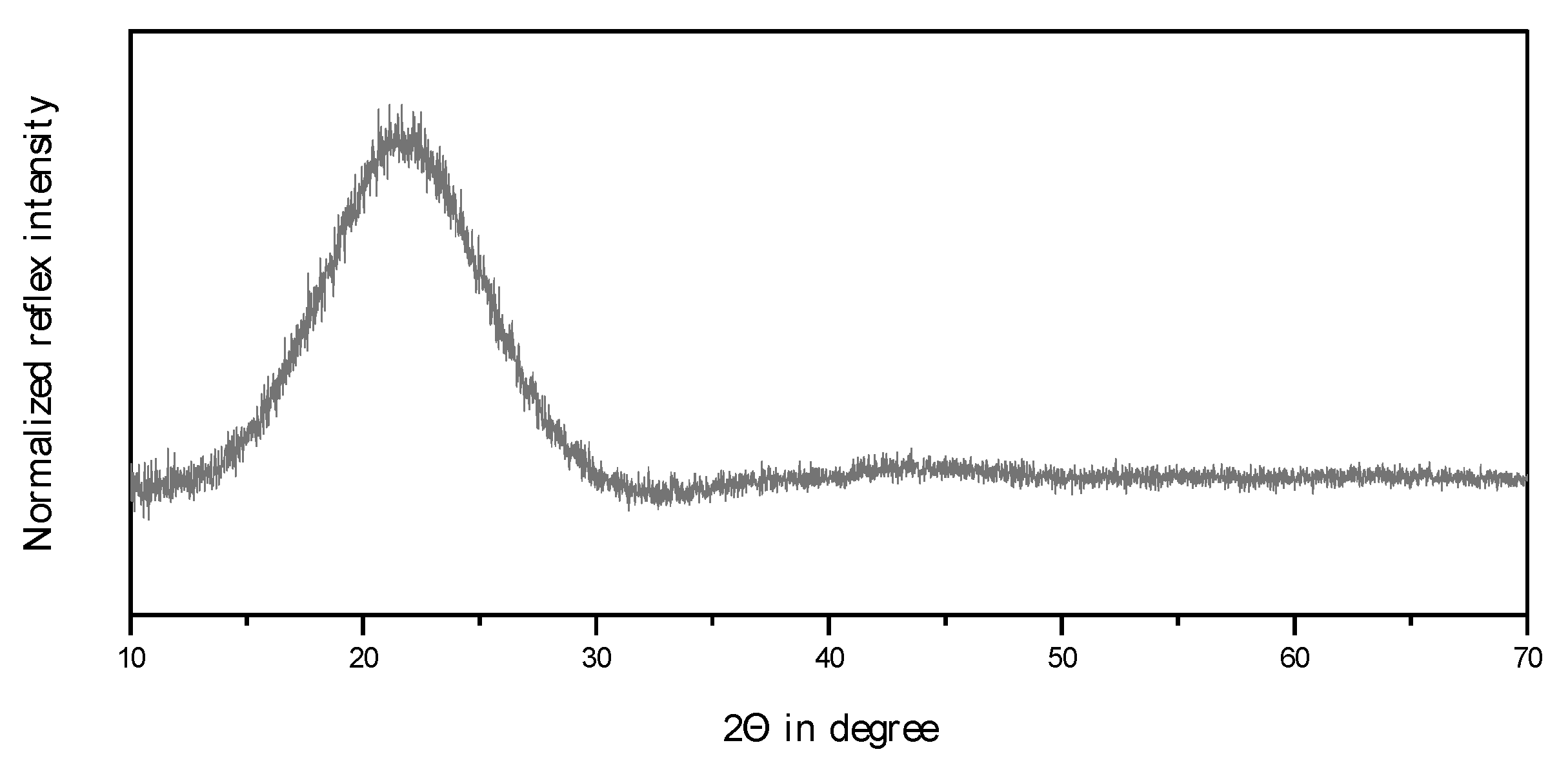
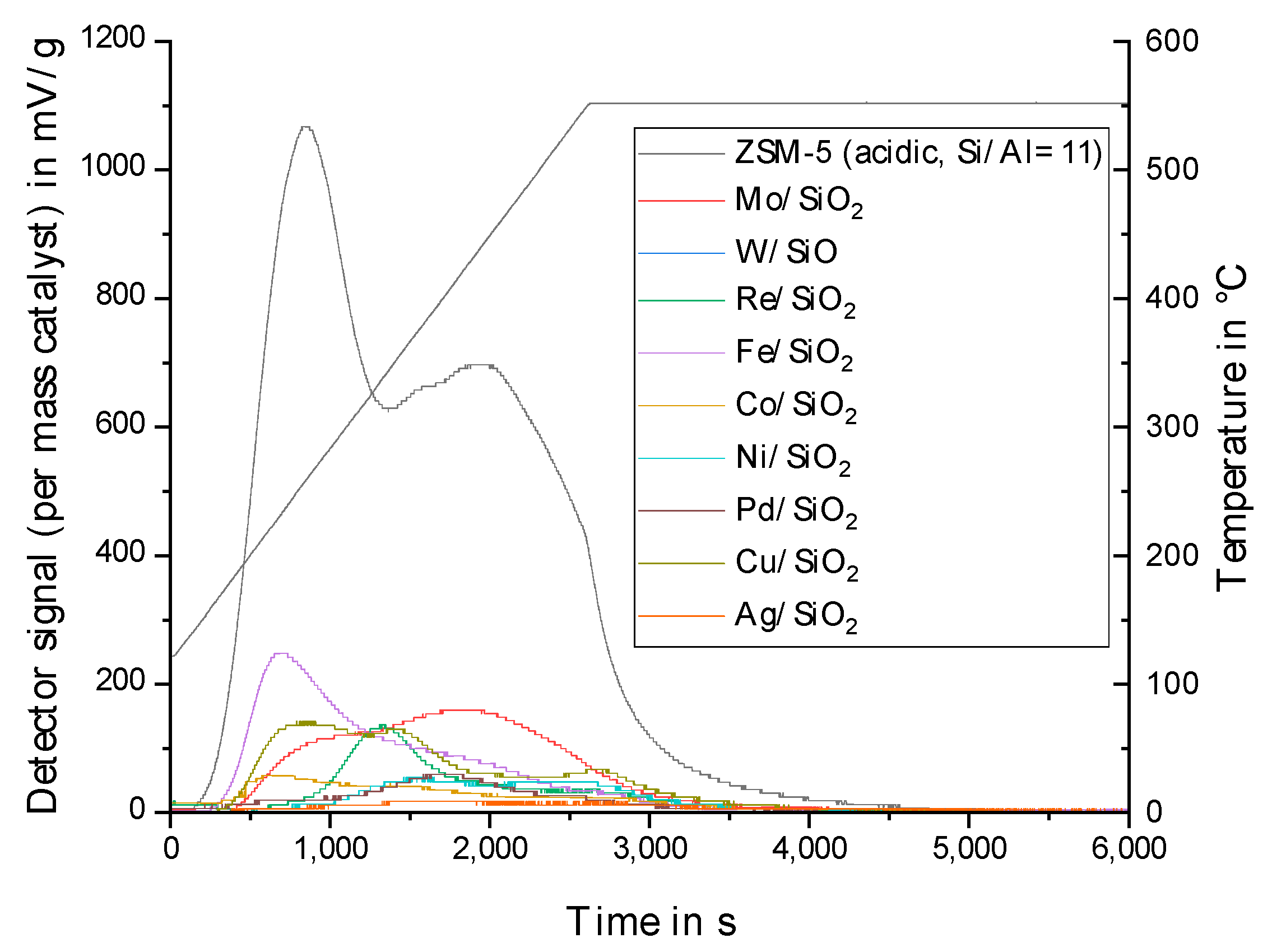
2.1. Choice of the Inert Silica Carrier Material
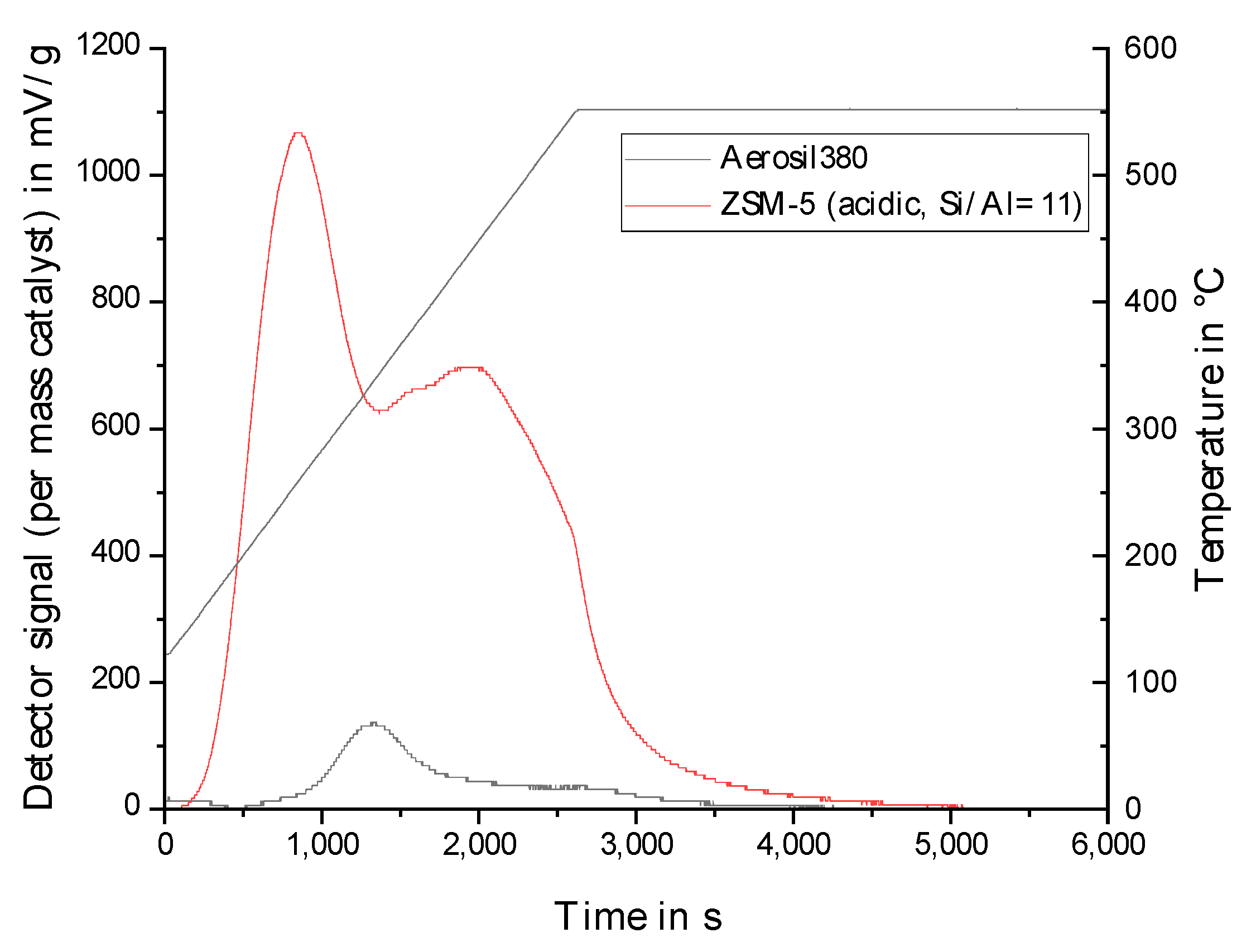
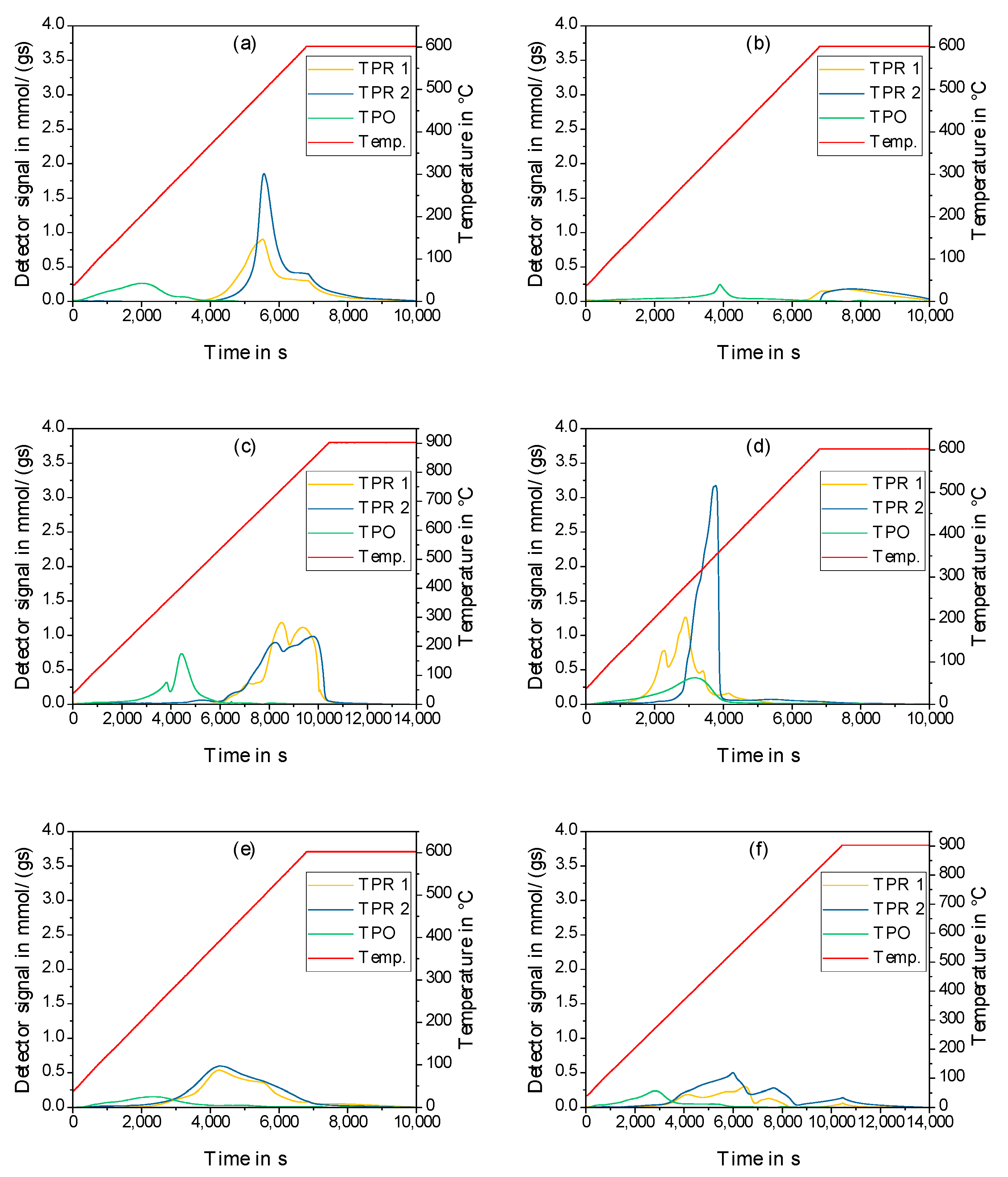
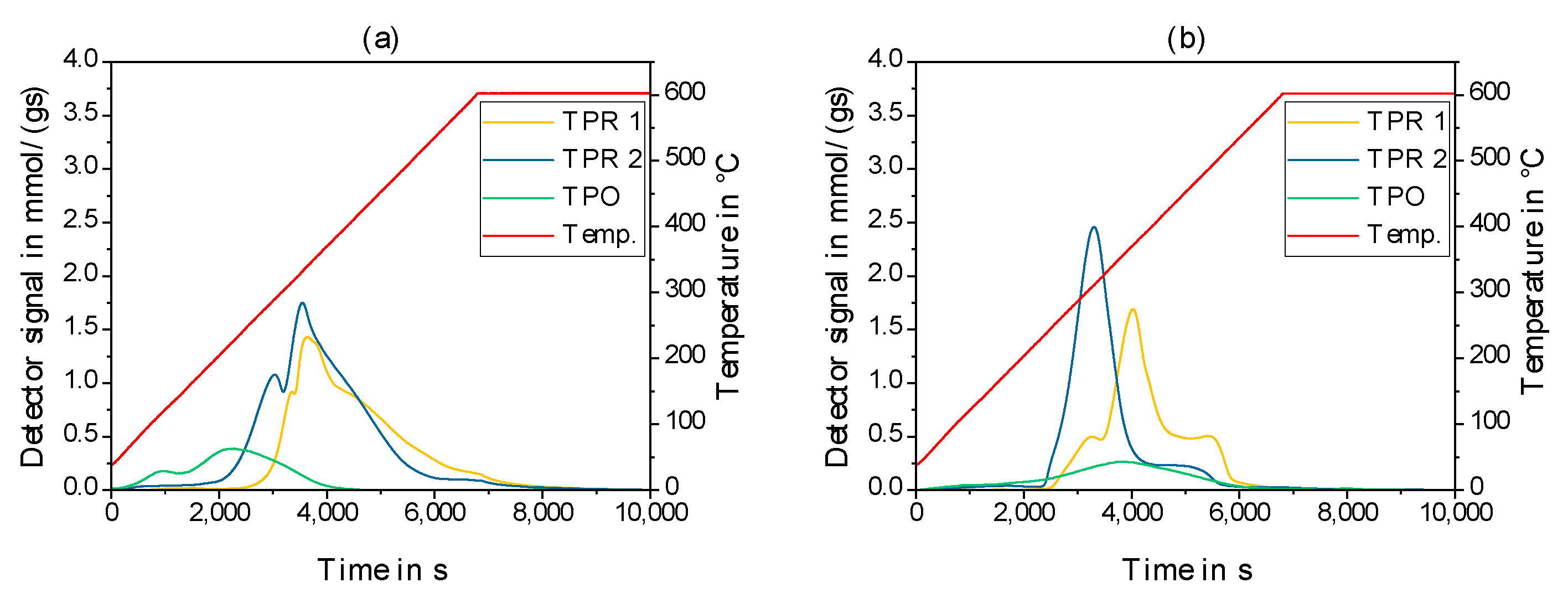
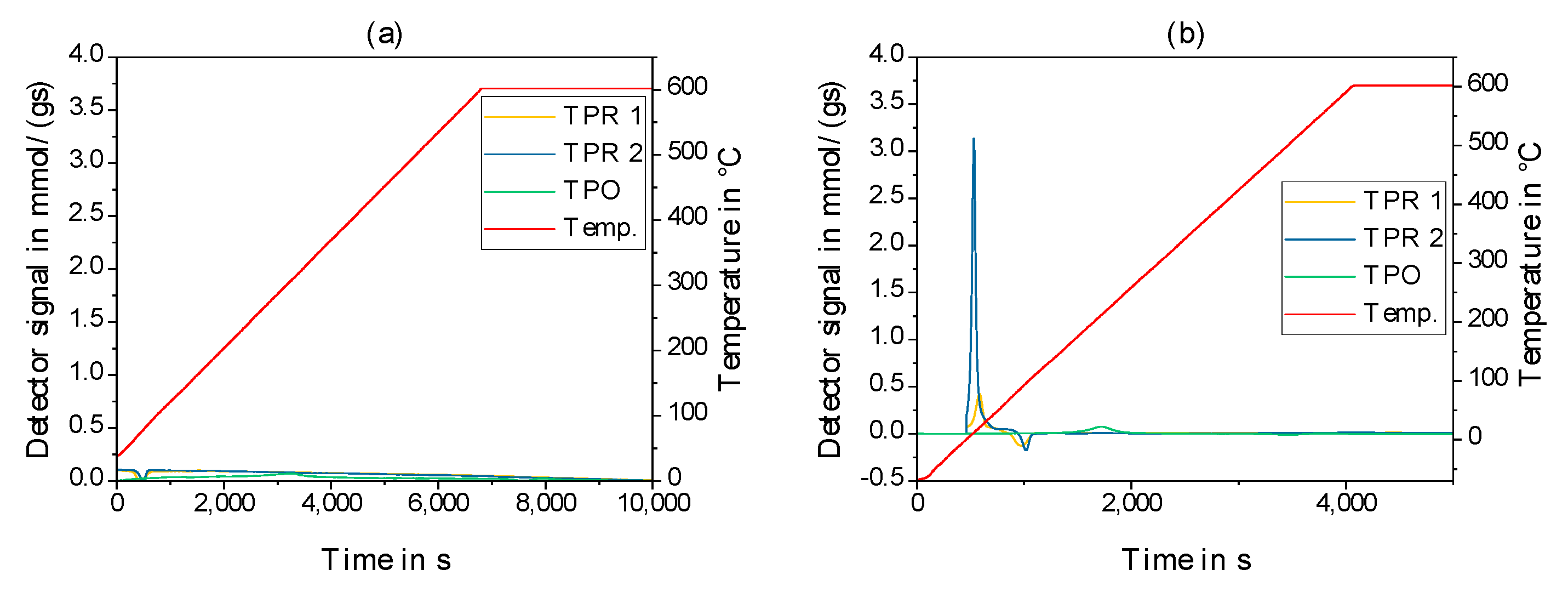
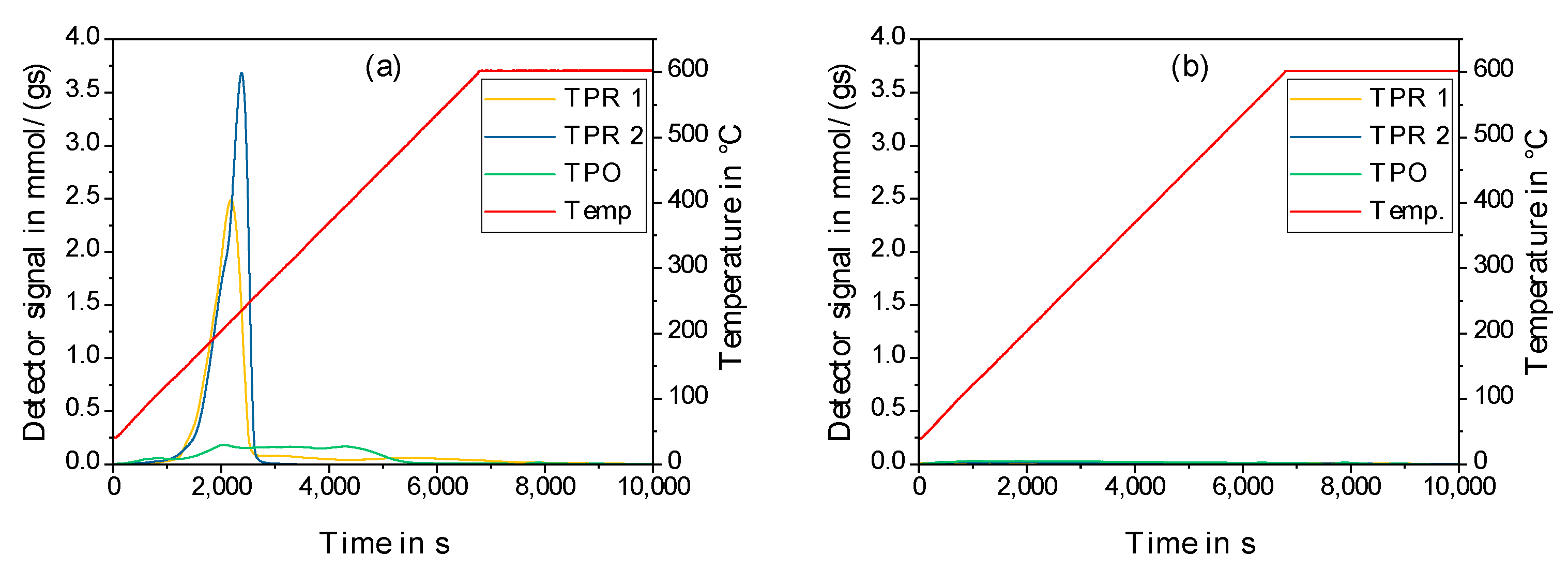
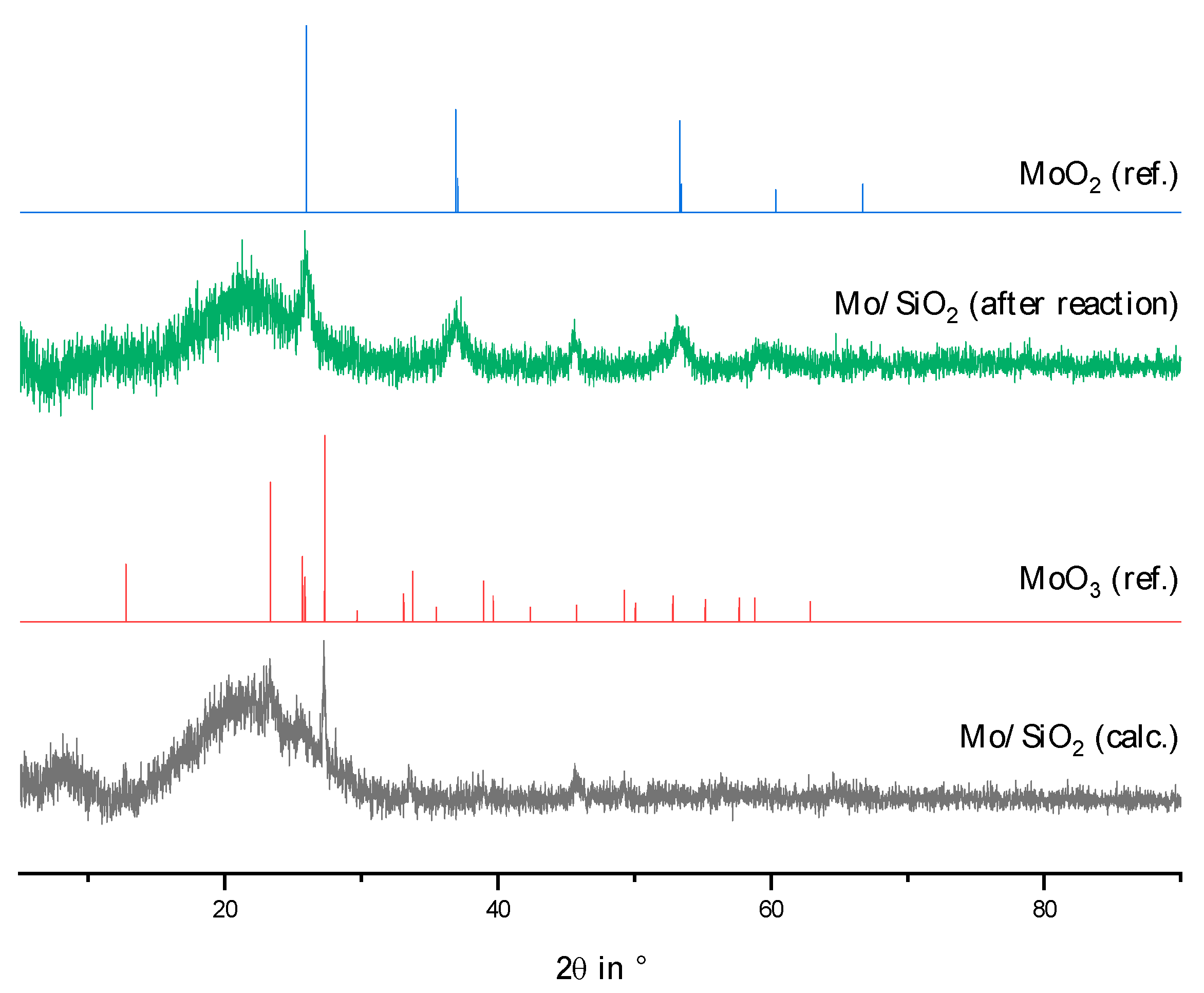
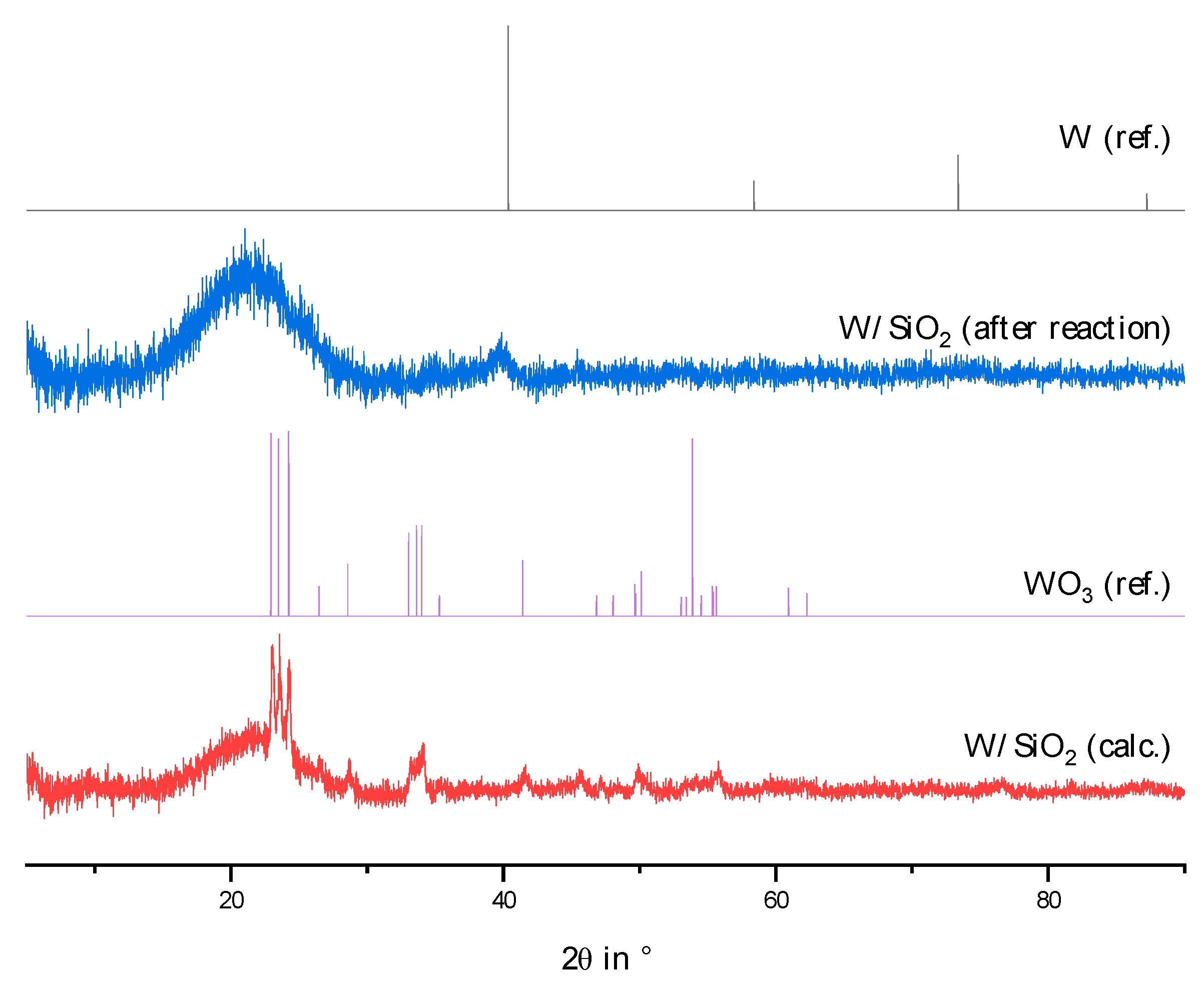
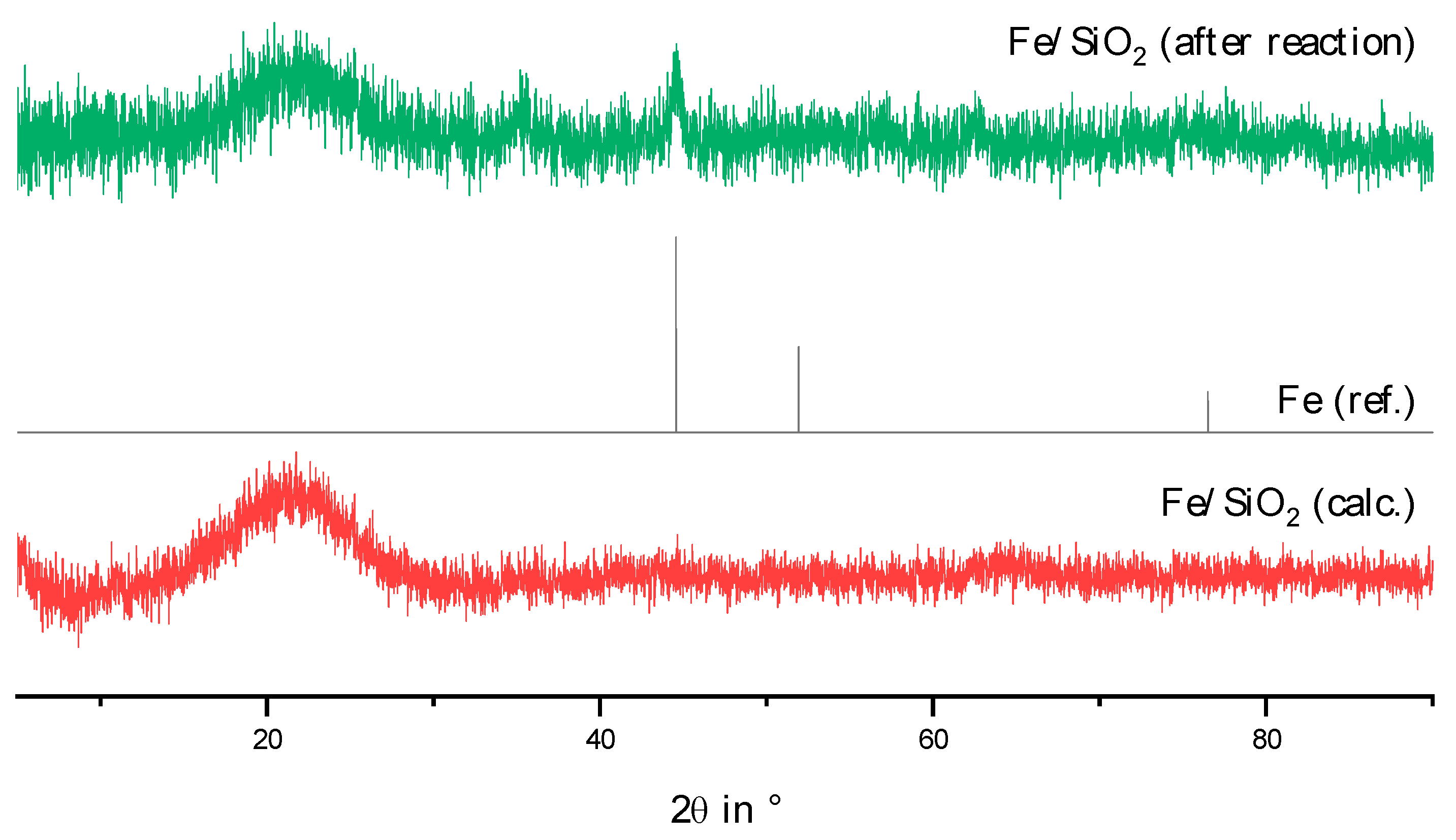
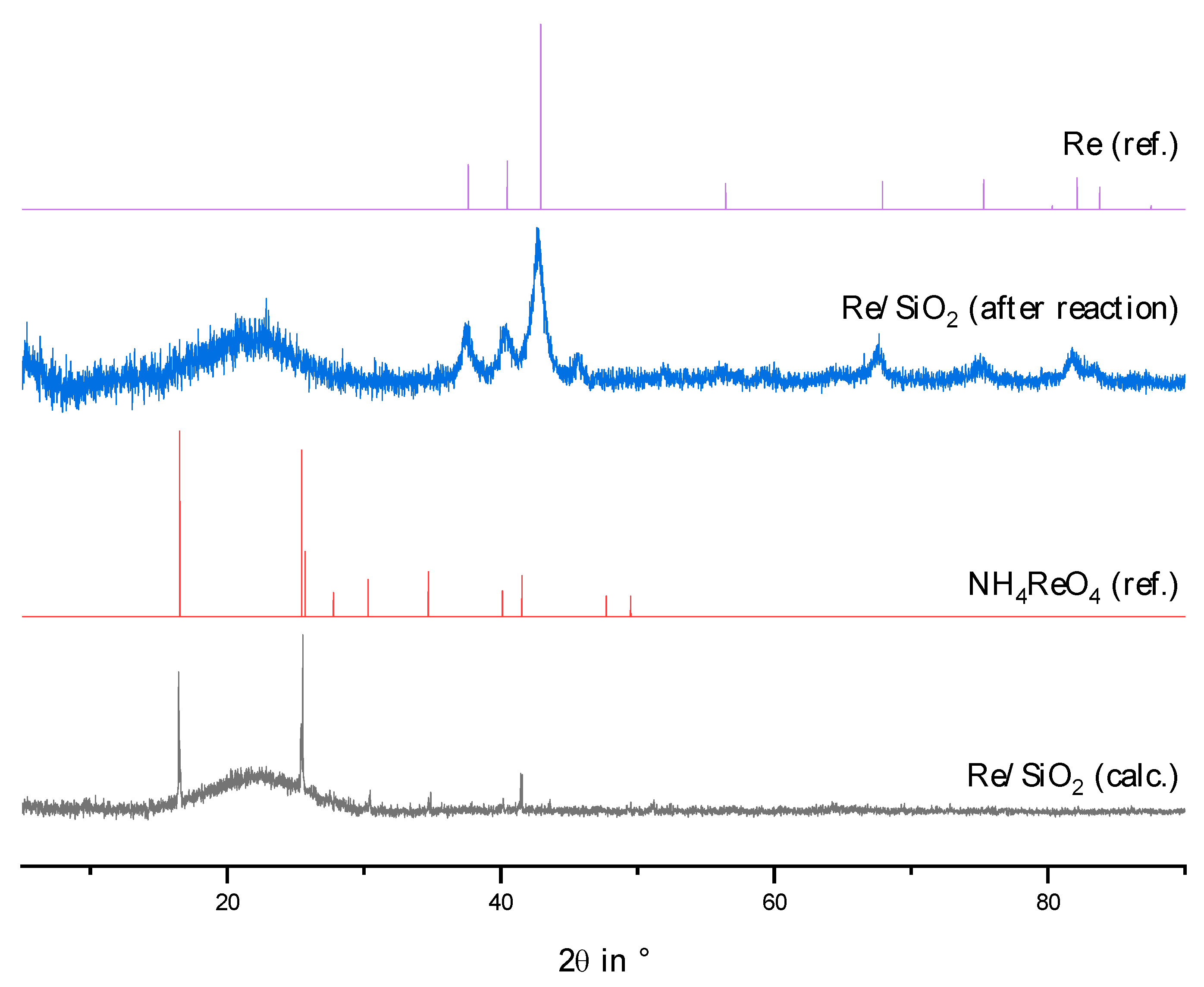
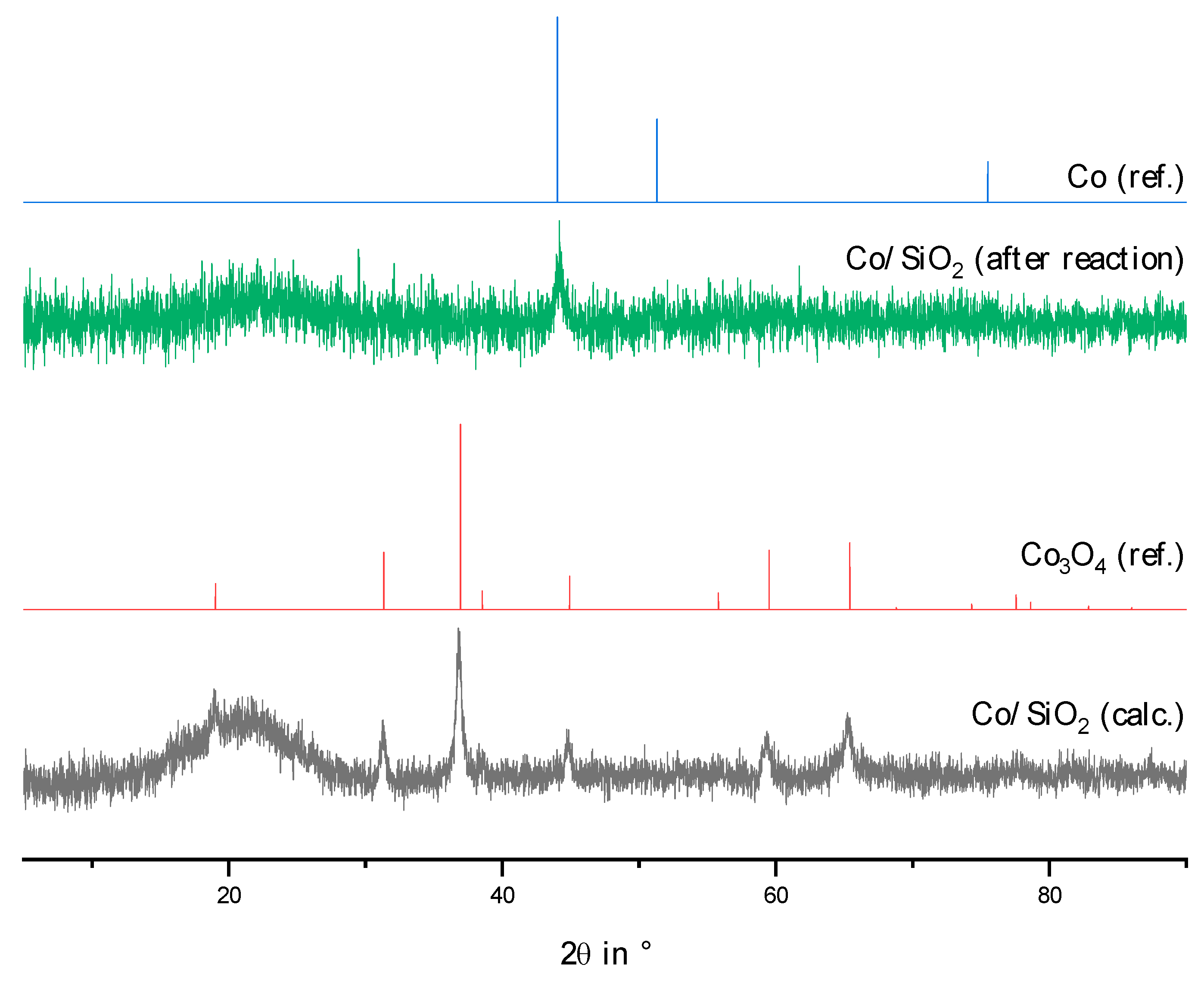
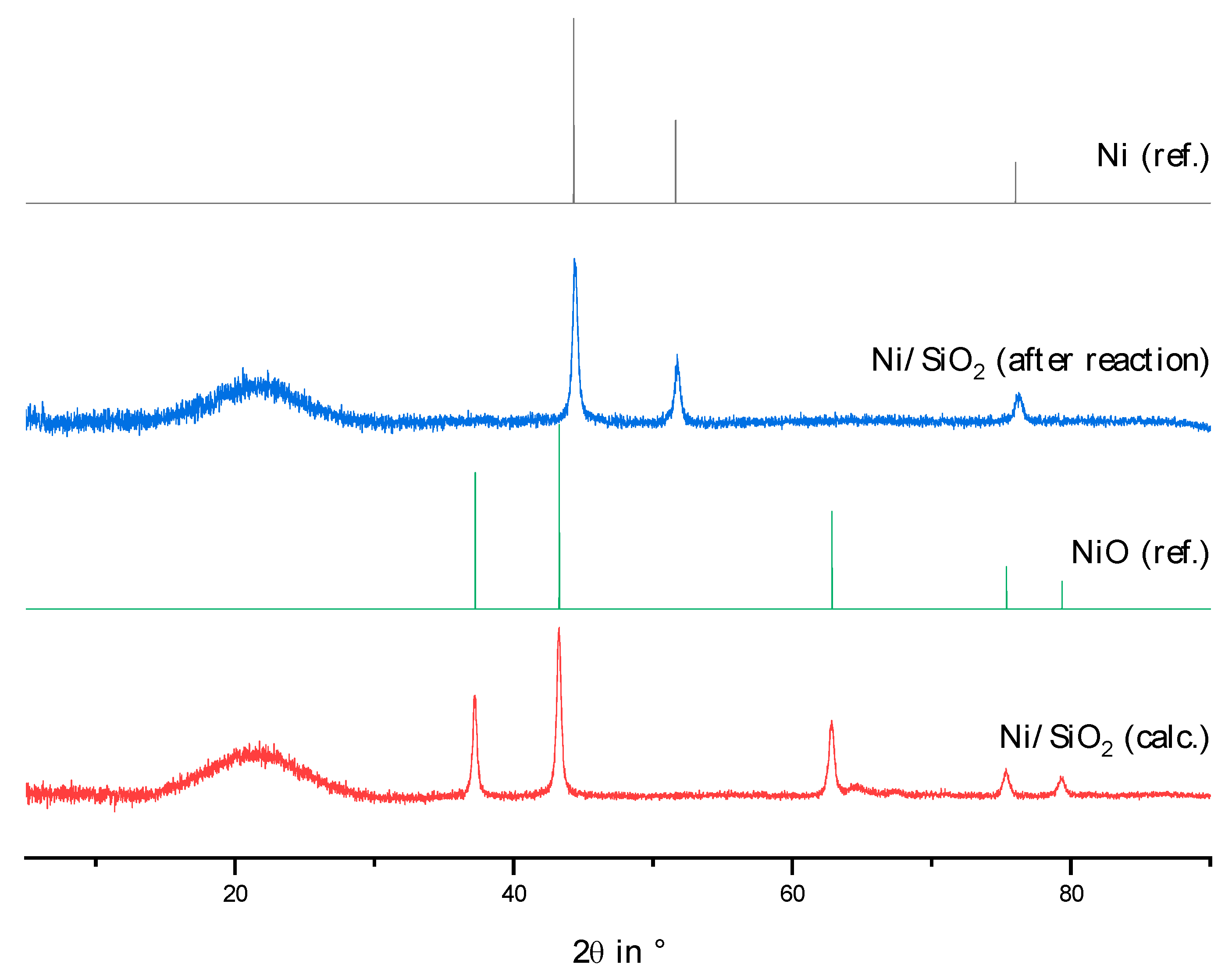
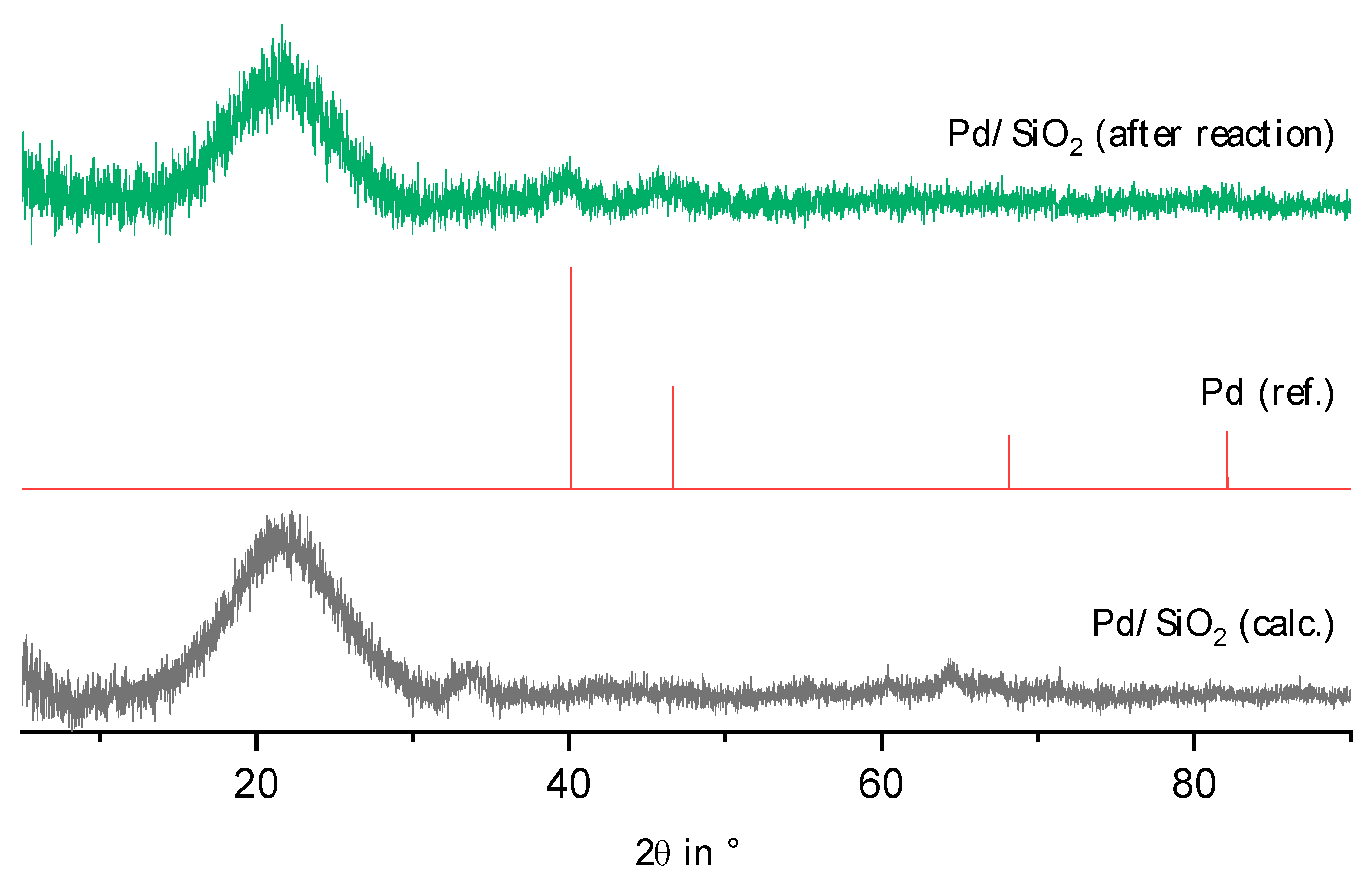
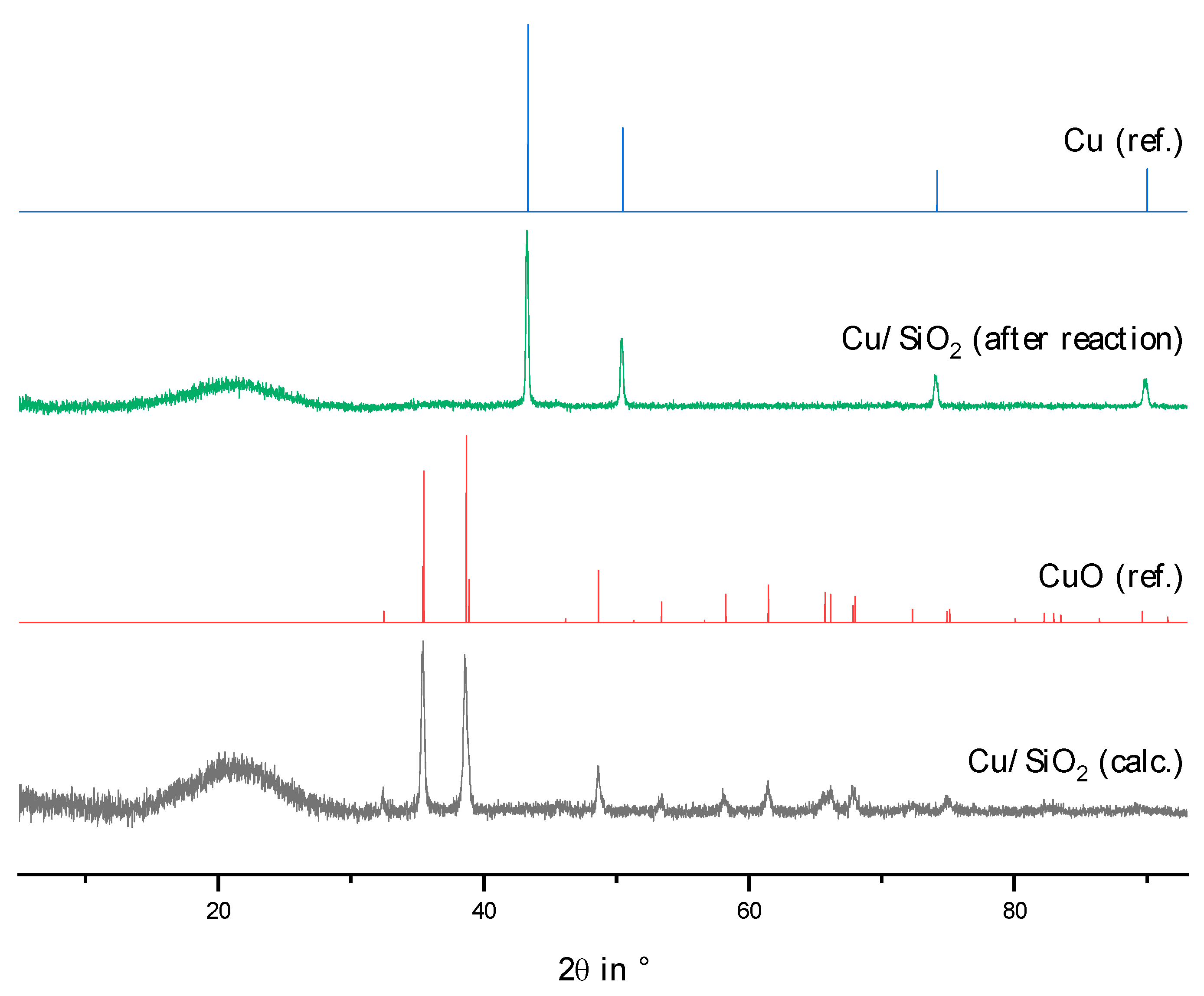
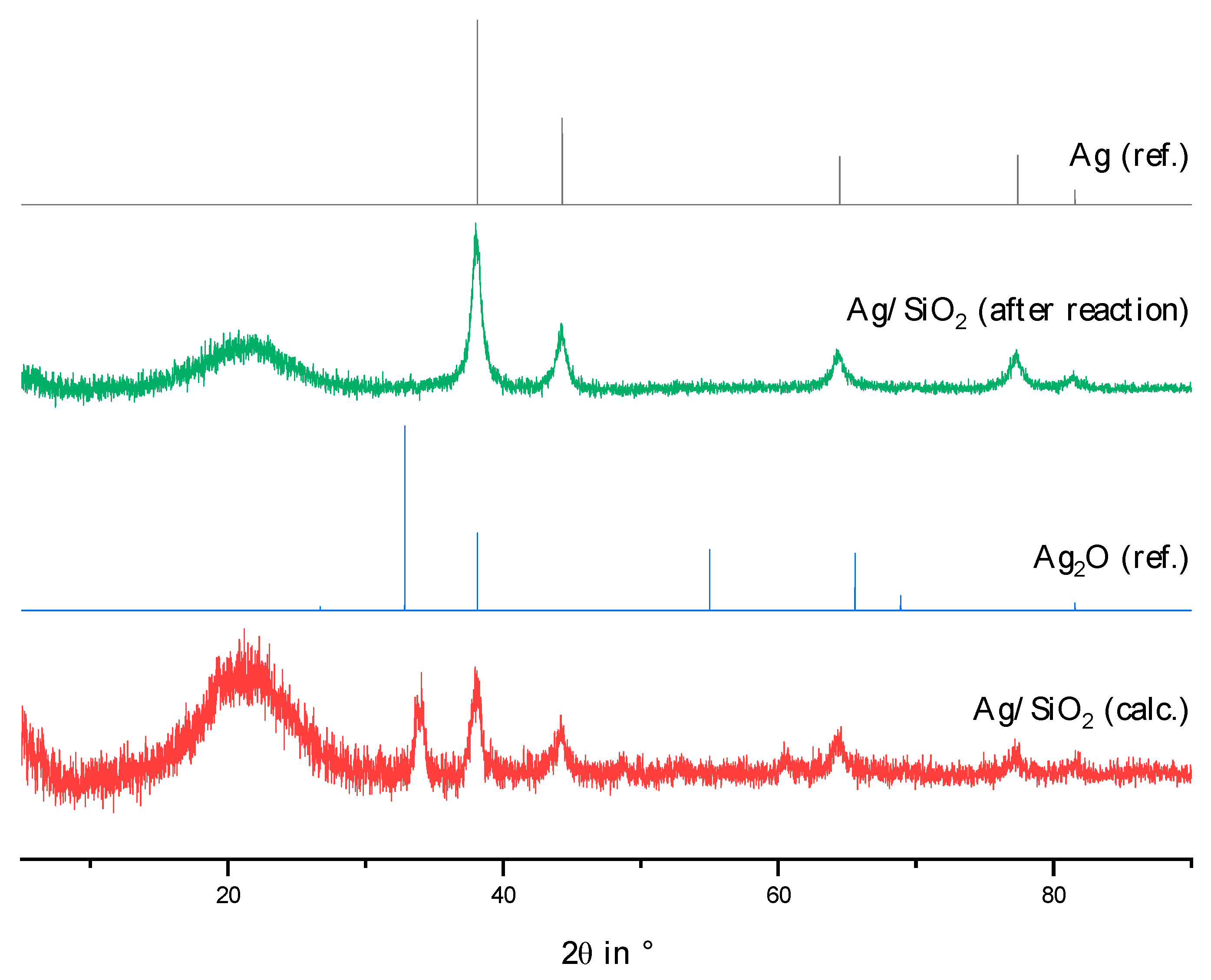
2.2. Physico-Chemical Characteristics of Different Transition Metal/Silica Catalyst Materials
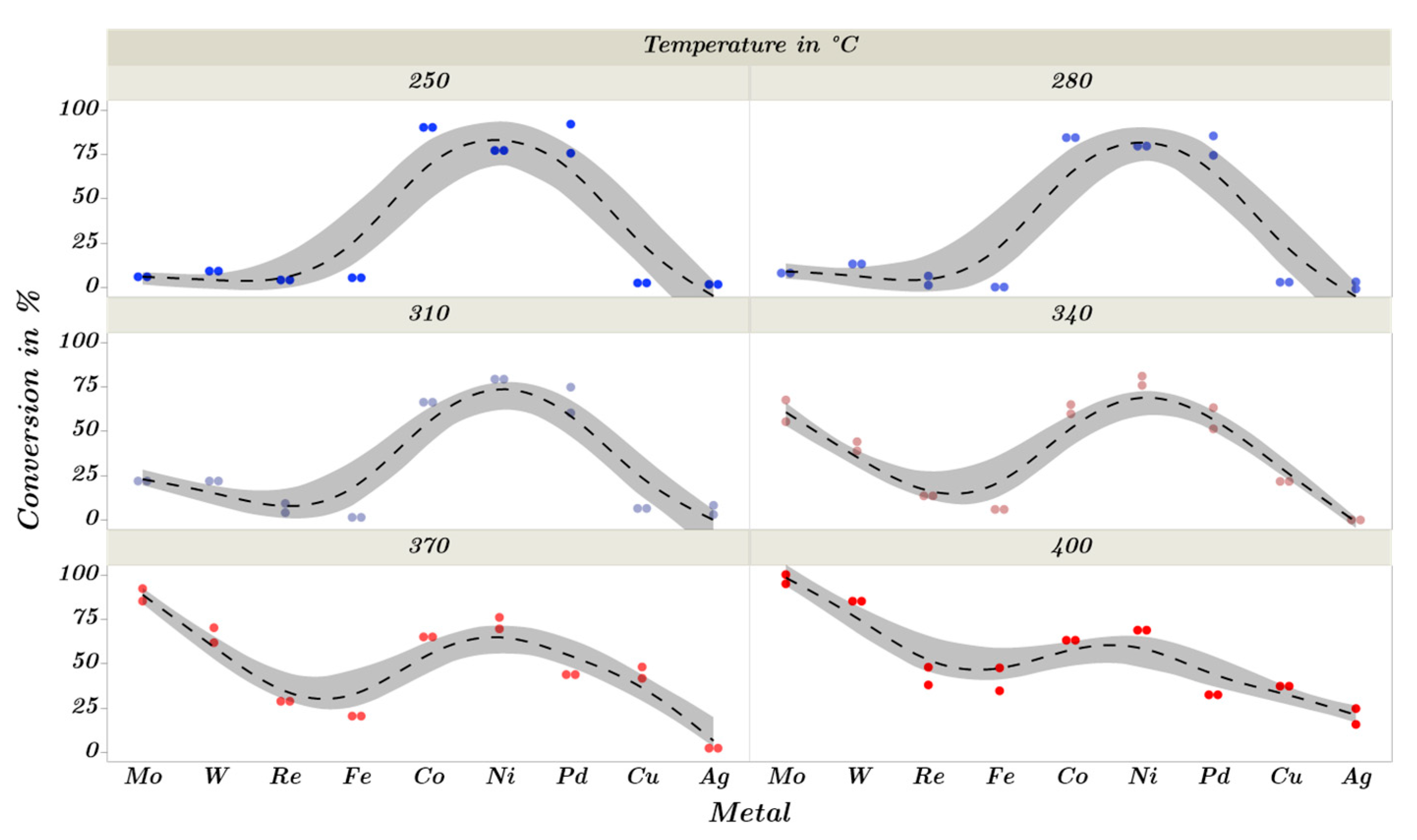
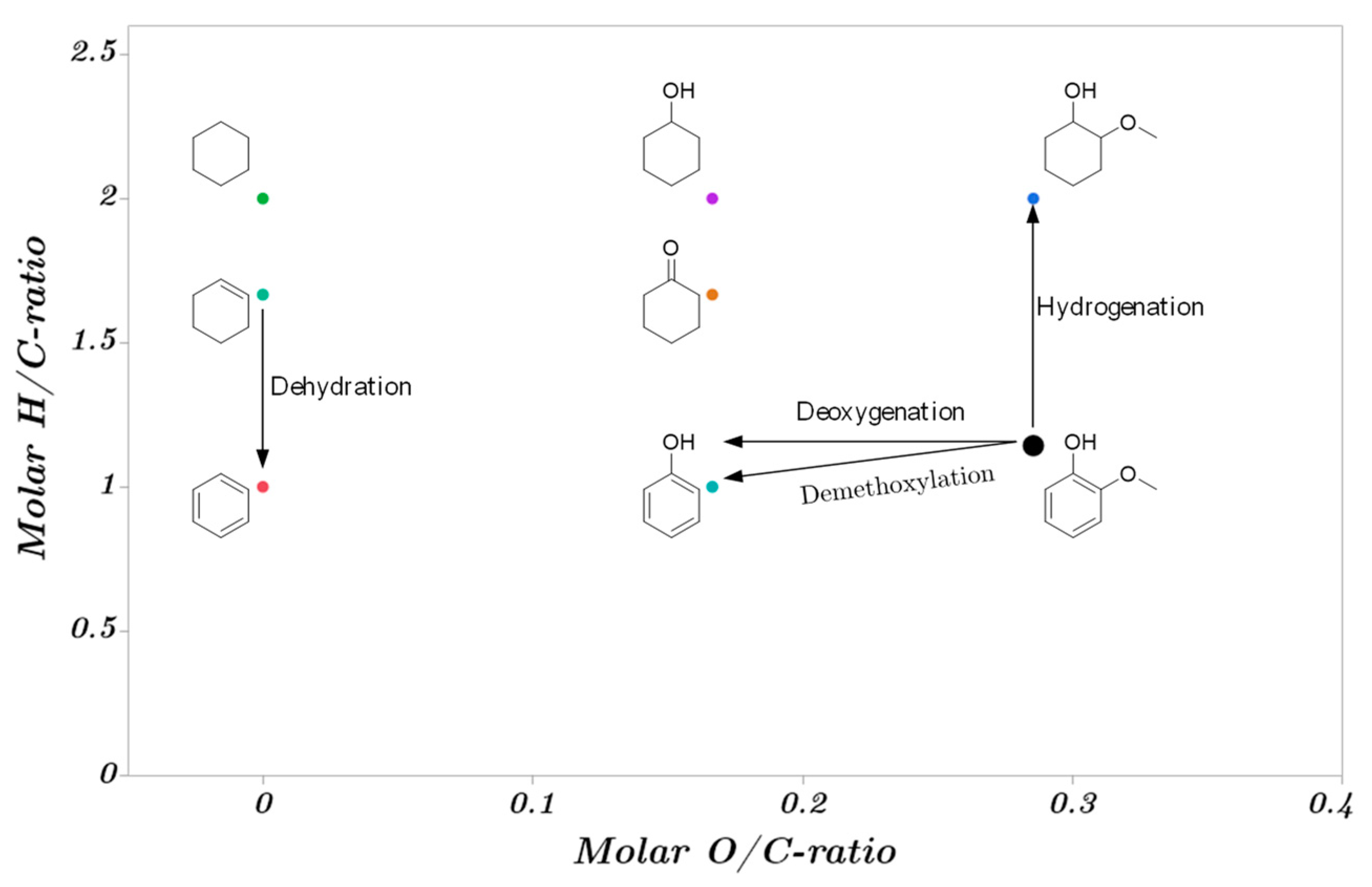
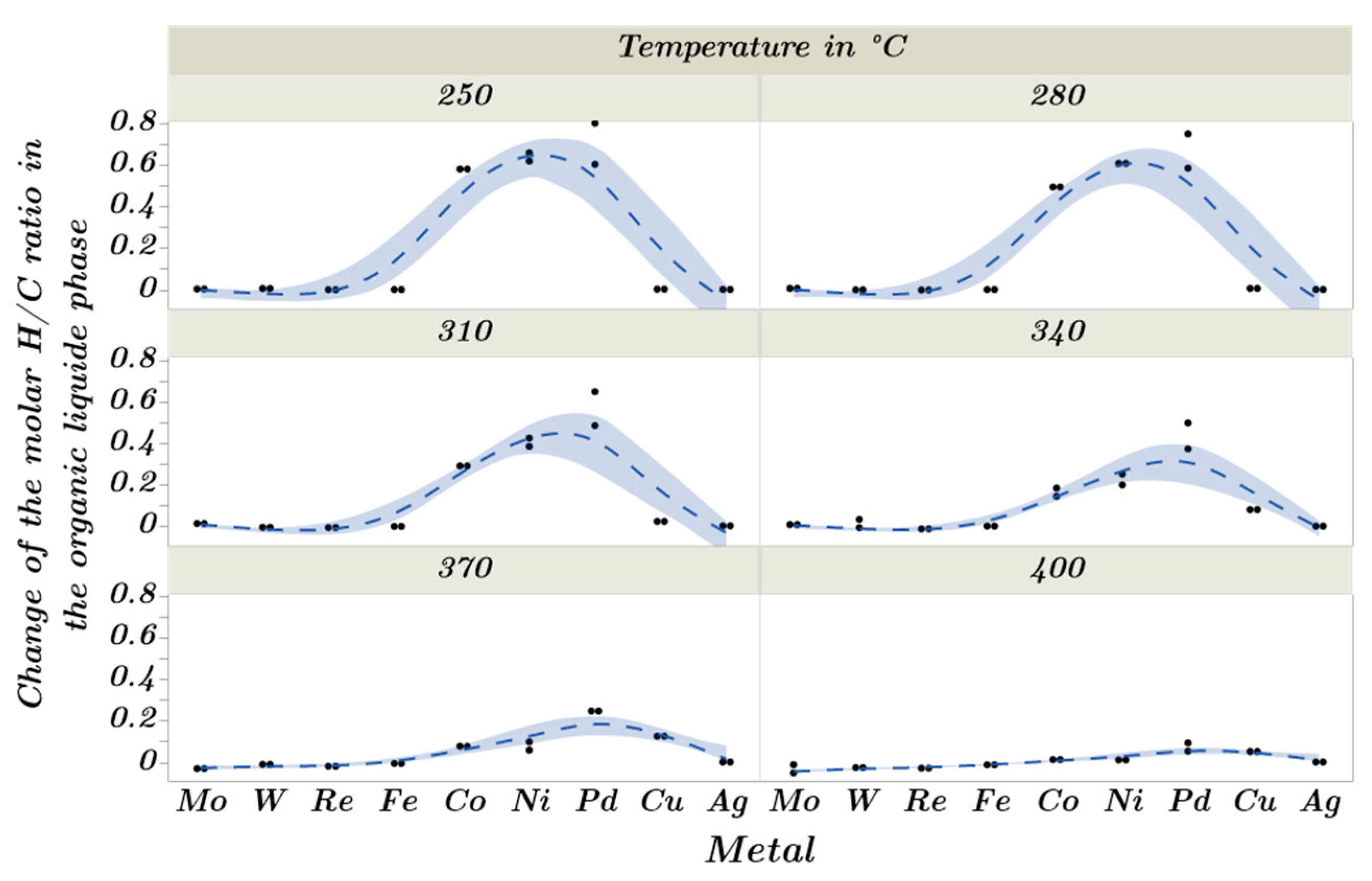
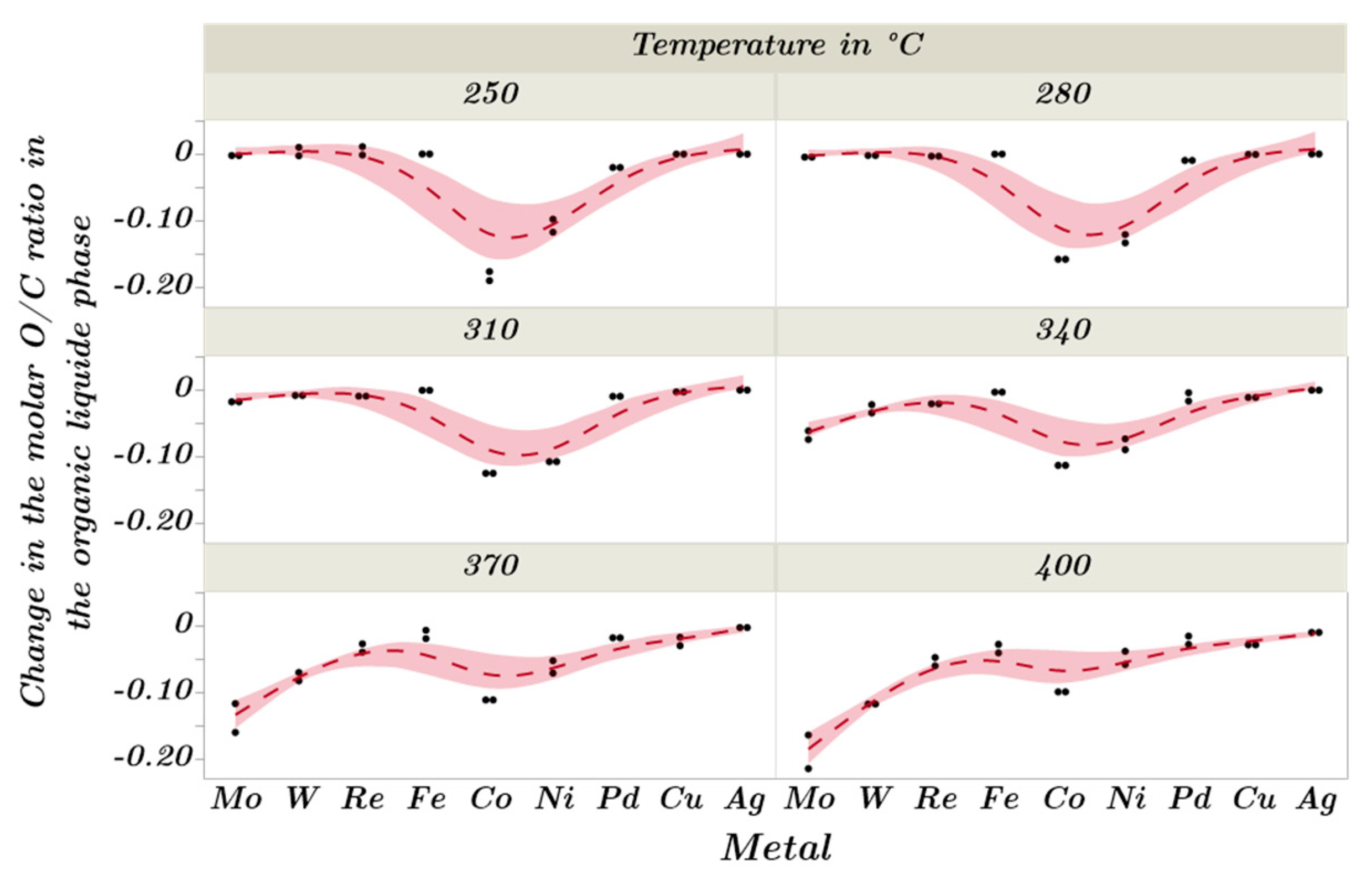
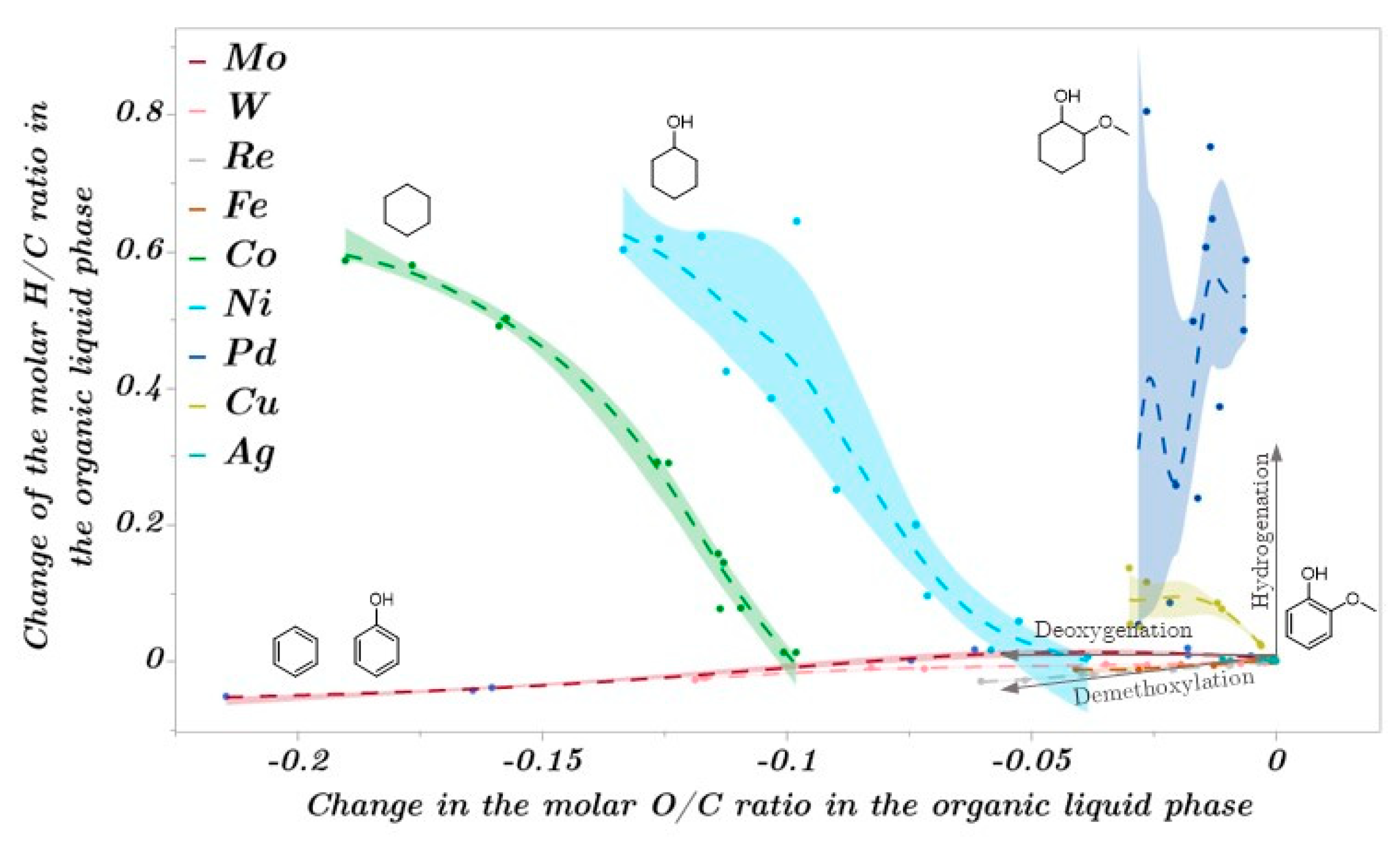
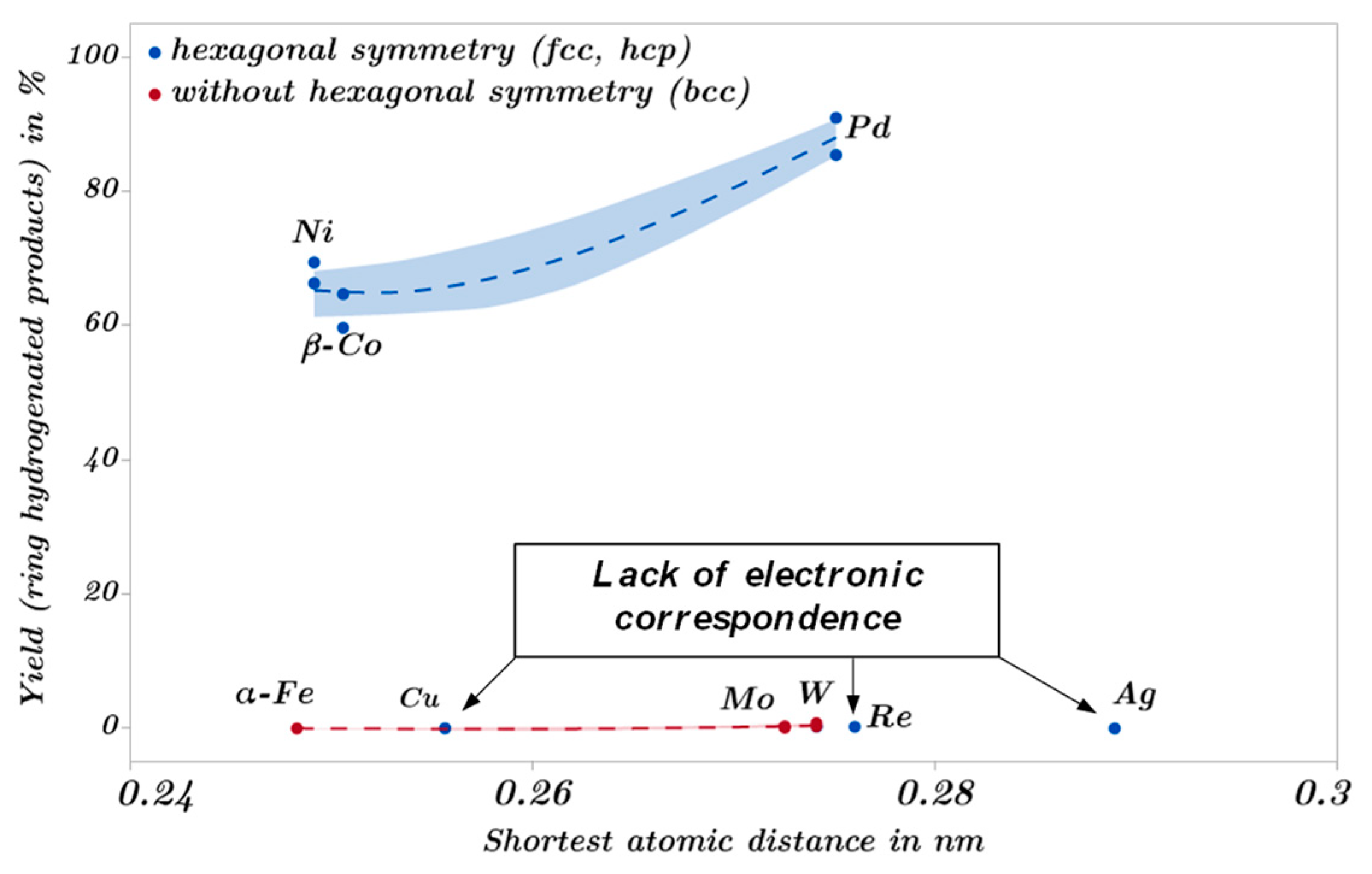
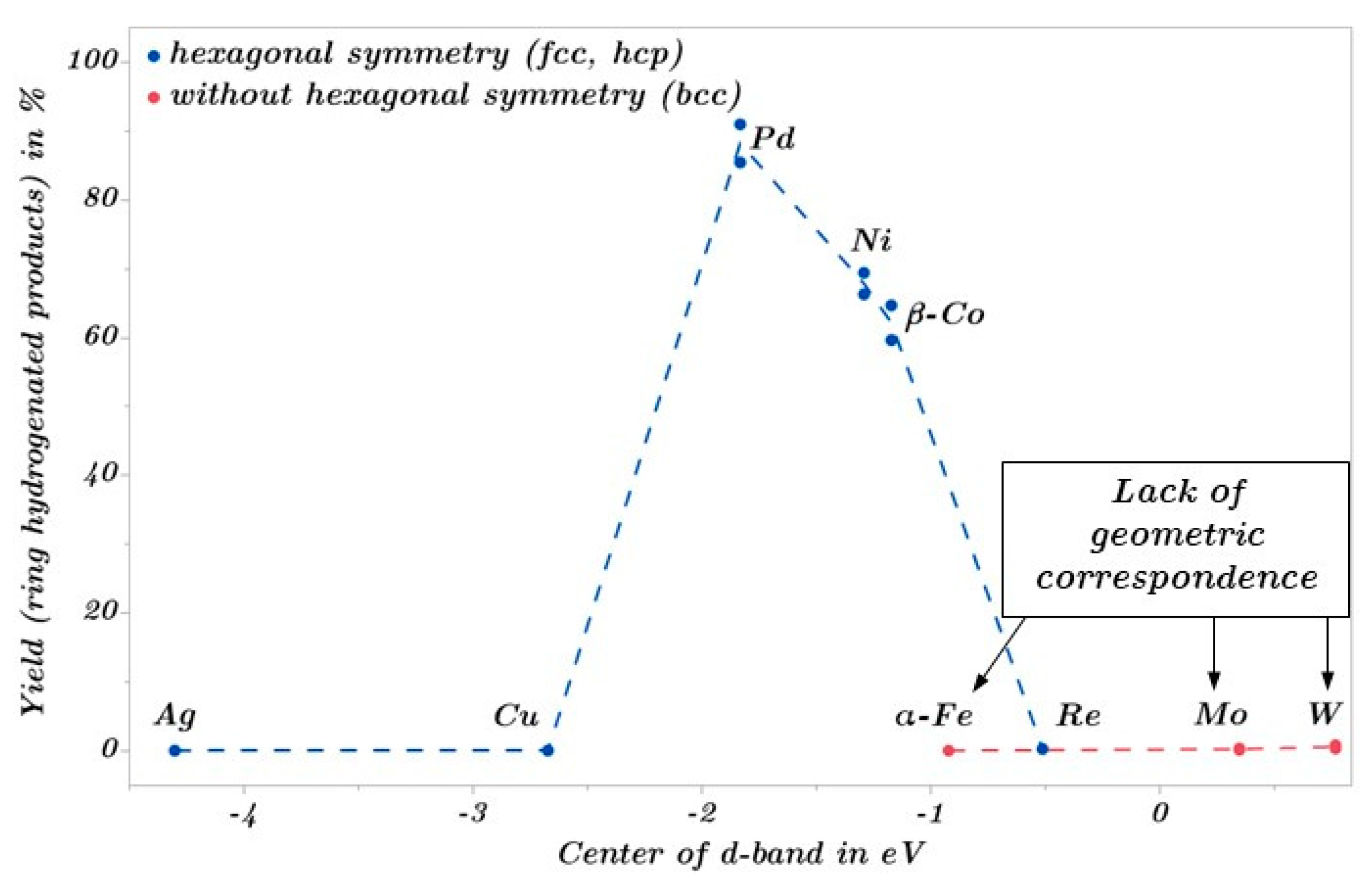
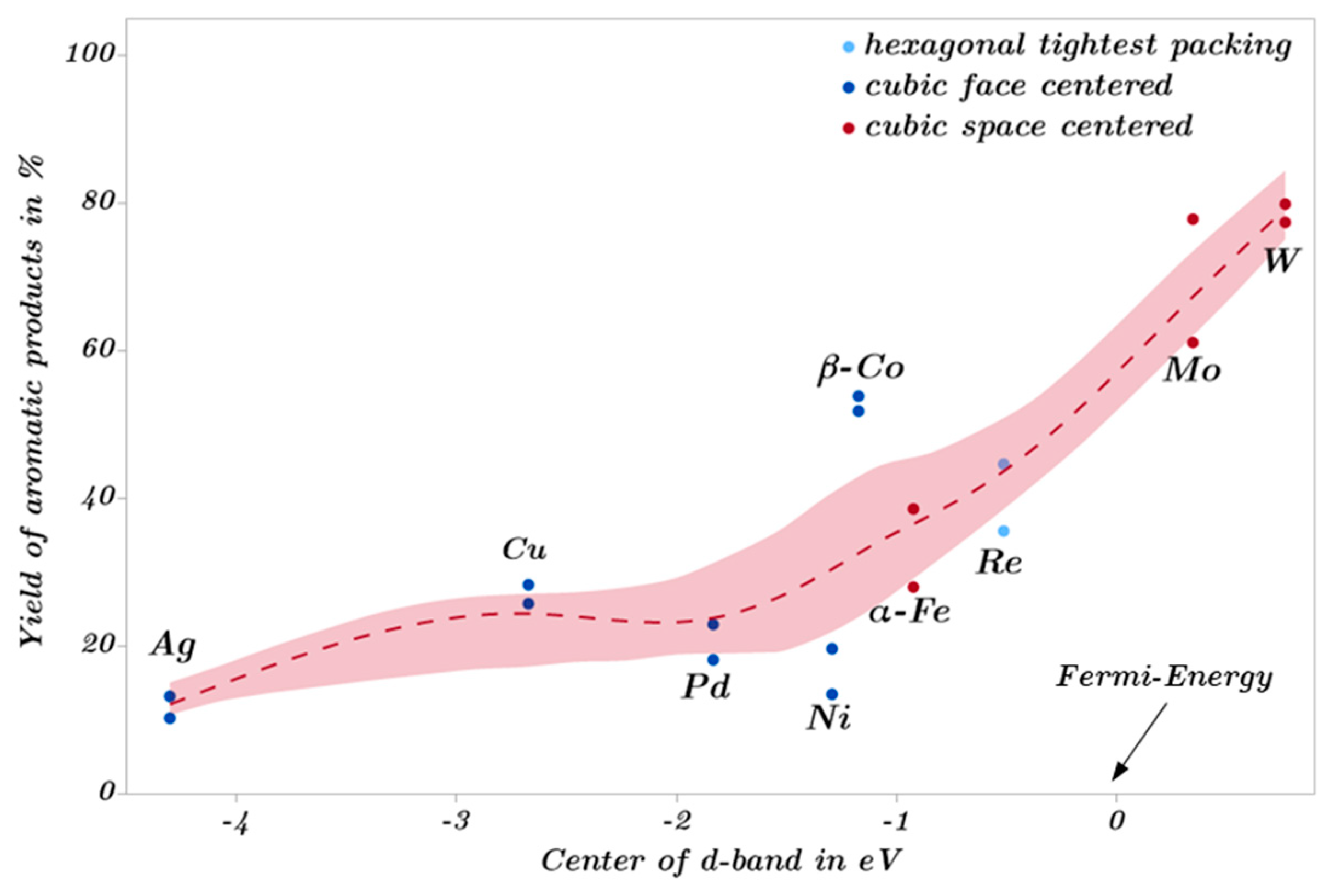
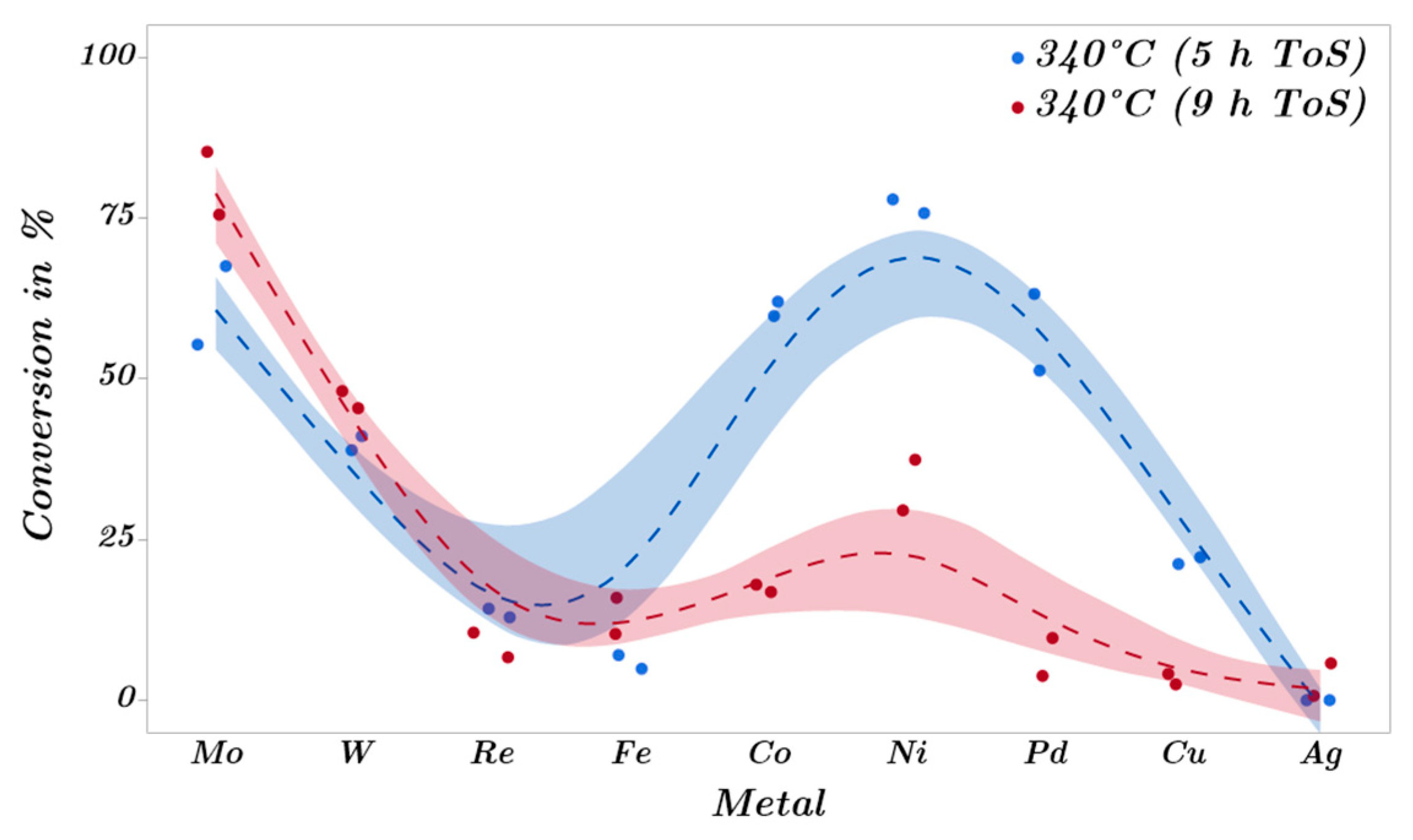
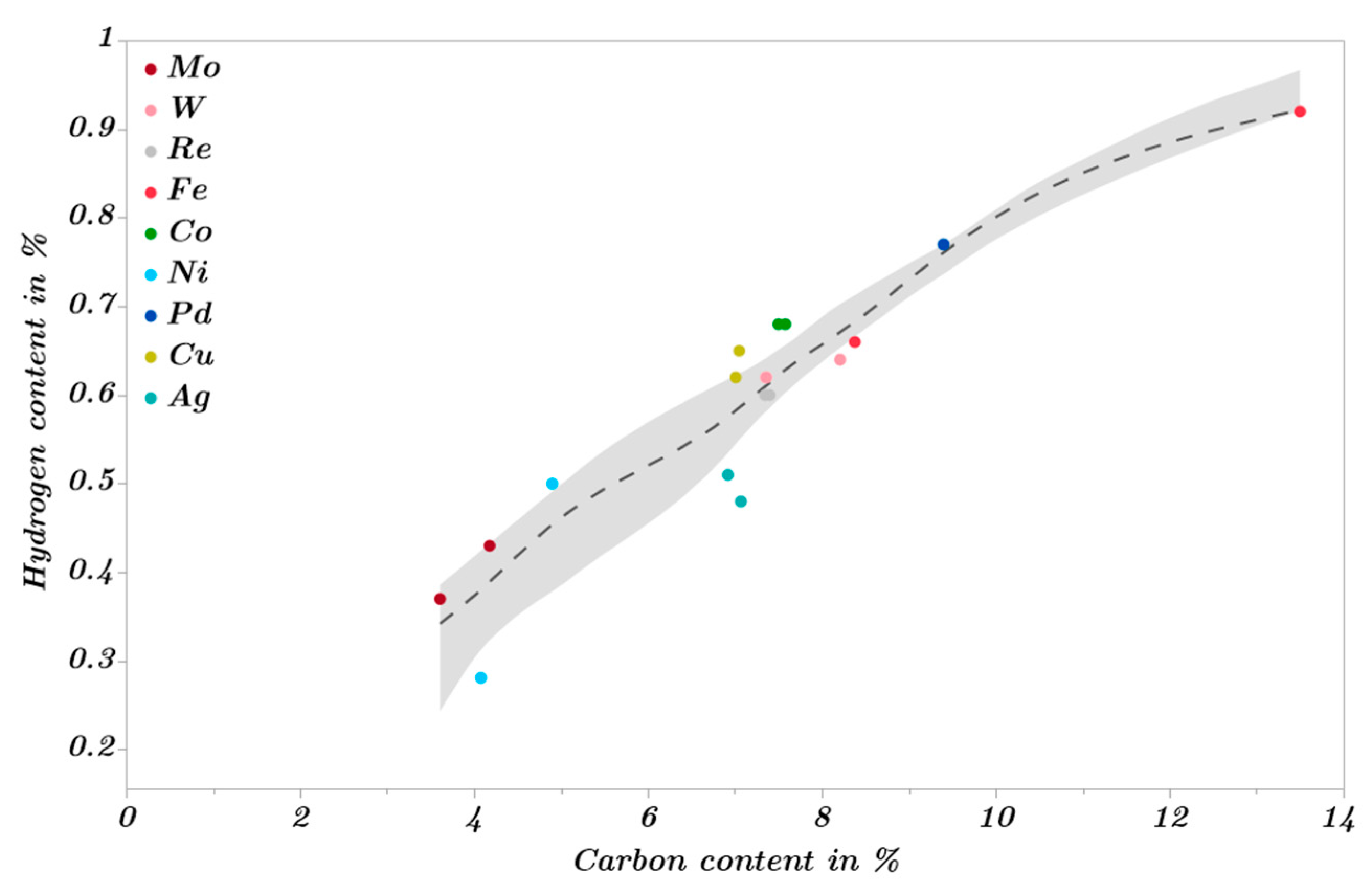
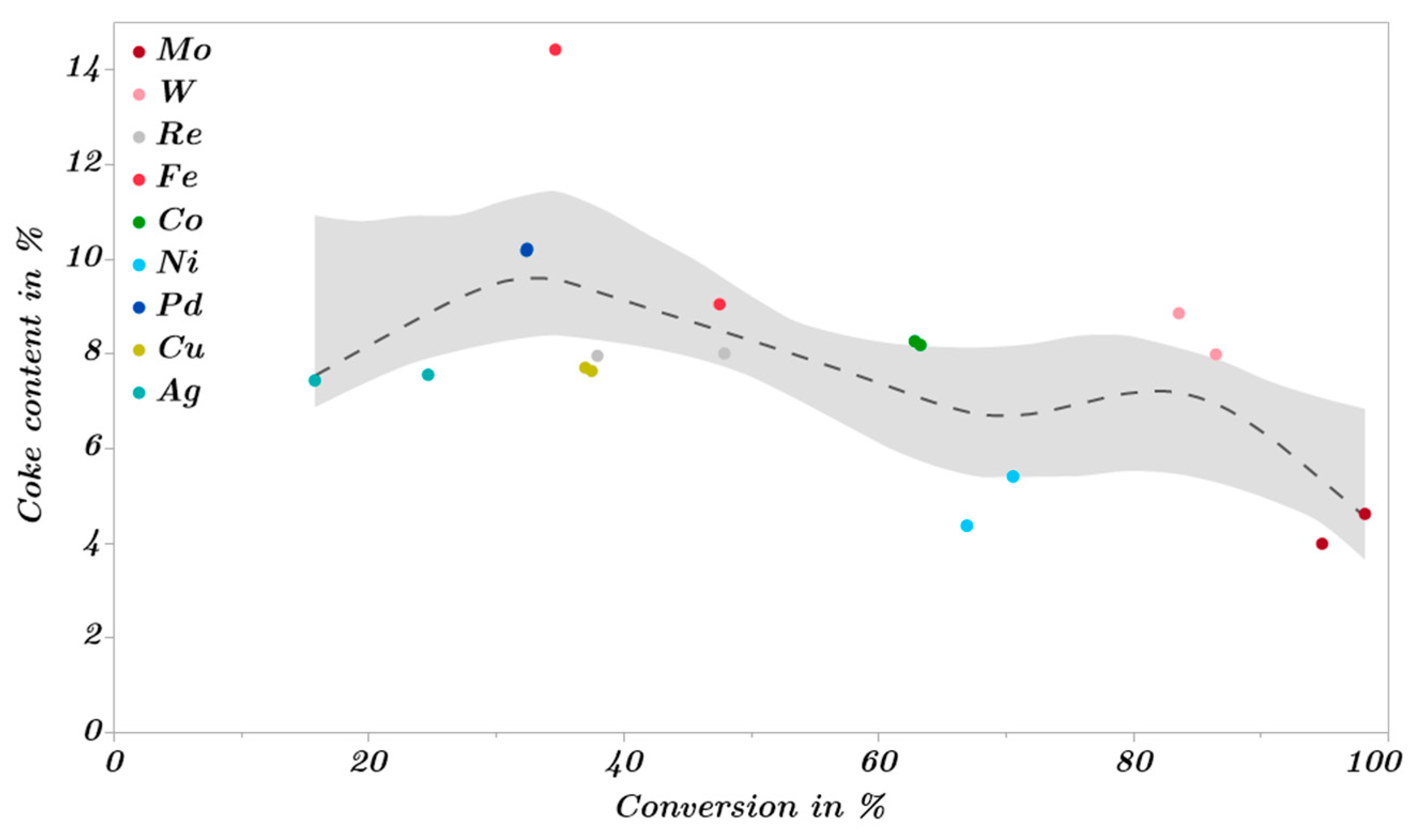
2.3. Catalytic Trends during the Metal Screening by Volcano Plots and Pseudo van Krevelen Diagram
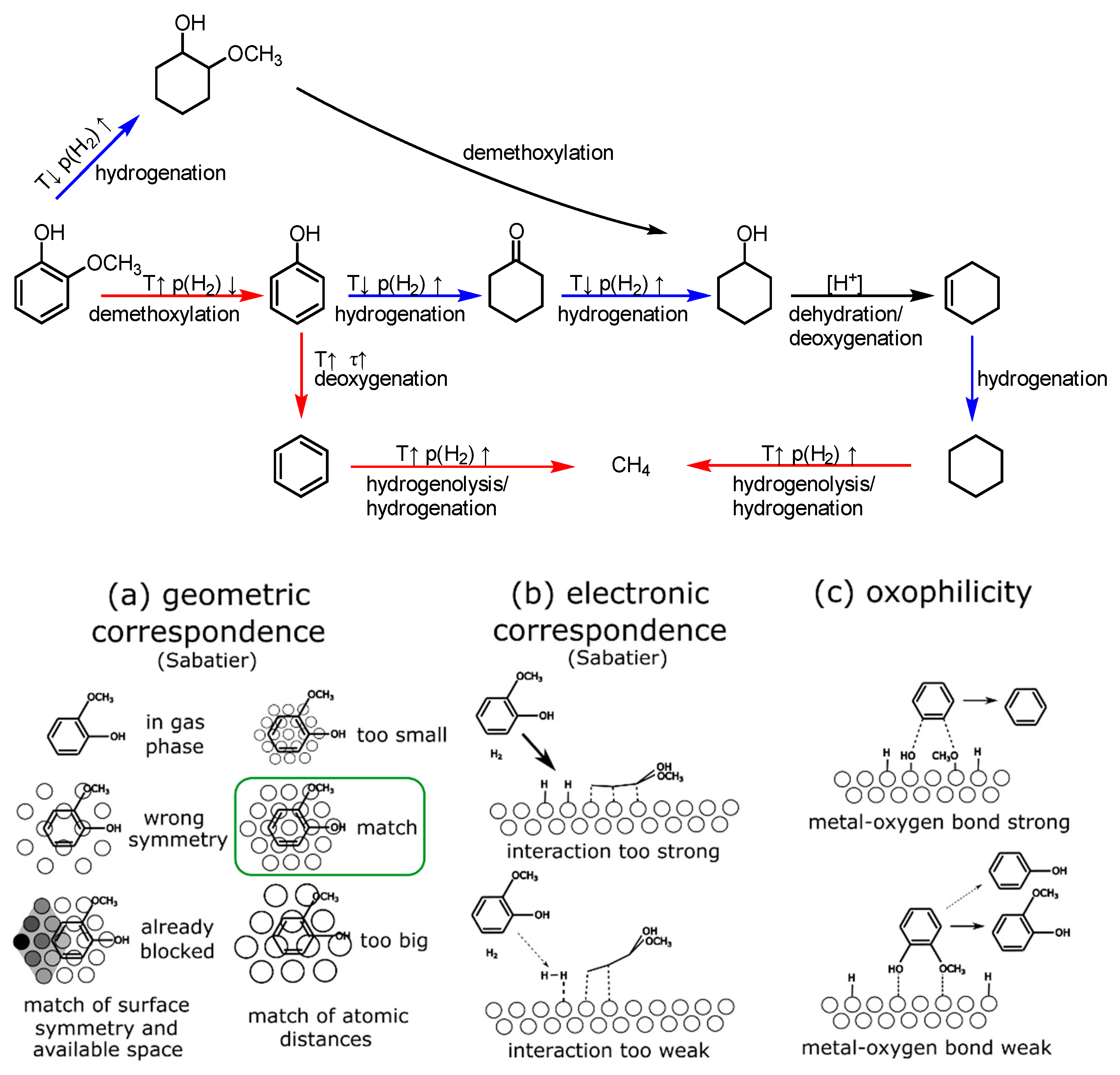
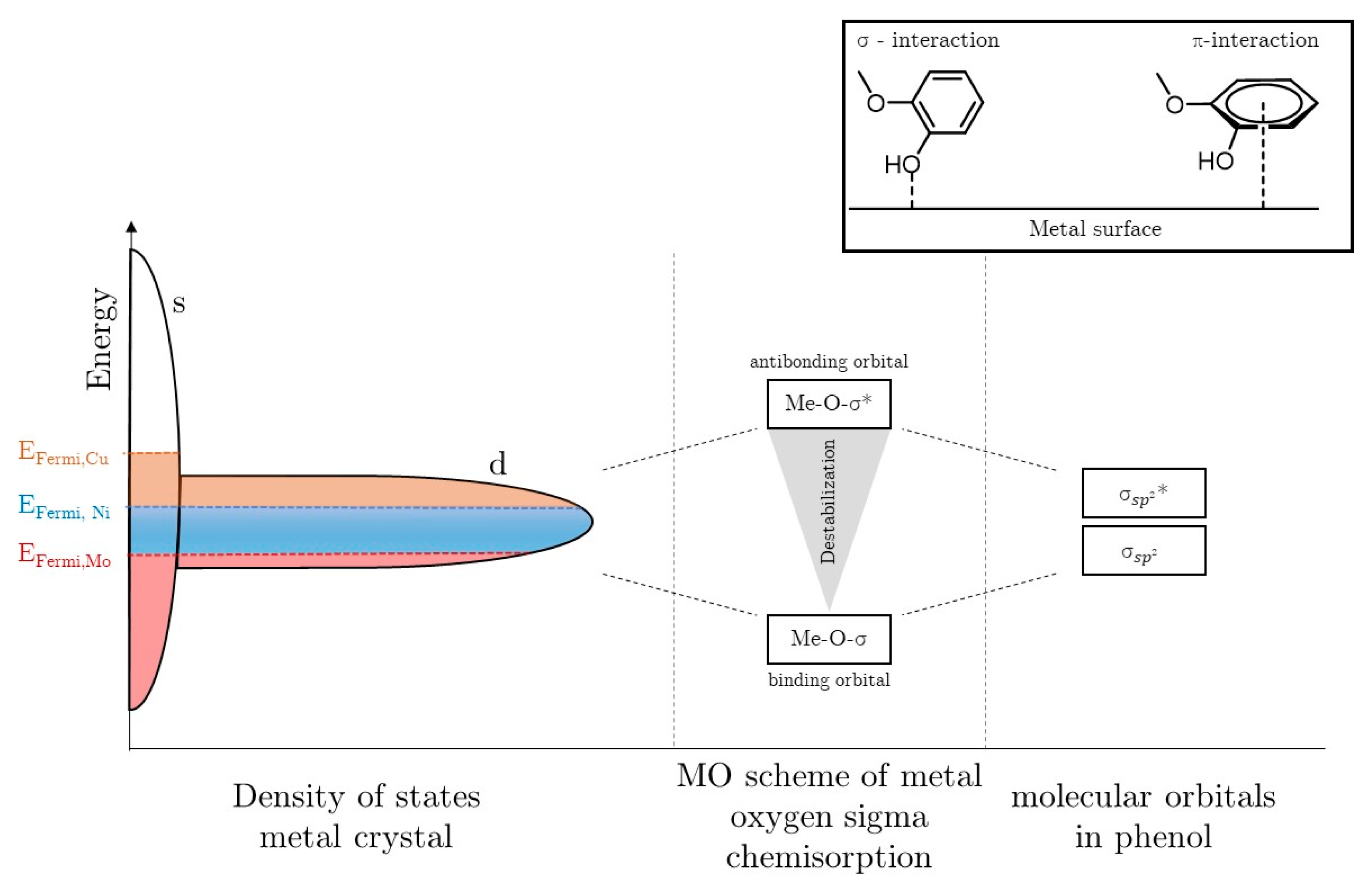
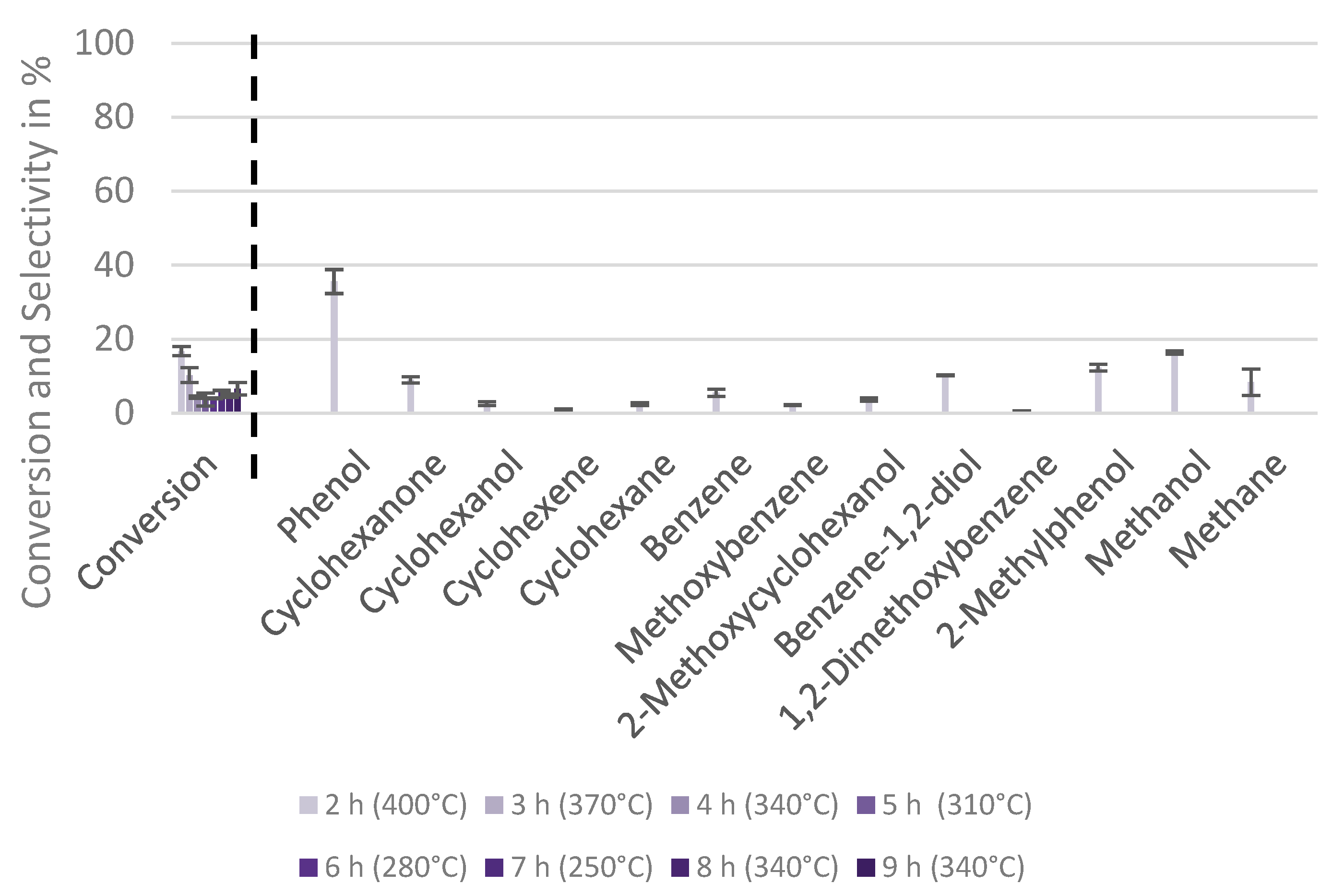
2.4. Mechanistic Conclusions for Hydrogenation, Deoxygenation and Deactivation Processes
3. Conclusions
4. Materials and Methods
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A











Appendix B

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metal | Electron Configuration of Isolated Atom | Metal Crystal Band Lineup [25] | Minimum Energy Configuration [85] | Center of the d-Band [76] in eV | D-Character [25] in % [a] | One-Electron Orbital Energy [85] | |||
|---|---|---|---|---|---|---|---|---|---|
| Calculations | Spectroscopy | ||||||||
| s | d | s | d | ||||||
| Mo | 4d55s1 | - | 4d4,6185s1,735 | 0.35 | 43 | 0.4421 | 0.6700 | 0.532 | 0.672 |
| W | 4f145d46s2 | - | 5d46s2 | 0.77 | 43 | 0.5308 | 0.5776 | 0.621 | 0.557 |
| Re | 4f145d56s2 | - | 5d5,1276s1,873 | −0.51 | 46 | 0.5485 | 0.6691 | 0.626 | 0.644 |
| Fe | 3d64s2 | 3d7,054s0,95 | 3d6,3704s1,630 | −0.92 | 39.7 | 0.4983 | 1.0242 | 0.5812 | 0.8567 |
| Co | 3d74s2 | 3d8,254s0,75 | 3d7,4254s1,575 | −1.17 | 39.5 | 0.5104 | 1.0781 | 0.595 | 0.891 |
| Ni | 3d84s2 | 3d9,454s0,55 | 3d8,4694s1,531 | −1.29 | 40 | 0.5225 | 1.1332 | 0.6045 | 0.9503 |
| Pd | 4d10 | 4d9,645s0,36 | 4d9,6675s0,333 | −1.83 | 46 | 0.4129 | 0.7478 | 0.5466 | 0.7041 |
| Cu | 3d104s1 | 3d104s1 | 3d9,5114s1,1489 | −2.67 | - | 0.5340 | 1.1849 | 0.6192 | 0.9897 |
| Ag | 4d105s1 | 4d105s1 | 4d105s1 | −4.30 | - | 0.4737 | 1.0228 | 0.5569 | 0.9394 |
| Metal | Covalent Radius [25,63,68] in nm | Lattice Type [25,60,68] | Shortest Atomic Distances [25] in nm | Bond Energy [86,87,88] | ||
|---|---|---|---|---|---|---|
| M-O in eV | M-C in eV | M-H in eV | ||||
| Mo | 0.1363 | cubic space centered | 0.27251 | −4.73 | - | −2.30 |
| W | 0.1370 | cubic space centered | 0.27409 | −5.64 | −8.59 | −3.30 |
| Re | 0.1371 | hexagonal tightest packing | 0.2741; 0.2760 [a] | −4.94 | - | −2.91 |
| α-Fe | 0.1241 | cubic space centered [b] | 0.24823 | −5.55 | −8.02 | −2.91 |
| γ-Fe | - | cubic face centered [b] | - | −5.55 | −8.02 | −2.91 |
| α-Co | 0.1253 | hexagonal tightest packing [c] | 0.25061 | −5.58 | −6.73 | −2.43 |
| β-Co | - | cubic face centered [c] | - | −5.58 | −6.73 | −2.43 |
| Ni | 0.1246 | cubic face centered | 0.24916 | −5.46 | −6.46 | −2.52 |
| Pd | 0.1376 | cubic face centered | 0.27511 | −3.60 | −5.07 | −1.73 |
| Cu | 0.132 | cubic face centered | 0.25560 | −4.73 | - | −2.43 |
| Ag | 0.145 | cubic face centered | 0.28894 | −3.64 | - | −2.04 |
| Chemicals | Formula | Purity | Manufacturer |
|---|---|---|---|
| Nickel(II)-nitrate-hexahydrate | Ni(NO3)26 H2O | 99% | Acros Organics (Geel, Belgium) |
| Iron (III)-nitrate-nonahydrate | Fe(NO3)3·9 H2O | >99% | Acros Organics (Geel, Belgium) |
| Cobalt(II)-nitrate-hexahydrate | Co(NO3)2·6 H2O | 99% | Acros Organics (Geel, Belgium) |
| Ammoniumheptamolybdate-tetrahydrate | (NH4)6Mo7O24·4 H2O | 99% | Merck (Darmstadt, Germany) |
| Silvernitrate | Ag(NO3) | >99.9 | Thermo Fisher Scientific (Waltham, MA, USA) |
| Ammoniumperrhenate | NH4ReO4 | >99.9% | Thermo Fisher Scientific (Waltham, MA, USA) |
| Palladiumnitrate-dihydrate | Pd(NO3)2·2 H2O | >99% | Thermo Fisher Scientific (Waltham, MA, USA) |
| Ammoniumparawolframate-tetrahydrate | (NH4)10(H2W12O42) ·4H2O | >99% | Merck (Darmstadt, Germany) |
| Copper(II)-nitrate-trihydrate | Cu(NO3)2·3 H2O | >99% | Fluka (St. Gallen, Switzerland) |
| Aerosil380 | SiO2 | >99.8% | Evonik (Essen, Germany) |
| HZSM-5 (HCZP 27) | H+x[(AlO2)−x(SiO2)y] | - | Clariant (Gendorf, Germany) |
References
- Tran, N.; Uemura, Y.; Trinh, T.; Ramli, A. Hydrodeoxygenation of Guaiacol over Pd–Co and Pd–Fe Catalysts: Deactivation and Regeneration. Processes 2021, 9, 430. [Google Scholar] [CrossRef]
- Zhou, H.; Wang, H.; Perras, F.A.; Naik, P.; Pruski, M.; Sadow, A.D.; Slowing, I.I. Two-step conversion of Kraft lignin to nylon precursors under mild conditions. Green Chem. 2020, 22, 4676–4682. [Google Scholar] [CrossRef]
- Güvenatam, B.; Kurşun, O.; Heeres, E.H.; Pidko, E.A.; Hensen, E.J. Hydrodeoxygenation of mono- and dimeric lignin model compounds on noble metal catalysts. Catal. Today 2014, 233, 83–91. [Google Scholar] [CrossRef]
- Li, W.; Wang, H.; Wu, X.; Betancourt, L.E.; Tu, C.; Liao, M.; Cui, X.; Li, F.; Zheng, J.; Li, R. Ni/hierarchical ZSM-5 zeolites as promising systems for phenolic bio-oil upgrading: Guaiacol hydrodeoxygenation. Fuel 2020, 274, 117859. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, Y.; Yang, G.; Deng, P.; Lu, X.; Ma, R.; Fu, Y.; Zhu, W. Low-Temperature Catalytic Hydrogenolysis of Guaiacol to Phenol over Al-Doped SBA-15 Supported Ni Catalysts. ChemCatChem 2020, 12, 4930–4938. [Google Scholar] [CrossRef]
- López, M.; Palacio, R.; Mamede, A.-S.; Fernández, J.J.; Royer, S. Hydrodeoxygenation of guaiacol into cyclohexane over mesoporous silica supported Ni–ZrO2 catalyst. Microporous Mesoporous Mater. 2020, 309, 110452. [Google Scholar] [CrossRef]
- Liu, W.; You, W.; Sun, W.; Yang, W.; Korde, A.; Gong, Y.; Deng, Y. Ambient-pressure and low-temperature upgrading of lignin bio-oil to hydrocarbons using a hydrogen buffer catalytic system. Nat. Energy 2020, 5, 759–767. [Google Scholar] [CrossRef]
- Roldugina, E.A.; Naranov, E.R.; Maximov, A.L.; Karakhanov, E.A. Hydrodeoxygenation of guaiacol as a model compound of bio-oil in methanol over mesoporous noble metal catalysts. Appl. Catal. A Gen. 2018, 553, 24–35. [Google Scholar] [CrossRef]
- Chen, C.; Chen, G.; Yang, F.; Wang, H.; Han, J.; Ge, Q.; Zhu, X. Vapor phase hydrodeoxygenation and hydrogenation of m-cresol on silica supported Ni, Pd and Pt catalysts. Chem. Eng. Sci. 2015, 135, 145–154. [Google Scholar] [CrossRef]
- Han, G.-H.; Lee, M.W.; Park, S.; Kim, H.J.; Ahn, J.-P.; Seo, M.; Lee, K.-Y. Revealing the factors determining the selectivity of guaiacol HDO reaction pathways using ZrP-supported Co and Ni catalysts. J. Catal. 2019, 377, 343–357. [Google Scholar] [CrossRef]
- Chen, C.; Zhou, M.; Liu, P.; Sharma, B.K.; Jiang, J. Flexible NiCo-based catalyst for direct hydrodeoxygenation of guaiacol to cyclohexanol. New J. Chem. 2020, 44, 18906–18916. [Google Scholar] [CrossRef]
- Liu, X.; Jia, W.; Xu, G.; Zhang, Y.; Fu, Y. Selective Hydrodeoxygenation of Lignin-Derived Phenols to Cyclohexanols over Co-Based Catalysts. ACS Sustain. Chem. Eng. 2017, 5, 8594–8601. [Google Scholar] [CrossRef]
- Wang, S.; Xu, D.; Chen, Y.; Zhou, S.; Zhu, D.; Wen, X.; Yang, Y.; Li, Y. Hydrodeoxygenation of anisole to benzene over an Fe2P catalyst by a direct deoxygenation pathway. Catal. Sci. Technol. 2020, 10, 3015–3023. [Google Scholar] [CrossRef]
- Sirous-Rezaei, P.; Jae, J.; Ha, J.-M.; Ko, C.H.; Kim, J.M.; Jeon, J.-K.; Park, Y.-K. Mild hydrodeoxygenation of phenolic lignin model compounds over a FeReO x/ZrO2 catalyst: Zirconia and rhenium oxide as efficient dehydration promoters. Green Chem. 2018, 20, 1472–1483. [Google Scholar] [CrossRef]
- Olcese, R.; Bettahar, M.M.; Malaman, B.; Ghanbaja, J.; Tibavizco, L.; Petitjean, D.; Dufour, A. Gas-phase hydrodeoxygenation of guaiacol over iron-based catalysts. Effect of gases composition, iron load and supports (silica and activated carbon). Appl. Catal. B Environ. 2013, 129, 528–538. [Google Scholar] [CrossRef]
- Olcese, R.N.; Bettahar, M.; Petitjean, D.; Malaman, B.; Giovanella, F.; Dufour, A. Gas-phase hydrodeoxygenation of guaiacol over Fe/SiO2 catalyst. Appl. Catal. B Environ. 2012, 115–116, 63–73. [Google Scholar] [CrossRef]
- Remón, J.; Ochoa, E.; Foguet, C.; Pinilla, J.L.; Suelves, I. Towards a sustainable bio-fuels production from lignocellulosic bio-oils: Influence of operating conditions on the hydrodeoxygenation of guaiacol over a Mo2C/CNF catalyst. Fuel Process. Technol. 2019, 191, 111–120. [Google Scholar] [CrossRef]
- Shetty, M.; Anderson, E.M.; Green, W.H.; Román-Leshkov, Y. Kinetic analysis and reaction mechanism for anisole conversion over zirconia-supported molybdenum oxide. J. Catal. 2019, 376, 248–257. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, J.; Zhang, Q.; Liu, Q.; Li, Y.; Chen, L.; Wang, C.; Ma, L. Hydrodeoxygenation of lignin-derived phenolic compounds into aromatic hydrocarbons under low hydrogen pressure using molybdenum oxide as catalyst. Catal. Today 2019, 319, 41–47. [Google Scholar] [CrossRef]
- Shi, Y.; Cao, Y.; Duan, Y.; Chen, H.; Chen, Y.; Yang, M.; Wu, Y. Upgrading of palmitic acid to iso-alkanes over bi-functional Mo/ZSM-22 catalysts. Green Chem. 2016, 18, 4633–4648. [Google Scholar] [CrossRef]
- Cheng, J.; Li, T.; Huang, R.; Zhou, J.; Cen, K. Optimizing catalysis conditions to decrease aromatic hydrocarbons and increase alkanes for improving jet biofuel quality. Bioresour. Technol. 2014, 158, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Thallada, B. Hydrodeoxygenation of lignin derived phenolics over a hydrous ruthenium oxide based catalyst(s): Role of surface water molecules and acidity of the support. Sustain. Energy Fuels 2021, 5, 3802–3817. [Google Scholar] [CrossRef]
- Prabhudesai, V.S.; Gurrala, L.; Vinu, R. Catalytic Hydrodeoxygenation of Lignin-Derived Oxygenates: Catalysis, Mechanism, and Effect of Process Conditions. Energy Fuels 2022, 36, 1155–1188. [Google Scholar] [CrossRef]
- Wu, X.; Ge, Q.; Zhu, X. Vapor phase hydrodeoxygenation of phenolic compounds on group 10 metal-based catalysts: Reaction mechanism and product selectivity control. Catal. Today 2021, 365, 143–161. [Google Scholar] [CrossRef]
- Reschetilowski, W. Einführung in Die Heterogene Katalyse; Springer: Berlin/Heidelberg, Germany, 2015; ISBN 978-3-662-46983-5. [Google Scholar]
- Fechete, I. Paul Sabatier—The father of the chemical theory of catalysis. Comptes Rendus Chim. 2016, 19, 1374–1381. [Google Scholar] [CrossRef] [Green Version]
- Balandin, A.A. Modern State of the Multiplet Theor of Heterogeneous Catalysis; Elsevier: Amsterdam, The Netherlands, 1969; pp. 1–210. ISBN 9780120078196. [Google Scholar]
- Nguyen, L.T.; Phan, D.-P.; Sarwar, A.; Tran, M.H.; Lee, O.K.; Lee, E.Y. Valorization of industrial lignin to value-added chemicals by chemical depolymerization and biological conversion. Ind. Crops Prod. 2021, 161, 113219. [Google Scholar] [CrossRef]
- Qu, L.; Jiang, X.; Zhang, Z.; Zhang, X.; Song, G.; Wang, H.; Yuan, Y.; Chang, Y. A review of hydrodeoxygenation of bio-oil: Model compounds, catalysts, and equipment. Green Chem. 2021, 23, 9348–9376. [Google Scholar] [CrossRef]
- Jing, Y.; Dong, L.; Guo, Y.; Liu, X.; Wang, Y. Chemicals from Lignin: A Review of Catalytic Conversion Involving Hydrogen. ChemSusChem 2020, 13, 4181–4198. [Google Scholar] [CrossRef] [PubMed]
- Shu, R.; Li, R.; Lin, B.; Wang, C.; Cheng, Z.; Chen, Y. A review on the catalytic hydrodeoxygenation of lignin-derived phenolic compounds and the conversion of raw lignin to hydrocarbon liquid fuels. Biomass Bioenergy 2020, 132, 105432. [Google Scholar] [CrossRef]
- Tomishige, K.; Nakagawa, Y.; Tamura, M. Design of supported metal catalysts modified with metal oxides for hydrodeoxygenation of biomass-related molecules. Curr. Opin. Green Sustain. Chem. 2020, 22, 13–21. [Google Scholar] [CrossRef]
- Hammer, B.; Norskov, J.K. Why gold is the noblest of all the metals. Nature 1995, 376, 238–240. [Google Scholar] [CrossRef]
- Hammer, B.; Nørskov, J.K. Electronic factors determining the reactivity of metal surfaces. Surf. Sci. 1995, 343, 211–220. [Google Scholar] [CrossRef]
- Wang, X.; Zhu, S.; Wang, S.; Wang, J.; Fan, W.; Lv, Y. Ni nanoparticles entrapped in nickel phyllosilicate for selective hydrogenation of guaiacol to 2-methoxycyclohexanol. Appl. Catal. A Gen. 2018, 568, 231–241. [Google Scholar] [CrossRef]
- Zhou, H.; Wang, H.; Sadow, A.D.; Slowing, I.I. Toward hydrogen economy: Selective guaiacol hydrogenolysis under ambient hydrogen pressure. Appl. Catal. B Environ. 2020, 270, 118890. [Google Scholar] [CrossRef]
- Shafaghat, H.; Rezaei, P.S.; Daud, W.M.A.W. Catalytic hydrodeoxygenation of simulated phenolic bio-oil to cycloalkanes and aromatic hydrocarbons over bifunctional metal/acid catalysts of Ni/HBeta, Fe/HBeta and NiFe/HBeta. J. Ind. Eng. Chem. 2016, 35, 268–276. [Google Scholar] [CrossRef]
- Xiang, M.; Wu, D. Highly selective catalytic conversion of lignin-derived phenolic compounds to cycloalkanes over a hierarchically structured zeolite catalyst. J. Mater. Sci. 2019, 54, 2940–2959. [Google Scholar] [CrossRef]
- Teles, C.A.; Rabelo-Neto, R.C.; de Lima, J.R.; Mattos, L.V.; Resasco, D.E.; Noronha, F.B. The Effect of Metal Type on Hydrodeoxygenation of Phenol Over Silica Supported Catalysts. Catal. Lett. 2016, 146, 1848–1857. [Google Scholar] [CrossRef]
- Hong, D.-Y.; Miller, S.J.; Agrawal, P.K.; Jones, C.W. Hydrodeoxygenation and coupling of aqueous phenolics over bifunctional zeolite-supported metal catalysts. Chem. Commun. 2010, 46, 1038–1040. [Google Scholar] [CrossRef]
- Yan, P.; Bryant, G.; Li, M.M.J.; Mensah, J.; Kennedy, E.; Stockenhuber, M. Shape selectivity of zeolite catalysts for the hydrodeoxygenation of biocrude oil and its model compounds. Microporous Mesoporous Mater. 2020, 309, 110561. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, J.; Yi, X.; Zheng, A.; Deng, F.; Chen, C.; Ji, Y.; Liu, F.; Meng, X.; Xiao, F.-S. Mesoporous ZSM-5 Zeolite-Supported Ru Nanoparticles as Highly Efficient Catalysts for Upgrading Phenolic Biomolecules. ACS Catal. 2015, 5, 2727–2734. [Google Scholar] [CrossRef]
- Evonik. Analysenzertifikat-Aerosil380; Evonik: Essen, Germany, 2020. [Google Scholar]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef] [Green Version]
- Zhao, D.; Feng, J.; Huo, Q.; Melosh, N.; Fredrickson, G.H.; Chmelka, B.F.; Stucky, G.D. Triblock copolymer syntheses of mesoporous silica with periodic 50 to 300 angstrom pores. Science 1998, 279, 548–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of Gases in Multimolecular Layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Barrett, E.P.; Joyner, L.G.; Halenda, P.P. The Determination of Pore Volume and Area Distributions in Porous Substances. I. Computations from Nitrogen Isotherms. J. Am. Chem. Soc. 1951, 73, 373–380. [Google Scholar] [CrossRef]
- Evonik Industries. Aerosil: Pyrogene Kieselsäuren; Evonik: Essen, Germany, Technical Overview-Auflage 8.
- Hadley, P. Molekül- und Festkörperphysik: Vorlesungsskriptum; Technische Universität Graz: Graz, Austria, 2013. [Google Scholar]
- Institut für Arbeitsschutz der Deutschen Gesetzlichen Unfallversicherung. Eintrag zu Guajacol. Available online: https://gestis.dguv.de/data?name=492497 (accessed on 5 August 2022).
- Ambursa, M.M.; Juan, J.C.; Yahaya, Y.; Taufiq-Yap, Y.H.; Lin, Y.-C.; Lee, H.V. A Review on Catalytic Hydrodeoxygenation of Lignin to Transportation Fuels by Using Nickel-Based Catalysts. In Renewable and Sustainable Energy Reviews; Elsevier Ltd.: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Ouedraogo, A.S.; Bhoi, P.R. Recent progress of metals supported catalysts for hydrodeoxygenation of biomass derived pyrolysis oil. J. Clean. Prod. 2020, 253, 119957. [Google Scholar] [CrossRef]
- Kim, S.; Kwon, E.E.; Kim, Y.T.; Jung, S.; Kim, H.J.; Huber, G.W.; Lee, J. Recent advances in hydrodeoxygenation of biomass-derived oxygenates over heterogeneous catalysts. Green Chem. 2019, 21, 3715–3743. [Google Scholar] [CrossRef]
- Shi, Y.; Xing, E.; Wu, K.; Wang, J.; Yang, M.; Wu, Y. Recent progress on upgrading of bio-oil to hydrocarbons over metal/zeolite bifunctional catalysts. Catal. Sci. Technol. 2017, 7, 2385–2415. [Google Scholar] [CrossRef]
- Raikwar, D.; Munagala, M.; Majumdar, S.; Shee, D. Hydrodeoxygenation of guaiacol over Mo, W and Ta modified supported nickel catalysts. Catal. Today 2019, 325, 117–130. [Google Scholar] [CrossRef]
- Regalbuto, J.R.; Ha, J.-W. A corrected procedure and consistent interpretation for temperature programmed reduction of supported MoO3. Catal. Lett. 1994, 29, 189–207. [Google Scholar] [CrossRef]
- Sicius, H. Chromgruppe: Elemente der Sechsten Nebengruppe: Eine Reise Durch das Periodensystem; Springer Spektrum: Wiesbaden, Germany, 2016; ISBN 9783658135430. [Google Scholar]
- Mos, Y.M.; Vermeulen, A.C.; Buisman, C.J.; Weijma, J. X-Ray Diffraction of Iron Containing Samples: The Importance of a Suitable Configuration. Geomicrobiol. J. 2018, 35, 511–517. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.-H.; Wan, H.-J.; Yang, Y.; Xiang, H.-W.; Li, Y.-W. Study on the iron–silica interaction of a co-precipitated Fe/SiO2 Fischer–Tropsch synthesis catalyst. Catal. Commun. 2006, 7, 733–738. [Google Scholar] [CrossRef]
- Sicius, H. Eisengruppe: Eine Reise Durch das Periodensystem; Springer Fachmedien Wiesbaden GmbH: Wiesbaden, Germany, 2017; ISBN 9783658155612. [Google Scholar]
- Cardarelli, F. Materials Handbook: A Concise Desktop Reference, 2nd ed.; Springer: London, UK, 2008; ISBN 978-1-84628-668-1. [Google Scholar]
- Sicius, H. Nickelgruppe: Elemente der Zehnten Nebengruppe: Eine Reise Durch das Periodensystem; Springer Spektrum: Wiesbaden/Heidelberg, Germany, 2017; ISBN 9783658168070. [Google Scholar]
- Sicius, H. Kupfergruppe: Elemente der Ersten Nebengruppe: Eine Reise Durch das Periodensystem; Springer Spektrum: Wiesbaden, Germany, 2017; ISBN 9783658172053. [Google Scholar]
- Scherrer, P. Bestimmung der Größe und der inneren Struktur von Kolloidteilchen mittels Röntgenstrahlen. In Nachrichten von der Gesellschaft der Wissenschaften zu Göttingen, Mathematisch-Physikalische Klasse; Springer Spektrum: Wiesbaden/Heidelberg, Germany, 1918; pp. 98–100. [Google Scholar]
- Mile, B.; Stirling, D.; Zammitt, M.A.; Lovell, A.; Webb, M. The location of nickel oxide and nickel in silica-supported catalysts: Two forms of “NiO” and the assignment of temperature-programmed reduction profiles. J. Catal. 1988, 114, 217–229. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, K.; Chen, J.; Zhu, L.; Wang, S. Mild hydrogenation of bio-oil and its derived phenolic monomers over Pt–Ni bimetal-based catalysts. Appl. Energy 2020, 275, 115154. [Google Scholar] [CrossRef]
- Li, X.; Nisa, M.; Chen, Y.; Li, Z. Co-Based Catalysts Supported on Silica and Carbon Materials: Effect of Support Property on Cobalt Species and FischerTropsch Synthesis Performance. Ind. Eng. Chem. Res. 2019, 58, 3459–3467. [Google Scholar] [CrossRef]
- Sicius, H. Cobaltgruppe: Eine Reise durch das Periodensystem; Springer Fachmedien Wiesbaden: Wiesbaden, Germany, 2016; ISBN 9783658163464. [Google Scholar]
- Haynes, W.M. CRC Handbook of Chemistry and Physics; CRC: Boca Raton, FL, USA, 2016. [Google Scholar]
- van Krevelen, D.W. Organic geochemistry—Old and new. Org. Geochem. 1984, 6, 1–10. [Google Scholar] [CrossRef]
- Krevelen, V. Graphical-statistical method for the study of structure and reaction processes of coal. Fuel 1950, 29, 269–284. [Google Scholar]
- Vollmer, S.; Witte, G.; Wöll, C. Copper supported on porous activated carbon obtained by wetness impregnation: Effect of preparation conditions on the ozonation catalyst’s characteristics. Catal Lett 2001, 77, 97–101. [Google Scholar] [CrossRef]
- Domke, W. Werkstoffkunde und Werkstoffprüfung, 10th ed.; Cornelsen-Velhagen u. Klasing: Düsseldorf/Bielefeld, Germany, 1986; ISBN 3-590-81220-6. [Google Scholar]
- Duong, N.; Tan, Q.; Resasco, D.E. Controlling phenolic hydrodeoxygenation by tailoring metal–O bond strength via specific catalyst metal type and particle size selection. Comptes Rendus Chim. 2018, 21, 155–163. [Google Scholar] [CrossRef]
- Greeley, J.; Nørskov, J.K. A general scheme for the estimation of oxygen binding energies on binary transition metal surface alloys. Surf. Sci. 2005, 592, 104–111. [Google Scholar] [CrossRef]
- Gates, B.C.; Knozinger, H. Impact of Surface Science on Catalysis; Academic Press: San Diego, CA, USA, 2000; ISBN 9780120078455. [Google Scholar]
- Bhattacharjee, S.; Waghmare, U.V.; Lee, S.-C. An improved d-band model of the catalytic activity of magnetic transition metal surfaces. Sci. Rep. 2016, 6, 35916. [Google Scholar] [CrossRef] [Green Version]
- Ranaware, V.; Verma, D.; Insyani, R.; Riaz, A.; Kim, S.M.; Kim, J. Highly-efficient and magnetically-separable ZnO/Co@N-CNTs catalyst for hydrodeoxygenation of lignin and its derived species under mild conditions. Green Chem. 2019, 21, 1021–1042. [Google Scholar] [CrossRef]
- Popov, A.; Kondratieva, E.; Goupil, J.M.; Mariey, L.; Bazin, P.; Gilson, J.P.; Travert, A.; Maugé, F. Bio-oils hydrodeoxygenation: Adsorption of phenolic molecules on oxidic catalyst supports. J. Phys. Chem. C 2010, 114, 15661–15670. [Google Scholar] [CrossRef]
- Zhao, J.; He, Y.; Wang, F.; Zheng, W.; Huo, C.; Liu, X.; Jiao, H.; Yang, Y.; Li, Y.; Wen, X. Suppressing Metal Leaching in a Supported Co/SiO2 Catalyst with Effective Protectants in the Hydroformylation Reaction. ACS Catal. 2020, 10, 914–920. [Google Scholar] [CrossRef]
- Blanco, E.; Dongil, A.B.; Escalona, N. Synergy between Ni and Co Nanoparticles Supported on Carbon in Guaiacol Conversion. Nanomaterials 2020, 10, 2199. [Google Scholar] [CrossRef]
- Putz, H.; Brandenburg, K. Match!—Phase Analysis Using Powder Diffraction; Crystal Impact: Bonn, Germany.
- De Boer, J.H.; Lippens, B.C.; Linsen, B.G.; Broekhoff, J.; van den Heuvel, A.; Osinga, T.J. Thet-curve of multimolecular N2-adsorption. J. Colloid Interface Sci. 1966, 21, 405–414. [Google Scholar] [CrossRef]
- Kretzschmar, N.; Seifert, M.; Busse, O.; Weigand, J.J. Prediction of Retention Indices and Response Factors of Oxygenates for GC-FID by Multilinear Regression. Data 2022, 7, 133. [Google Scholar] [CrossRef]
- Mann, J.B.; Meek, T.L.; Knight, E.T.; Capitani, J.F.; Allen, L.C. Configuration Energies of the d-Block Elements. J. Am. Chem. Soc. 2000, 122, 5132–5137. [Google Scholar] [CrossRef]
- Frese, K.W. Calculation of surface binding energy for hydrogen, oxygen, and carbon atoms on metallic surfaces. Surf. Sci. 1987, 182, 85–97. [Google Scholar] [CrossRef]
- Lee, J.; Xu, Y.; Huber, G.W. High-throughput screening of monometallic catalysts for aqueous-phase hydrogenation of biomass-derived oxygenates. Appl. Catal. B Environ. 2013, 140–141, 98–107. [Google Scholar] [CrossRef]
- Petrova, N.V.; Yakovkin, I.N. Binding energies for oxygen on transition metal surfaces. Surf. Rev. Lett. 2009, 16, 291–296. [Google Scholar] [CrossRef]























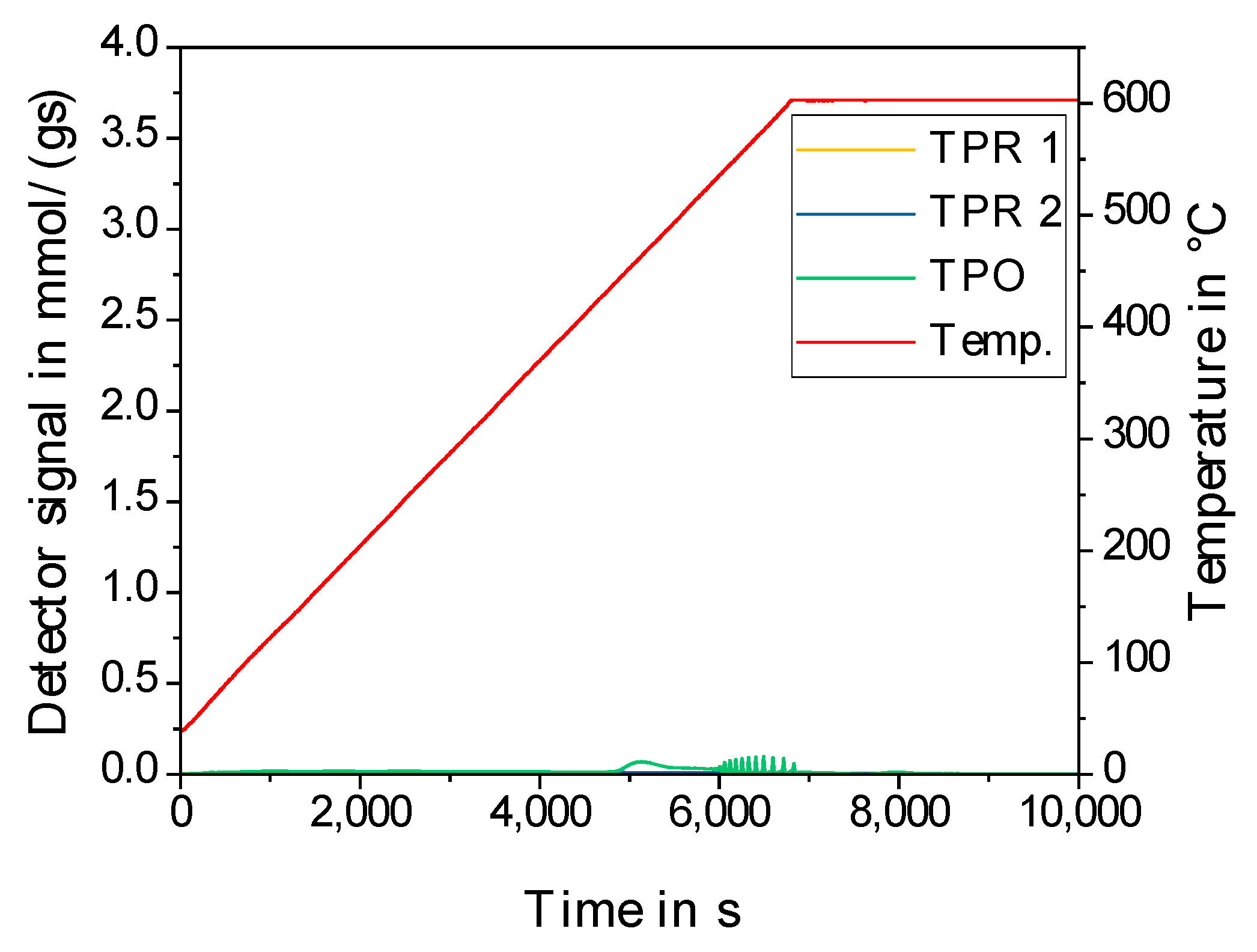
| Results of P-XRD | Physisorption of Nitrogen | Results of TPR 2 c | ||||
|---|---|---|---|---|---|---|
| Phase Analysis | Crystal Size after TPROR in nm | Specific Surface Area | Exp. in µmol (H2)/g | Theor. in µmol (H2)/g | ||
| After Calcination (Oxidized) | After Catalytic Test (Reduced) | B.E.T. in m2/g | B.J.H. in m2/g | |||
| MoO3 | MoO2 | 207 | 250 | 190 | 1710 | 2980 |
| WO3 | (W) a | n.d. a | 305 | 260 | 970 c | 1590 |
| NH4ReO4 | Re | 26 | 290 | 260 | 2015 | 2060 |
| n.d. b | Fe | n.d. b | 305 | 270 | 1430 | 2575 |
| Co3O4 | Co | 149 | 310 | 200 | 3990 | 2183 |
| NiO | Ni | 27 d | 310 | 285 | 2345 | 1660 |
| n.d. | Pd | 6 e | 325 | 275 | 0 | 90 |
| CuO | Cu | 42 | 285 | 255 | 1950 | 1535 |
| Ag2O + Ag | Ag | 33 | 240 | 190 | 0 | 460 |
| Step | Ramp in K/min | TTarget in °C | Dwell Time in h |
|---|---|---|---|
| 1 | 1.1 | 120 | 1 |
| 2 | 1.1 | 540 | 12 |
| Phase | Duration in min | TStart in °C | TZiel in °C | Ramp in K/h | Dwell Time in min | Gas |
|---|---|---|---|---|---|---|
| Drying | 120 | RT | 120 | 180 | 80 | N2 |
| Heating | 180 | 120 | 550 | 180 | 37 | N2 |
| Oxidation | 120 | 550 | 550 | 0 | 120 | Syn. Air |
| Flushing N2 | 15 | 550 | 550 | 0 | 15 | N2 |
| Reduction | 120 | 550 | 550 | 0 | 120 | H2 |
| Cooling | 180 | 550 | RT | - | - | H2 |
| ToS in h | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|---|---|---|
| Temperatur in °C | 250 | 280 | 310 | 340 | 370 | 400 | 340 | 340 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kretzschmar, N.; Busse, O.; Seifert, M. Impact of Geometric and Electronic Factors on Selective Hydro-Deoxygenation of Guaiacol by Surface-Rich Metal/Silica Catalysts. Catalysts 2023, 13, 425. https://doi.org/10.3390/catal13020425
Kretzschmar N, Busse O, Seifert M. Impact of Geometric and Electronic Factors on Selective Hydro-Deoxygenation of Guaiacol by Surface-Rich Metal/Silica Catalysts. Catalysts. 2023; 13(2):425. https://doi.org/10.3390/catal13020425
Chicago/Turabian StyleKretzschmar, Nils, Oliver Busse, and Markus Seifert. 2023. "Impact of Geometric and Electronic Factors on Selective Hydro-Deoxygenation of Guaiacol by Surface-Rich Metal/Silica Catalysts" Catalysts 13, no. 2: 425. https://doi.org/10.3390/catal13020425
APA StyleKretzschmar, N., Busse, O., & Seifert, M. (2023). Impact of Geometric and Electronic Factors on Selective Hydro-Deoxygenation of Guaiacol by Surface-Rich Metal/Silica Catalysts. Catalysts, 13(2), 425. https://doi.org/10.3390/catal13020425