
Transient Absorption Spectrum Analysis for Photothermal Catalysis Perovskite Materials

Abstract
:1. Introduction
2. Results and Discussion
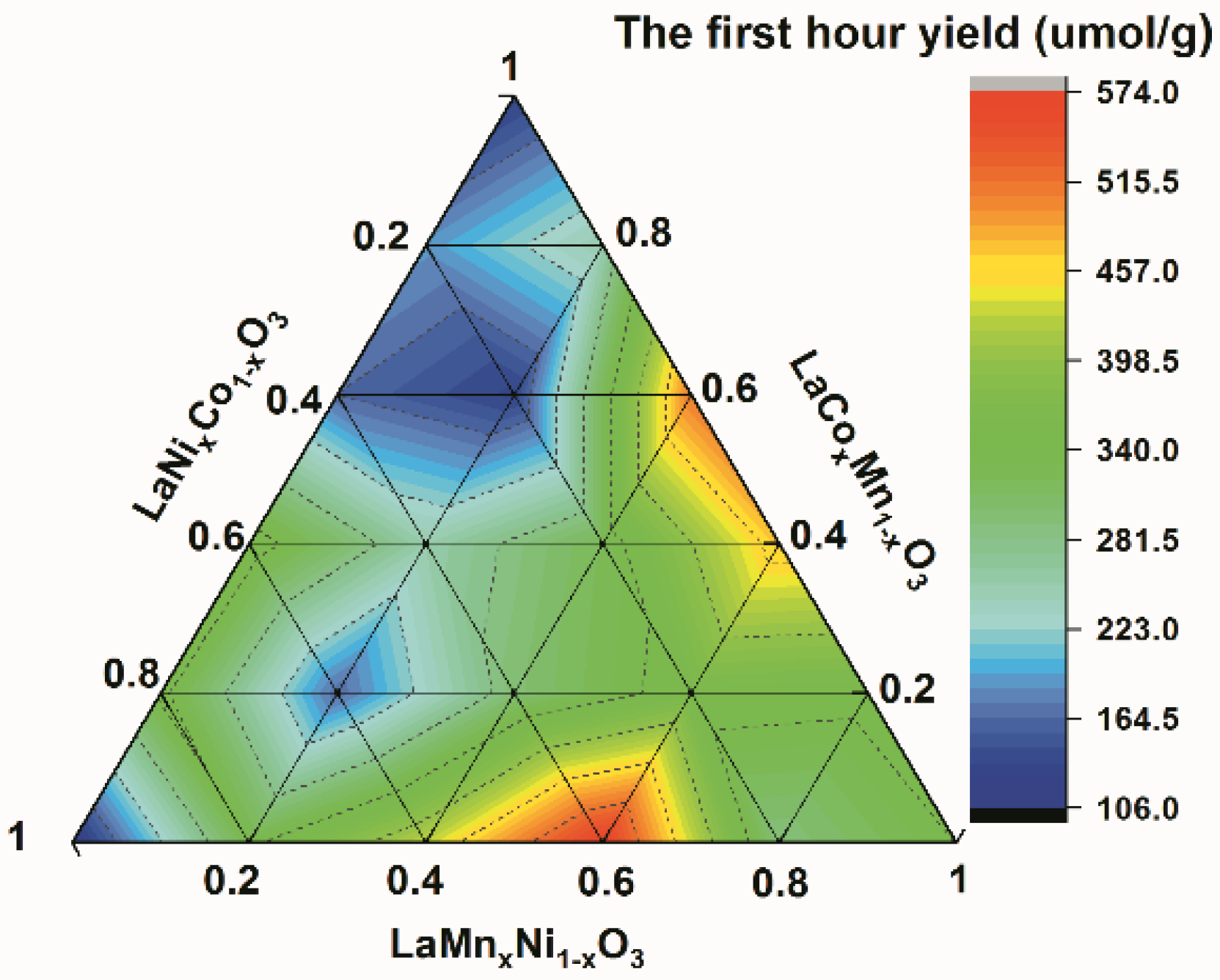
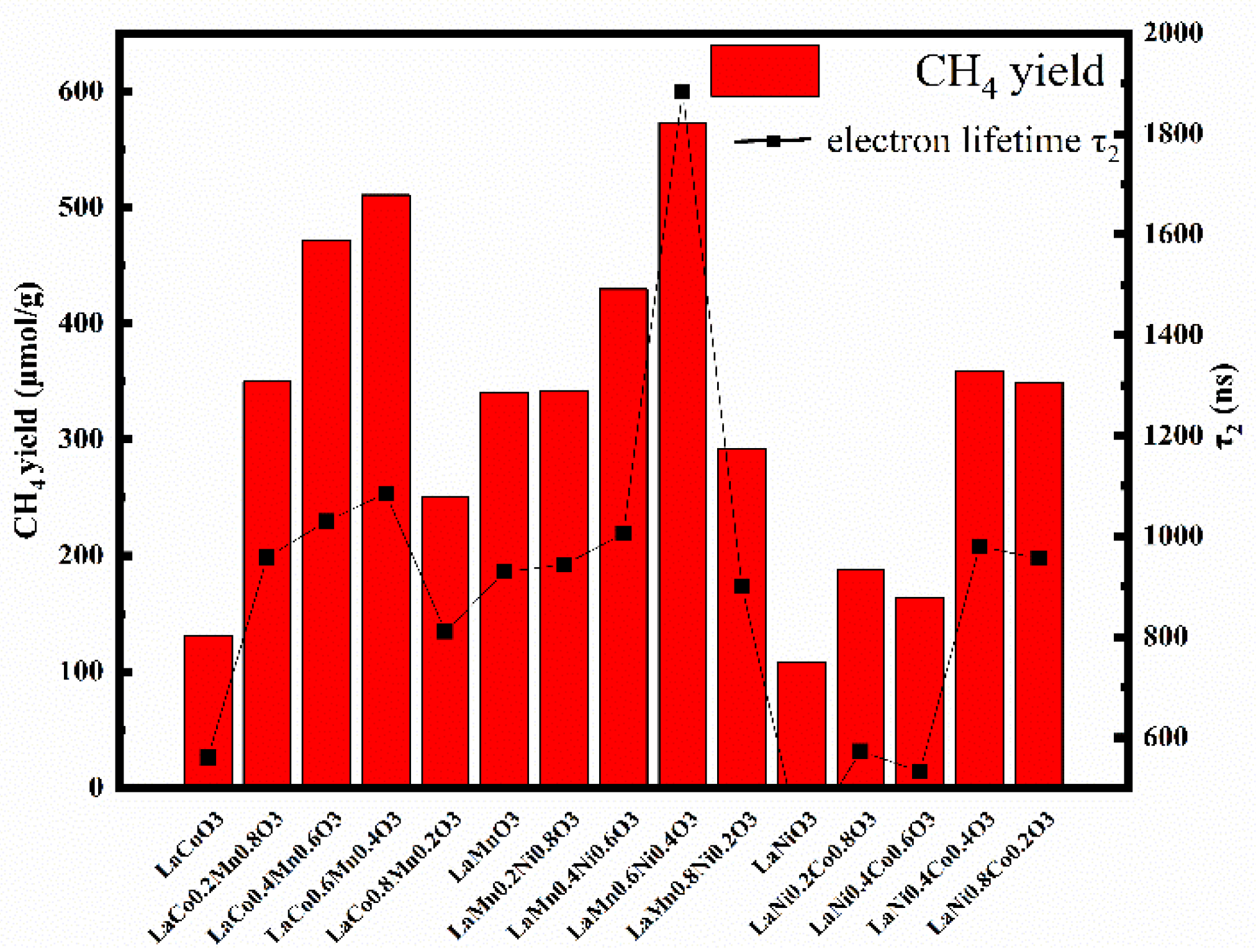
2.1. Basic Material Properties and Catalysis Performance
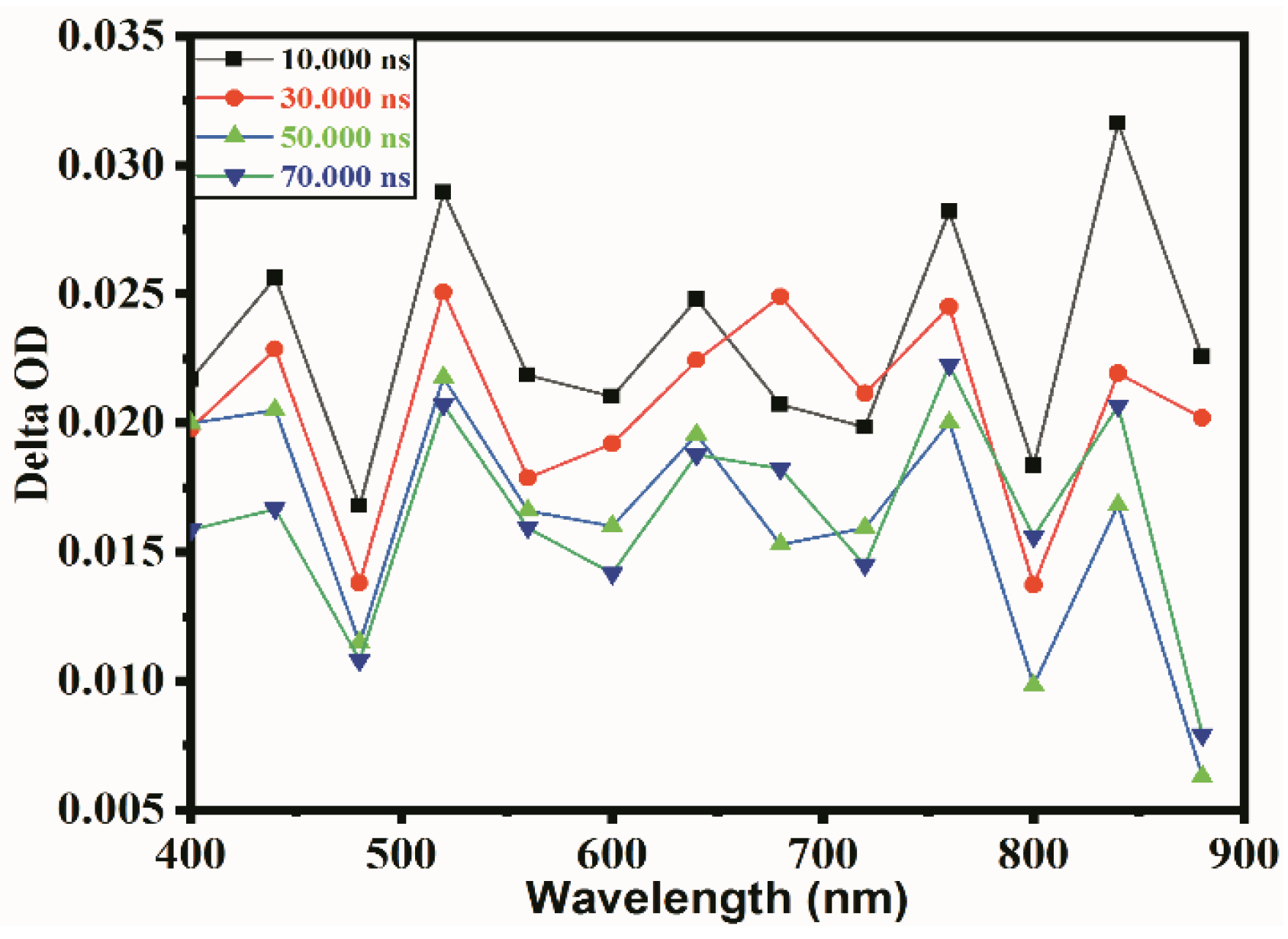
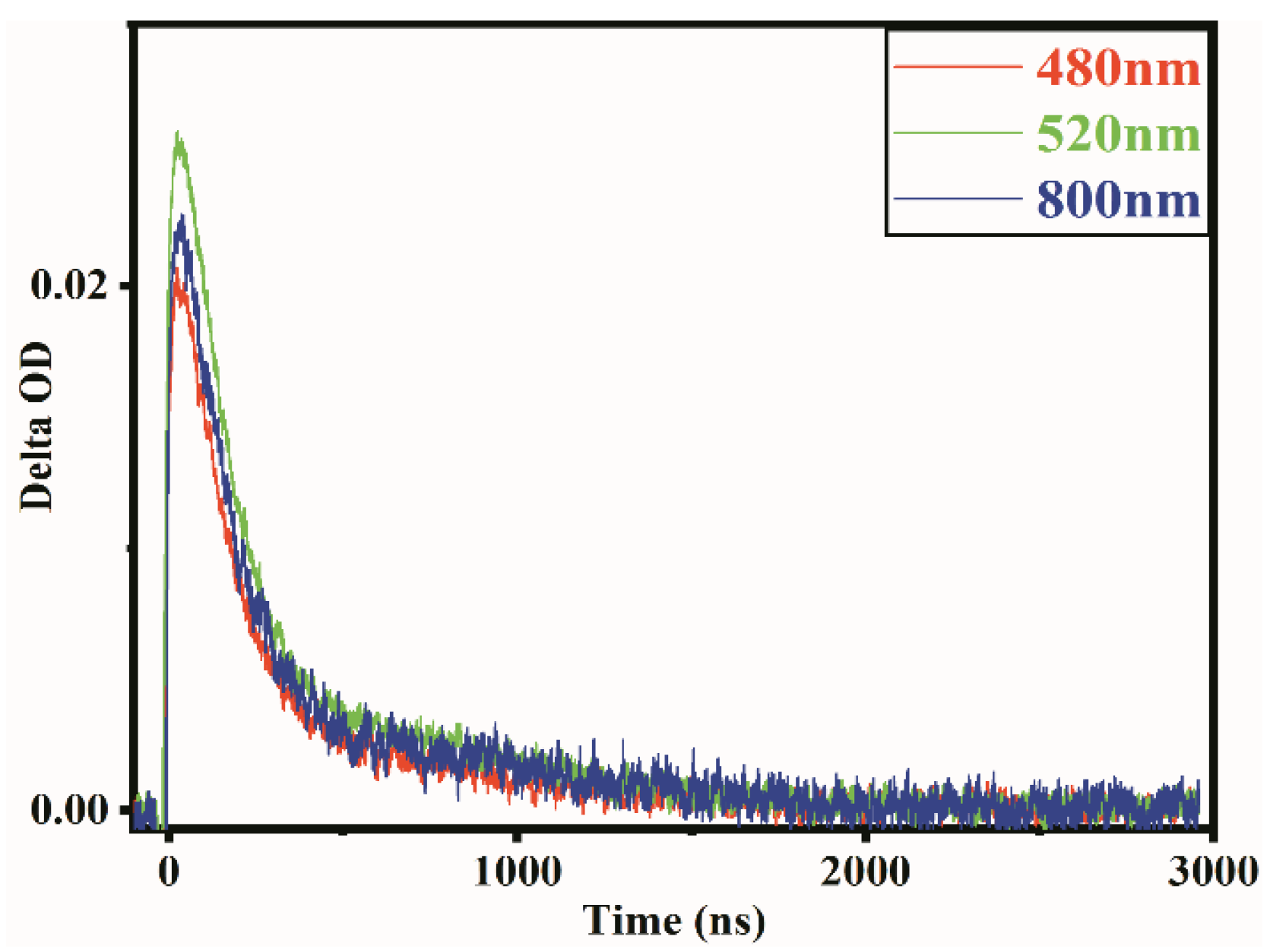
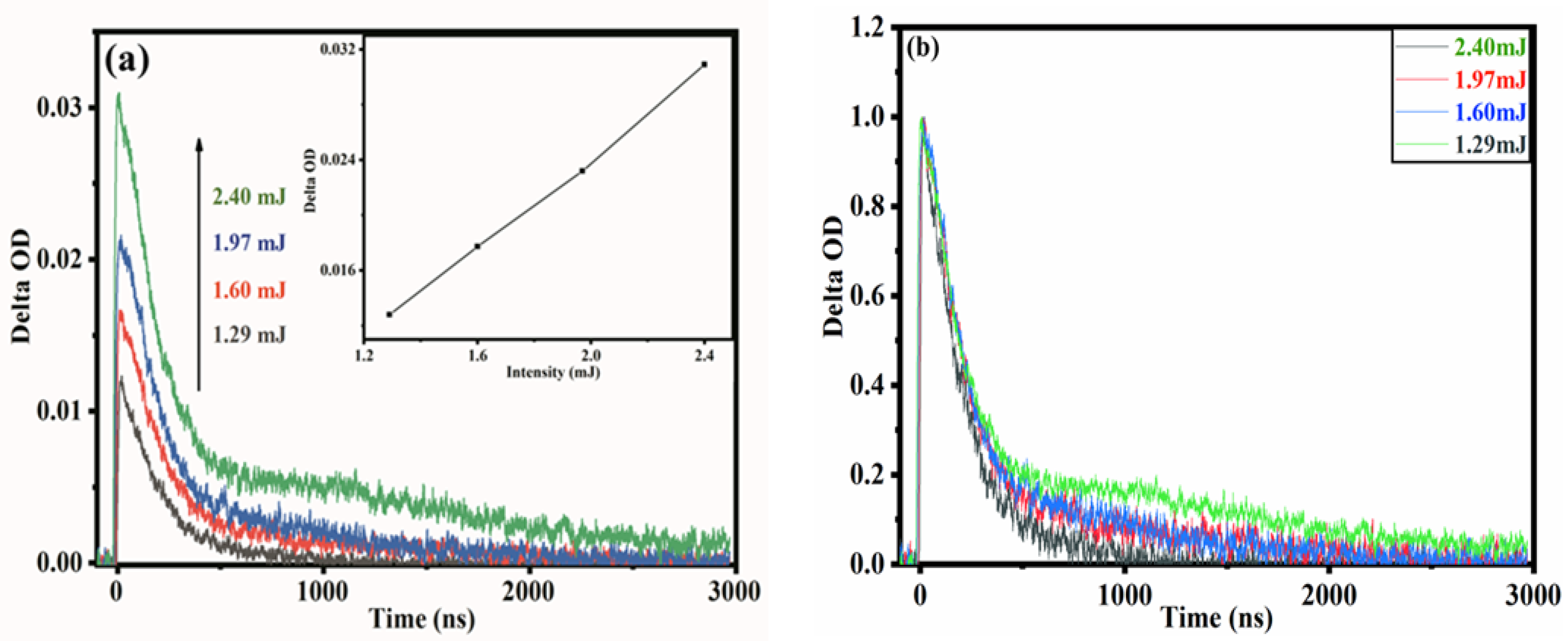
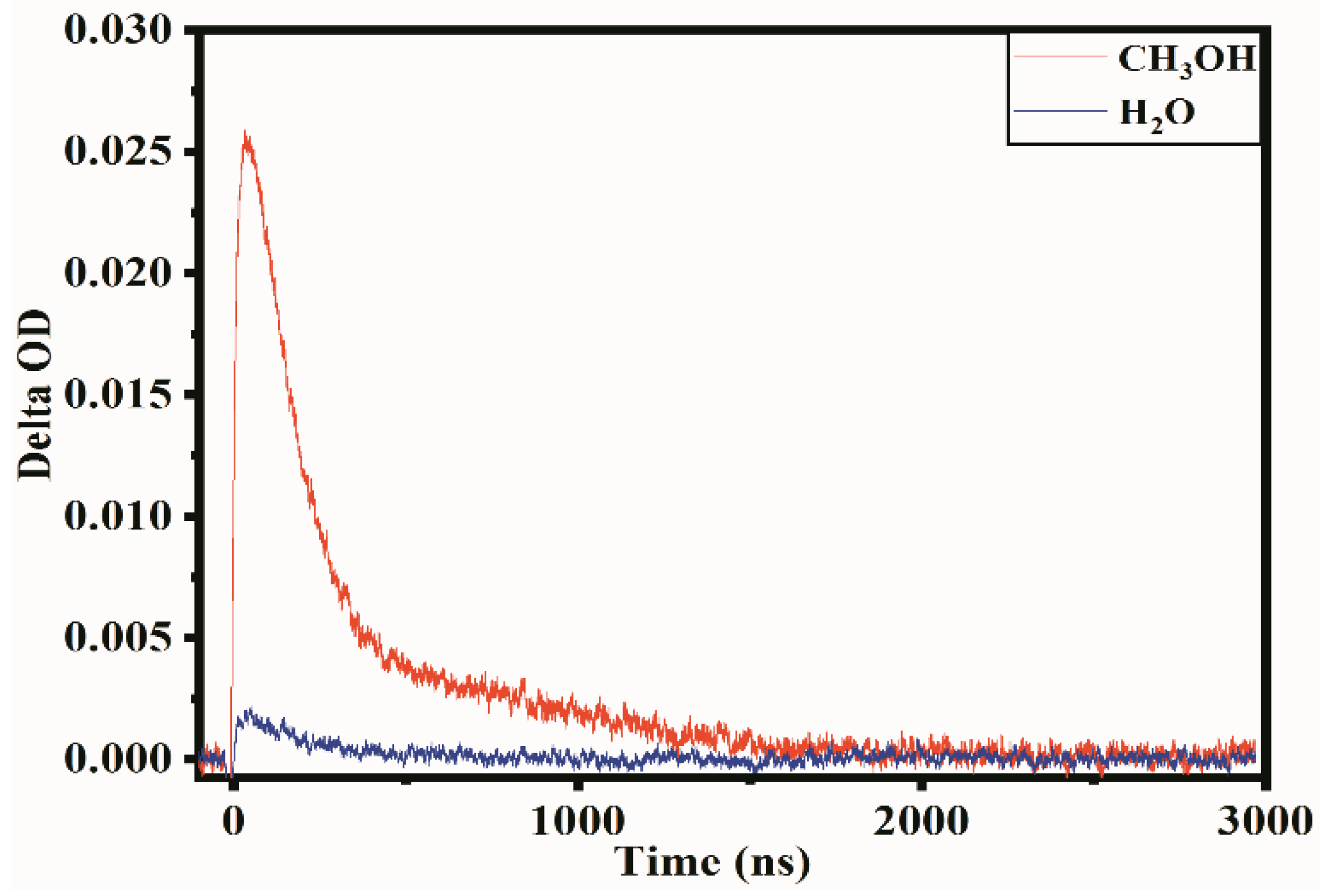
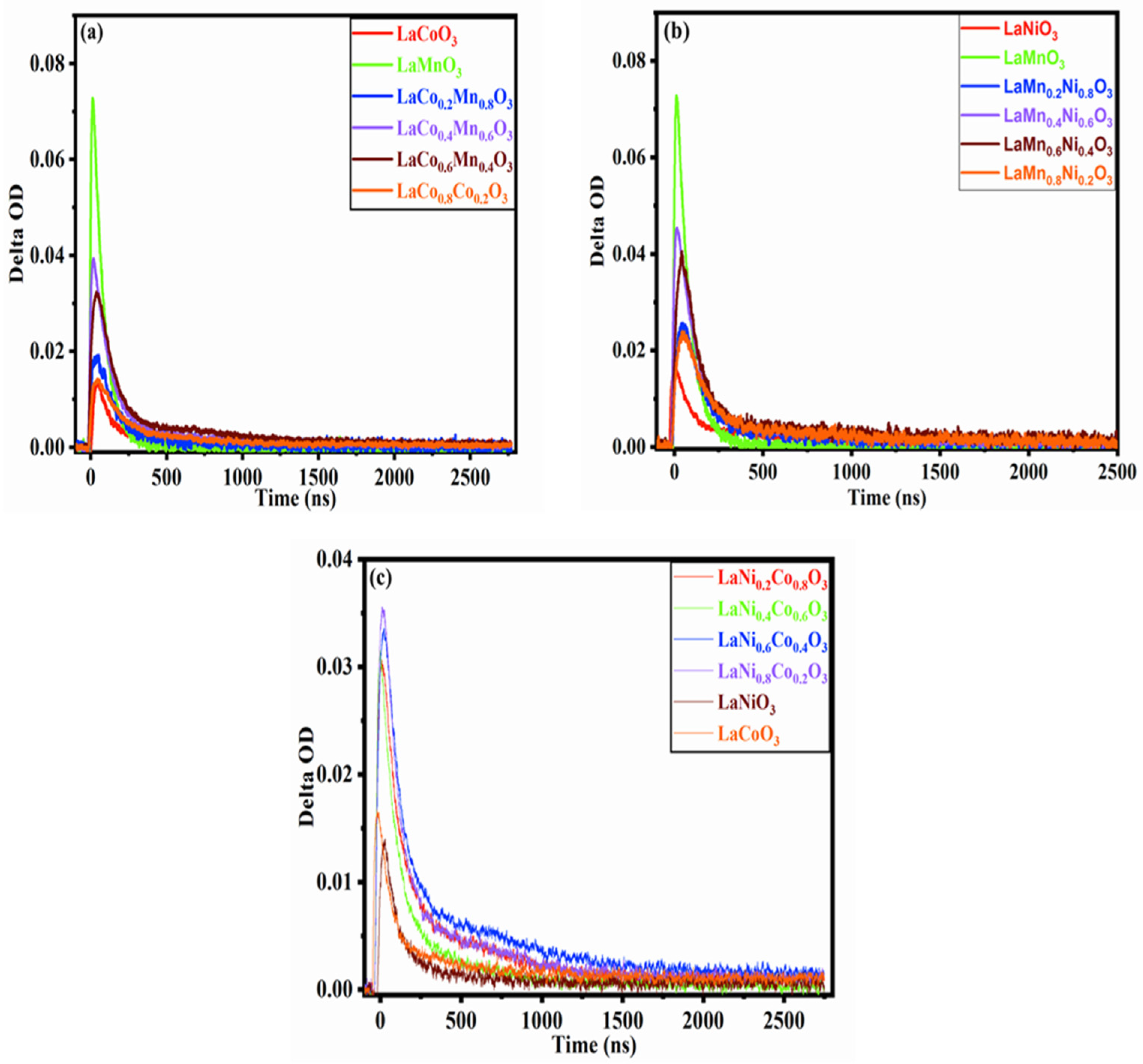
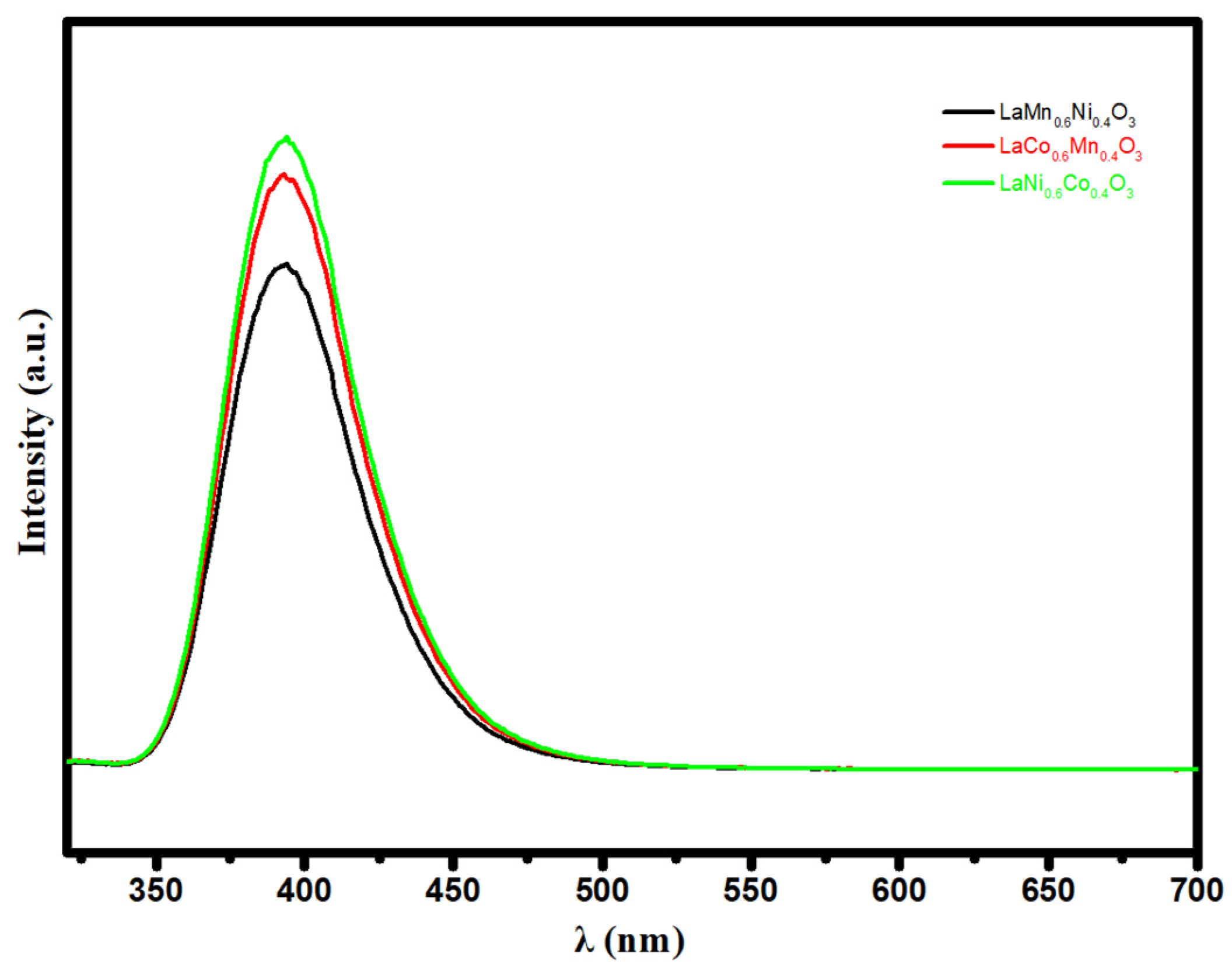
2.2. Transient Absorption Spectrum Analysis
3. Materials and Methods
3.1. Materials
3.2. Catalytic Measurements
3.3. Transient Absorption Spectrum Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Stolarczyk, J.K.; Bhattacharyya, S.; Polavarapu, L.; Feldmann, J. Challenges and Prospects in Solar Water Splitting and CO2 Reduction with Inorganic and Hybrid Nanostructures. ACS Catal. 2018, 8, 3602–3635. [Google Scholar] [CrossRef]
- Shi, R.; Waterhouse, G.I.N. Recent Progress in Photocatalytic CO2 Reduction Over Perovskite Oxides. Sol. RRL 2017, 1, 1700126. [Google Scholar] [CrossRef]
- Neaţu, Ş.; Maciá-Agulló, J.A.; Concepción, P.; Garcia, H. Gold–Copper Nanoalloys Supported on TiO2 as Photocatalysts for CO2 Reduction by Water. J. Am. Chem. Soc. 2014, 136, 15969–15976. [Google Scholar] [CrossRef]
- Abdullah, H.; Khan, M.M.R.; Ong, H.R.; Yaakob, Z. Modified TiO2 Photocatalyst for CO2 Photocatalytic Reduction: An Overview. J. CO2 Util. 2017, 22, 15–32. [Google Scholar] [CrossRef]
- Li, K.; Peng, B.; Peng, T. Recent Advances in Heterogeneous Photocatalytic CO2 Conversion to Solar Fuels. ACS Catal. 2016, 6, 7485–7527. [Google Scholar] [CrossRef]
- Zeng, S.; Kar, P.; Thakur, U.K.; Shankar, K. A Review on Photocatalytic CO2 Reduction Using Perovskite Oxide Nanomaterials. Nanotechnology 2018, 29, 052001. [Google Scholar] [CrossRef]
- Li, Y.; Wang, W.-N.; Zhan, Z.; Woo, M.-H.; Wu, C.-Y.; Biswas, P. Photocatalytic Reduction of CO2 with H2O on Mesoporous Silica Supported Cu/TiO2 Catalysts. Appl. Catal. B: Environ. 2010, 100, 386–392. [Google Scholar] [CrossRef]
- Ye, L.; Wu, D.; Chu, K.H.; Wang, B.; Xie, H.; Yip, H.Y.; Wong, P.K. Phosphorylation of G-C3N4 for Enhanced Photocatalytic CO2 Reduction. Chem. Eng. J. 2016, 304, 376–383. [Google Scholar] [CrossRef]
- Matsubara, Y.; Grills, D.C.; Kuwahara, Y. Thermodynamic Aspects of Electrocatalytic CO2 Reduction in Acetonitrile and with an Ionic Liquid as Solvent or Electrolyte. ACS Catal. 2015, 5, 6440–6452. [Google Scholar] [CrossRef]
- Izumi, Y. Recent Advances in the Photocatalytic Conversion of Carbon Dioxide to Fuels with Water and/or Hydrogen Using Solar Energy and Beyond. Coord. Chem. Rev. 2013, 257, 171–186. [Google Scholar] [CrossRef] [Green Version]
- Qian, R.; Zong, H.; Schneider, J.; Zhou, G.; Zhao, T.; Li, Y.; Yang, J.; Bahnemann, D.W.; Pan, J.H. Charge Carrier Trapping, Recombination and Transfer during TiO2 Photocatalysis: An Overview. Catal. Today 2019, 335, 78–90. [Google Scholar] [CrossRef]
- Zhu, H.; Zhang, P.; Dai, S. Recent Advances of Lanthanum-Based Perovskite Oxides for Catalysis. ACS Catal. 2015, 5, 6370–6385. [Google Scholar] [CrossRef]
- Yan, J.-Q.; Zhou, J.-S.; Goodenough, J.B. Ferromagnetism in LaCoO3. Phys. Rev. B 2004, 70, 014402. [Google Scholar] [CrossRef]
- Abdolrahmani, M.; Parvari, M.; Habibpoor, M. Effect of Copper Substitution and Preparation Methods on the LaMnO3±δ Structure and Catalysis of Methane Combustion and CO Oxidation. Chin. J. Catal. 2010, 31, 394–403. [Google Scholar] [CrossRef]
- Dhinesh Kumar, R.; Thangappan, R.; Jayavel, R. Enhanced Visible Light Photocatalytic Activity of LaMnO3 Nanostructures for Water Purification. Res. Chem. Intermed. 2018, 44, 4323–4337. [Google Scholar] [CrossRef]
- Gao, J.; Jia, L.; Fang, W.; Li, Q.; Song, H. Methanation of Carbon Dioxide over the LaNiO3 Perovskite Catalysts Activated under the Reactant Stream. J. Fuel Chem. Technol. 2009, 37, 573–577. [Google Scholar] [CrossRef]
- Xie, K.; Umezawa, N.; Zhang, N.; Reunchan, P.; Zhang, Y.; Ye, J. Self-Doped SrTiO3−δ Photocatalyst with Enhanced Activity for Artificial Photosynthesis under Visible Light. Energy Environ. Sci. 2011, 4, 4211–4219. [Google Scholar] [CrossRef]
- Xu, L.; Ha, M.N.; Guo, Q.; Wang, L.; Ren, Y.; Sha, N.; Zhao, Z. Photothermal Catalytic Activity of Combustion Synthesized LaCoxFe1−xO3 (0 ≤ x ≤ 1) Perovskite for CO2 Reduction with H2O to CH4 and CH3OH. RSC Adv. 2017, 7, 45949–45959. [Google Scholar] [CrossRef] [Green Version]
- Zheng, D.; Wei, G.; Xu, L.; Guo, Q.; Hu, J.; Sha, N.; Zhao, Z. LaNixFe1−xO3 (0 ≤ x ≤1) as Photothermal Catalysts for Hydrocarbon Fuels Production from CO2 and H2O. J. Photochem. Photobiol. A Chem. 2019, 377, 182–189. [Google Scholar] [CrossRef]
- Wei, G.; Zheng, D.; Xu, L.; Guo, Q.; Hu, J.; Sha, N.; Zhao, Z. Photothermal Catalytic Activity and Mechanism of LaNixCo1−xO3 (0 ≦ x ≦ 1) Perovskites for CO2 Reduction to CH4 and CH3OH with H2O. Mater. Res. Express 2019, 6, 086221. [Google Scholar] [CrossRef]
- Ha, M.N.; Lu, G.; Liu, Z.; Wang, L.; Zhao, Z. 3DOM-LaSrCoFeO6−δ as a Highly Active Catalyst for the Thermal and Photothermal Reduction of CO2 with H2O to CH4. J. Mater. Chem. A 2016, 4, 13155–13165. [Google Scholar] [CrossRef]
- Xie, S.; Zhang, Q.; Liu, G.; Wang, Y. Photocatalytic and Photoelectrocatalytic Reduction of CO2 Using Heterogeneous Catalysts with Controlled Nanostructures. Chem. Commun. 2016, 52, 35–59. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Wang, T.; Gong, J. CO2 Photo-Reduction: Insights into CO2 Activation and Reaction on Surfaces of Photocatalysts. Energy Environ. Sci. 2016, 9, 2177–2196. [Google Scholar] [CrossRef]
- Habisreutinger, S.N.; Schmidt-Mende, L.; Stolarczyk, J.K. Photocatalytic Reduction of CO2 on TiO2 and Other Semiconductors. Angew. Chem. Int. Ed. 2013, 52, 7372–7408. [Google Scholar] [CrossRef]
- Iwata, K.; Takaya, T.; Hamaguchi, H.; Yamakata, A.; Ishibashi, T.; Onishi, H.; Kuroda, H. Carrier Dynamics in TiO2 and Pt/TiO2 Powders Observed by Femtosecond Time-Resolved Near-Infrared Spectroscopy at a Spectral Region of 0.9–1.5 μm with the Direct Absorption Method. J. Phys. Chem. B 2004, 108, 20233–20239. [Google Scholar] [CrossRef]
- Cowan, A.J.; Barnett, C.J.; Pendlebury, S.R.; Barroso, M.; Sivula, K.; Grätzel, M.; Durrant, J.R.; Klug, D.R. Activation Energies for the Rate-Limiting Step in Water Photooxidation by Nanostructured α-Fe2O3 and TiO2. J. Am. Chem. Soc. 2011, 133, 10134–10140. [Google Scholar] [CrossRef]
- Cowan, A.J.; Tang, J.; Leng, W.; Durrant, J.R.; Klug, D.R. Water Splitting by Nanocrystalline TiO2 in a Complete Photoelectrochemical Cell Exhibits Efficiencies Limited by Charge Recombination. J. Phys. Chem. C 2010, 114, 4208–4214. [Google Scholar] [CrossRef]
- Tang, J.; Durrant, J.R.; Klug, D.R. Mechanism of Photocatalytic Water Splitting in TiO2. Reaction of Water with Photoholes, Importance of Charge Carrier Dynamics, and Evidence for Four-Hole Chemistry. J. Am. Chem. Soc. 2008, 130, 13885–13891. [Google Scholar] [CrossRef]
- Tang, J.; Cowan, A.J.; Durrant, J.R.; Klug, D.R. Mechanism of O2 Production from Water Splitting: Nature of Charge Carriers in Nitrogen Doped Nanocrystalline TiO2 Films and Factors Limiting O2 Production. J. Phys. Chem. C 2011, 115, 3143–3150. [Google Scholar] [CrossRef]
- Pesci, F.M.; Cowan, A.J.; Alexander, B.D.; Durrant, J.R.; Klug, D.R. Charge Carrier Dynamics on Mesoporous WO3 during Water Splitting. J. Phys. Chem. Lett. 2011, 2, 1900–1903. [Google Scholar] [CrossRef]
- Sachs, M.; Pastor, E.; Kafizas, A.; Durrant, J.R. Evaluation of Surface State Mediated Charge Recombination in Anatase and Rutile TiO2. J. Phys. Chem. Lett. 2016, 7, 3742–3746. [Google Scholar] [CrossRef] [Green Version]
- Vasileff, A.; Xu, C.; Jiao, Y.; Zheng, Y.; Qiao, S.-Z. Surface and Interface Engineering in Copper-Based Bimetallic Materials for Selective CO2 Electroreduction. Chem 2018, 4, 1809–1831. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Lim, C.; Zhou, M.; He, Z.; Sun, X.; Li, X.; Ye, Y.; Tan, T.; Zhang, H.; Yang, C.; et al. Activating Lattice Oxygen in Perovskite Oxide by B-Site Cation Doping for Modulated Stability and Activity at Elevated Temperatures. Adv. Sci. 2021, 8, 2102713. [Google Scholar] [CrossRef]
- Kim, B.-J.; Fabbri, E.; Abbott, D.F.; Cheng, X.; Clark, A.H.; Nachtegaal, M.; Borlaf, M.; Castelli, I.E.; Graule, T.; Schmidt, T.J. Functional Role of Fe-Doping in Co-Based Perovskite Oxide Catalysts for Oxygen Evolution Reaction. J. Am. Chem. Soc. 2019, 141, 5231–5240. [Google Scholar] [CrossRef] [Green Version]
- Usubharatana, P.; McMartin, D.; Veawab, A.; Tontiwachwuthikul, P. Photocatalytic Process for CO2 Emission Reduction from Industrial Flue Gas Streams. Ind. Eng. Chem. Res. 2006, 45, 2558–2568. [Google Scholar] [CrossRef]
- Tamaki, Y.; Furube, A.; Murai, M.; Hara, K.; Katoh, R.; Tachiya, M. Direct Observation of Reactive Trapped Holes in TiO2 Undergoing Photocatalytic Oxidation of Adsorbed Alcohols: Evaluation of the Reaction Rates and Yields. J. Am. Chem. Soc. 2006, 128, 416–417. [Google Scholar] [CrossRef] [PubMed]
- Baldoví, H.G.; Neaţu, Ş.; Khan, A.; Asiri, A.M.; Kosa, S.A.; Garcia, H. Understanding the Origin of the Photocatalytic CO2 Reduction by Au- and Cu-Loaded TiO2: A Microsecond Transient Absorption Spectroscopy Study. J. Phys. Chem. C 2015, 119, 6819–6827. [Google Scholar] [CrossRef]
- Katoh, R.; Murai, M.; Furube, A. Transient Absorption Spectra of Nanocrystalline TiO2 Films at High Excitation Density. Chem. Phys. Lett. 2010, 500, 309–312. [Google Scholar] [CrossRef]
- Bahnemann, D.W.; Hilgendorff, M.; Memming, R. Charge Carrier Dynamics at TiO2 Particles: Reactivity of Free and Trapped Holes. J. Phys. Chem. B 1997, 101, 4265–4275. [Google Scholar] [CrossRef]
- Baldoví, H.G.; Ferrer, B.; Álvaro, M.; García, H. Microsecond Transient Absorption Spectra of Suspended Semiconducting Metal Oxide Nanoparticles. J. Phys. Chem. C 2014, 118, 9275–9282. [Google Scholar] [CrossRef]
- Yoshihara, T.; Katoh, R.; Furube, A.; Tamaki, Y.; Murai, M.; Hara, K.; Murata, S.; Arakawa, H.; Tachiya, M. Identification of Reactive Species in Photoexcited Nanocrystalline TiO2 Films by Wide-Wavelength-Range (400–2500 nm) Transient Absorption Spectroscopy. J. Phys. Chem. B 2004, 108, 3817–3823. [Google Scholar] [CrossRef]
- Ma, Y.; Pendlebury, S.R.; Reynal, A.; Le Formal, F.; Durrant, J.R. Dynamics of Photogenerated Holes in Undoped BiVO4 Photoanodes for Solar Water Oxidation. Chem. Sci. 2014, 5, 2964–2973. [Google Scholar] [CrossRef] [Green Version]
- Le Formal, F.; Pendlebury, S.R.; Cornuz, M.; Tilley, S.D.; Grätzel, M.; Durrant, J.R. Back Electron–Hole Recombination in Hematite Photoanodes for Water Splitting. J. Am. Chem. Soc. 2014, 136, 2564–2574. [Google Scholar] [CrossRef] [PubMed]
- Shirai, K.; Fazio, G.; Sugimoto, T.; Selli, D.; Ferraro, L.; Watanabe, K.; Haruta, M.; Ohtani, B.; Kurata, H.; Di Valentin, C.; et al. Water-Assisted Hole Trapping at the Highly Curved Surface of Nano-TiO2 Photocatalyst. J. Am. Chem. Soc. 2018, 140, 1415–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.A.; Maity, P.; Al-Oufi, M.; Al-Howaish, I.K.; Idriss, H. Electron Transfer of the Metal/Semiconductor System in Photocatalysis. J. Phys. Chem. C 2018, 122, 16779–16787. [Google Scholar] [CrossRef]
- Mishra, A.; Bhattacharjee, S. Effect of A- or B-Site Doping of Perovskite Calcium Manganite on Structure, Resistivity, and Thermoelectric Properties. J. Am. Ceram. Soc. 2017, 100, 4945–4953. [Google Scholar] [CrossRef]
- Kobosko, S.M.; DuBose, J.T.; Kamat, P.V. Perovskite Photocatalysis. Methyl Viologen Induces Unusually Long-Lived Charge Carrier Separation in CsPbBr3 Nanocrystals. ACS Energy Lett. 2020, 5, 221–223. [Google Scholar] [CrossRef] [Green Version]
- Lai, T.-H.; Katsumata, K.; Hsu, Y.-J. In Situ Charge Carrier Dynamics of Semiconductor Nanostructures for Advanced Photoelectrochemical and Photocatalytic Applications. Nanophotonics 2020, 10, 777–795. [Google Scholar] [CrossRef]
- Pendlebury, S.R.; Wang, X.; Le Formal, F.; Cornuz, M.; Kafizas, A.; Tilley, S.D.; Grätzel, M.; Durrant, J.R. Ultrafast Charge Carrier Recombination and Trapping in Hematite Photoanodes under Applied Bias. J. Am. Chem. Soc. 2014, 136, 9854–9857. [Google Scholar] [CrossRef] [Green Version]
- Bhosale, S.S.; Kharade, A.K.; Jokar, E.; Fathi, A.; Chang, S.; Diau, E.W.-G. Mechanism of Photocatalytic CO2 Reduction by Bismuth-Based Perovskite Nanocrystals at the Gas–Solid Interface. J. Am. Chem. Soc. 2019, 141, 20434–20442. [Google Scholar] [CrossRef]
- Hung, P.-H.; Vequizo, J.J.M.; Wu, R.-A.; Yamakata, A.; Tseng, W.J. Effect of CuFe2O4 Ferrite on Photocatalysis and Carrier Dynamics of Electrospun α-Fe2O3 Nanofibers by Time-Resolved Transient Absorption Spectroscopy. Ceram. Int. 2019, 45, 15676–15680. [Google Scholar] [CrossRef]
- Baiyee, Z.M.; Chen, C.; Ciucci, F. A DFT+U Study of A-Site and B-Site Substitution in BaFeO3−δ. Phys. Chem. Chem. Phys. 2015, 17, 23511–23520. [Google Scholar] [CrossRef]
- Alay-e-Abbas, S.M.; Javed, F.; Abbas, G.; Amin, N.; Laref, A. Density Functional Theory Evaluation of Ceramics Suitable for Hybrid Advanced Oxidation Processes: A Case Study for Ce4+-Doped BaZrO3. J. Phys. Chem. C 2019, 123, 6044–6053. [Google Scholar] [CrossRef]
- Phanichphant, S.; Nakaruk, A.; Chansaenpak, K.; Channei, D. Evaluating the Photocatalytic Efficiency of the BiVO4/RGO Photocatalyst. Sci. Rep. 2019, 9, 16091. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wang, Y.; Cheng, Y.; Liu, Z.; Guo, Q.; Ha, M.N.; Zhao, Z. Hydrogen-Treated Mesoporous WO3 as a Reducing Agent of CO2 to Fuels (CH4 and CH3OH) with Enhanced Photothermal Catalytic Performance. J. Mater. Chem. A 2016, 4, 5314–5322. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | τ1 (ns) | τ2 (ns) |
|---|---|---|
| LaCoO3 | 70 | 560 |
| LaCo0.2Mn0.8O3 | 105 | 958 |
| LaCo0.4Mn0.6O3 | 104 | 1030 |
| LaCo0.6Mn0.4O3 | 107 | 1085 |
| LaCo0.8Mn0.2O3 | 159 | 810 |
| LaMnO3 | 72 | 929 |
| LaMn0.2Ni0.8O3 | 106 | 943 |
| LaMn0.4Ni0.6O3 | 103 | 1005 |
| LaMn0.6Ni0.4O3 | 95 | 1884 |
| LaMn0.8Ni0.2O3 | 121 | 900 |
| LaNiO3 | 83 | 383 |
| LaNi0.2Co0.8O3 | 89 | 573 |
| LaNi0.4Co0.6O3 | 101 | 532 |
| LaNi0.6Co0.4O3 | 96 | 980 |
| LaNi0.8Co0.2O3 | 92 | 956 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, J.; Liu, L.; Nian, H.; Guo, Q.; Sha, N.; Zhao, Z. Transient Absorption Spectrum Analysis for Photothermal Catalysis Perovskite Materials. Catalysts 2023, 13, 452. https://doi.org/10.3390/catal13030452
Tian J, Liu L, Nian H, Guo Q, Sha N, Zhao Z. Transient Absorption Spectrum Analysis for Photothermal Catalysis Perovskite Materials. Catalysts. 2023; 13(3):452. https://doi.org/10.3390/catal13030452
Chicago/Turabian StyleTian, Jindan, Lili Liu, Hongqiang Nian, Qiangsheng Guo, Na Sha, and Zhe Zhao. 2023. "Transient Absorption Spectrum Analysis for Photothermal Catalysis Perovskite Materials" Catalysts 13, no. 3: 452. https://doi.org/10.3390/catal13030452
APA StyleTian, J., Liu, L., Nian, H., Guo, Q., Sha, N., & Zhao, Z. (2023). Transient Absorption Spectrum Analysis for Photothermal Catalysis Perovskite Materials. Catalysts, 13(3), 452. https://doi.org/10.3390/catal13030452