

CO Oxidation Reaction by Platinum Clusters on the Surface of Multiwalled Carbon Nanotubes: Experimental and Theoretical Study of Kinetics in a Wide Range of O2/CO Ratios

 , , , ,
, , , ,  , and
, and
Abstract
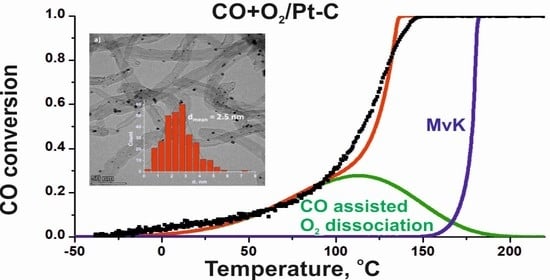
:
1. Introduction
2. Results and Discussion
2.1. Oxidation State of Platinum and Its Structure in the Pt-C Catalyst
2.2. Experimental Study of the Kinetics of the CO + O2 Reaction on Bulk (Pt Foil) and Highly Dispersed Platinum (Pt-C)
2.3. Results of the Mathematical Modelling: Comparison with the Data of the Kinetic Experiments
2.3.1. Mechanism and Mathematical Model of the Reaction over Bulk Pt Foil
2.3.2. Mathematical Modelling of the Dynamics of CO Oxidation over Pt-C
3. Materials and Methods
3.1. Synthesis
Synthesis of the MWCNTs and the Catalysts
3.2. Physicochemical Methods
3.3. Catalytic Properties
3.3.1. Temperature-Programmed Reaction (TPR-CO + O2)
3.3.2. Isothermal Experiments
3.3.3. Temperature-Programmed Desorption of CO (TPD-CO)
3.3.4. Temperature-Programmed Reaction with CO (TPR-CO)
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | O2/CO | T10, °C | T50, °C | T90, °C |
|---|---|---|---|---|
| Pt foil | 0.5 | 300 | 350 | 375 |
| 1.25 | 275 | 315 | 325 | |
| 2.5 | 265 | 295 | 305 | |
| 5 | 255 | 280 | 285 | |
| Pt-C-Ox | 5 | 25 | 115 | 150 |
| Pt-C-Red | 5 | 5 | 90 | 125 |



| T, °C | CO, μmol/g | CO2, μmol/g | CO/Pt | CO2/Pt | |
|---|---|---|---|---|---|
| TPR-CO | 20 | 88 | 88 | 0.28 | 0.28 |
| TPD-CO | −25 | 4 | 0.013 | ||
| 125, 175, 250 | 25 | 0.08 | |||
| 300 | 3.6 | 0.012 | |||
| TPR-CO + O2 | 10, 32 | 10 | 0.0324 |


Appendix A.1. Absence of Mass Transport and Heat Transfer during Kinetic Measurements
- robs = observed reaction rate, mol/kgcat·s;
- N = reaction order—1;
- dp = catalyst particle diameter—0.375 mm;
- ρp = bulk density of catalyst bed—236.6 kg/m3;
- Deff = effective diffusivity, m2/s;
- = bulk gas concentration of CO, mol/m3;
- Sh is the Sherwood number;
- DAB = 3.7 × 10−5 m2 s−1 = the binary gas diffusivity.
Appendix A.2. The Absence of Heat Transfer was Checked by Mears’ Criterion [45]
| CCO, ppm | robs, mol/kgcat·s | Deff, m2/s | ϕ | ΔC(CO) | CM | |
|---|---|---|---|---|---|---|
| 50 | 3.07 × 10−5 | 9.68 × 10−7 | 2.94 × 10−3 | 0.13 | 3.06 × 10−6 | 3.6 × 10−4 |
| 1600 | 9.82 × 10−4 | 9.68 × 10−7 | 6.54 × 10−2 | 0.13 | 9.81 × 10−5 | 1.1 × 10−2 |
Appendix A.3. Calculation of Adiabatic Heating
| ΔTad = QpC/(cpρ) = 0.5 °C at 25 °C and CO concentration 50 ppm, conversion 90% |
| Where Qp [kJ/mol]—heat of reaction = 283 |
| C [mol/m3]—reacted CO concentration = 0.002 |
| cp [kJ/(kg deg)]—specific heat = 1.043 |
| ρ [kg/m3]—gas density—1.123 |
| ΔTad = QpC/(cpρ) = 16 °C at 25 °C and CO concentration 1600 ppm, conversion 95% |
| Where Qp [kJ/mol]—heat of reaction = 283 |
| C [mol/m3]—reacted CO concentration = 0.068 |
| cp [kJ/(kg deg)]—specific heat = 1.043 |
| ρ [kg/m3]—gas density = 1.123 |
References
- Etim, U.J.; Bai, P.; Gazit, O.M.; Zhong, Z. Low-Temperature Heterogeneous Oxidation Catalysis and Molecular Oxygen Activation. Chem. Rev. 2021, 1–187. [Google Scholar] [CrossRef]
- Royer, S.; Duprez, D. Catalytic Oxidation of Carbon Monoxide over Transition Metal Oxides. ChemCatChem 2011, 3, 24–65. [Google Scholar] [CrossRef]
- Kota, A.S.; Dadi, R.K.; Luss, D.; Balakotaiah, V. Analysis of light-off during oxidation of reactant mixtures on Pt/Al2O3 using micro-kinetic models. Chem. Eng. Sci. 2017, 166, 320–333. [Google Scholar] [CrossRef]
- Haneda, M.; Nakamura, Y.; Yamada, T.; Minami, S.; Kato, N.; Iwashina, K.; Endo, Y.; Nakahara, Y.; Iwachido, K. Comprehensive study of the light-off performance and surface properties of engine-aged Pd-based three-way catalysts. Catal. Sci. Technol. 2021, 11, 912–922. [Google Scholar] [CrossRef]
- Wu, X.; Xu, L.; Weng, D. The thermal stability and catalytic performance of Ce-Zr promoted Rh-Pd/[gamma]-Al2O3 automotive catalysts. Appl. Surf. Sci. 2004, 221, 375–383. [Google Scholar] [CrossRef]
- Aldridge, J.K.; Smith, L.R.; Morgan, D.J.; Carley, A.F.; Humphreys, M.; Clarke, M.J.; Wormald, P.; Taylor, S.H.; Hutchings, G.J. Ambient Temperature CO Oxidation Using Palladium–Platinum Bimetallic Catalysts Supported on Tin Oxide/Alumina. Catalysts 2020, 10, 1223. [Google Scholar] [CrossRef]
- Razon, L.F.; Schmitz, R.A. Intrinsically Unstable Behavior during the Oxidation of Carbon Monoxide on Platinum. Chem. Rev. 1986, 28, 89–164. [Google Scholar] [CrossRef]
- Aghalayam, P.; Park, Y.K.; Vlachos, D.G. A detailed surface reaction mechanism for CO oxidation on Pt. Proc. Combust. Inst. 2000, 28, 1331–1339. [Google Scholar] [CrossRef]
- Engel, T.; Ertl, G. Elementary Steps in the Catalytic Oxidation of Carbon Monoxide on Platinum Metals. In Advances in Catalysis; Eley, D.D., Pines, H., Weez, P.B., Eds.; Academic Press: Cambridge, MA, USA, 1979; Volume 28, pp. 1–78. [Google Scholar]
- Hori, G.K.; Schmidt, L.D. Transient kinetics in CO oxidation on platinum. J. Catal. 1975, 38, 335–350. [Google Scholar] [CrossRef]
- Fischer-Wolfarth, J.H.; Farmer, J.A.; Flores-Camacho, J.M.; Genest, A.; Yudanov, I.V.; Rösch, N.; Campbell, C.T.; Schauermann, S.; Freund, H.J. Particle-size dependent heats of adsorption of CO on supported Pd nanoparticles as measured with a single-crystal microcalorimeter. Phys. Rev. B Condens. Matter. 2010, 81, 241416. [Google Scholar] [CrossRef] [Green Version]
- Yudanov, I.V.; Matveev, A.V.; Neyman, K.M.; Rösch, N. How the C−O Bond Breaks during Methanol Decomposition on Nanocrystallites of Palladium Catalysts. J. Am. Chem. Soc. 2008, 130, 9342–9352. [Google Scholar] [CrossRef] [PubMed]
- DeRita, L.; Dai, S.; Lopez-Zepeda, K.; Pham, N.; Graham, G.W.; Pan, X.; Christopher, P. Catalyst Architecture for Stable Single Atom Dispersion Enables Site-Specific Spectroscopic and Reactivity Measurements of CO Adsorbed to Pt Atoms, Oxidized Pt Clusters, and Metallic Pt Clusters on TiO2. J. Am. Chem. Soc. 2017, 139, 14150–14165. [Google Scholar] [CrossRef] [PubMed]
- Mason, M.G. Electronic structure of supported small metal clusters. Phys. Rev. B 1983, 27, 748–762. [Google Scholar] [CrossRef]
- Bayindir, Z.; Duchesne, P.N.; Cook, S.C.; MacDonald, M.A.; Zhang, P. X-ray spectroscopy studies on the surface structural characteristics and electronic properties of platinum nanoparticles. J. Chem. Phys. 2009, 131, 244716. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sauter, E.; Nefedov, A.; Heißler, S.; Maurer, F.; Casapu, M.; Grunwaldt, J.-D.; Wang, Y.; Wöll, C. Dynamic Structural Evolution of Ceria-Supported Pt Particles: A Thorough Spectroscopic Study. J. Phys. Chem. C 2022, 126, 9051–9058. [Google Scholar] [CrossRef]
- Wong, S.; Flytzani-Stephanopoulos, M.; Chen, M.; Hutchinson, T.E.; Schmidt, L.D. Morphologies of Pt catalyst surfaces. J. Vac. Sci. Technol. 1977, 14, 452–455. [Google Scholar] [CrossRef]
- Kibis, L.; Zadesenets, A.; Garkul, I.; Korobova, A.; Kardash, T.; Slavinskaya, E.; Stonkus, O.; Korenev, S.; Podyacheva, O.; Boronin, A. Pd-Ce-Ox/MWCNTs and Pt-Ce-Ox/MWCNTs Composite Materials: Morphology, Microstructure, and Catalytic Properties. Materials 2022, 15, 7485. [Google Scholar] [CrossRef]
- Altass, H.M.; Morad, M.; Khder, A.E.-R.S.; Mannaa, M.A.; Jassas, R.S.; Alsimaree, A.A.; Ahmed, S.A.; Salama, R.S. Enhanced Catalytic Activity for CO Oxidation by Highly Active Pd Nanoparticles Supported on Reduced Graphene Oxide /Copper Metal Organic Framework. J. Taiwan Inst. Chem. Eng. 2021, 128, 194–208. [Google Scholar] [CrossRef]
- Salama, R.S.; Mannaa, M.A.; Altass, H.M.; Ibrahim, A.A.; Khder, A.E.-R.S. Palladium supported on mixed-metal–organic framework (Co–Mn-MOF-74) for efficient catalytic oxidation of CO. RSC Adv. 2021, 11, 4318–4326. [Google Scholar] [CrossRef]
- Altass, H.M.; Ahmed, S.A.; Salama, R.S.; Moussa, Z.; Jassas, R.S.; Alsantali, R.I.; Al-Rooqi, M.M.; Ibrahim, A.A.; Khder, M.A.; Morad, M.; et al. Low Temperature CO Oxidation Over Highly Active Gold Nanoparticles Supported on Reduced Graphene Oxide@Mg-BTC Nanocomposite. Catal. Lett. 2023, 153, 876–886. [Google Scholar] [CrossRef]
- Wojciechowski, B.W.; Asprey, S.P. Kinetic studies using temperature-scanning: The oxidation of carbon monoxide. Appl. Catal. A: Gen. 2000, 190, 1–24. [Google Scholar] [CrossRef]
- Allian, A.D.; Takanabe, K.; Fujdala, K.L.; Hao, X.; Truex, T.J.; Cai, J.; Buda, C.; Neurock, M.; Iglesia, E. Chemisorption of CO and Mechanism of CO Oxidation on Supported Platinum Nanoclusters. J. Am. Chem. Soc. 2011, 133, 4498–4517. [Google Scholar] [CrossRef] [PubMed]
- Kibis, L.S.; Korobova, A.N.; Zadesenets, A.V.; Romanenko, A.V.; Kardash, T.Y.; Stonkus, O.A.; Korenev, S.V.; Podyacheva, O.Y.; Slavinskaya, E.M.; Boronin, A.I. Catalysts for Low-Temperature CO Oxidation Based on Platinum, CeO2, and Carbon Nanotubes. Dokl. Phys. Chem. 2022, 505, 115–121. [Google Scholar] [CrossRef]
- Peuckert, M.; Bonzel, H.P. Characterization of oxidized platinum surfaces by X-ray photoelectron spectroscopy. Surf. Sci. 1984, 145, 239–259. [Google Scholar] [CrossRef]
- Kaushik, V.K. Identification of Mixed Platinum States and Electronic Effects of Support on Platinum in Supported Catalysts. Z. Für Phys. Chem. 1991, 173, 105–113. [Google Scholar] [CrossRef]
- Kalinkin, A.V.; Smirnov, M.Y.; Nizovskii, A.I.; Bukhtiyarov, V.I. X-ray photoelectron spectra of platinum compounds excited with monochromatic AgLα irradiation. J. Electron Spectrosc. Relat. Phenom. 2010, 177, 15–18. [Google Scholar] [CrossRef]
- Svintsitskiy, D.A.; Kibis, L.S.; Stadnichenko, A.I.; Koscheev, S.V.; Zaikovskii, V.I.; Boronin, A.I. Highly Oxidized Platinum Nanoparticles Prepared through Radio-Frequency Sputtering: Thermal Stability and Reaction Probability towards CO. ChemPhysChem 2015, 16, 3318–3324. [Google Scholar] [CrossRef]
- Eiswirth, M.; Ertl, G. Kinetic oscillations in the catalytic CO oxidation on a Pt(110) surface. Surf. Sci. 1986, 177, 90–100. [Google Scholar] [CrossRef]
- Vishnevskii, A.L.; Savchenko, V.I. Self-oscillations in the rate of CO oxidation on Pt(110). React. Kinet. Catal. Lett. 1989, 38, 167–173. [Google Scholar] [CrossRef]
- Van Spronsen, M.A.; Frenken, J.W.M.; Groot, I.M.N. Observing the oxidation of platinum. Nat. Commun. 2017, 8, 429. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Park, S.; Chan, H.Y.H.; Weaver, M.J. Surface Oxidation of Platinum-Group Transition Metals in Ambient Gaseous Environments: Role of Electrochemical versus Chemical Pathways. J. Phys. Chem. B 2000, 104, 8250–8258. [Google Scholar] [CrossRef]
- Lashina, E.A.; Chumakova, N.A.; Chumakov, G.A.; Boronin, A.I. Chaotic dynamics in the three-variable kinetic model of CO oxidation on platinum group metals. Chem. Eng. J. 2009, 154, 82–87. [Google Scholar] [CrossRef]
- Van Spronsen, M.A.; Frenken, J.W.M.; Groot, I.M.N. Surface science under reaction conditions: CO oxidation on Pt and Pd model catalysts. Chem. Soc. Rev. 2017, 46, 4347–4374. [Google Scholar] [CrossRef] [Green Version]
- Sales, B.C.; Turner, J.E.; Maple, M.B. Oscillatory oxidation of CO over Pt, Pd and Ir catalysts: Theory. Surf. Sci. 1982, 114, 381–394. [Google Scholar] [CrossRef]
- Garcia-Martinez, F.; García-Fernández, C.; Simonovis, J.P.; Hunt, A.; Walter, A.; Waluyo, I.; Bertram, F.; Merte, L.R.; Shipilin, M.; Pfaff, S.; et al. Catalytic Oxidation of CO on a Curved Pt(111) Surface: Simultaneous Ignition at All Facets through a Transient CO-O Complex. Angew. Chem. Int. Ed. 2020, 59, 20037–20043. [Google Scholar] [CrossRef]
- Stakheev, A.Y.; Bokarev, D.A.; Prosvirin, I.P.; Bukhtiyarov, V.I. Chapter 8—Particle-Size Effect in Catalytic Oxidation Over Pt Nanoparticles. In Advanced Nanomaterials for Catalysis and Energy; Sadykov, V.A., Ed.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 295–320. [Google Scholar]
- Podyacheva, O.Y.; Suboch, A.N.; Yashnik, S.A.; Salnikov, A.V.; Cherepanova, S.V.; Kibis, L.S.; Simenyuk, G.Y.; Romanenko, A.I.; Ismagilov, Z.R. Effect of Structure and Surface State of Nitrogen Doped Carbon Nanotubes on Their Functional and Catalytic Properties. J. Struct. Chem. 2021, 62, 771–781. [Google Scholar] [CrossRef]
- Vasilchenko, D.; Tkachev, S.; Baidina, I.; Korenev, S. Speciation of Platinum(IV) in Nitric Acid Solutions. Inorg. Chem. 2013, 52, 10532–10541. [Google Scholar] [CrossRef]
- Vasilchenko, D.; Topchiyan, P.; Berdyugin, S.; Filatov, E.; Tkachev, S.; Baidina, I.; Komarov, V.; Slavinskaya, E.; Stadnichenko, A.; Gerasimov, E. Tetraalkylammonium Salts of Platinum Nitrato Complexes: Isolation, Structure, and Relevance to the Preparation of PtOx/CeO2 Catalysts for Low-Temperature CO Oxidation. Inorg. Chem. 2019, 58, 6075–6087. [Google Scholar] [CrossRef]
- Larciprete, R.; Gardonio, S.; Petaccia, L.; Lizzit, S. Atomic oxygen functionalization of double walled C nanotubes. Carbon 2009, 47, 2579–2589. [Google Scholar] [CrossRef]
- Kibis, L.S.; Zadesenets, A.V.; Garkul, I.A.; Korobova, A.N.; Kardash, T.Y.; Fedorova, E.A.; Slavinskaya, E.M.; Stonkus, O.A.; Korenev, S.V.; Podyacheva, O.Y.; et al. Pd-Ce-Ox species on MWCNTs surface: Probing the structure-activity correlation in low-temperature CO oxidation. Appl. Surf. Sci. 2023, 611, 155750. [Google Scholar] [CrossRef]
- Boronin, A.I.; Slavinskaya, E.M.; Figueroba, A.; Stadnichenko, A.I.; Kardash, T.Y.; Stonkus, O.A.; Fedorova, E.A.; Muravev, V.V.; Svetlichnyi, V.A.; Bruix, A.; et al. CO oxidation activity of Pt/CeO2 catalysts below 0 °C: Platinum loading effects. Appl. Catal. B Environ. 2021, 286, 119931. [Google Scholar] [CrossRef]
- Carlsson, P.-A.; Zhdanov, V.; Skoglundh, M. Self-sustained kinetic oscillations in CO oxidation over silica-supported Pt. Phys. Chem. Chem. Phys. 2006, 8, 2703–2706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.-H.; Wang, Y.; Jia, A.-P.; Wang, S.-Y.; Luo, M.-F.; Lu, J.-Q. Oxygen vacancy promoted CO oxidation over Pt/CeO2 catalysts: A reaction at Pt–CeO2 interface. Appl. Surf. Sci. 2014, 314, 725–734. [Google Scholar] [CrossRef]













| Free Centre | Adsorbed CO | Adsorbed O | Oxide |
|---|---|---|---|
| * | CO* | O* | Ox |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lashina, E.; Slavinskaya, E.; Kibis, L.; Stadnichenko, A.; Stonkus, O.; Zhuravlev, D.; Zadesenets, A.; Korenev, S.; Podyacheva, O.; Boronin, A. CO Oxidation Reaction by Platinum Clusters on the Surface of Multiwalled Carbon Nanotubes: Experimental and Theoretical Study of Kinetics in a Wide Range of O2/CO Ratios. Catalysts 2023, 13, 568. https://doi.org/10.3390/catal13030568
Lashina E, Slavinskaya E, Kibis L, Stadnichenko A, Stonkus O, Zhuravlev D, Zadesenets A, Korenev S, Podyacheva O, Boronin A. CO Oxidation Reaction by Platinum Clusters on the Surface of Multiwalled Carbon Nanotubes: Experimental and Theoretical Study of Kinetics in a Wide Range of O2/CO Ratios. Catalysts. 2023; 13(3):568. https://doi.org/10.3390/catal13030568
Chicago/Turabian StyleLashina, Elena, Elena Slavinskaya, Lidiya Kibis, Andrey Stadnichenko, Olga Stonkus, Daniil Zhuravlev, Andrey Zadesenets, Sergey Korenev, Olga Podyacheva, and Andrei Boronin. 2023. "CO Oxidation Reaction by Platinum Clusters on the Surface of Multiwalled Carbon Nanotubes: Experimental and Theoretical Study of Kinetics in a Wide Range of O2/CO Ratios" Catalysts 13, no. 3: 568. https://doi.org/10.3390/catal13030568
APA StyleLashina, E., Slavinskaya, E., Kibis, L., Stadnichenko, A., Stonkus, O., Zhuravlev, D., Zadesenets, A., Korenev, S., Podyacheva, O., & Boronin, A. (2023). CO Oxidation Reaction by Platinum Clusters on the Surface of Multiwalled Carbon Nanotubes: Experimental and Theoretical Study of Kinetics in a Wide Range of O2/CO Ratios. Catalysts, 13(3), 568. https://doi.org/10.3390/catal13030568