1. Introduction
During the past decades, photopolymerization has been an active research field, mainly supported by the development of 3D printing but also by the necessity to replace in the near future the historical UV photo-initiating systems with visible light photo-initiating systems in the industry [
1,
2,
3,
4,
5,
6,
7,
8,
9,
10,
11]. Indeed, UV photopolymerization is more and more the focus of safety concerns, originating from the numerous drawbacks of UV light. Notably, UV light can cause eye damage and skin cancers [
12,
13,
14,
15]. Parallel to this, molecular oxygen can be converted to ozone during the polymerization process, constituting an additional drawback of this approach [
15]. In addition, photopolymerization constitutes an appealing polymerization technique, which exhibits numerous specificities and advantages compared to traditional thermal polymerization. In order to illustrate this, the possibility of polymerizing without solvents to obtain efficient spatial and temporal control during the polymerization process can be cited as relevant examples [
16,
17,
18,
19,
20,
21,
22,
23,
24,
25,
26,
27,
28,
29]. Development of photo-initiating systems is not new since the first report mentioning a photoinduced electron transfer between triethanolamine and electron-accepting dyes (xanthenes, acridines, thiazines) was reported as soon as 1954 by Oster and coworkers [
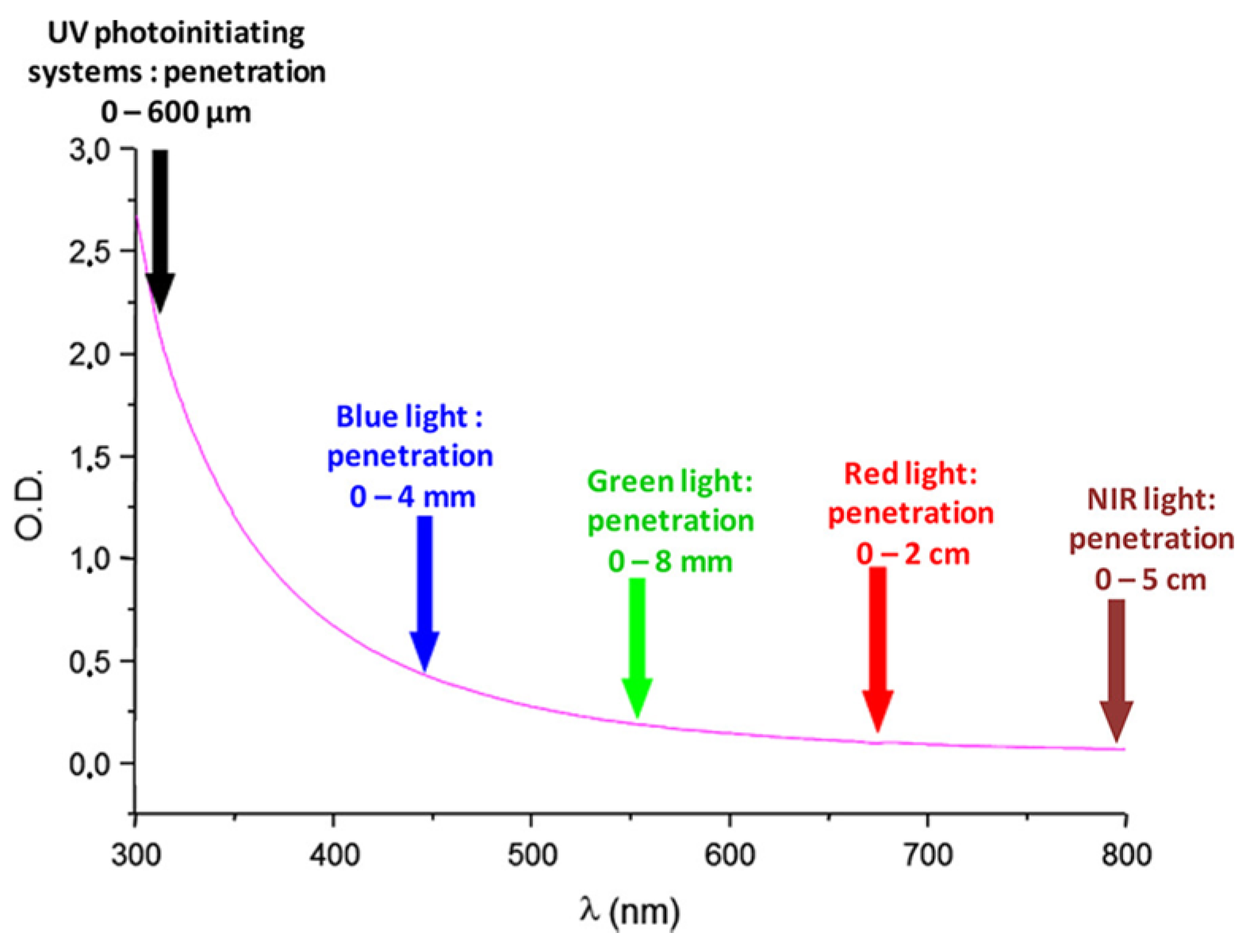
30]. Since 1954, photopolymerization has greatly evolved, enabling now polymerization in safer and energy-saving conditions. As a breakthrough, visible light photopolymerization has emerged as a promising alternative to the historical UV photopolymerization. As the main advantage of visible light photopolymerization, a higher light penetration within the photocurable resins can be obtained than in the UV range (See
Figure 1). Indeed, if the light penetration is limited to a few hundred micrometers in the UV range, this value can increase up to 4 mm at 450 nm and even reach 5 cm at 800 nm [
31]. In these conditions, photopolymerization becomes capable of polymerizing thick samples and is not limited anymore to the polymerization of thin samples, as in the past when UV light was used [
32]. However, light penetration within the photocurable resins is an important issue of photopolymerization, and some clarifications should be given. For instance, the light penetration is strongly related to the molar extinction coefficient of the photoinitiator at the wavelength used for irradiation and on the photoinitiator concentration in the system. Only in the case of photo-bleachable initiators can the light penetrate deeply. This concerns the light of any wavelengths, with the exception that short-wavelength UV may also be absorbed by monomers.
Figure 1 depicts the light penetration only in the light-scattering system, which cannot be generalized to all photocurable systems because most photocurable resins are transparent and not light-scattering. Consequently, the statement that the main advantage of visible light photopolymerization is the higher light penetration within the photocurable resins than in the case of UV light is not applicable to all resins. In complement to this first point, one of the main drawbacks of visible light photopolymerization remains the difficulty of obtaining colorless coatings [
33]. Indeed, UV photoinitiators are colorless compounds, enabling the ability to obtain easily colorless polymers. Conversely, visible light photopolymerization makes use of dyes absorbing in the visible range, and these dyes are often responsible for the final color of the polymers. In order to address this issue, major efforts have been devoted to developing photo-bleachable photo-initiating systems, with more or less success [
33].
This effort is also supported by the wide range of applications using photopolymerization. Notably, 3D and 4D printing, microelectronics, dentistry, coatings, solvent-free paints, adhesives and varnishes can be cited as the main applications of photopolymerization [
1,
2,
3,
4,
5,
6,
7,
8,
9,
10]. Another drawback of visible light photopolymerization is that visible light photons are less energetic than UV photons, so more reactive photo-initiating systems have to be developed in order to overcome this lower energy. In the search for highly reactive photo-initiating systems, numerous structures have been examined as potential candidates capable of addressing the reactivity issue and carbazoles [
34,
35,
36,
37,
38,
39,
40,
41,
42,
43,
44,
45,
46,
47], dihydroanthraquinones [
48], camphorquinone [
49,
50], chalcones [
9,
51,
52,
53,
54,
55,
56,
57,
58,
59,
60,
61,
62,
63], naphthalimides [
64,
65,
66,
67,
68,
69,
70,
71,
72,
73,
74,
75,
76,
77,
78,
79,
80,
81,
82], benzophenones [
83,
84,
85,
86,
87,
88,
89,
90], silyl glyoximides [
91], phenothiazines [
92,
93,
94,
95,
96,
97,
98,
99,
100,
101,
102,
103], thioxanthones [
28,
104,
105,
106,
107,
108,
109,
110,
111,
112,
113,
114,
115,
116], curcumin [
117,
118,
119,
120], pyrenes [
121,
122,
123,
124,
125,
126,
127,
128,
129], iodonium salts [
64,
130,
131,
132,
133,
134,
135,
136], push-pull dyes [
137,
138,
139], copper complexes [
140,
141,
142,
143], iron complexes [
144,
145] zinc complexes [
146] iridium complexes [
147,
148] and
N-heterocyclic carbene boranes [
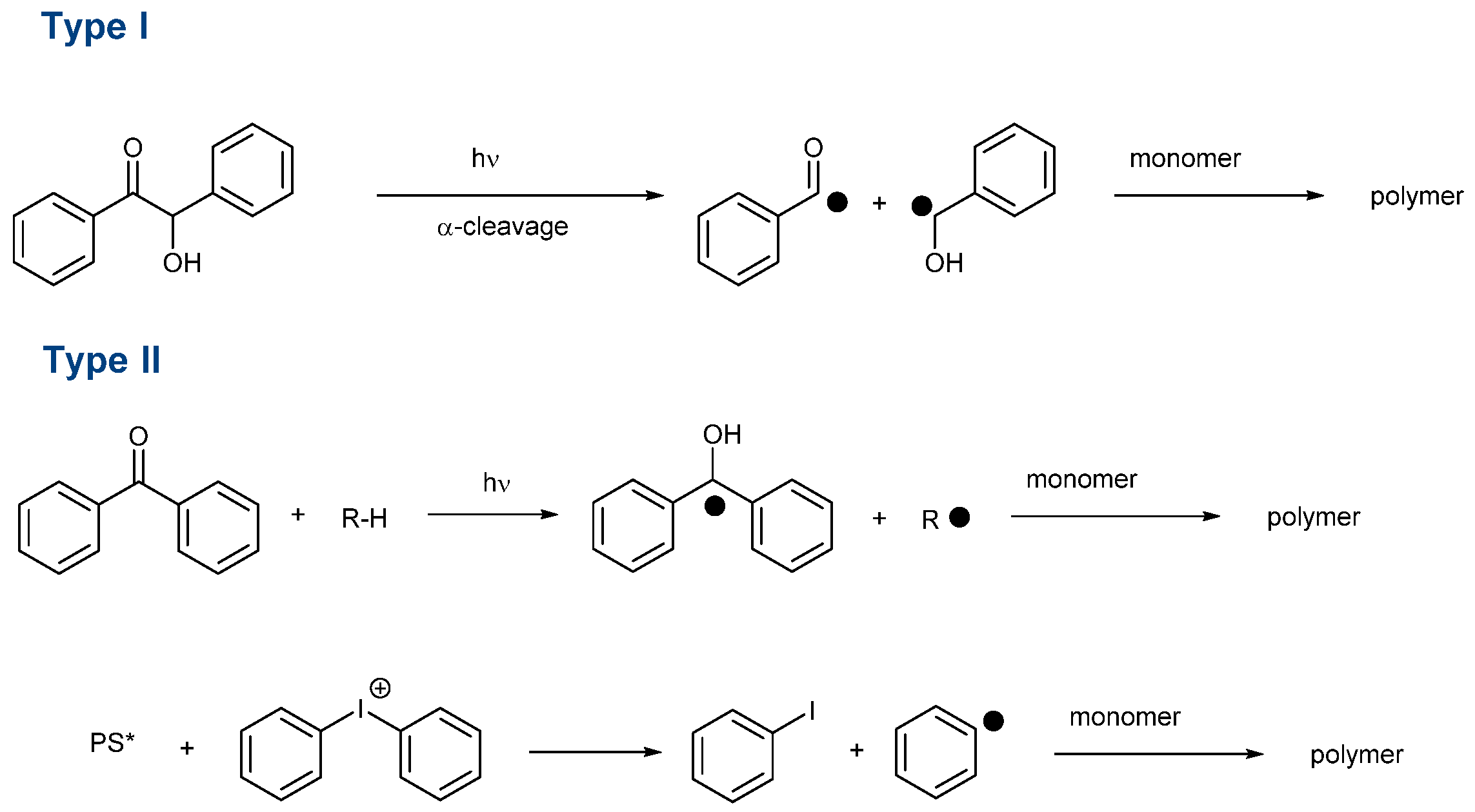
29] can be cited among the most extensively studied structures of the past decade. Beyond the simple selection of the chromophore, the way how to generate initiating species is important. Notably, photoinitiators can be divided into two main categories, namely, Type I and Type II photoinitiators. In the case of Type I photoinitiators, these structures can generate reactive species by homolytic cleavage of a specific bond (See
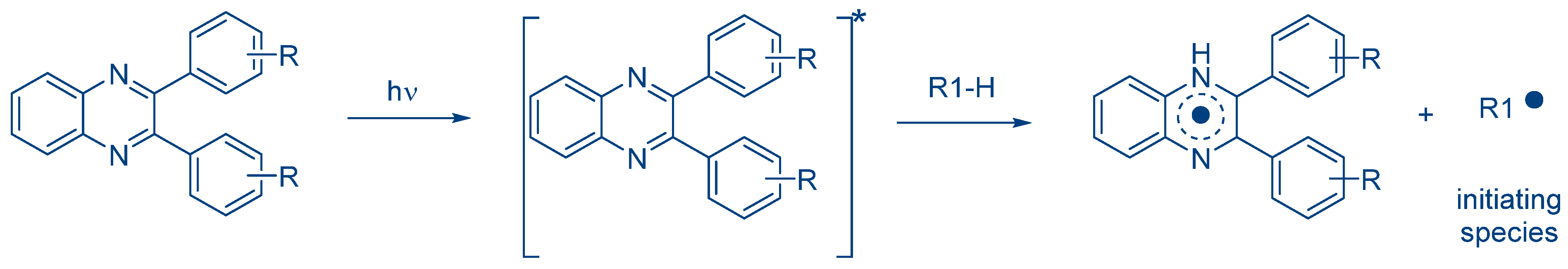
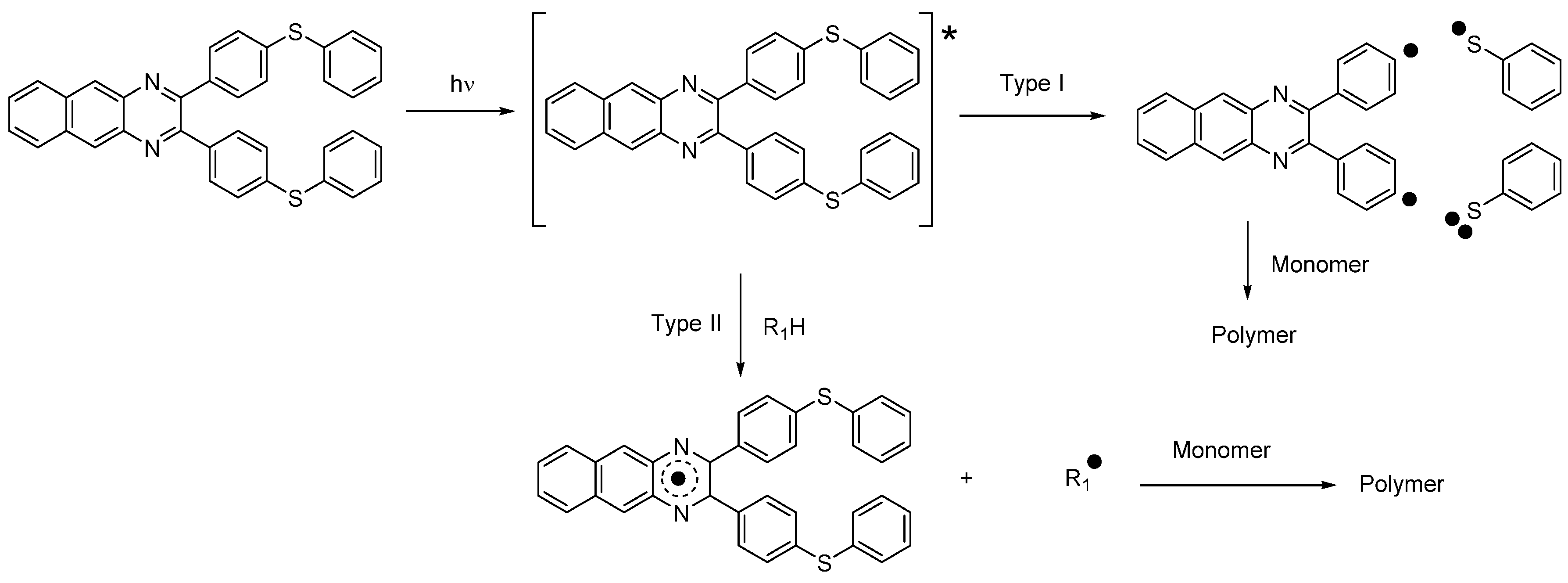
Scheme 1) [
149,
150,
151,
152,
153,
154,
155,
156,
157,
158].
In this first family,
O-acyl-α-oximino ketones, acetophenones, acylgermanes, benzoin ether derivatives, α-aminoketones benzyl ketals, acylphosphine oxides, aminoalkyl phenones, α-hydroxyalkyl ketones, hydroxylalkylphenones and oxime esters can cleave homolytically, generating initiating radicals [
103,
151,
153,
158,
159,
160,
161]. As the main interest of these structures, Type I photoinitiators can be used as mono-component systems. As a drawback, the photodecomposition is irreversible, so the concentration continuously decreases during the polymerization process. Conversely, Type II photoinitiators are unable to generate initiating species without additives. However, a few exceptions exist. For instance, free radical polymerization of ether acrylates (such as poly(ethylene glycol) diacrylates) is possible with benzophenone, thioxanthone and different chalcones, the monomer itself acting as a co-initiator because -CH
2O- groups are good hydrogen donors [
162]. Type II photoinitiators are typically used for the sensitization of onium salts [
163,
164,
165,
166,
167,
168]. A photoinduced electron transfer from the photosensitizer towards the onium salts is the key step to generating initiating radicals. However, Type II photoinitiators are also combined with hydrogen donors, leading to the formation of a ketyl radical with hydrogen abstraction and an additional radical issued from the hydrogen donor (See
Scheme 1) [
105,
110,
169,
170,
171,
172,
173,
174]. Considering that Type II photoinitiators are bimolecular photoinitiators, the introduction of a sacrificial amine can contribute to regenerating the photosensitizer in its initial redox state during the polymerization process so that this latter can be introduced in a catalytic amount. This point is important, considering that the photosensitizer is responsible for the final color of the polymer. By decreasing its content, less colored coatings can be obtained. The search for new structures is also motivated by the recent interest in developing photo-initiating systems activable with sunlight [
175,
176,
177,
178,
179,
180,
181,
182,
183,
184] or capable of initiating a polymerization process in water [
22,
60,
116,
185,
186,
187,
188,
189,
190,
191,
192,
193,
194,
195,
196,
197,
198,
199,
200]. With the aim of polymerizing in energy-saving conditions, the use of light-emitting diodes (LEDs) is now generalized in photopolymerization due to their low costs, long operating lifetimes, compactness and their precise emission wavelengths. In the search for photo-initiating systems that can be activated under low light intensity, i.e., LEDs, quinoxalines have been identified as potential candidates for visible light photopolymerization. Quinoxalines are heterocyclic compounds in which two nitrogen atoms replace two carbons in the ring of naphthalene. Quinoxalines have been extensively studied for their biological properties. Indeed, quinoxalines are biologically active against bacteria, viruses, cancer, leishmania, tuberculosis, malaria, depression and fungi [
201]. Nevertheless, quinoxalines were also used for the design of light-emitting materials for OLEDs [
202,
203], semiconductors and charge transport materials for solar cells [
204,
205,
206,
207], the design of building blocks for covalent organic frameworks [
208]. Recently, different quinoxaline derivatives were also proposed as fluorescent probes for near-infrared II (NIR-II, 1000–1700 nm) fluorescent imaging [
209].
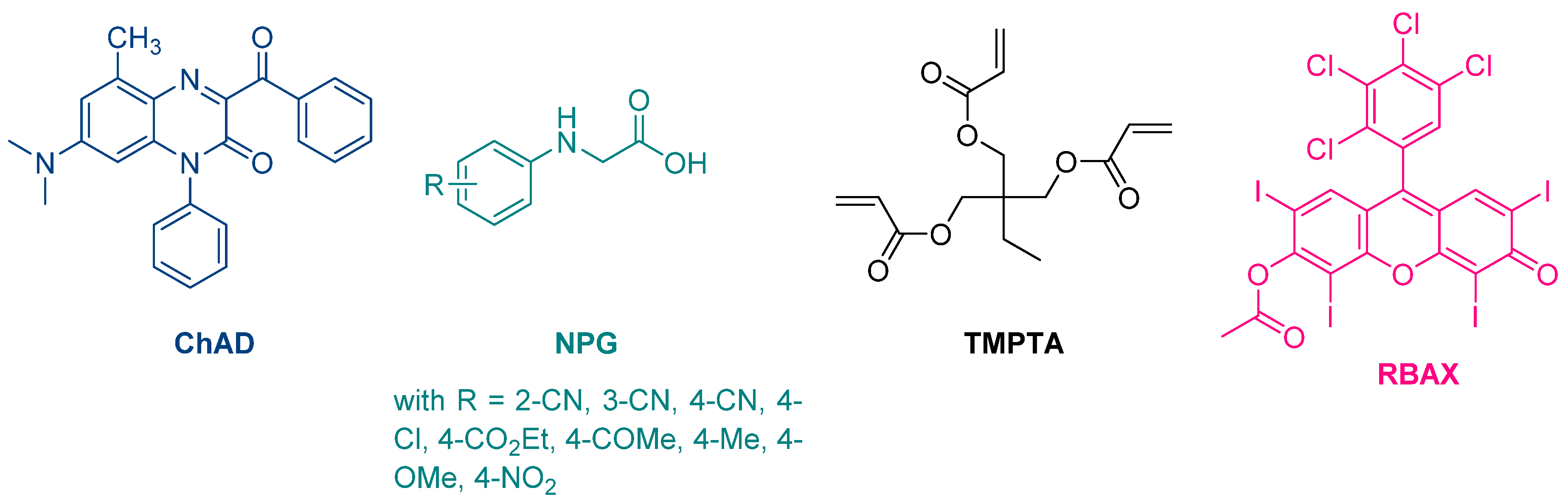
The first report mentioning the use of quinoxalines as photoinitiators of polymerization was reported in 1999 by Pączkowski and coworkers. By using 3-benzoyl-7-diethylamino-5-methyl-1-phenyl-1
H-quinoxalin-2-one (ChAD) as an electron acceptor and
N-phenylglycine derivatives (NPG) as electron donors, the free radical polymerization of trimethylolpropane triacrylate (TMPTA) was carried out, using an argon ion laser (emission between 351 and 361 nm, 25.5 mW/cm
2) or a He/Ne laser as the light sources (See
Figure 2) [
210,
211]. The best monomer conversion was obtained using (4-methoxyphenyl)glycine as the electron donor. Noticeably, efficiencies of the different photo-initiating systems based on quinoxalines were lower than that of a Rose Bengal derivative, namely RBAX, previously reported in the literature.
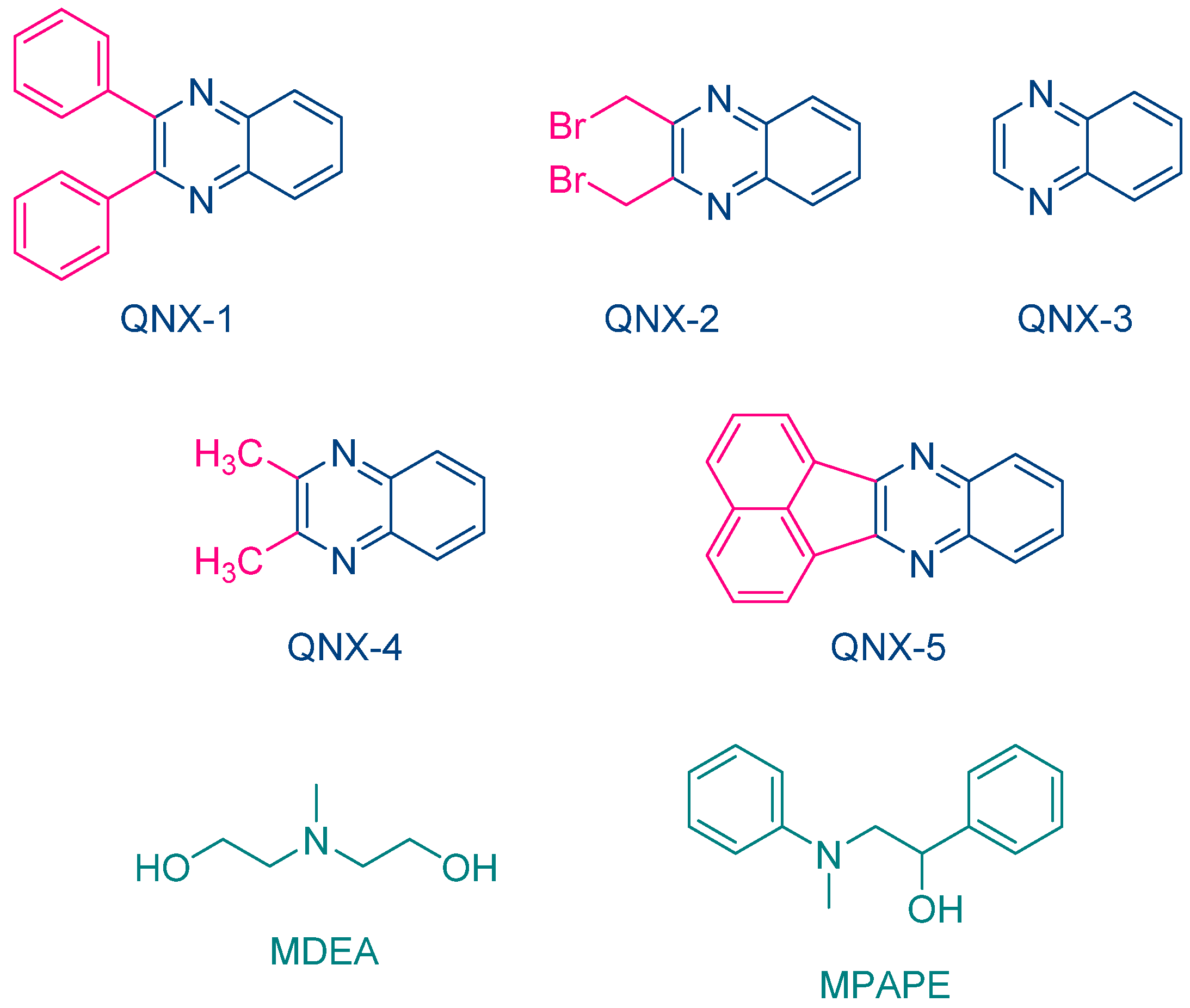
The same year, Aydin and coworkers proposed a series of quinoxalines (QNX-1-QNX-5) that proved to be excellent photoinitiators in combination with electron donors such as
N-methyldiethanolamine (MDEA) and 2-(
N-methyl-
N-phenylamino)-1-phenylethanol (MPAPE). By using these two co-initiators, the polymerization of methyl methacrylate (MMA) was carried out upon irradiation with a UV light (λ = 350 nm) (See
Figure 3) [
212]. A few years later, this strategy was extended to an unusual co-initiator in photopolymerization, namely benzaldehyde [
213].
Since these pioneering works, numerous quinoxaline derivatives have been proposed, enabling the initiation of free radical polymerizations and cationic polymerizations in the UV and visible range. It has to be noticed that quinoxalines were also investigated as chromophores for two-photon polymerization [
214]. Triphenylamine-modified quinoxalines were notably reported as exhibiting a two-photon value higher than 160 GM in the 780–820 nm range, greatly higher than the values reported for most of the benzil derivatives investigated as photoinitiators for two-photon initiated polymerization. Recently, a dipyrido[3,2-d:2′,3′-f]quinoxaline (dpq) ligand was also used for the design of an iridium catalyst used for both the photogeneration of hydrogen from water and as a photocatalyst for the polymerization of methyl methacrylate [
215]. However, the drawback of this approach is the toxicity of iridium and the scarcity of this metal on Earth. Quinoxalines used in two-photon polymerization and transition metal complexes comprising quinoxaline units will not be further detailed in this review. In this work, an overview of the different quinoxaline derivatives reported to date as one-photon initiators is given. Over the years, numerous derivatives have been proposed for UV-induced or visible light-induced photopolymerization. In order to evidence the polymerization efficiencies of these systems, comparisons with reference compounds will be provided.
2. Quinoxalines as Photoinitiators of Polymerization
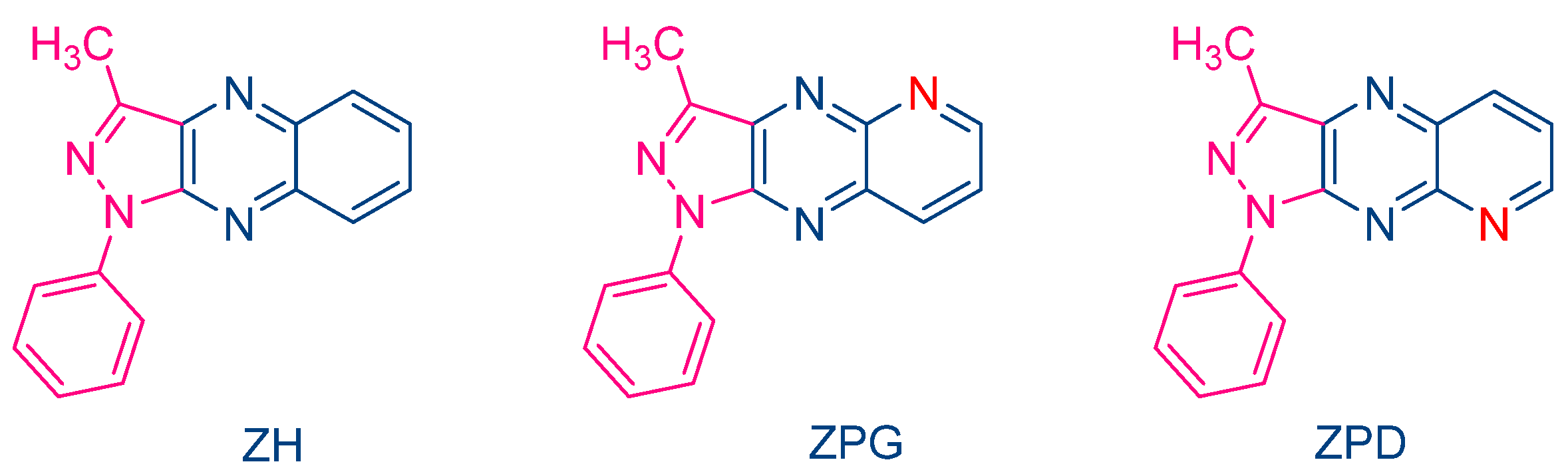
In 2000, Pączkowski and coworkers examined two dyes containing pyrazoloquinoxaline moieties, i.e., ZPG and ZPD, and investigated the photochemical mechanism involved during the free radical polymerization (FRP) of TMPTA (See
Figure 4) [
216]. Noticeably, similar absorption maxima were found for ZH (λ
max = 409 nm in ethyl acetate) and ZPG/ZPD (λ
max = 415 nm in ethyl acetate). The photo-initiating abilities of these two dyes were compared with that of the quinoxaline derivative ZH. While the FRP of TMPTA was carried out upon irradiation with an argon ion laser, ZPG and ZPD greatly outperformed the monomer conversion obtained with ZH. To support this, the authors evidenced that the intersystem crossing between the singlet excited state and the triplet excited state was more efficient for ZPG and ZPD than for ZH, enabling these dyes to interact more efficiently with the electron donor
N-phenylglycine (NPG) and facilitating the generation of radicals. Application of the Rehm–Weller equation also revealed the free energy change to be more positive for ZH than for ZPD and ZPG so that the rate constant of electron transfer between ZH and NPG was expected to be slower than with ZPD and ZPG. However, the authors also suggested a competitive back electron transfer between ZH and NPG, adversely affecting the photo-initiating ability of this dye.
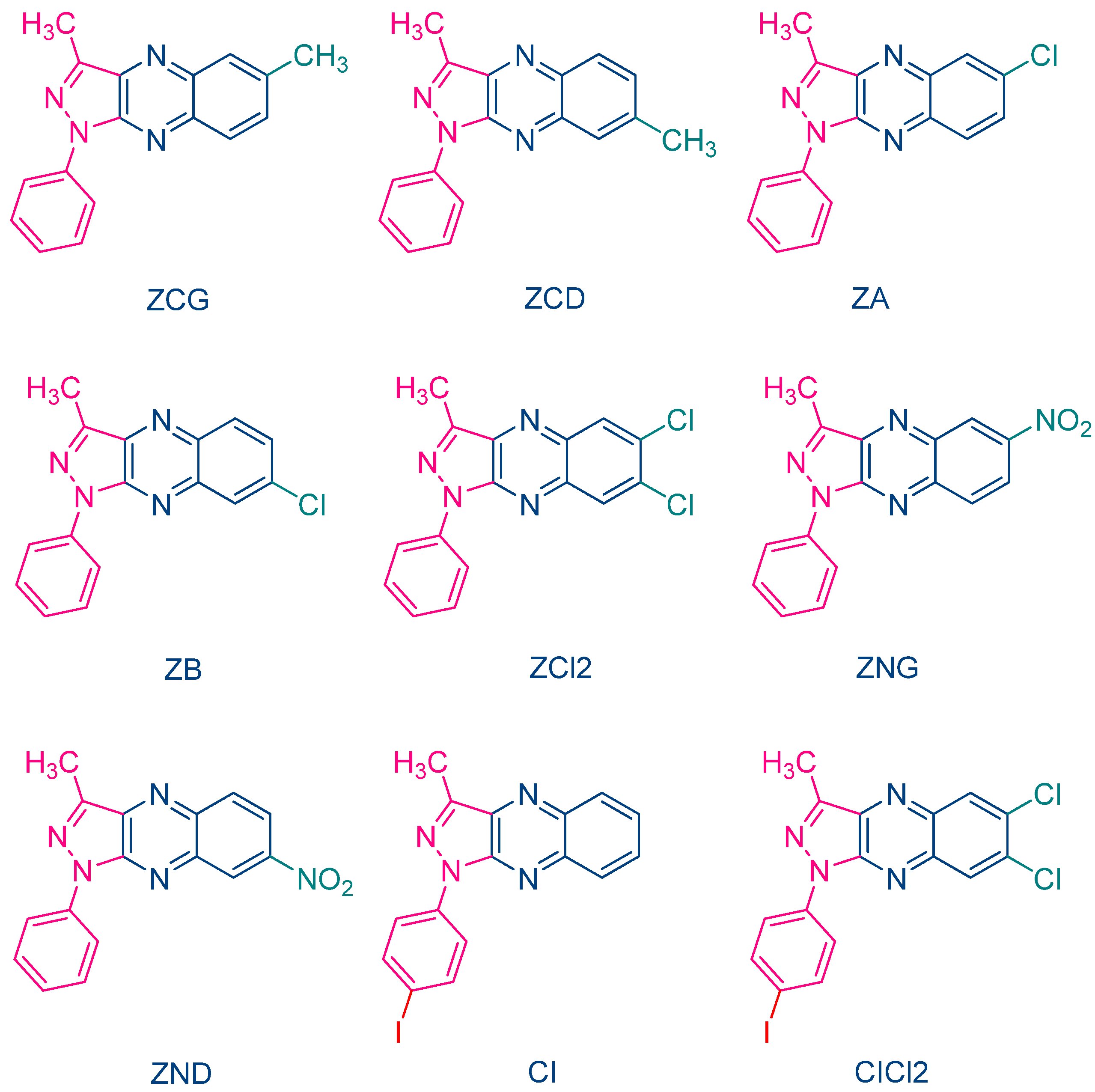
The occurrence of a back electron transfer was confirmed with a series of ten pyrazoloquinoxalines, including the previously studied quinazoline dye ZH (See
Figure 5) [
217]. Examination of their UV-visible absorption spectra in ethyl acetate revealed that these structures were relatively insensitive to the substitution pattern. Thus, absorption maxima ranging between 404 nm for ZCD and 435 nm for ZND were determined. It has to be noticed that the positions of the absorption maxima were affected by the substitution pattern. Thus, for the nitro-substituted quinoxaline, positions of the absorption maxima varied from 417 nm for ZNG and up to 435 nm for ZND, in which the nitro group was in a conjugated position with the rest of the molecule (See
Table 1). In this series of dyes, the authors could establish a linear relationship between the monomer conversion and the efficiency of singlet oxygen formation, evidencing that the electron transfer between NPG and the different dyes was occurring via the triplet state. By introducing heavy atom in ZCl2, CI and CICl2, quantum yields of the triplet state formation were greatly improved, enhancing the photo-initiating ability.
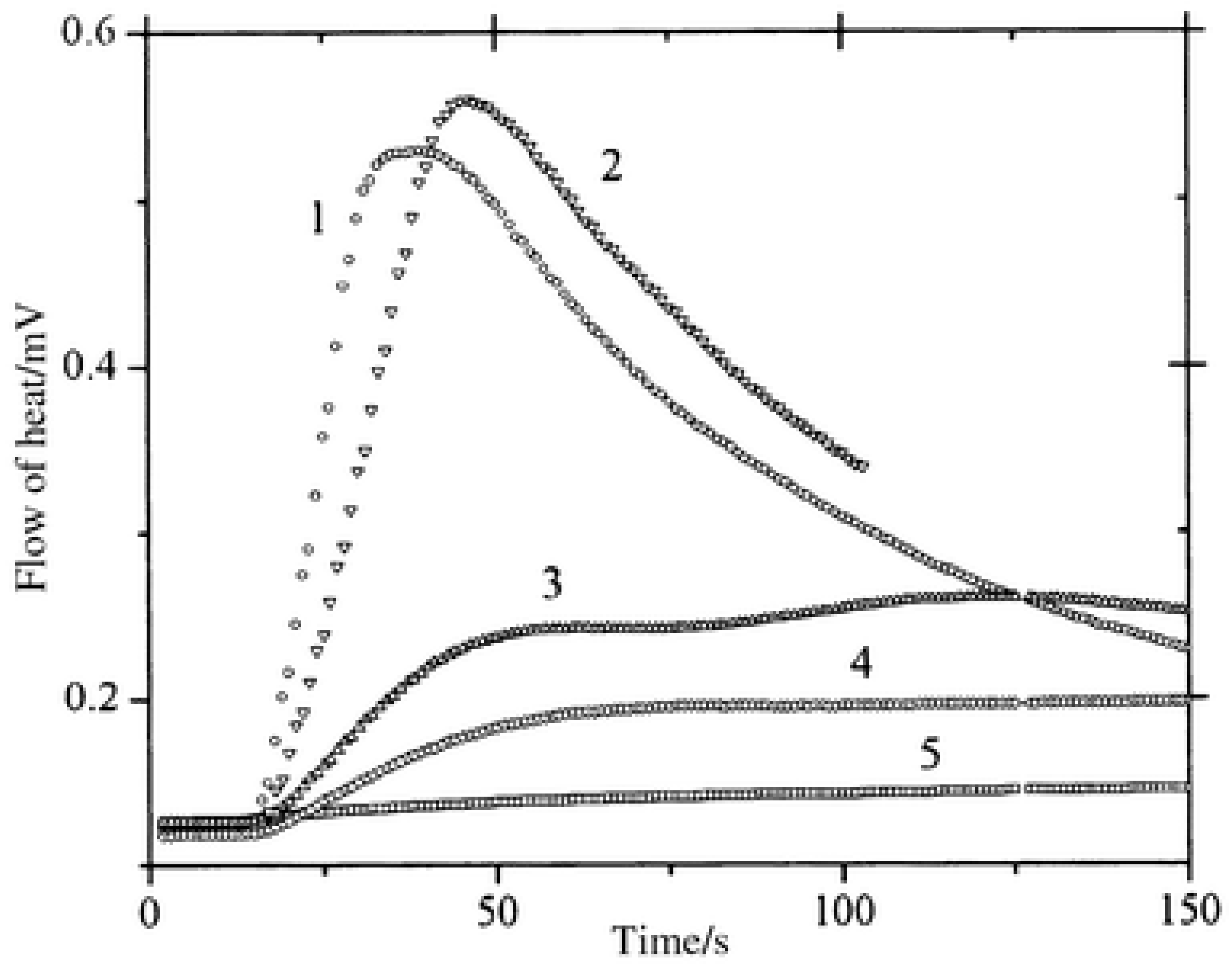
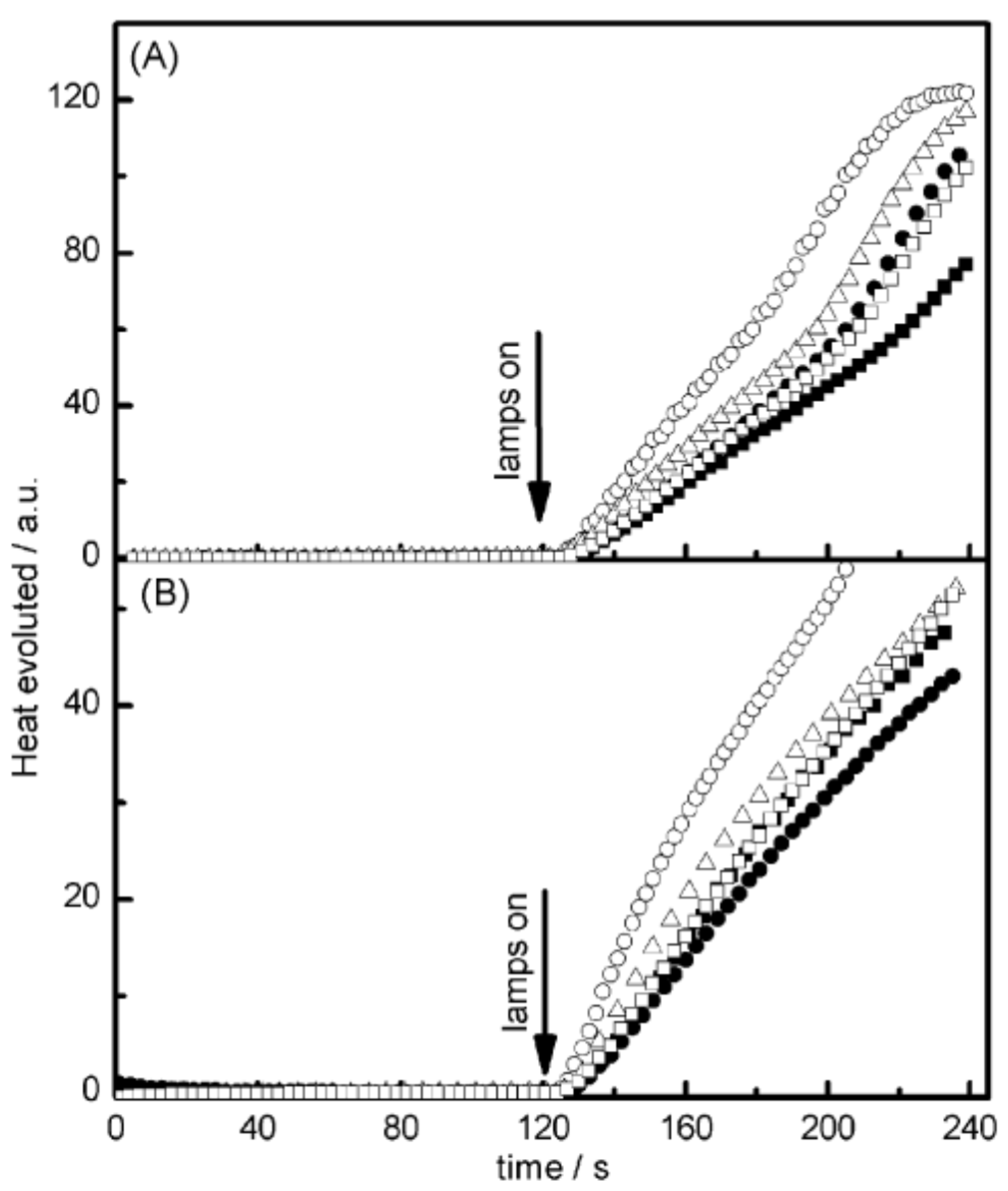
In benefiting from these elongated excited state lifetimes, interactions between the dyes and NPG in the excited state were favored, improving the polymerization efficiency. As shown in
Figure 6, halogenated quinoxalines polymerized TMPTA within 50 s, contrarily to the non-halogenated dyes for which a three-fold elongation of the polymerization time was necessary. To monitor the polymerization process, photo-DSC was used. In this case, access to the monomer conversion was not directly possible. Using photo-DSC, only the heat flow can be determined. Heat flow is proportional to the polymerization rate, which is the derivative of the conversion versus time function. In the present case, the highest heat flows were obtained for CICl2 and CI, the two compounds bearing halogens.
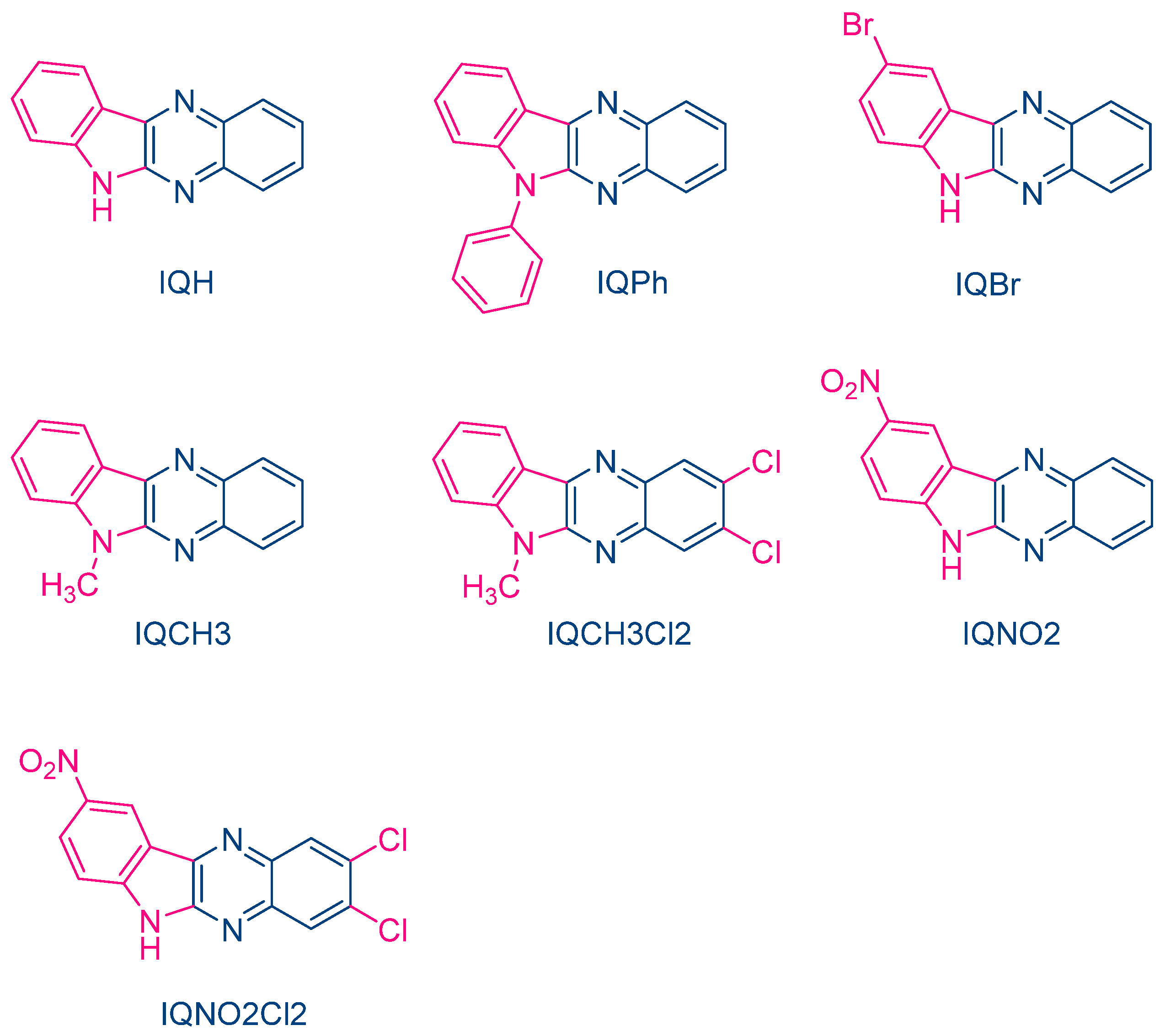
In 2004, quinoxaline derivatives were tested for the first time in lower light intensity (no use of lasers as the light sources as in the previous works) since dental lamps were used for the polymerization experiments [
218]. A series of seven dyes were examined, differing by the substitution pattern and the alkylation or not of the nitrogen groups (See
Figure 7). All dyes exhibited an absorption centered in the near UV-visible range, the absorption maxima peaking between 386 nm for IQH and up to 409 nm for IQNO2Cl2 (See
Table 2). Noticeably, in this series of dyes, the lowest monomer conversions were obtained for the nitro derivatives, namely IQNO2 and IQNO2Cl2. These counter-performances were assigned to the photoreduction of the nitro groups to nitroso groups, constituting efficient free radical scavengers and good inhibitors of free radical polymerization [
219]. Polymerization efficiency was also strongly related to the electron donors used. Among the five electron donors tested, namely
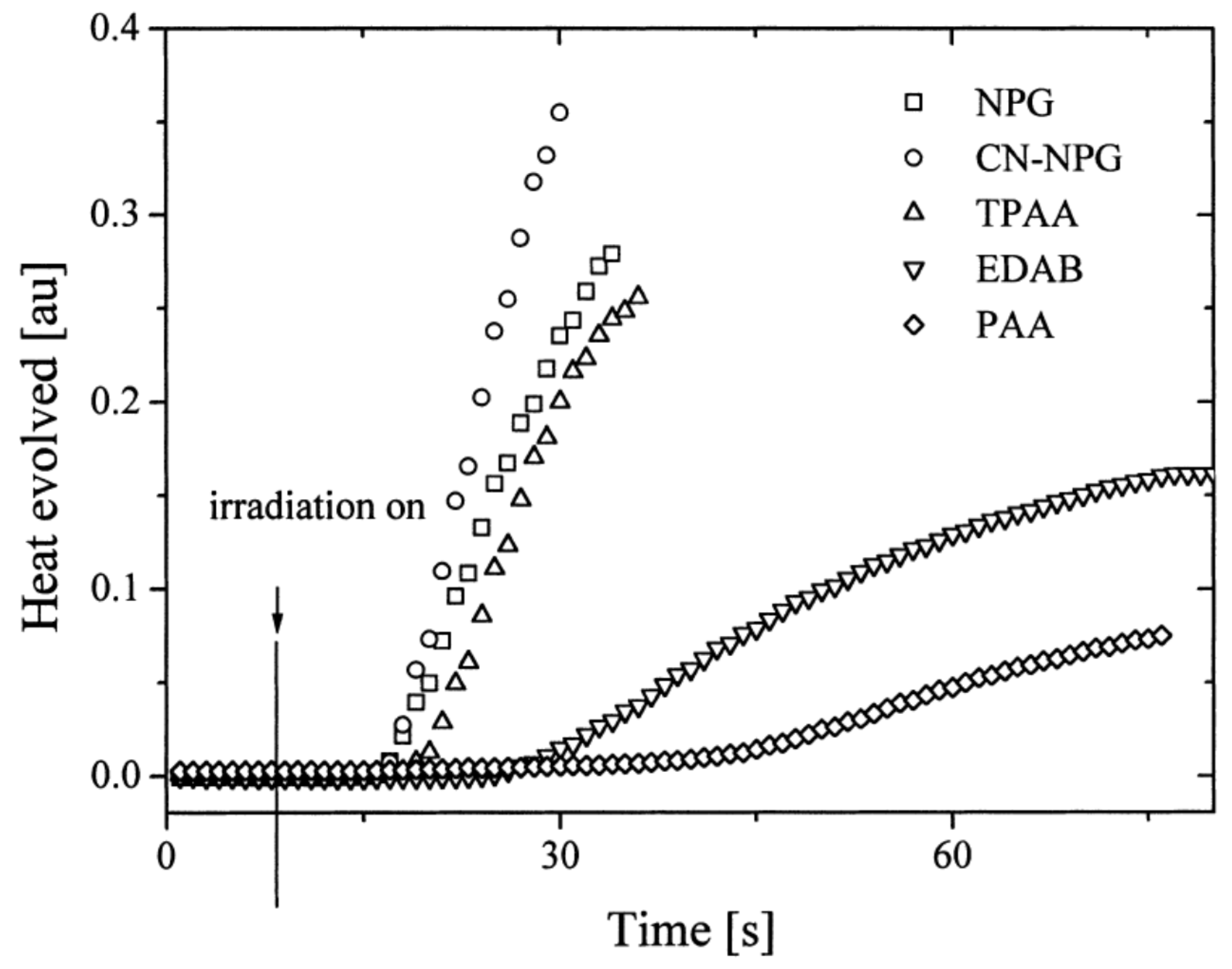
N-phenylglycine (NPG),
N-(4-cyanophenyl)glycine (CN-NPG), (phenylthio)acetic acid (TPAA), ethyl 4-dimethylaminobenzoate (EDAB) and phenoxyacetic acid (PAA), the best monomer conversions were obtained with NPG and CN-NPG while using IQCH3 as the dye (See
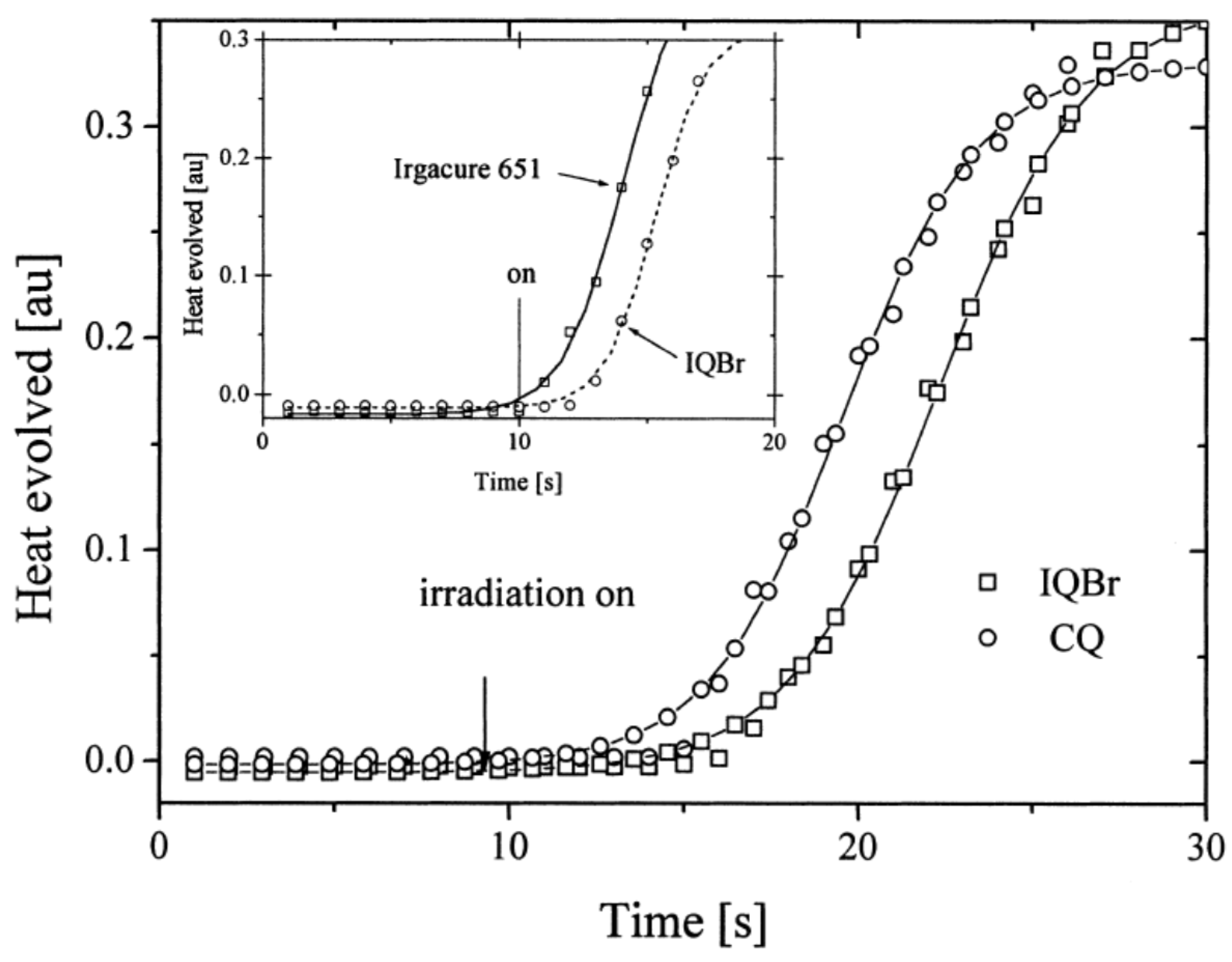
Figure 8). Using the best electron donors (NPG and CN-NPG), the highest monomer conversions were obtained with IQBr and IQCH3Cl2 bearing halogens. Here again, the beneficial effect of heavy atoms on quinoxalines was demonstrated. Noticeably, no direct correlations could be established between the rates of the electron transfer and the polymerization rates determined for the different dyes. This unexpected result was assigned to differences in molar extinction coefficients between dyes and the diffusion effects of radicals within the resins affecting the polymerization efficiency.
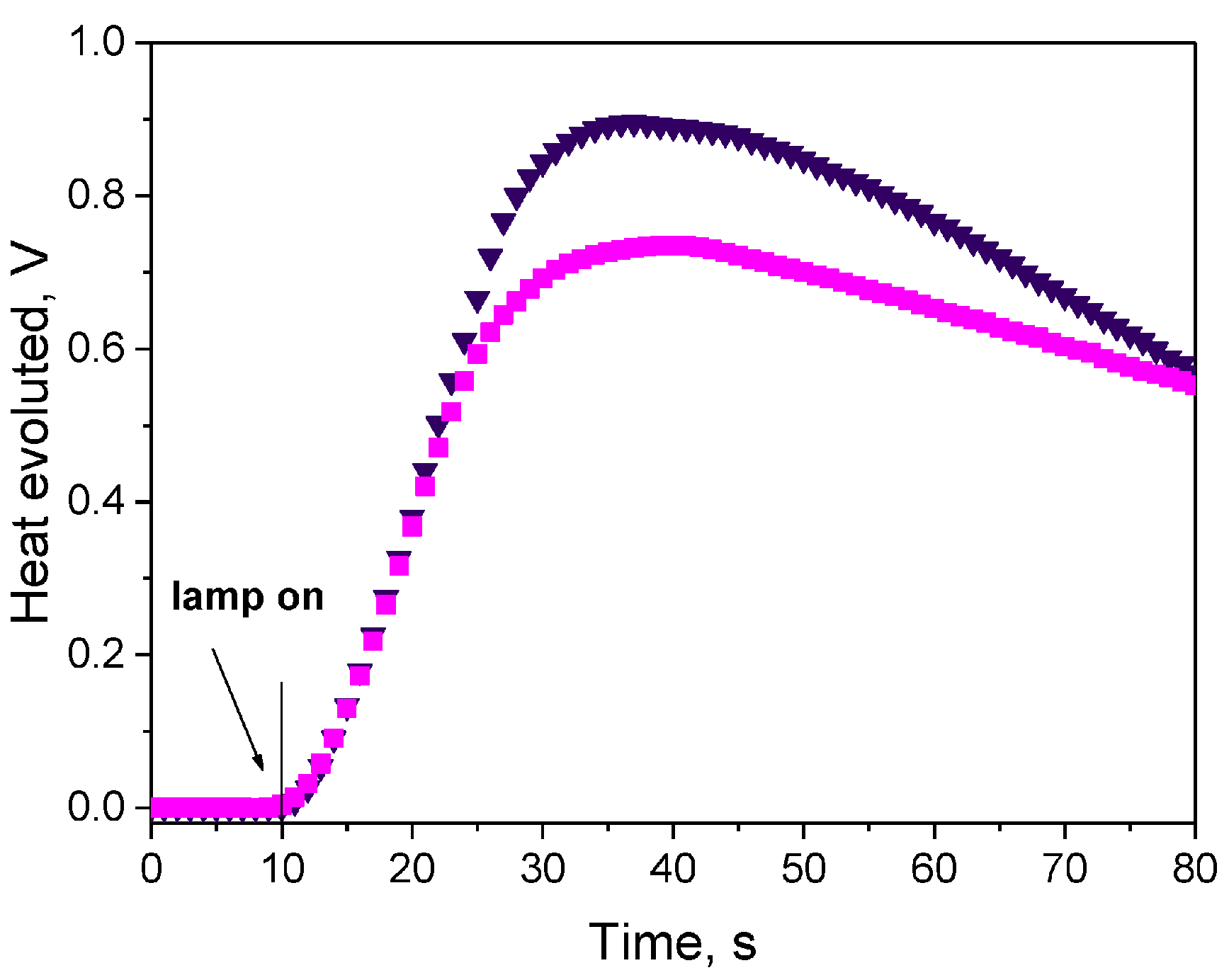
Finally, a comparison of the photo-initiating ability of IQBr with that of camphorquinone (CQ) and 2,2-dimethoxy-2-phenylacetophenone (DMPA, Irgacure 651) revealed IQBr furnished similar polymerization rates to these benchmark photoinitiators (See
Figure 9).
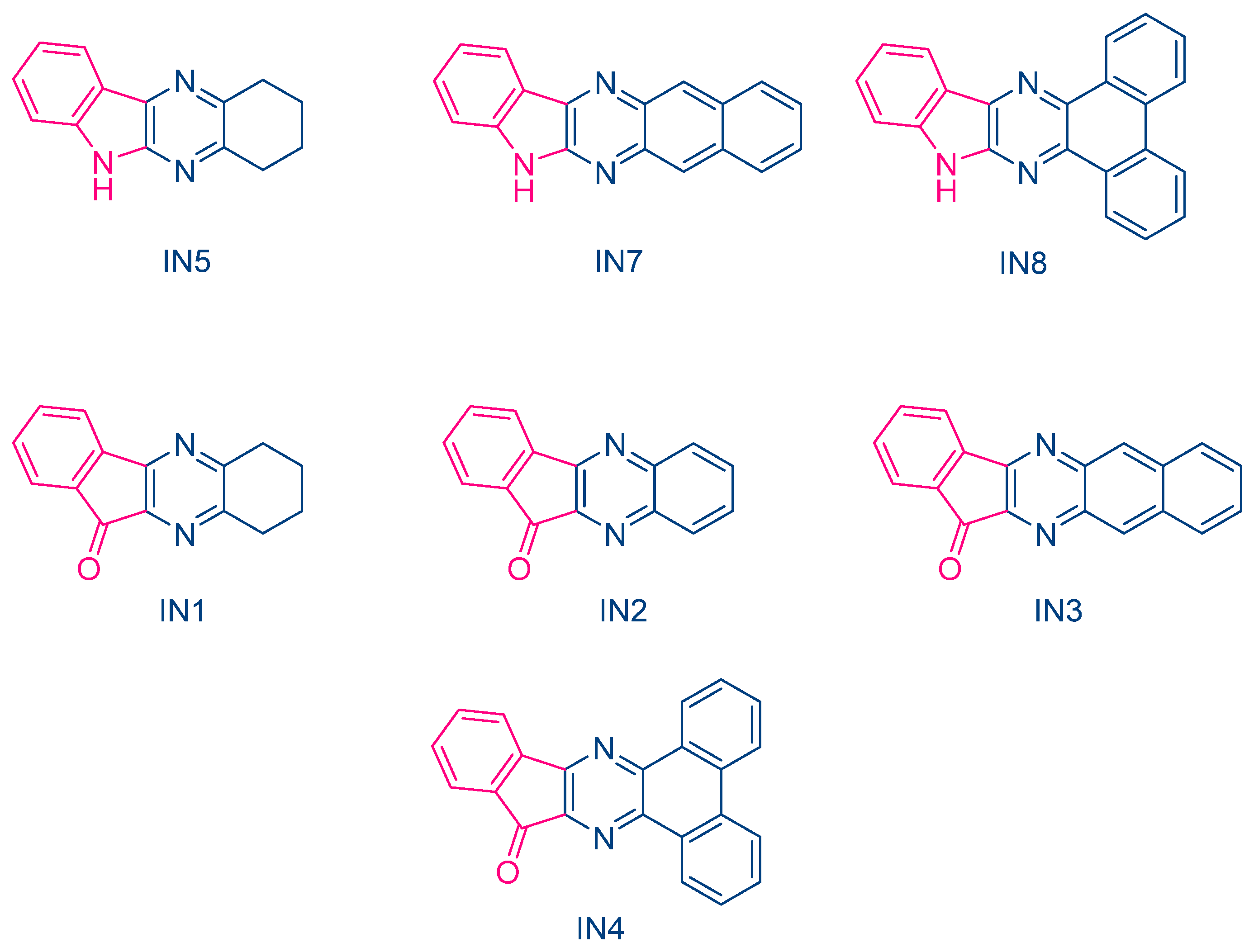
In 2023, Jędrzejewska and coworkers revisited IQH in the context of a comparative study between indenono- and indoloquinoxaline derivatives IN1-IN5, IQH, IN7 and IN8 (See
Figure 10) [
220,
221]. The different dyes were used as electron acceptors for (phenylthio)acetic acid (PTAA) [
222], and the resulting photoredox pairs were used as photo-initiating systems for dental applications. The mechanism of radical generation is presented in
Scheme 2.
From the absorption viewpoint, all dyes IN1-IN5, IQH, IN7 and IN8 showed an intense absorption band centered in the near UV range (See
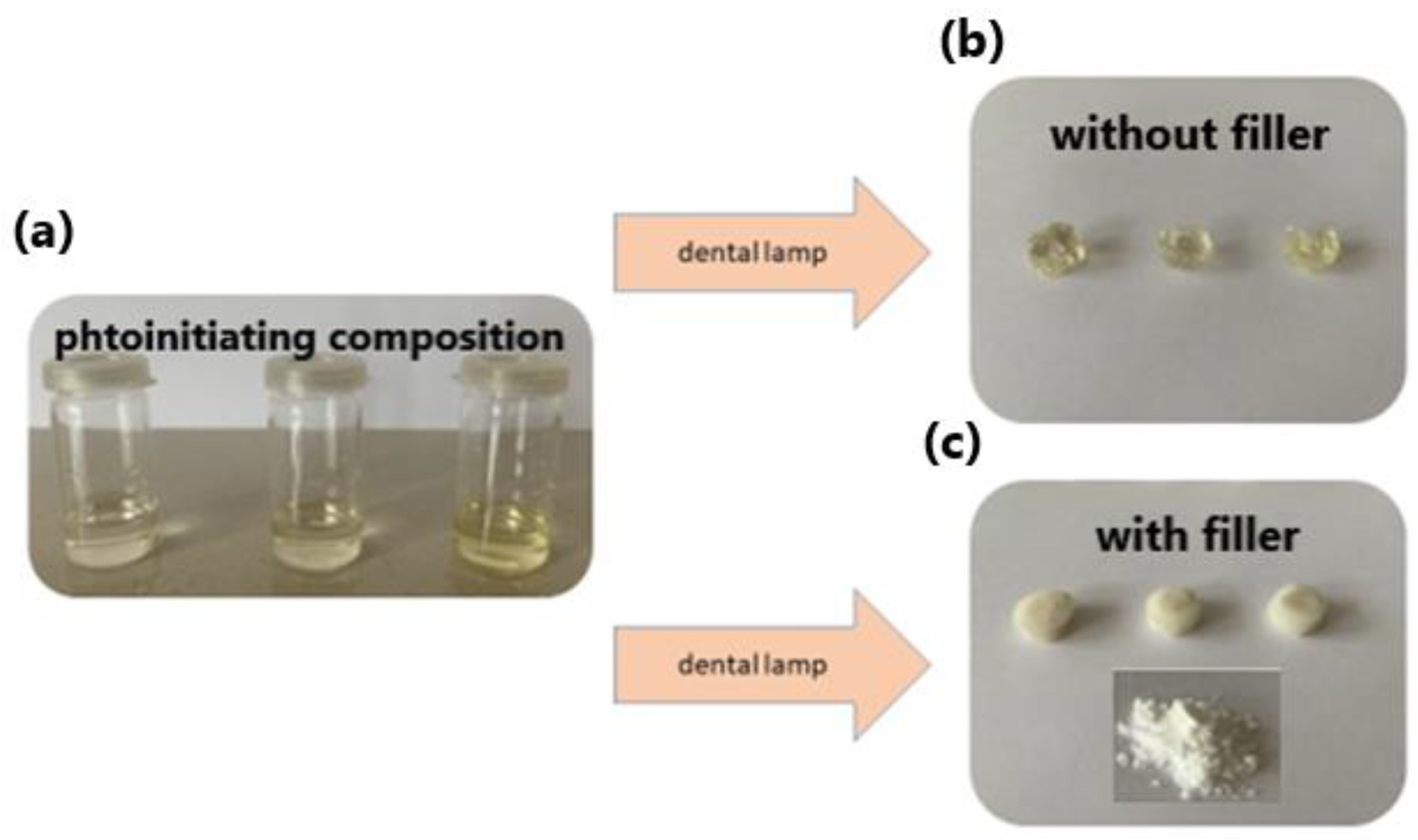
Table 3). Only IN8 exhibited an absorption peak located in the visible range, peaking at 417 nm and attributable to the presence of the phenanthrene moiety extending the aromaticity of this dye. In fact, IN4 and IN8 exhibited the most red-shifted absorption for the two series of dyes, namely the indenono- and indoloquinoxaline series. This redshift was beneficial for the FRP experiments. Indeed, when paired with PTAA, the highest polymerization rates were obtained with these two dyes due to a better match between the emission of the dental lamp emitting at 400 nm and the absorption maxima of these chromophores. In fact, the photo-initiating abilities of these photoredox pairs were comparable to that of camphorquinone (CQ), a benchmark photoinitiator commonly used in dentistry. Interestingly, in the context of dental fillings, an increase of the temperature lower than 5 °C was evidenced, which did not exceed the temperature tolerance threshold for the tooth pulp.
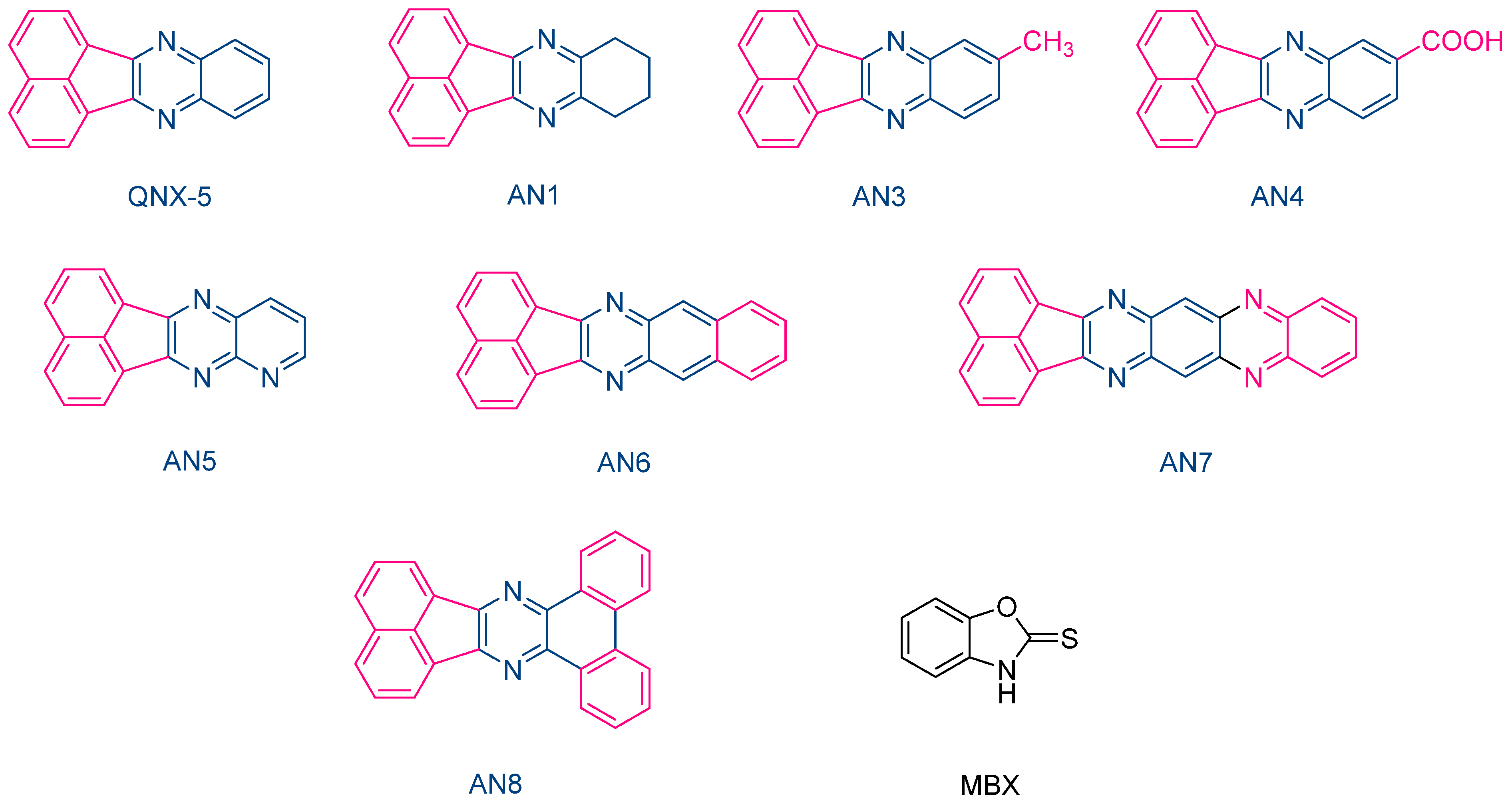
The same performances were also obtained with another series of photoredox pairs based on acenaphthoquinoxalines and 2-mercaptobenzoxazole (MBX) used as the co-initiator (See
Figure 11) [
223]. In this series of dyes (QNX5, AN1, AN3-AN-8) and during the FRP of TMPTA, the lowest heat increase was obtained with the AN6/MBX combination. In addition, the temperature increase with the other dyes remained in the tolerance threshold for dental applications. Due to their strong absorption located in the UV range (See
Table 4), colorless dental fillings could be obtained with these different acenaphthoquinoxalines, making these dyes suitable candidates for dental applications.
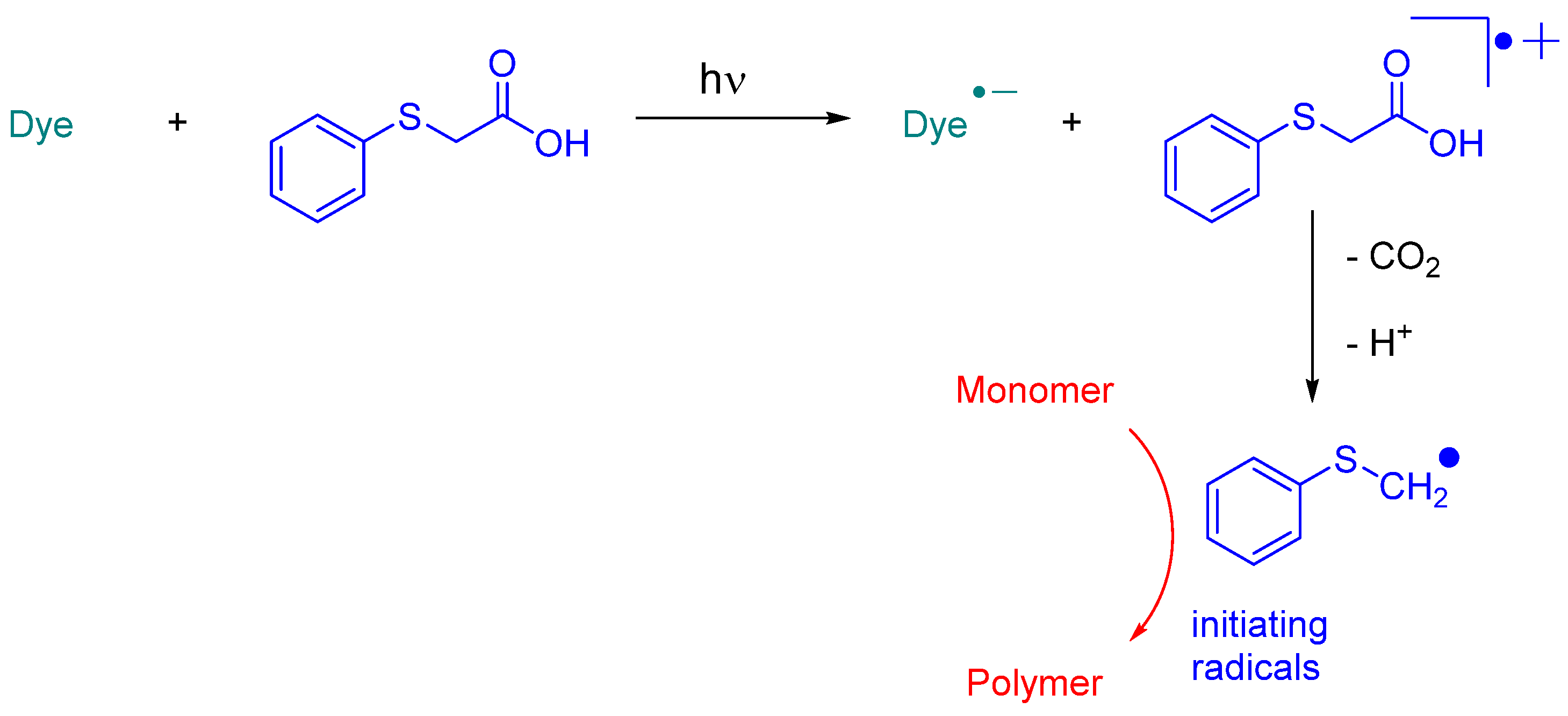
While NPG was extensively used as an electron donor for quinoxaline derivatives, other compounds were also examined, as exemplified with
N-methyldiethanolamine (MDEA) that was used as an electron donor for UV photopolymerization experiments (See
Figure 12) [
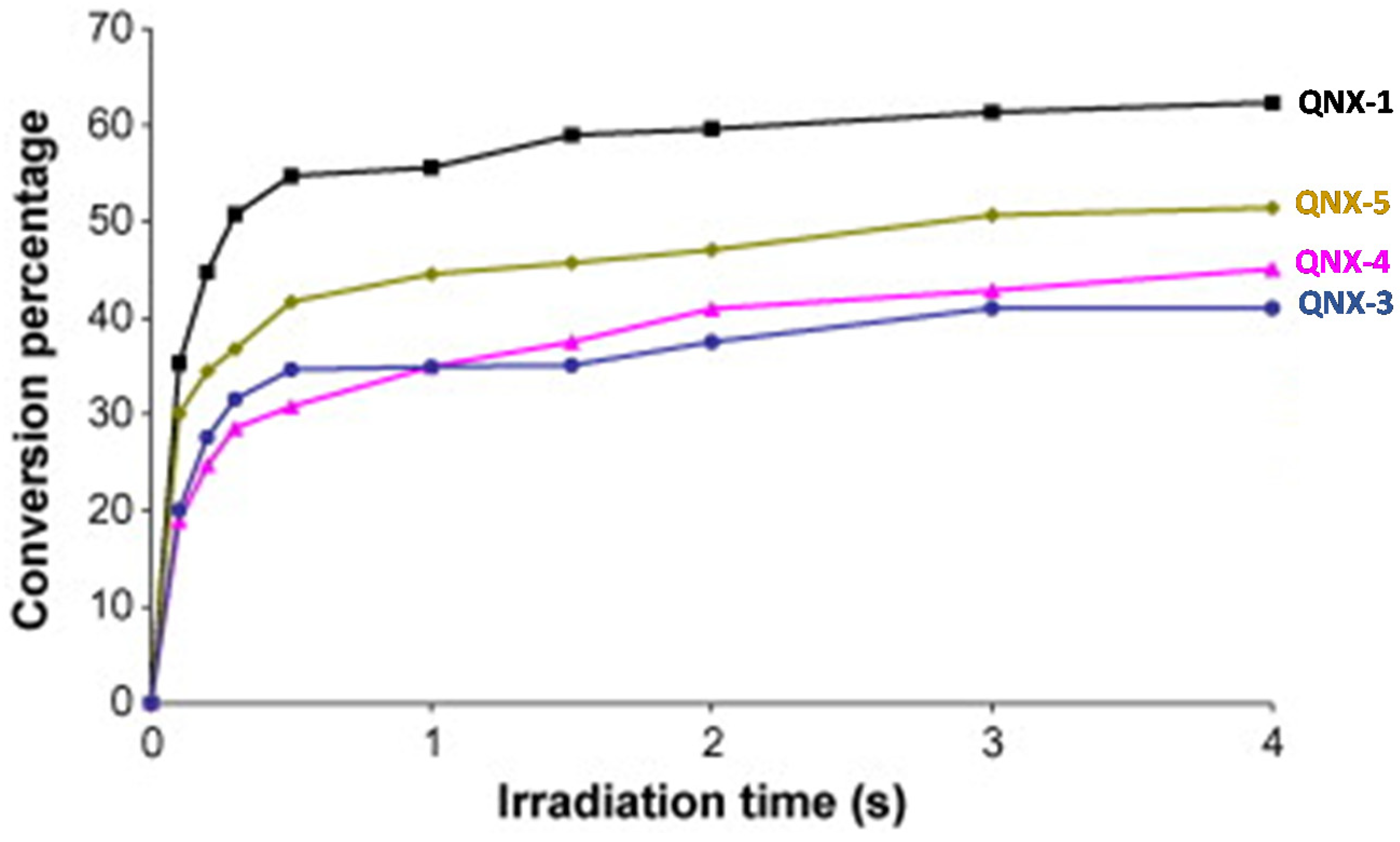
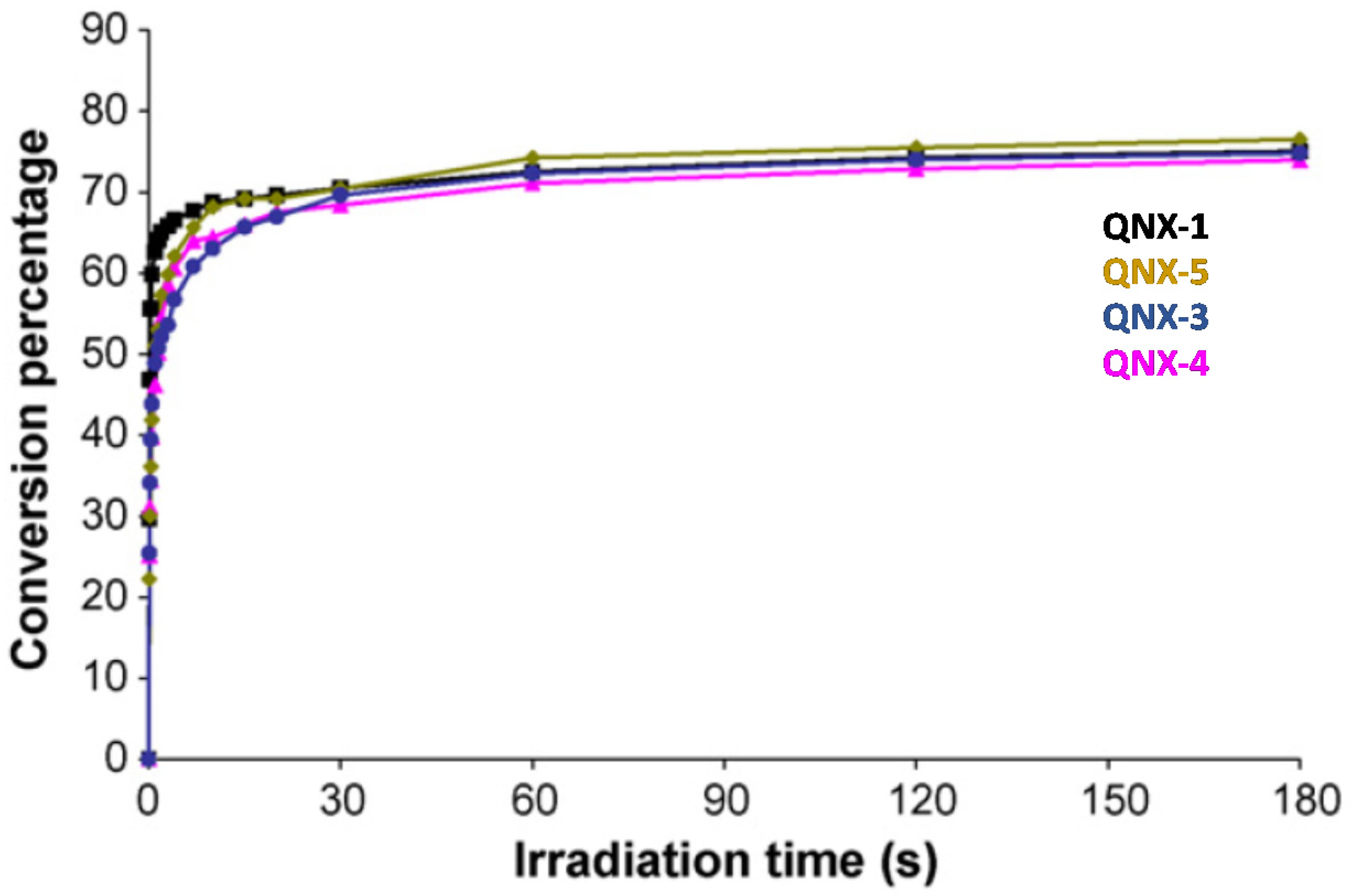
224]. 2,3-Diphenylquinoxaline (QNX-1), quinoxaline (QNX-3), 2,3-dimethylquinoxaline (QNX-4) and acenaphthoquinoxaline (QNX-5) were tested as UV photoinitiators.
Interestingly, by using a polychromatic light, very fast polymerization processes were evidenced since a full curing of TMPTA was obtained within 4 s of irradiation (See
Figure 13). The highest final monomer conversion was obtained for QNX-1, with a conversion of 60% after 4 s. Noticeably, the highest monomer conversions were obtained for the most polyaromatic structures (QNX-1, QNX-5), certainly attributable to higher molar extinction coefficients in the visible range. Contrarily to the different work previously mentioned in this review, the TMPTA conversion could be monitored by Real-Time Fourier Transform Infrared (RT-FTIR) spectroscopy, giving direct access to the monomer conversion. To conduct this, modification of the IR peak at ca 6120 cm
−1 was monitored, corresponding to a characteristic peak of TMPTA. The polymerization profiles were established from the difference between the initial peak area before irradiation and the peak area after irradiation for a given time t [
225,
226].
This trend of reactivity was confirmed during the FRP of other monomers, such as a 75% P-3038 epoxyacrylate (EA) and 25% tripropyleneglycol diacrylate (TPGDA) (3/1
v/
v) blend (See
Figure 14). However, by elongating the irradiation time to 180 s, QNX-5 furnished a similar monomer conversion to QNX-1. Clearly, the difference in monomer conversions is directly related to differences in polymerization rates at early irradiation time.
In the case of 5,12-dihydroquinoxalino[2,3
b]quinoxalines (i.e., fluoflavins), these dyes were interesting candidates to induce the reductive decomposition of alkoxypyridinium salts (See
Figure 15) [
227]. The fluoflavin dyes/alkoxypyridinium salts combinations proved to be excellent two-component systems to initiate the FRP of TMPTA under visible light. It has to be noticed that previously to this work, cyanines [
228], ketocoumarins [
229] and acridinedione dyes [
230] were also used as sensitizers capable of inducing the decomposition of alkoxypyridinium salts by photoinduced electron transfer. From the mechanistic viewpoint, upon excitation of the dye, a photoinduced electron transfer from fluoflavins towards the alkoxypyridinium salt can occur, generating an alkoxypyridinium radical that immediately decomposes, generating initiating alkoxy radicals (RO•) (See
Scheme 3). Fluoflavin dyes were relatively insensitive to the substitution pattern since absorption maxima located at 420 nm were determined for all dyes in 1-methyl-2-pyrrolidone as the solvent (See
Table 5).
Examination of their photo-initiating abilities during the FRP of TMPTA upon irradiation with a visible light revealed QNX-7 and QNX-8 furnished the best polymerization rates (See
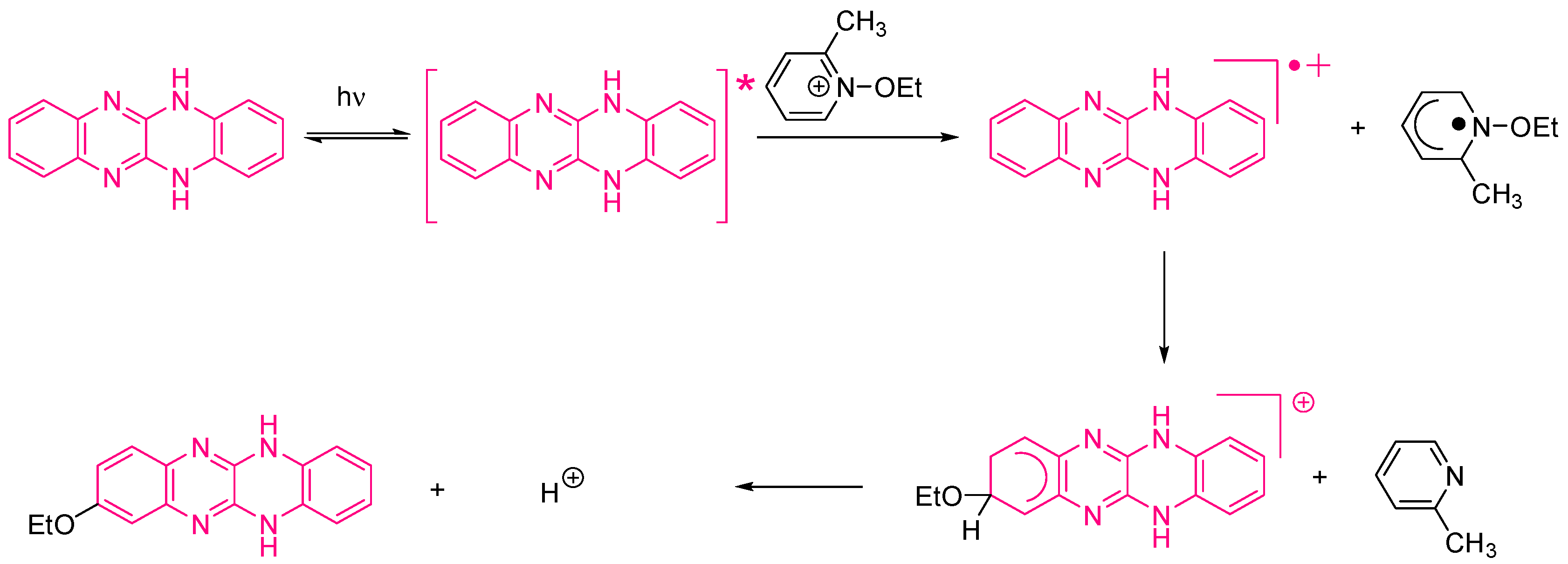
Figure 16). More precisely, QNX-7 was determined as outperforming all the other dyes, irrespective of the alkoxypyridinium salt. Experiments carried out with Py2 also furnish higher monomer conversion than Py1, once again evidencing the necessity to screen both the electron donors and acceptors in order to obtain the best combination. Interestingly, an efficient photobleaching of the TMPTA resins was observed during polymerization, assigned to the addition of alkoxy radicals on fluoflavins, according to the mechanism depicted in
Scheme 4. Precisely, a reaction between the fluoflavin radical cation and the ethoxyl radicals at the 9-position of the phenyl ring was proposed by analogy to previous works performed on anthracene [
231,
232]. After proton release, an ethoxy derivative of the fluoflavin dyes can be formed, enabling the ability to obtain an efficient bleaching of the final polymers.
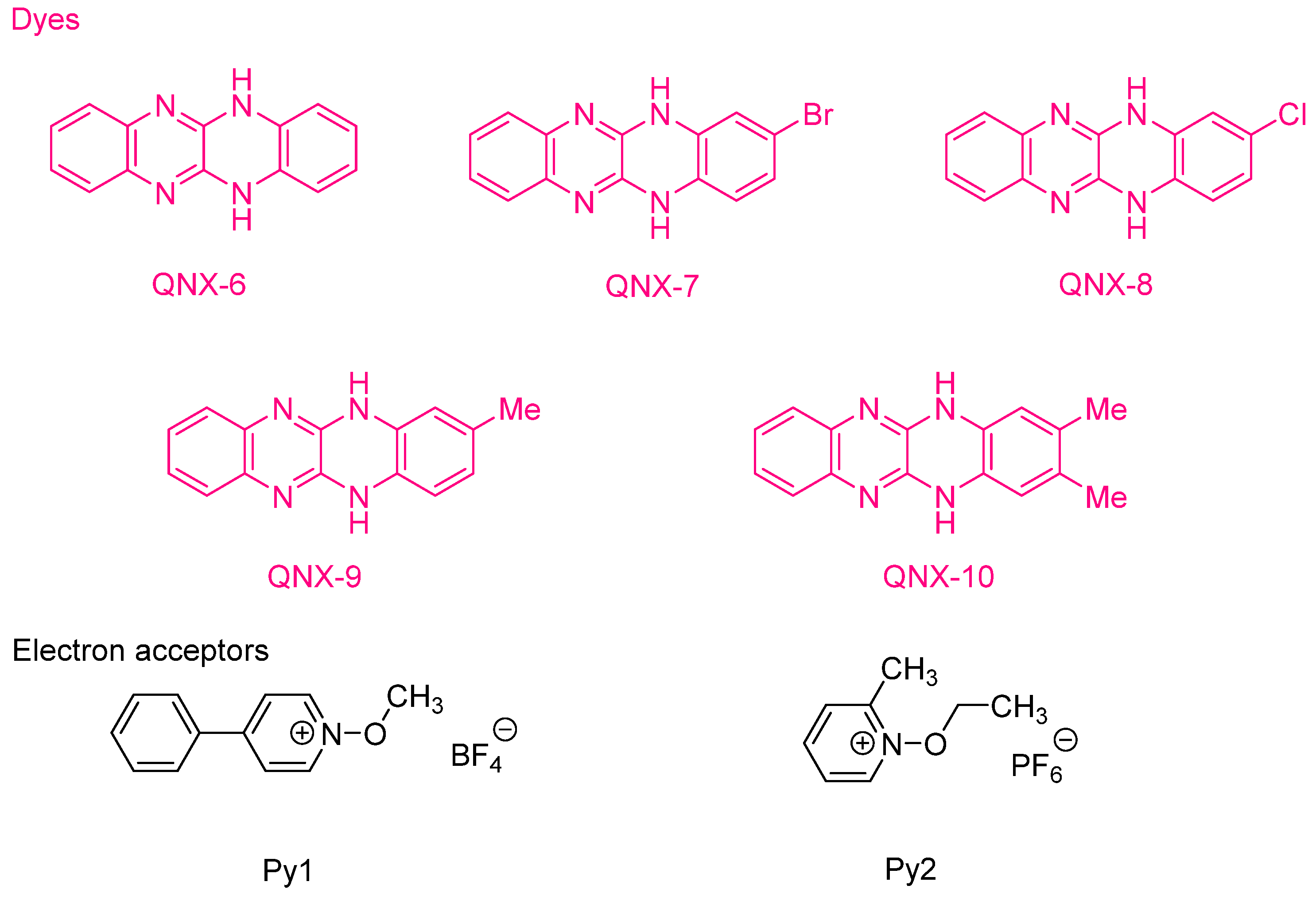
The possibility to initiate free-radical/cationic hybrid polymerizations (concomitant polymerization of TMPTA and 3,4-epoxycyclohexylmethyl 3′,4′-epoxycyclohexanecarboxylate (EPOX)) with fluoflavin derivatives was also examined. Using QNX-6-QNX-8 as the photosensitizers for triarylsulfonium hexafluoroantimonates, epoxide conversions higher than that of acrylates were observed with all photo-initiating systems [
233]. The lower acrylate conversion determined during hybrid photopolymerization was assigned to oxygen inhibition favoring the cationic polymerization of epoxides. Indeed, contrarily to the cationic polymerization, which is insensitive to oxygen inhibition, free radicals can react with molecular oxygen, generating unreactive peroxyl radicals. In 2009, the same authors examined a series of dyes analogues to fluoflavins, namely QNX-11-QNX-19, that were used as electron donors for the reductive photosensitization of
N-methoxy-4-phenylpyridinium tetrafluoroborate (Py1) and
N-ethoxy-2-methylpyridinium hexafluorophosphate (Py2) examined in the previous work and for diphenyliodonium hexafluorophosphate (Ph
2I
+PF
6−) (See
Figure 17) [
234]. These photoinitiators were advantageously used for the FRP of acrylates under visible light and the cationic polymerization (CP) of cyclohexene oxide (CHO).
Noticeably, the QNX-11-QNX-19 series exhibited similar absorption properties irrespective of the substitution pattern. An intense absorption band was notably detected in the visible region and located at approximately 415 nm (see
Table 6). Absorption maxima ranging from 409 nm for QNX-14 and QNX-17 and up to 421 nm for QNX-16 and QNX-19 were determined in 1-methyl-2-pyrrolidinone.
The most red-shifted absorptions were determined for the dyes bearing chlorine atoms, except for QNX-17, which exhibited the lowest absorption wavelength listed in
Table 6. Compared to the previous series of 5,12-dihydroquinoxalino[2,3-
b]quinoxalines, the presence of an additional nitrogen atom in 5,12-dihydroquinoxalino[2,3-
b]pyridopyrazines resulted in a slight blue shift of their absorption maxima (See
Table 5). Once again, faster polymerization processes and higher monomer conversions were obtained with Py2 and the different dyes during the FRP of TMPTA. The fastest polymerization process was obtained with QNX-11, irrespective of the electron acceptors. In all cases, relatively long inhibition times were determined. This is directly related to the inhibition effects of oxygen dissolved in the photocurable resins. Oxygen can quench the triplet states of the dyes, adversely affecting the polymerization process. Finally, the QNX-11-QNX-19/Ph
2I
+PF
6− combinations also initiated the CP of CHO. However, the polymerization process was slow since 30 min. of irradiation was required to obtain a full curing of the resin. Similarly, as in the case of FRP of TMPTA, the best performances during the CP of CHO were also obtained with QNX-14-QNX-16, i.e., for all dyes substituted with a bromine atom. The higher polymerization efficiency of these dyes is thus related to the heavy atom effect, elongating the excited state lifetimes of the dyes by favoring the triplet state pathway and thus improving the intermolecular interactions of the dyes with the different additives.
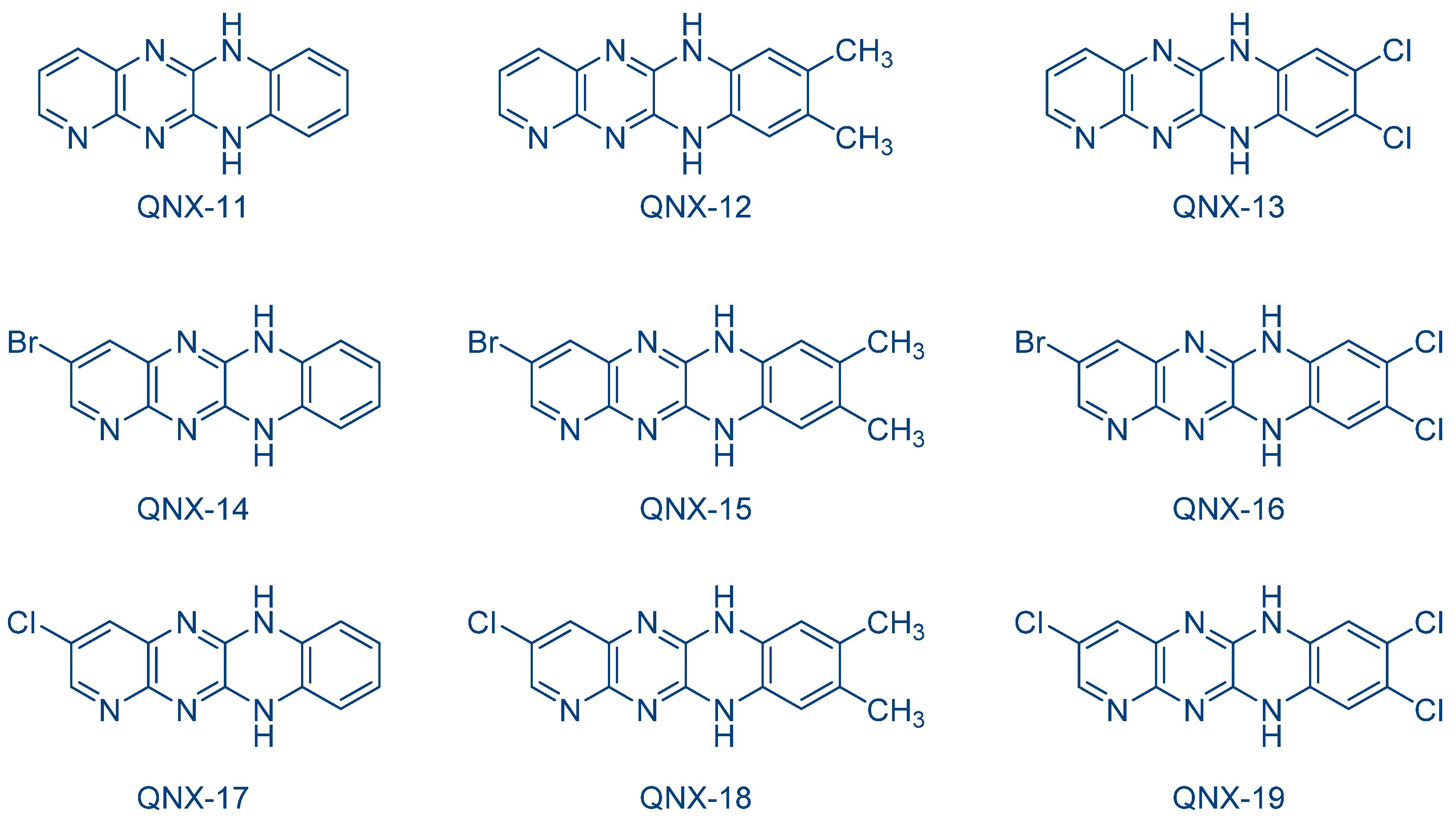
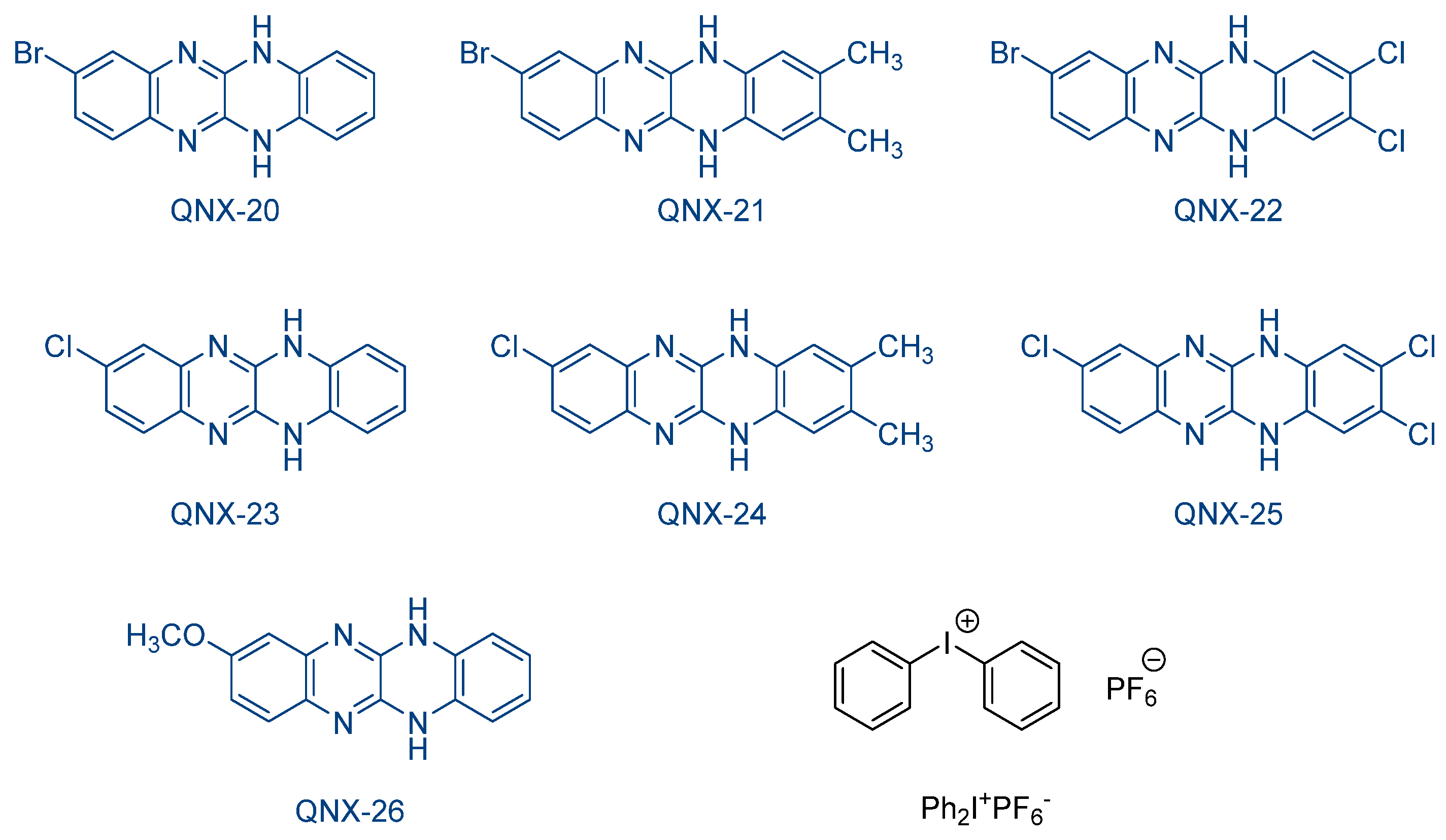
In order to investigate the heavy atom effects, a series of 8-halogeno-5,12-dihydroquinoxalino[2,3-
b]quinoxalines QNX-20-QNX-26 was examined by the same group (See
Figure 18) [
234]. As interesting features, all dyes absorb around 420 nm (See
Table 7), i.e., at the same position as the previous series (i.e., the QNX-11-QNX-19 series). Noticeably, in this series of dyes, QNX-20-QNX-22 bearing bromine atoms clearly outperformed the other dyes, irrespective of the electron acceptor. Notably, during the FRP of TMPTA, similar monomer conversions were obtained using
N-methoxy-4-phenylpyridinium tetrafluoroborate (Py1) or
N-ethoxy-2-methylpyridinium hexafluorophosphate (Py2) as the electron acceptors (See
Table 7). Thus, while conversions ranging between 21 and 24% were determined with QNX-20-QNX-22 as the photosensitizers and Py2 as the co-initiator, these values decreased to 1–18% for the non-brominated quinoxalines QNX24-WNX-26. No significant modification of the monomer conversion was obtained by replacing Py2 with Py1 as the co-initiator. During the CP of cyclohexene oxide (CHO), higher monomer conversions could be obtained with regards to the monomer conversions determined during the FRP of TMPTA. By combining QNX-20-QNX-22 with Ph
2I
+PF
6−, a CHO conversion of 97% was obtained with the two-component QNX-20/Ph
2I
+PF
6− combination, greatly higher than that obtained with QNX-23-QNX-26 as the photosensitizers (CHO conversions ranging between 29 and 63% after 240 s of irradiation with a visible light) (See
Table 8). These monomer conversions perfectly fit with the quantum yield of acid release determined upon photolysis. Indeed, the QNX-20/Ph
2I
+PF
6− system was determined as the photo-initiating system furnishing the highest quantum yield of acid release of the series.
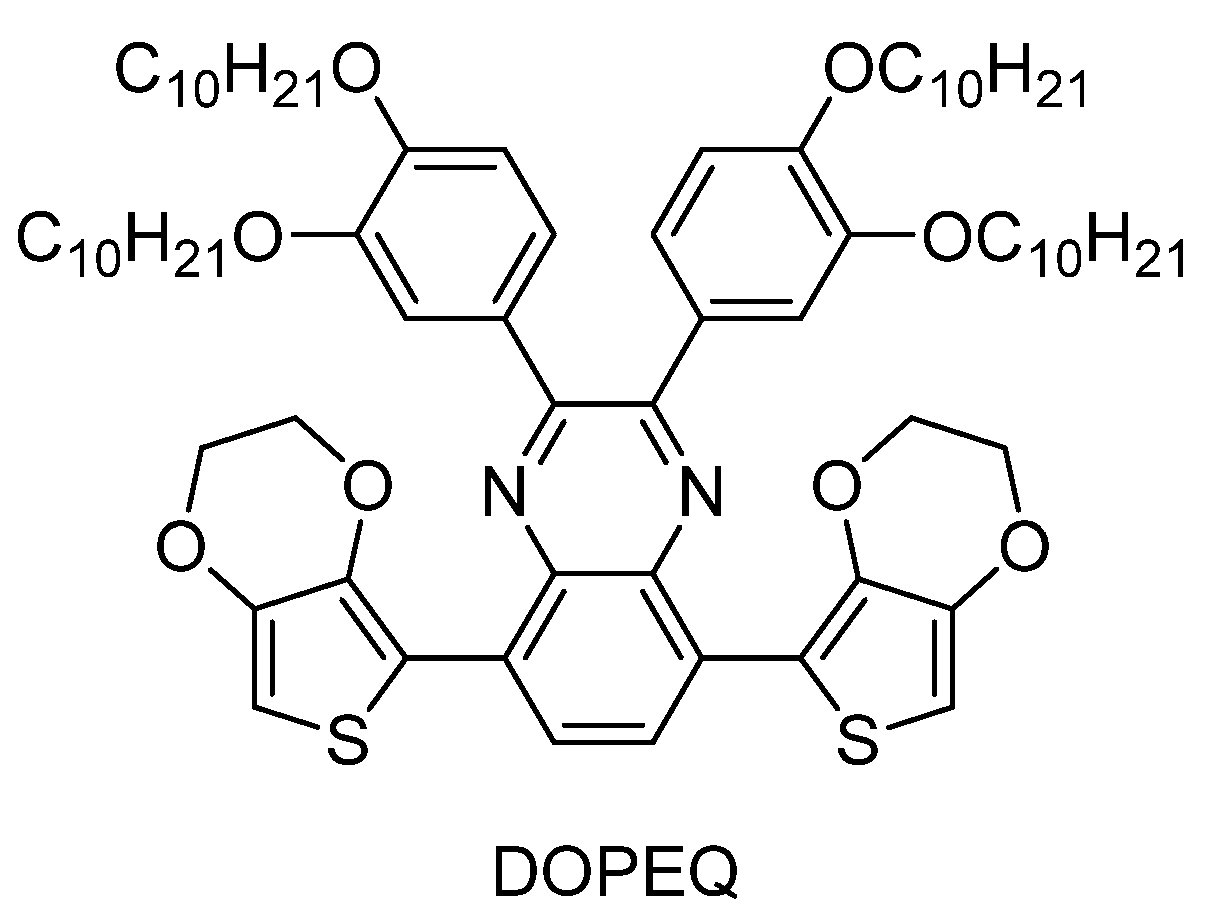
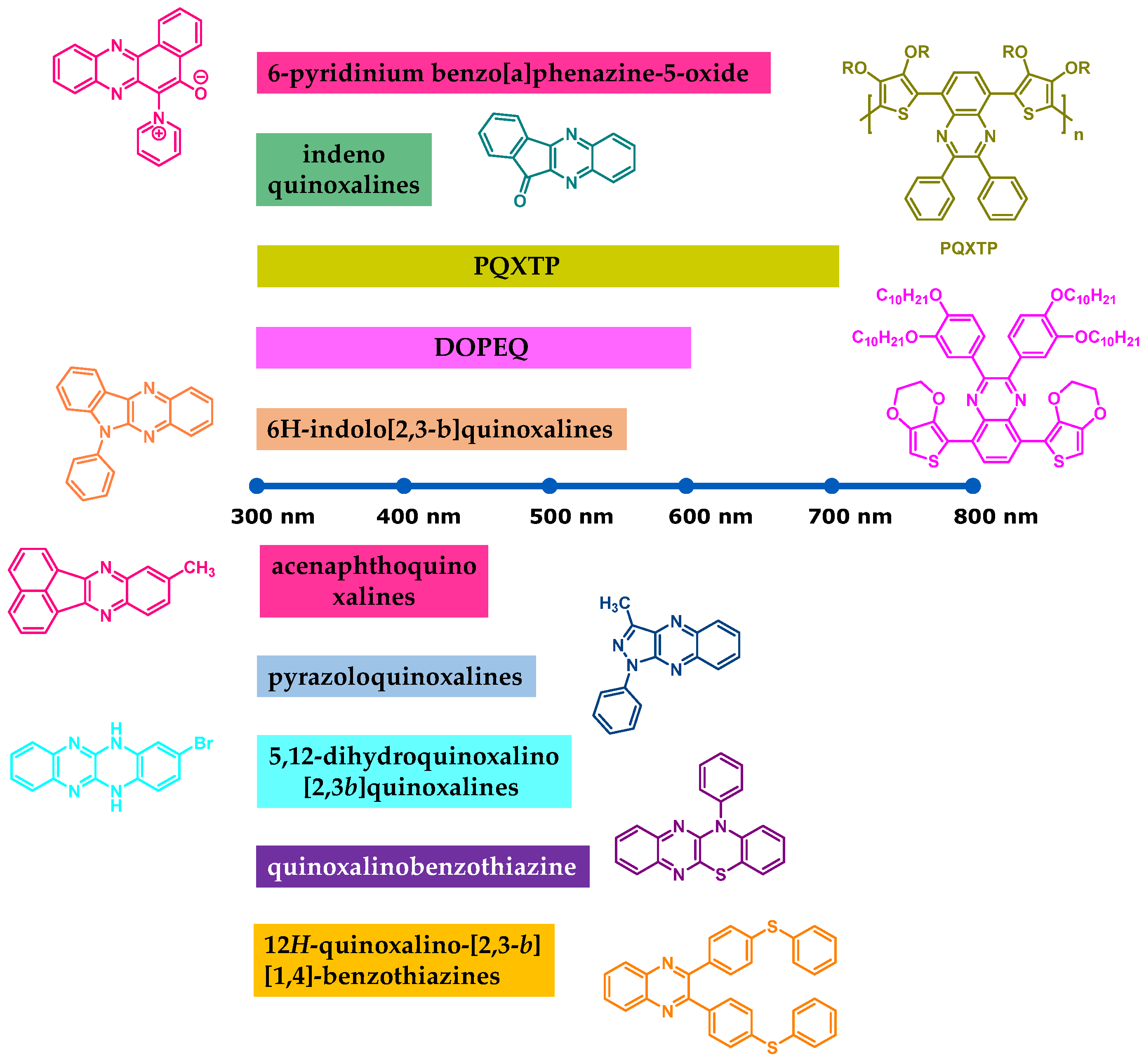
The design of quinoxaline derivatives absorbing in the 450–550 nm range is a real challenge, as these chromophores naturally absorb in the 350–420 nm range. In 2009, Toppare and coworkers succeeded in addressing this issue by developing an original strategy in order to redshift the absorption of a quinoxaline derivative (See
Figure 19) [
235]. Toppare and coworkers notably proposed 2,3-
bis(3,4-
bis(decyloxy)phenyl)-5,8-
bis(2,3-dihydrothieno[3,4-
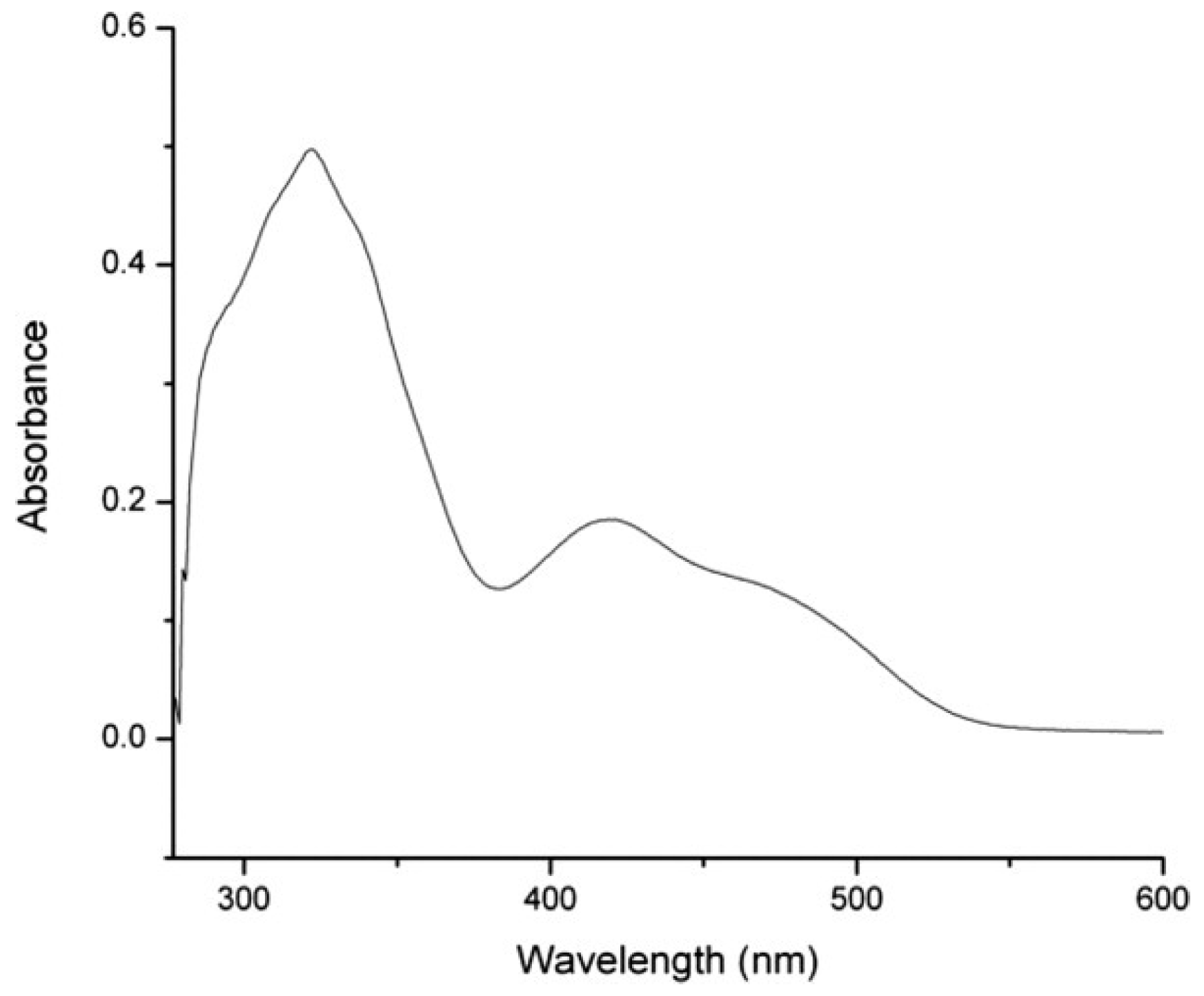
b][1,4]dioxin-5-yl)quinoxaline (DOPEQ) exhibiting two ethylenedioxythiophene (EDOT) groups introduced in lateral positions of the quinoxaline core to extend the conjugation. Four decyloxy groups were also introduced for solubility. Due to its extended conjugation, a broad absorption was found for DOPEQ, extending between 300 and 550 nm, with an absorption maximum located at 420 nm (See
Figure 20).
Cationic photopolymerization of CHO conducted with the two-component DOPEQ/Ph
2I
+PF
6− (0.1%/1%
w/
w) system using a UV light of low intensity (10 mW/cm
2) revealed the polymerization process to be very fast, the polymerization ended within 5 s. By using EPOX as a difunctional monomer, the polymerization ended after 20 s. In light of this remarkable reactivity, polymerization tests were carried out under sunlight because the absorption spectrum of DOPEQ was relatively broad. Using sunlight, polymerization of EPOX occurred within 5 min., evidencing the high reactivity of this two-component system (See
Figure 21).
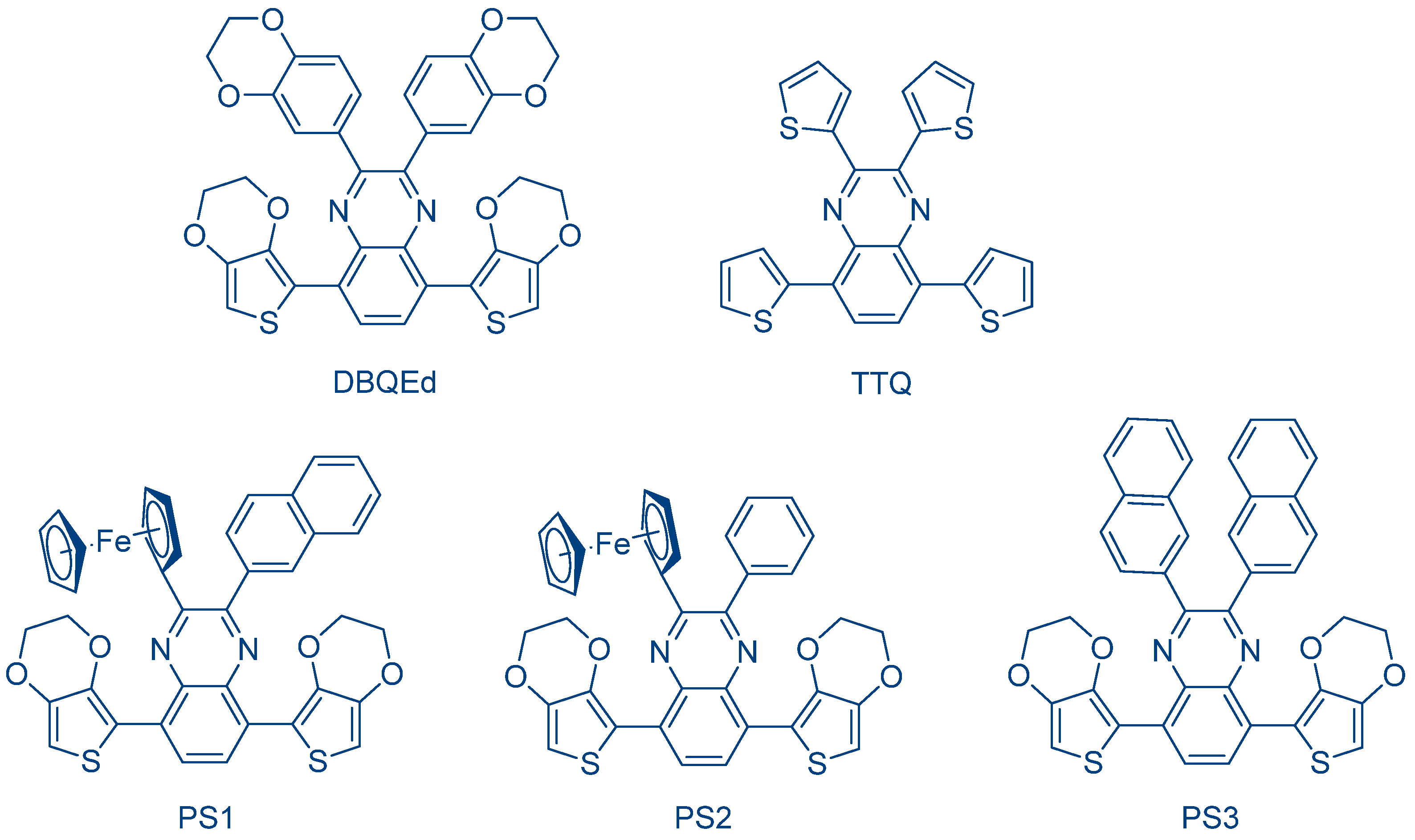
Following this work, other long wavelength photosensitizers were designed by the same authors on the basis of the DOPEQ scaffold, as exemplified with 2,3,5,8-tetra(2,3-dihydrobenzo[
b][1,4]dioxin-6-yl)quinoxaline (DBQEd) and 2,3,5,8-tetra(thiophen-2-yl)quinoxaline (TTQ) (See
Figure 22) [
236].
Here again, highly efficient photo-initiating systems were obtained using Ph2I+PF6− as the cationic initiator. Especially compared to DOPEQ, a reduction of the polymerization time under sunlight was obtained; EPOX was polymerized within 30 s using the two-component DBQEd/Ph2I+PF6− (0.1%/1% w/w) system and 2 min. with the two-component TTQ/Ph2I+PF6− (0.1%/1% w/w) system. The higher reactivity of DBQEd and TTQ compared to DOPEQ was assigned to the presence of electron-rich groups, reducing the oxidation potential of these two dyes and thus favoring the intermolecular electron transfer with the iodonium salt.
The introduction of metallocene, such as ferrocene, as peripheral groups for quinoxalines was also examined, as exemplified with PS1 and PS2 (See
Figure 22) [
237]. Due to the presence of ferrocene, broad absorption spectra extending between 400 and 600 nm were determined for the two dyes in dichloromethane. Especially, while similar extinction coefficients were determined for PS1 and PS2 comprising a ferrocene unit, a lower molar extinction coefficient was measured for PS3 bearing naphthalene groups (See
Figure 23). Excellent photo-initiating abilities were also demonstrated upon sunlight irradiation for PS1-PS3. During the CP of CHO, a full curing of the resin was obtained within 5 min. by using the two-component dye/Ph
2I
+PF
6− (0.1%/1%
w/
w) systems. For comparison, a full curing of CHO was obtained upon irradiation of the samples with a mercury lamp, with reaction times ranging between 10 and 30 s depending on the dyes.
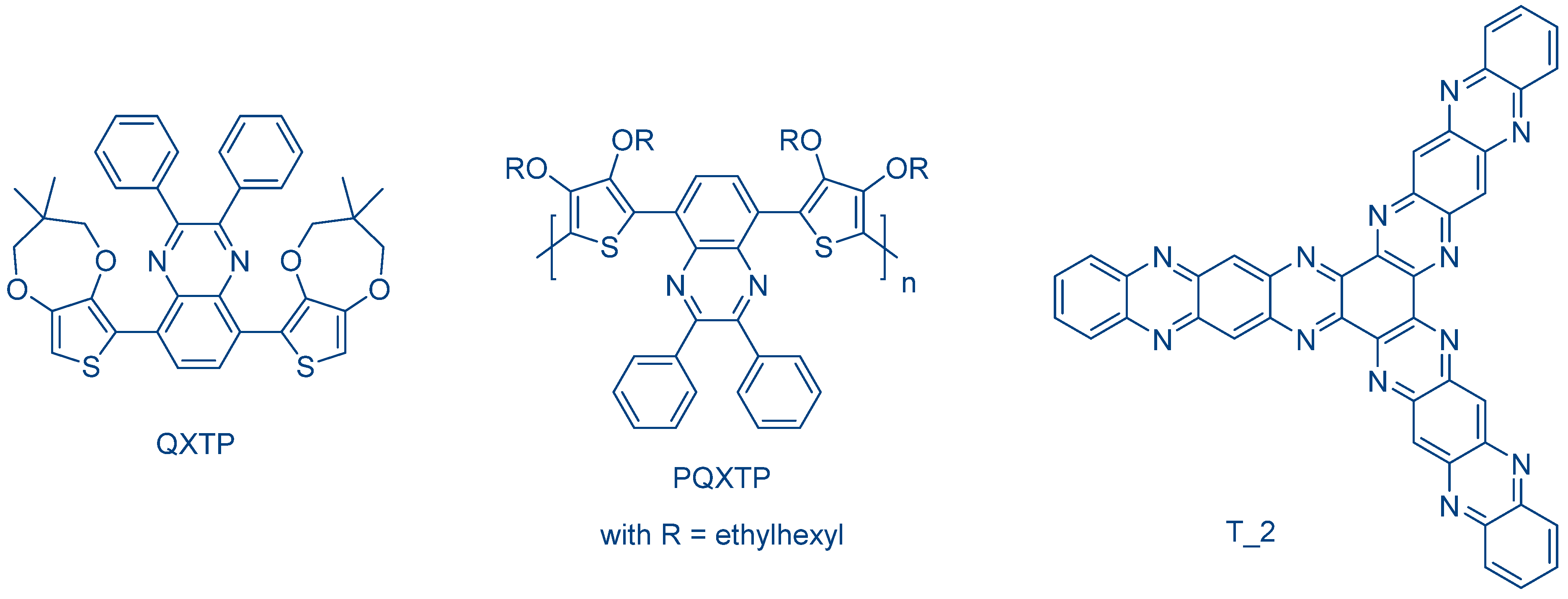
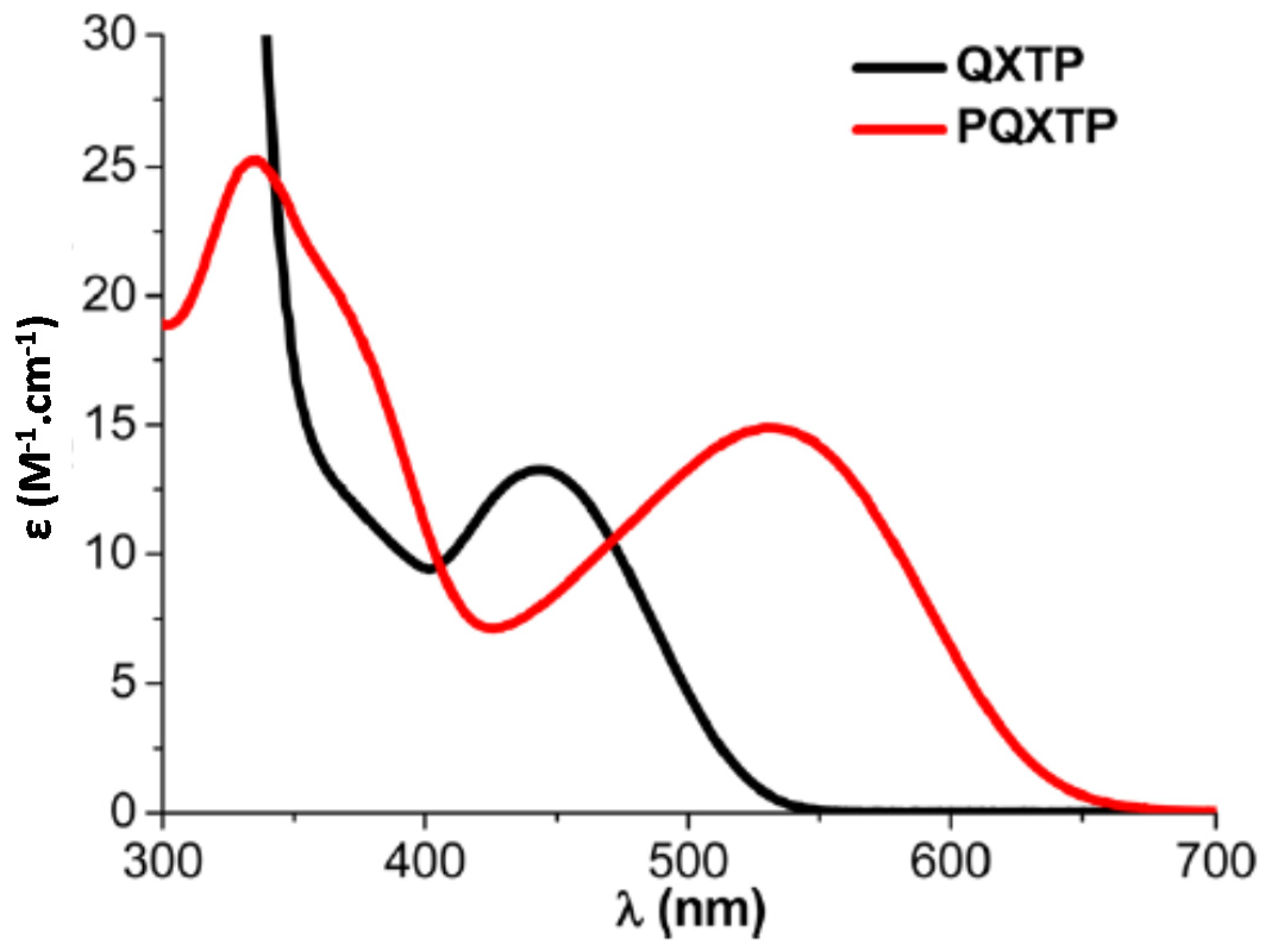
In 2013, Lalevée and coworkers investigated another thiophene derivative, i.e., QXTP and the polymeric PQXTP (See
Figure 24) [
238]. By extending the π-conjugation in PQXTP, a redshift of the absorption was obtained (See
Figure 25).
Interestingly, due to the broad absorptions of QXTP and PQXTP, polymerization experiments could be carried out with laser diodes emitting at 405, 457, 473, 532 and 625 nm and with a halogen lamp (See
Table 9). In order to perform the polymerization experiments, three component dye/Ph
2I
+PF
6−/NVK photo-initiating systems were used (where NVK stands for
N-vinylcarbazole). During the CP of EPOX, the direct correlation existing between the molar extinction coefficient at the irradiation wavelength and the monomer conversion was clearly evidenced. Thus, by irradiating at long wavelengths (532 and 635 nm), a severe reduction of the monomer conversions was observed with PQXTP. The polymerization of interpenetrated polymer networks (IPNs) by concomitantly polymerizing TMPTA and EPOX was also investigated.
The extractability of photosensitizers from the polymer films is a major issue, especially for applications such as food packaging and biomedical applications. Due to its high molecular weight, PTXQT was not extracted from the polymer network due to its polymeric structure. The same experiments performed with QXTP revealed that the extractability was lower than 0.01% for IPNs prepared under an inert atmosphere and 0.5% for IPNs prepared under air. The extractability issue was also addressed with another structure derived from quinoxalines, namely
tris(aza)pentacene T_2 [
129].
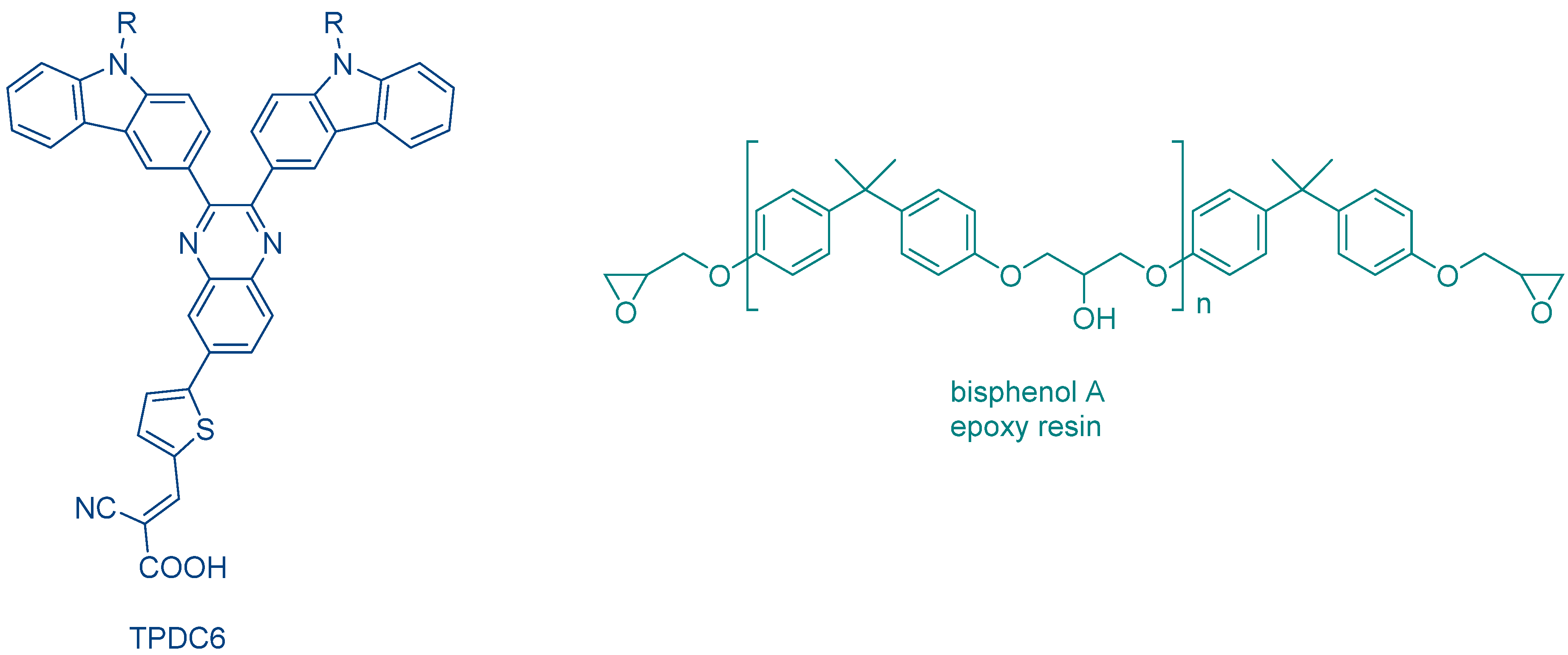
All the previous photoinitiators detailed in this review and absorbing at long wavelengths have been designed with thiophene units. In 2021, Yagci and coworkers introduced carbazole units as peripheral groups for TPDC6 (See
Figure 26) [
239]. From the electrochemical viewpoint, carbazole and thiophene exhibit similar electron-donating properties, so an excellent electron-donating chromophore could be prepared with carbazoles.
The absorption of TPDC6 was broad, extending between 400 and 550 nm. An absorption maximum peaking at 438 nm was also determined in dichloromethane. Due to the strong electron-donating properties of the carbazole groups, an efficient photoinduced electron transfer could occur with Ph2I+PF6−, so that the resulting photo-initiating system efficiently promoted the cationic polymerization of the epoxy resin (bisphenol A Epoxy resin) upon sunlight irradiation. Noticeably, while a monomer conversion of 83% could be obtained after 120 min. of irradiation, an inhibition time as long as 30 min. was evidenced.
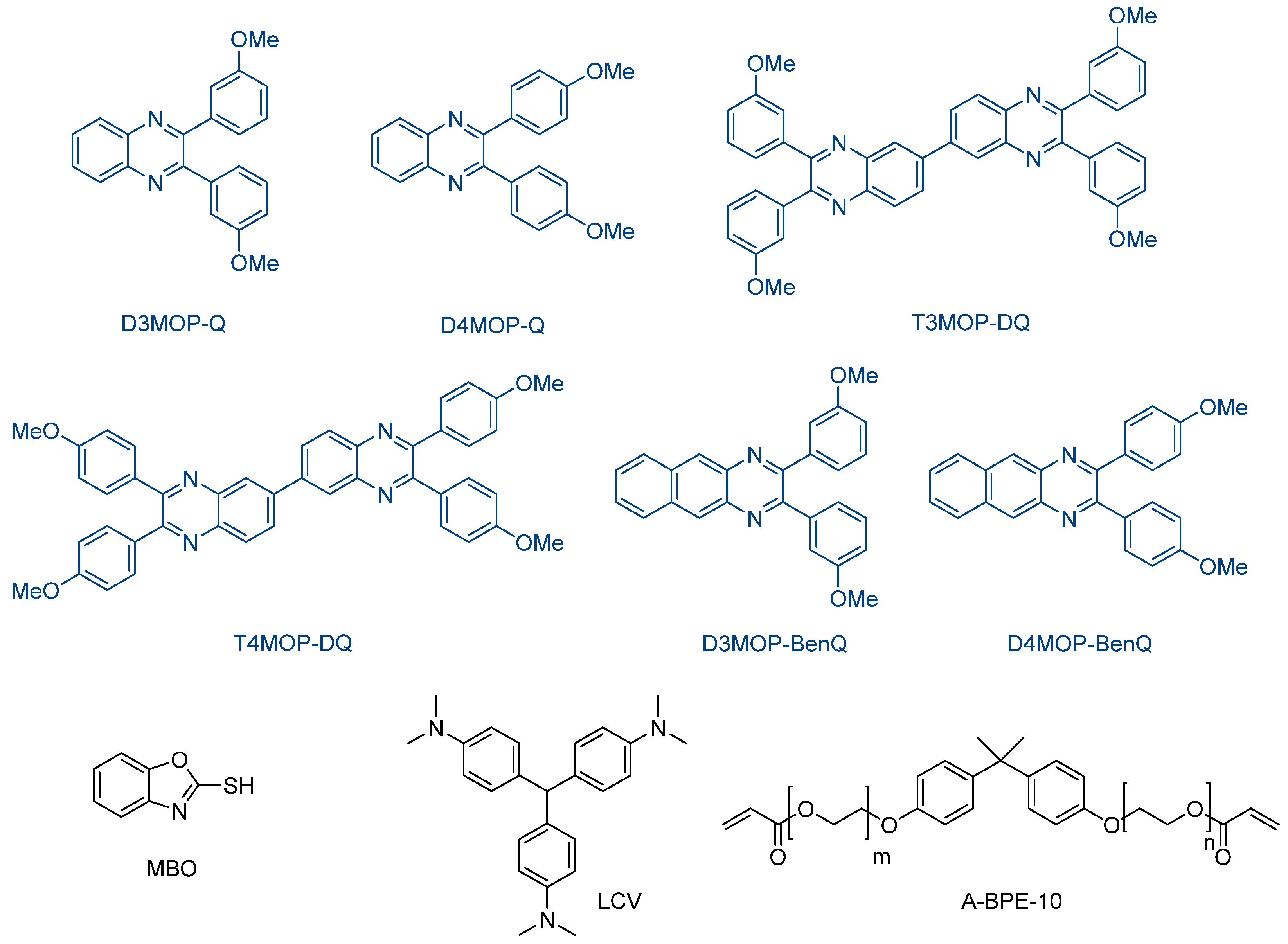
Still based on the diphenylquinoxaline scaffold, a series of methoxyphenylquinoxalines (MOPQs) was investigated for the polymerization of 2,2-
bis(4-(acryloxypolyethoxy)phenyl)propane (A-BPE-10) (See
Figure 27) [
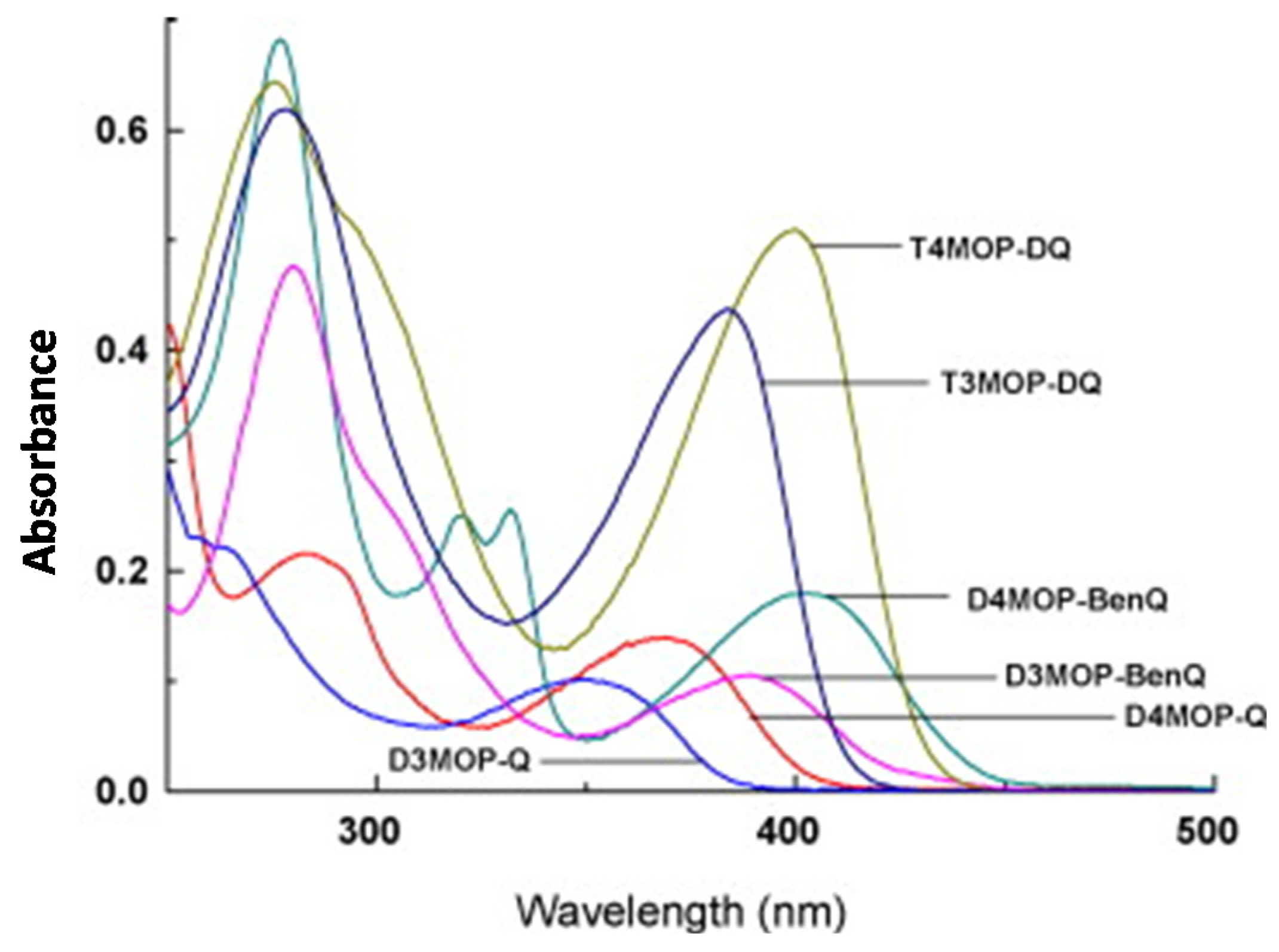
240]. Contrarily to DOPEQ, which exhibited an absorption spectrum in the visible range, absorption spectra of MOPQs remained strongly UV-centered, with absorption maxima ranging between 349 nm for D3MOP-Q and up to 402 nm for D4MOP-BenQ (See
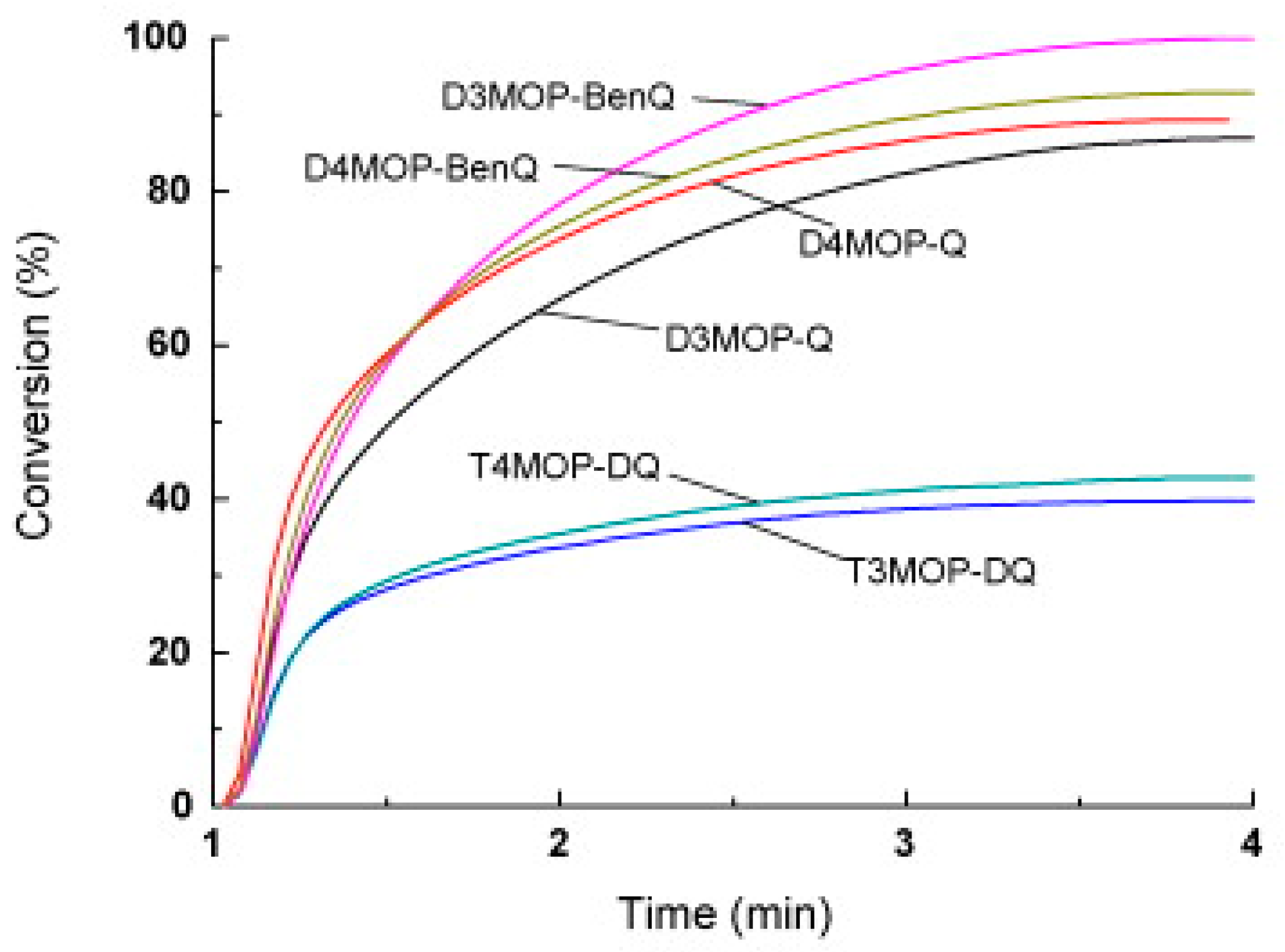
Figure 28). Among the four co-initiators examined in this work, namely leucocrystal violet (LCV), 2-mercaptobenzoxazole (MBO), NPG and MDEA, MBO proved to be the best co-initiator for the different MOPQ derivatives. In this series of dyes, the highest monomer conversion was obtained with D3MOP-BenQ when used as a photosensitizer for MBO and upon irradiation at 365 nm (See
Table 10 and
Figure 29).
Although T3MOP-DQ and T4MOP-DQ exhibited excellent absorption properties, only low monomer conversions were obtained with these two dyes, attributable to their low solubilities in the monomer, which may be caused by their extended and planar conjugated structures. To support the polymerization initiated by the different quinoxaline derivatives, a mechanism based on a hydrogen abstraction process was suggested by the authors, as depicted in
Scheme 5.
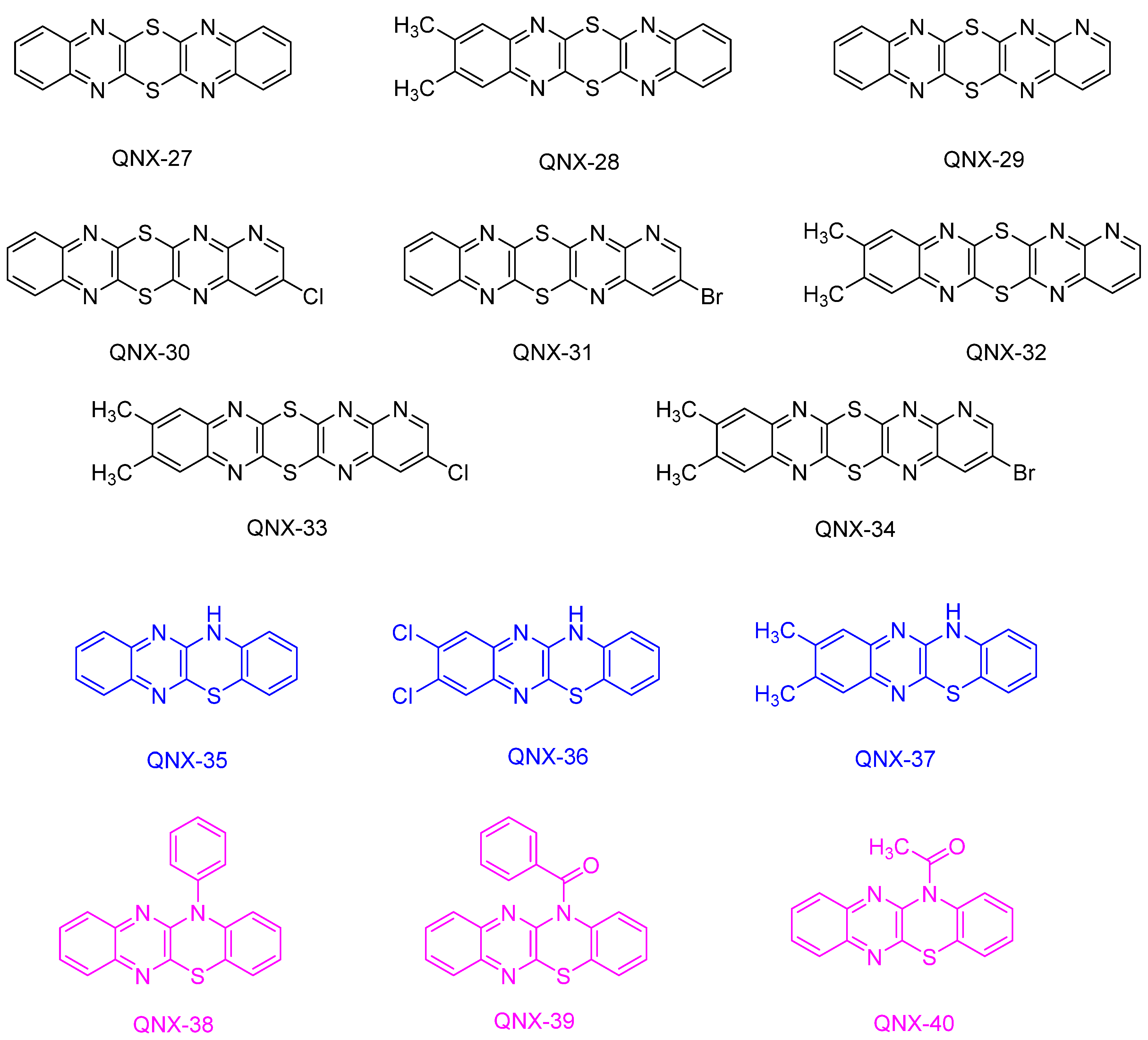
Several attempts were carried out to introduce heteroatoms into the structure of quinoxalines. Among these structures, several sulfur-containing dyes such as dithiinoquinoxalines QNX-27-QNX-34 [
234] and 12
H-quinoxalino-[2,3-
b][1,4]-benzothiazines QNX-35-QNX-37 [
241,
242,
243] were proposed (See
Figure 30).
From the absorption viewpoint, the presence of these sulfur atoms in the quinoxaline derivatives did not significantly modify their absorptions compared to the parent structures since absorption maxima ranging between 386 nm for QNX-27 and 406 nm for QNX-34 were determined. A higher influence was evidenced for 12
H-quinoxalino-[2,3-
b][1,4]-benzothiazines QNX-35-QNX-37 since an absorption maximum located at 442 nm was determined for QNX-37. Noticeably, upon substitution at the nitrogen atom of 12
H-quinoxalino-[2,3-
b][1,4]-benzothiazines QNX-38-QNX-40 with various groups, a blue shift of the absorption maximum was detected, as shown in
Table 11.
Interestingly, a good photobleaching of the polymer films was obtained with the different 12-substituted 12
H-quinoxalino-[2,3-
b][1,4]-benzothiazines, in combination with Ph
2I
+PF
6− during the FRP of acrylates [
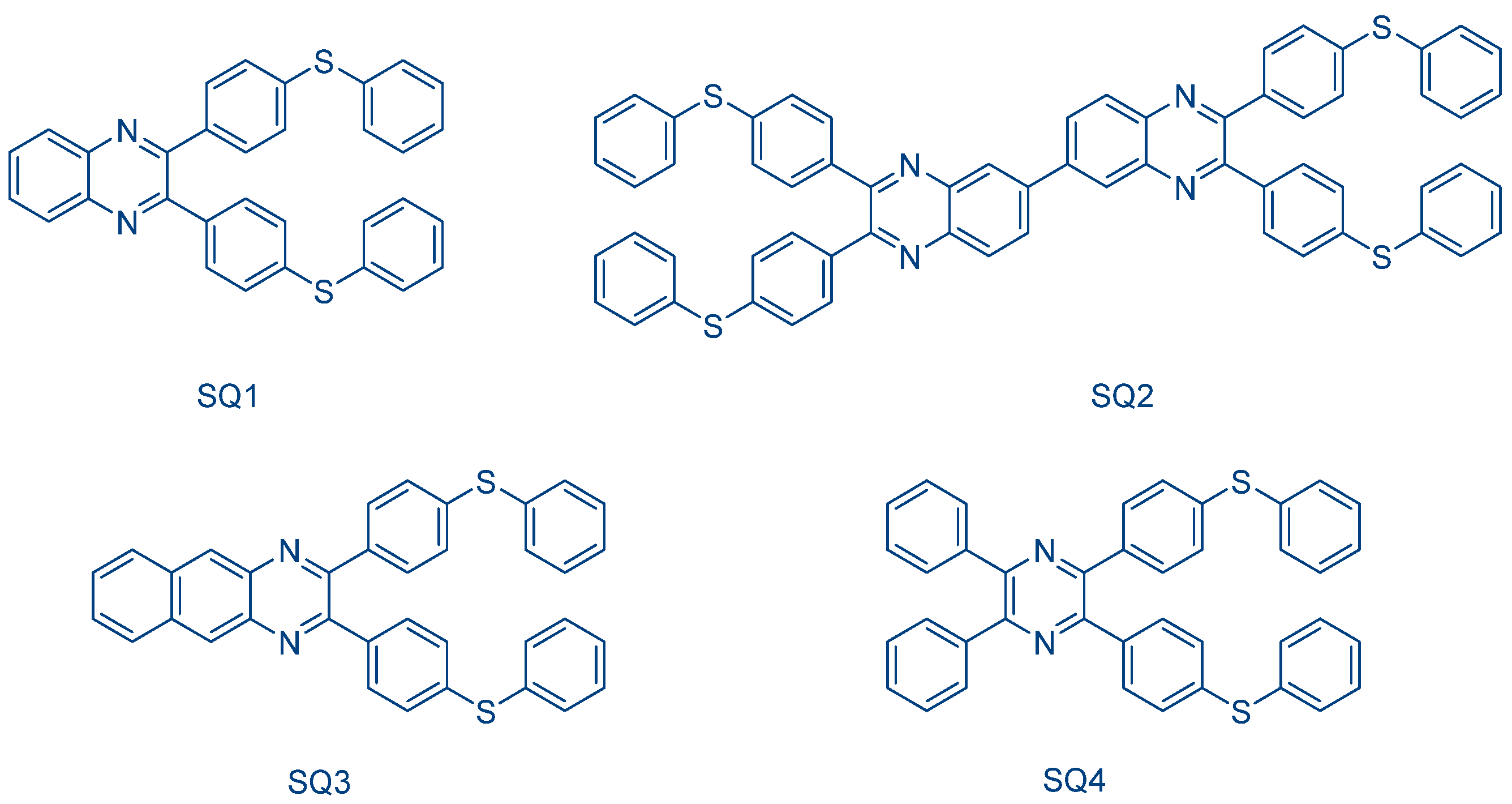
243]. In 2017, a series of quinoxalines bearing photocleavable groups and hydrogen-abstracting groups was proposed by Jiang and coworkers (See
Figure 31) [
244]. In depending on the reaction conditions, these photoinitiators can act as Type I photoinitiators and Type II photoinitiators in the presence of a hydrogen donor. The mechanism of photoinitiation in the two cases is depicted in
Scheme 6.
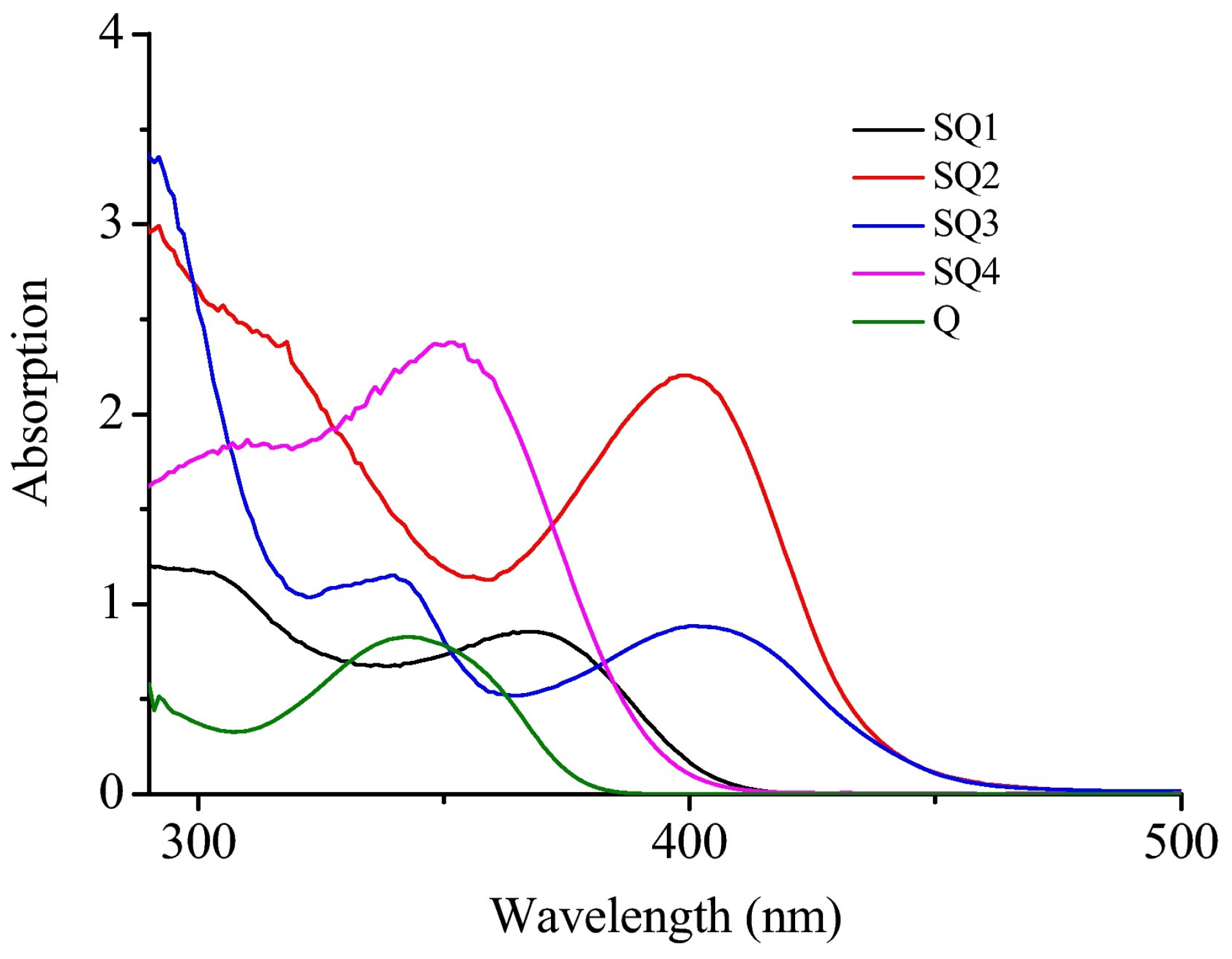
In this series of dyes, the most red-shifted absorptions were found for SQ2 and SQ3, exhibiting the most extended π-conjugation (See
Figure 32 and
Table 12). Absorption maxima peaking at 402 and 410 nm were, respectively, determined for SQ2 and SQ3. Considering that all dyes absorb in the 320–400 nm range, photopolymerization experiments were carried out at 350 nm.
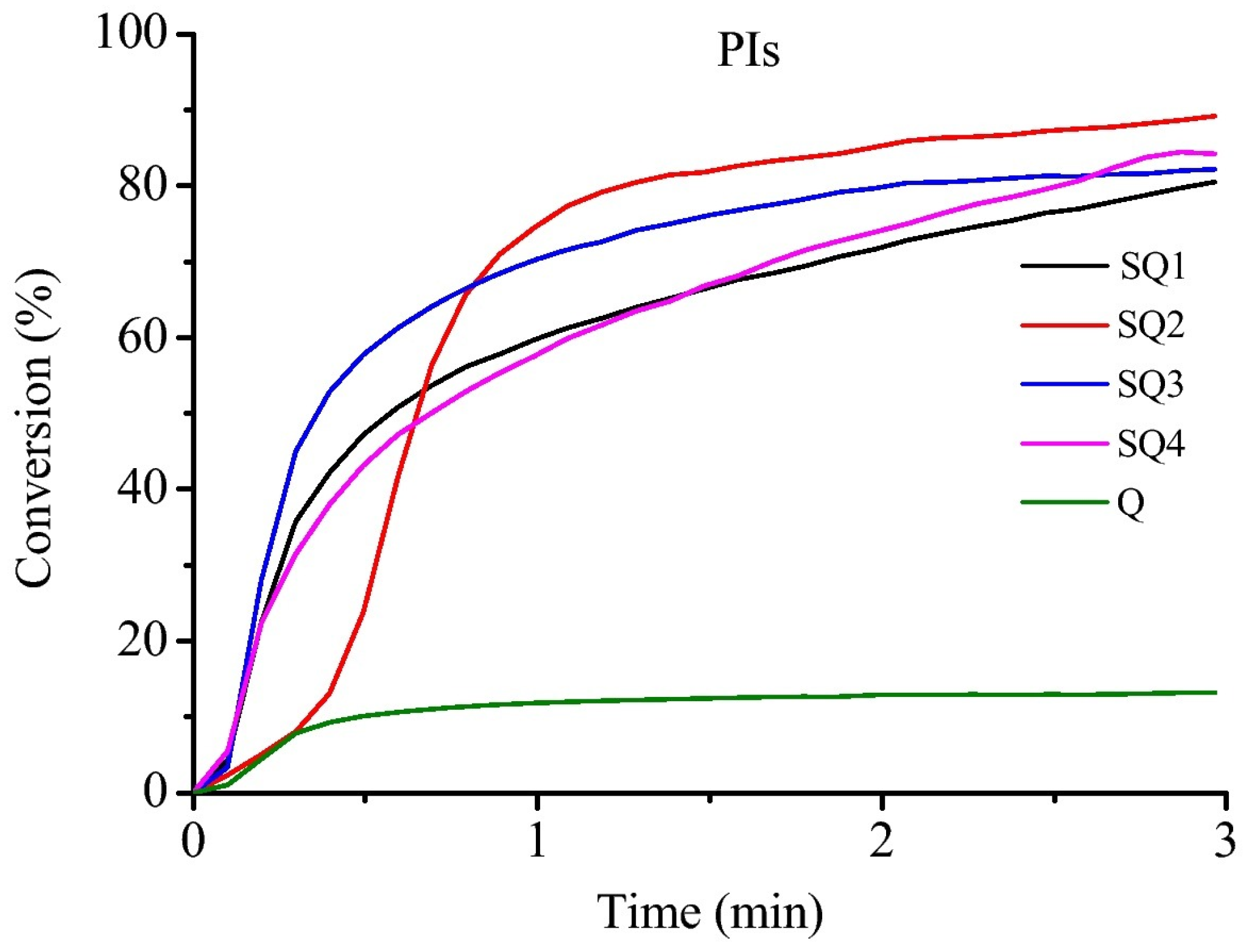
Photopolymerization of 1,6-hexanediol diacrylate (HDDA) using SQ1–SQ4 as Type I photoinitiators revealed all dyes furnished monomer conversions higher than 80% upon irradiation at 350 nm for 3 min. Despite a long inhibition time, SQ2 furnished the highest monomer conversion, around 90% (See
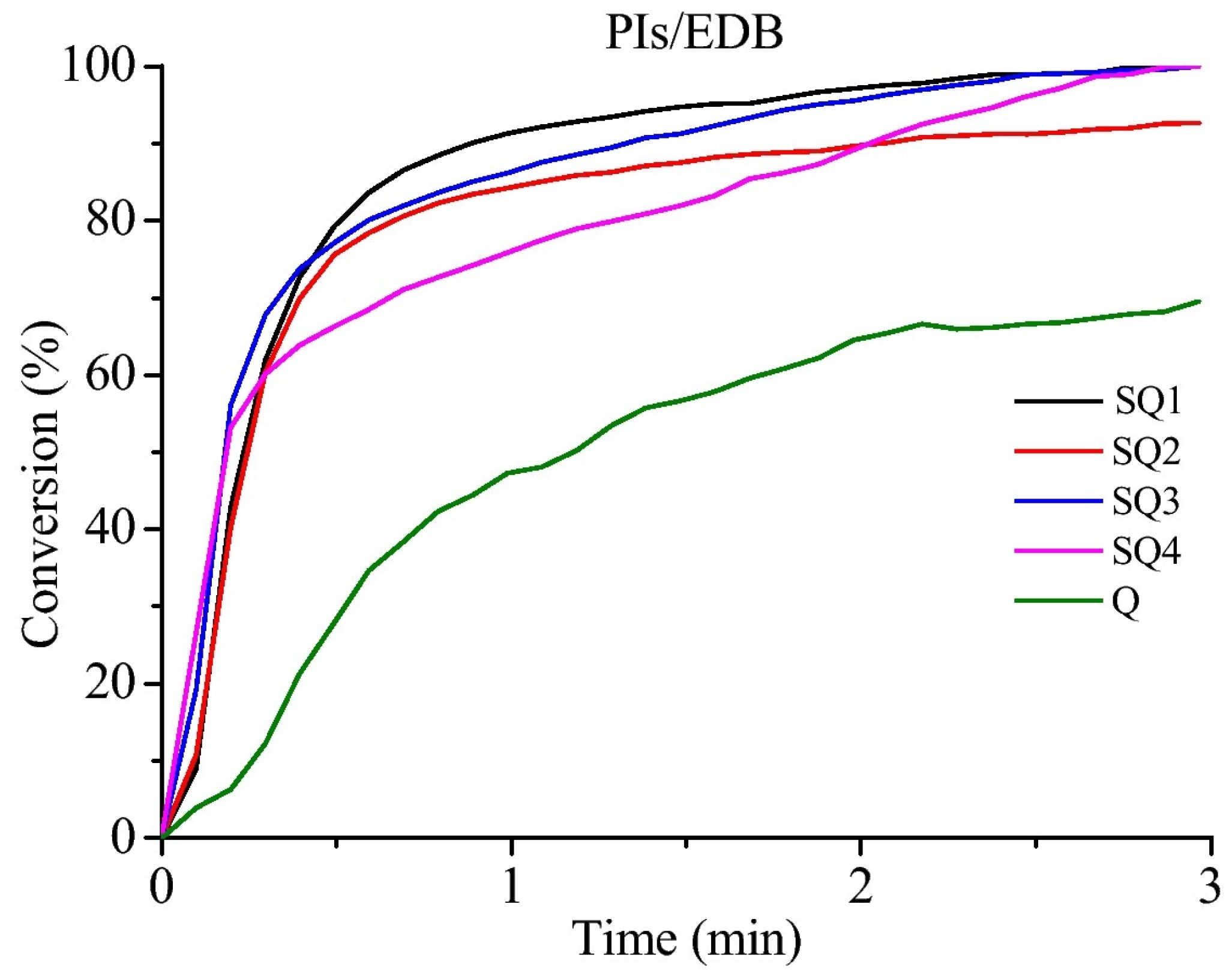
Figure 33). Examination of their photo-initiating abilities as Type II photoinitiators revealed that the HDDA conversion was greatly improved by using the two-component dye/EDB systems (See
Figure 34). In this case, monomer conversions higher than 90% were determined.
The higher monomer conversions obtained by using the two-component dye/EDB systems were assigned to synergistic effects resulting from the photocleavage of the thioether groups and the hydrogen abstraction mechanism of the quinoxaline groups, producing, in turn, more free radicals due to the concomitant occurrence of the Type I and Type II mechanism. Among the three hydrogen donors examined, i.e., EDB, MBO and MDEA, EDB proved to be the best co-initiator irrespective of the dye.
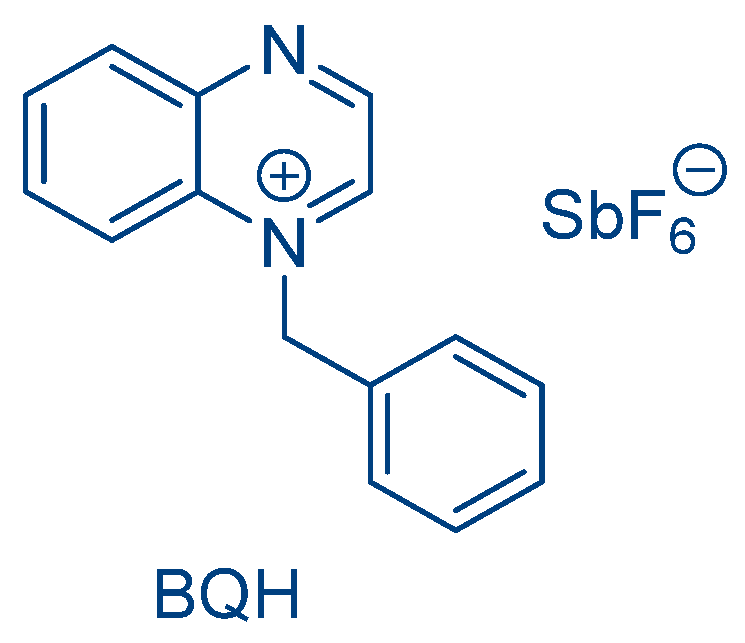
In 2014, a benzyl quinoxalinium hexafluoroantimonate (BQH) was reported for the first time as a Type I photo-latent initiator (See
Figure 35) [
245]. Upon irradiation with a UV light, the salt cleaved, generating a benzyl cation acting as the initiation species. However, due to the lack of absorption of the quinoxalinium salt in the visible range, this approach remained limited to UV photopolymerization.
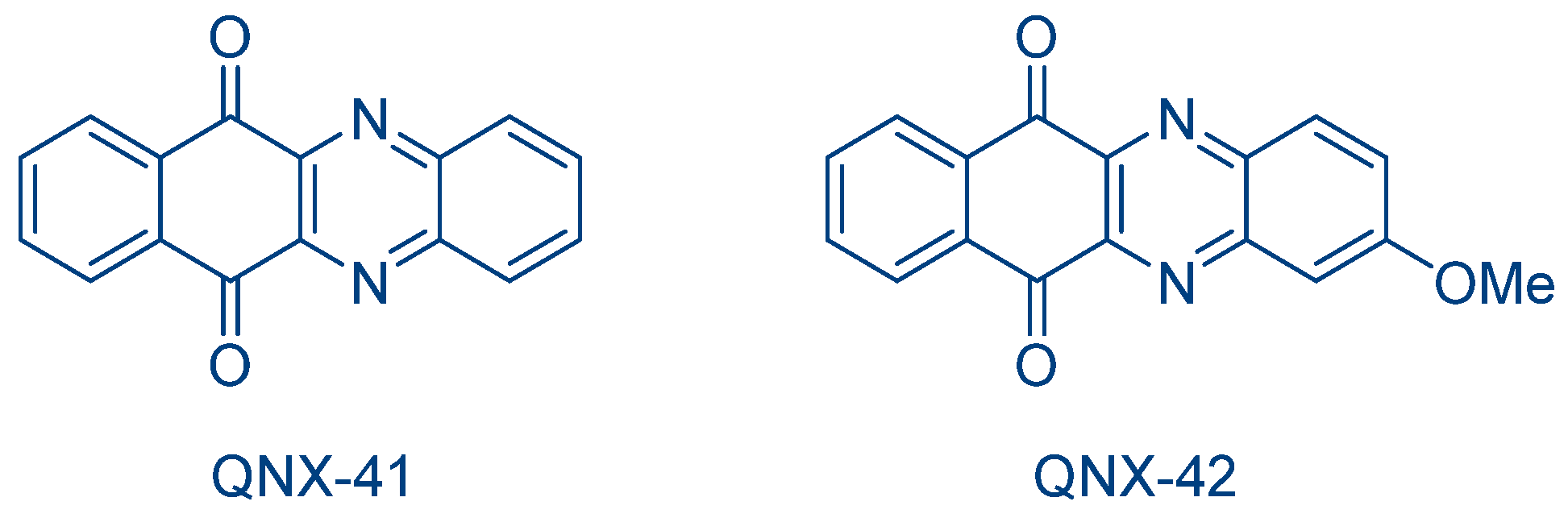
Quinoxaline derivatives were also extensively used as electron acceptors in photoredox pairs. In order to improve the electron-withdrawing ability of quinoxalines, naphthoquinone was incorporated in the structures of QNX-41 and QNX-42 (See
Figure 36) [
246]. Using phenylthiolacetic acid, with phenoxyacetic acid and
N-phenylglycine as the electron donors, the FRP of TMPTA was efficiently promoted. Among the most interesting results, QNX-41 and QNX-42 were used as electron donors despite the presence of the naphthoquinone moiety, and these dyes also induced the cationic polymerization of CHO using Py1, Py2 and Ph
2I
+PF
6− as the electron acceptors. A strong influence of the electron acceptor used was evidenced since a two-fold increase in the CHO conversion was obtained with Py1, compared to Py2 and Ph
2I
+PF
6− (See
Table 13).
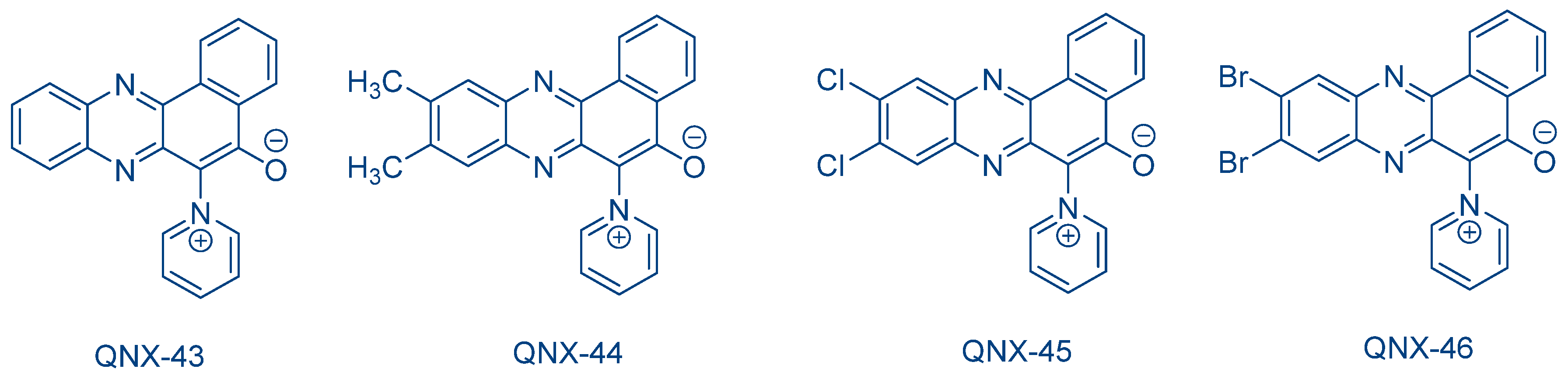
The ability of dyes to act indifferently as electron donors or electron acceptors was also demonstrated for a series of 6-pyridinium benzo[a]phenazine-5-oxide derivatives QNX-43-QNX-46 (See
Figure 37) [
247]. Here again, the different two-component systems initiated the FRP of TMPTA or the CP of CHO.
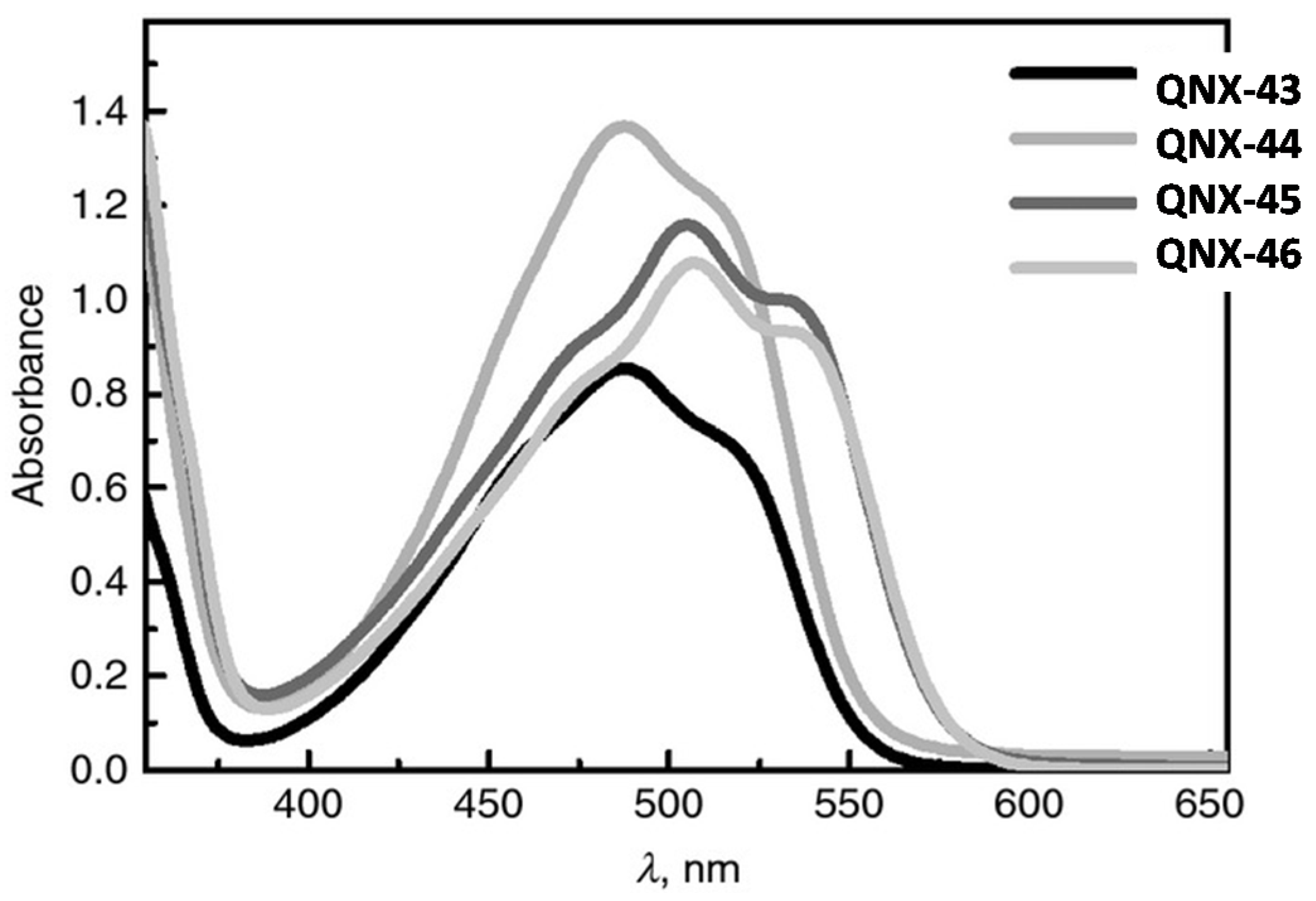
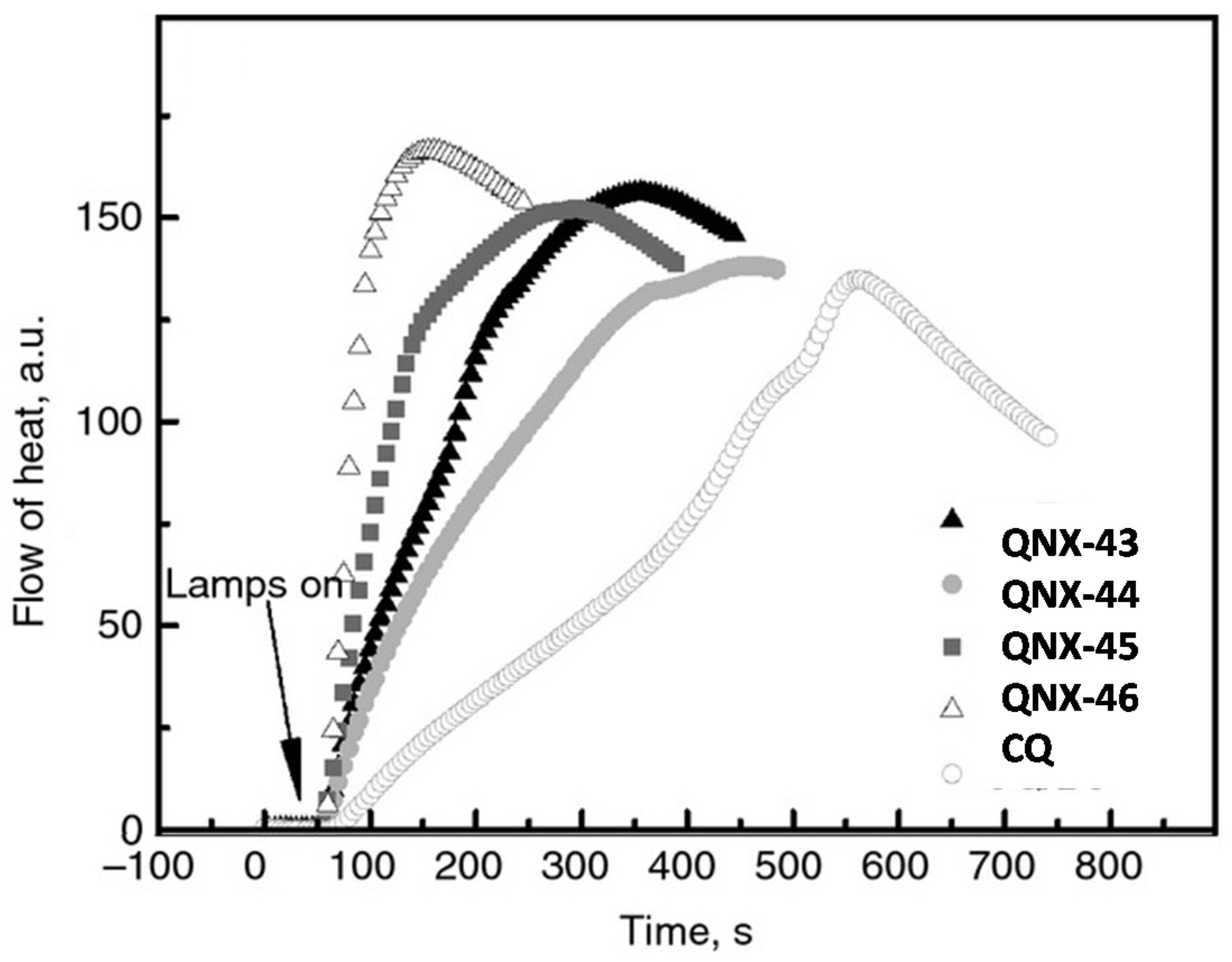
As shown in
Figure 38 and in
Table 14, the absorption of these dyes was broad, extending in the visible range between 400 and 600 nm. A xenon lamp could thus be used for the polymerization experiments. Once again, the presence of halogens and especially of a bromine atom on QNX-46 was beneficial for the polymerization rate. Notably, during the FRP of TMPTA and using the two-component dye/NPG systems, the highest polymerization rate was determined for QNX-46 (See
Figure 39). Following QNX-46, the second-best TMPTA polymerization rate was obtained with QNX-45 bearing chlorine atoms. Comparisons performed with camphorquinone used as a reference compound revealed that the new photo-initiating systems outperformed this reference compound.




















































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
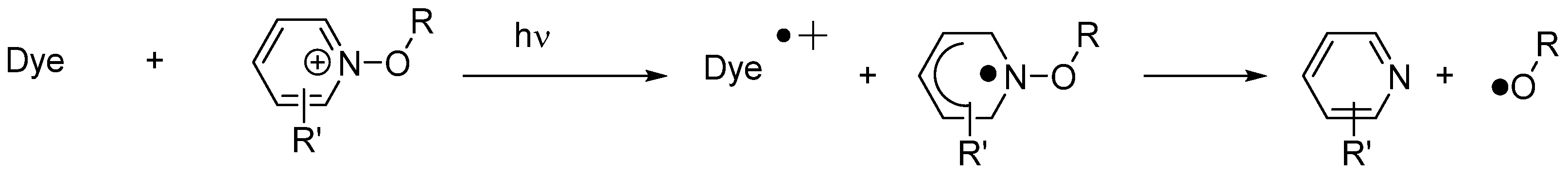{kind=link}
{kind=link}
{kind=link}
{kind=link}