
Measurements of Dioxygen Formation in Catalytic Electrochemical Water Splitting



Abstract
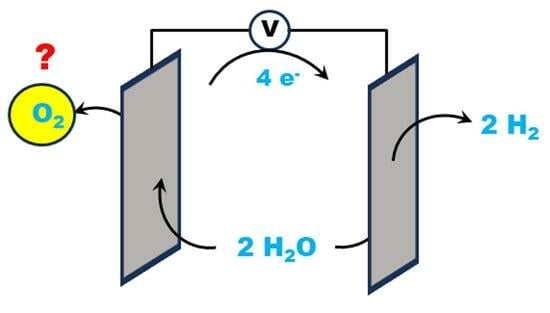
:
{kind=link}
1. Introduction
2. Discussion
2.1. Problems in Electrochemical Water Oxidation Studies
- Hydrogen Oxidation Reaction (HOR) is a common side reaction if electrolysis is performed in one compartment cell. Hydrogen may also diffuse through a membrane such as a glass frit.
- Catalytic oxidation of electrolyte or buffer ions. For instance, chloride anions from an electrolyte or acetate anions from acetate buffer are prone to oxidation.
- Catalytic oxidation of an organic ligand of a catalyst of OER or an electron transfer catalyst.
- Catalytic oxidation of anode material (such as graphite).
- Surface passivation can hinder the transport of reactants and products, leading to decreased catalytic activity and increased overpotentials.
2.2. Visual Observation of Gas Formation
2.3. Gas Cromatography (GC)
2.4. Volumetric Measurements of Dioxygen Yield
2.5. Use of Fluorescence Probe
2.6. Pseudo-Clark Generator/Collector Set-Up
2.7. Stirring of Solutions during Elecrochemical Experiments [22]
2.8. Rotating Ring-Disk-Electrode (RRDE)
3. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, Y.; Pei, J.; Yu, X.; Bi, L. Study on Catalytic Water Oxidation Properties of Polynuclear Manganese Containing Polyoxometalates. Catalysts 2022, 12, 160. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, Y.; Lu, X. Construction of NiFe-Layered Double Hydroxides Arrays as Robust Electrocatalyst for Oxygen Evolution Reaction. Catalysts 2023, 13, 586. [Google Scholar] [CrossRef]
- Huang, Y.; Chen, H.; Zhang, B. Constructing Molybdenum Phosphide@Cobalt Phosphide Heterostructure Nanoarrays on Nickel Foam as a Bifunctional Electrocatalyst for Enhanced Overall Water Splitting. Molecules 2023, 28, 3647. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Xie, Y.; Feng, C.; Hassan, A.; Wang, J. Nitrogen-Rich Porous Carbon Nanotubes Coated Co/Mo2N Composites Derived from Metal-Organic Framework as Efficient Bifunctional Oxygen Electrocatalysts. Catalysts 2023, 13, 801. [Google Scholar] [CrossRef]
- Abdullahi, I.M.; Nath, M. Molecular Cluster Complex of High-Valence Chromium Selenide Carbonyl as Effective Electrocatalyst for Water Oxidation. Catalysts 2023, 13, 721. [Google Scholar] [CrossRef]
- Khan, I. Pluronic-123 Assisted Synthesis of Cobalt Vanadate Microparticles (µ-CoV MPs) for Durable Electrochemical Oxygen Evolution Reaction in Seawater and Connate Water. Catalysts 2023, 13, 636. [Google Scholar] [CrossRef]
- Lee, J.; Shin, H.; Geum, S.; Lee, S.; Ok, K.M.; Do, J.; Kwon, S.J. Synthesis of Cobalt Complex Containing Trans-Cinnamate and Its Electrocatalytic Activity for Oxygen Evolution Reaction. Catalysts 2023, 13, 507. [Google Scholar] [CrossRef]
- Kim, T.-H.; Koo, K.-Y.; Park, C.-S.; Jeong, S.-U.; Kim, J.-E.; Lee, S.-H.; Kim, Y.-H.; Kang, K.-S. Effect of Fe on Calcined Ni(OH)2 Anode in Alkaline Water Electrolysis. Catalysts 2023, 13, 496. [Google Scholar] [CrossRef]
- Wan, X.; Wang, X.; Lu, D.; Xu, Y.; Liu, G.; Fu, Y.; Shui, T.; Wang, H.; Cheng, Z. Composition and Morphology Modulation of Bimetallic Nitride Nanostructures on Nickel Foams for Efficient Oxygen Evolution Electrocatalysis. Catalysts 2023, 13, 230. [Google Scholar] [CrossRef]
- Xu, X.; Yu, X.; Guo, K.; Dong, L.; Miao, X. Alkaline Media Regulated NiFe-LDH-Based Nickel and Iron Phosphides toward Robust Overall Water Splitting. Catalysts 2023, 13, 198. [Google Scholar] [CrossRef]
- Kushner-Lenhoff, M.N.; Blakemore, J.D.; Schley, N.D.; Crabtree, R.H.; Brudvig, G.W. Effects of Aqueous Buffers on Electrocatalytic Water Oxidation with an Iridium Oxide Material Electrodeposited in Thin Layers from an Organometallic Precursor. Dalton Trans. 2013, 42, 3617–3622. [Google Scholar] [CrossRef] [PubMed]
- Kanan, M.W.; Nocera, D.G. In Situ Formation of an Oxygen-Evolving Catalyst in Neutral Water Containing Phosphate and Co2+. Science 2008, 321, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- McCrory, C.C.; Jung, S.; Peters, J.C.; Jaramillo, T.F. Benchmarking heterogeneous electrocatalysts for the oxygen evolution reaction. J. Am. Chem. Soc. 2013, 135, 16977–16987. [Google Scholar] [CrossRef] [PubMed]
- Tahir, M.; Pan, L.; Idrees, F.; Zhang, X.; Wang, L.; Zou, J.J.; Wang, Z.L. Electrocatalytic oxygen evolution reaction for energy conversion and storage: A comprehensive review. Nano Energy 2017, 37, 136–157. [Google Scholar] [CrossRef]
- Hausmann, J.N.; Traynor, B.; Myers, R.J.; Driess, M.; Menezes, P.W. The pH of Aqueous NaOH/KOH Solutions: A Critical and Non-trivial Parameter for Electrocatalysis. ACS Energy Lett. 2021, 6, 3567–3571. [Google Scholar] [CrossRef]
- Bose, S.; Debgupta, J.; Ramsundar, R.M.; Das, S.K. Electrochemical Water Oxidation Catalyzed by an In Situ Generated α-Co(OH)2 Film on Zeolite-Y Surface. Chem. Eur. J. 2017, 23, 8051–8057. [Google Scholar] [CrossRef]
- Ravi, A.; Mulkapuri, S.; Das, S.K. Hydroxylated Polyoxometalate with Cu(II)- and Cu(I)-Aqua Complexes: A Bifunctional Catalyst for Electrocatalytic Water Splitting at Neutral pH. Inorg. Chem. 2023, 62, 12650–12663. [Google Scholar] [CrossRef]
- Surendranath, Y.; Dincǎ, M.; Nocera, D.G. Electrolyte-Dependent Electrosynthesis and Activity of Cobalt-Based Water Oxidation Catalysts. J. Am. Chem. Soc. 2009, 131, 2615–2620. [Google Scholar] [CrossRef]
- Guo, S.-X.; Liu, Y.; Lee, C.-Y.; Bond, A.M.; Zhang, J.; Geletii, Y.V.; Hill, C.L. Graphene-Supported [{Ru4O4(OH)2(H2O)4}(γ-SiW10O36)2]10− for Highly Efficient Electrocatalytic Water Oxidation. Energy Environ. Sci. 2013, 6, 2654–2663. [Google Scholar] [CrossRef]
- Lee, S.-H.A.; Zhao, Y.; Hernandez-Pagan, E.A.; Blasdel, L.; Youngblood, W.J.; Mallouk, T.E. Electron Transfer Kinetics in Water Splitting Dye-Sensitized Solar Cells Based on Core–Shell Oxide Electrodes. Faraday Discuss. 2012, 155, 165–176. [Google Scholar] [CrossRef]
- Fielden, J.; Sumliner, J.M.; Han, N.; Geletii, Y.V.; Xiang, X.; Musaev, D.G.; Lian, T.; Hill, C.L. Water Splitting with Polyoxometalate-Treated Photoanodes: Enhancing Performance Through Sensitizer Design. Chem. Sci. 2015, 6, 5531–5543. [Google Scholar] [CrossRef]
- Rotating Ring Disk Electrode Fundamentals. Available online: https://pineresearch.com/shop/kb/theory/hydrodynamic-electrochemistry/rotating-electrode-theory/ (accessed on 25 November 2023).
- Filimonenkov, I.S.; Istomin, S.Y.; Antipov, E.V.; Tsirlina, G.A.; Savinova, E.R. Rotating ring-disk electrode as a quantitative tool for the investigation of the oxygen evolution reaction. Electrochim. Acta 2018, 286, 304–312. [Google Scholar] [CrossRef]
- El-Sayed, H.A.; Weiß, A.; Olbrich, L.F.; Putro, G.P.; Gasteiger, H.A. OER catalyst stability investigation using RDE technique: A stability measure or an artifact? J. Electrochem. Soc. 2019, 166, F458–F464. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tiwari, C.K.; Geletii, Y.V. Measurements of Dioxygen Formation in Catalytic Electrochemical Water Splitting. Catalysts 2024, 14, 13. https://doi.org/10.3390/catal14010013
Tiwari CK, Geletii YV. Measurements of Dioxygen Formation in Catalytic Electrochemical Water Splitting. Catalysts. 2024; 14(1):13. https://doi.org/10.3390/catal14010013
Chicago/Turabian StyleTiwari, Chandan Kumar, and Yurii V. Geletii. 2024. "Measurements of Dioxygen Formation in Catalytic Electrochemical Water Splitting" Catalysts 14, no. 1: 13. https://doi.org/10.3390/catal14010013
APA StyleTiwari, C. K., & Geletii, Y. V. (2024). Measurements of Dioxygen Formation in Catalytic Electrochemical Water Splitting. Catalysts, 14(1), 13. https://doi.org/10.3390/catal14010013