Influence of Reduction Promoters on Stability of Cobalt/g-Alumina Fischer-Tropsch Synthesis Catalysts

Abstract
:1. Introduction
2. Results and Discussion


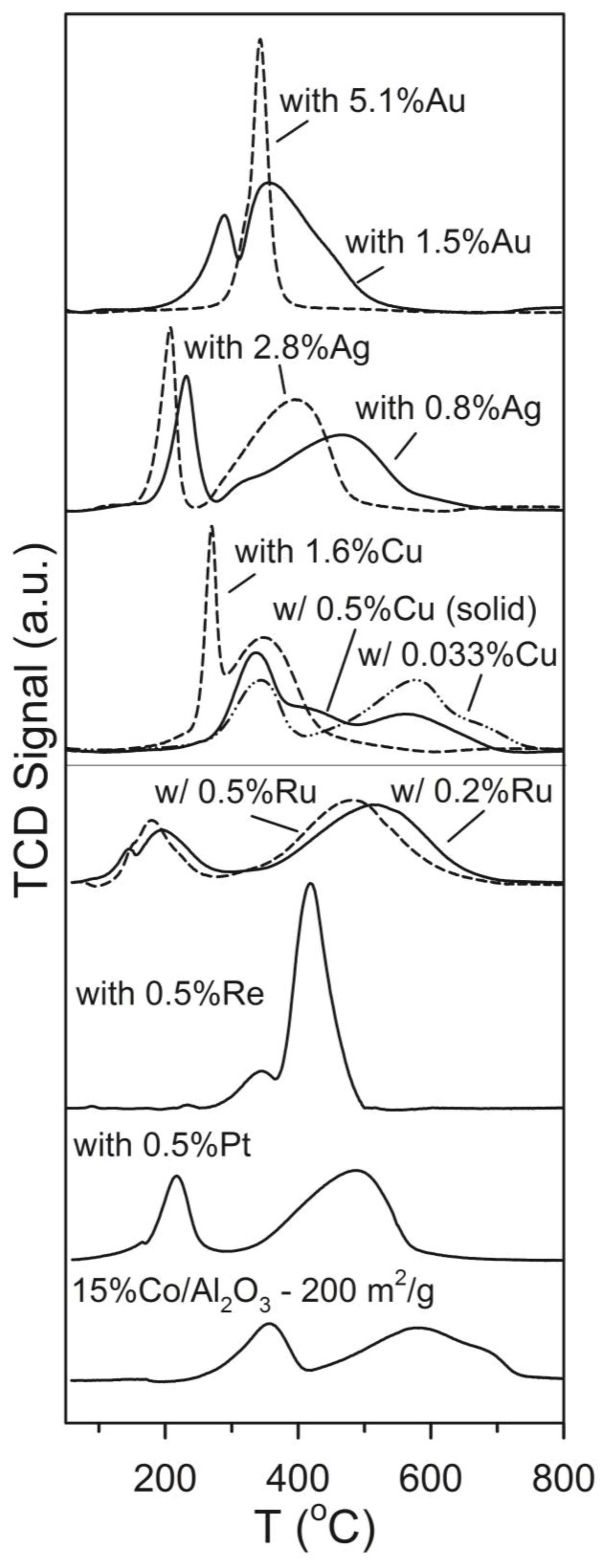
2.1. Influence of Promoter Choice and Loading on Catalyst Activity and Selectivity
2.1.1. Example #1—Copper

{kind=link}
{kind=link}
{kind=link}
{kind=link}
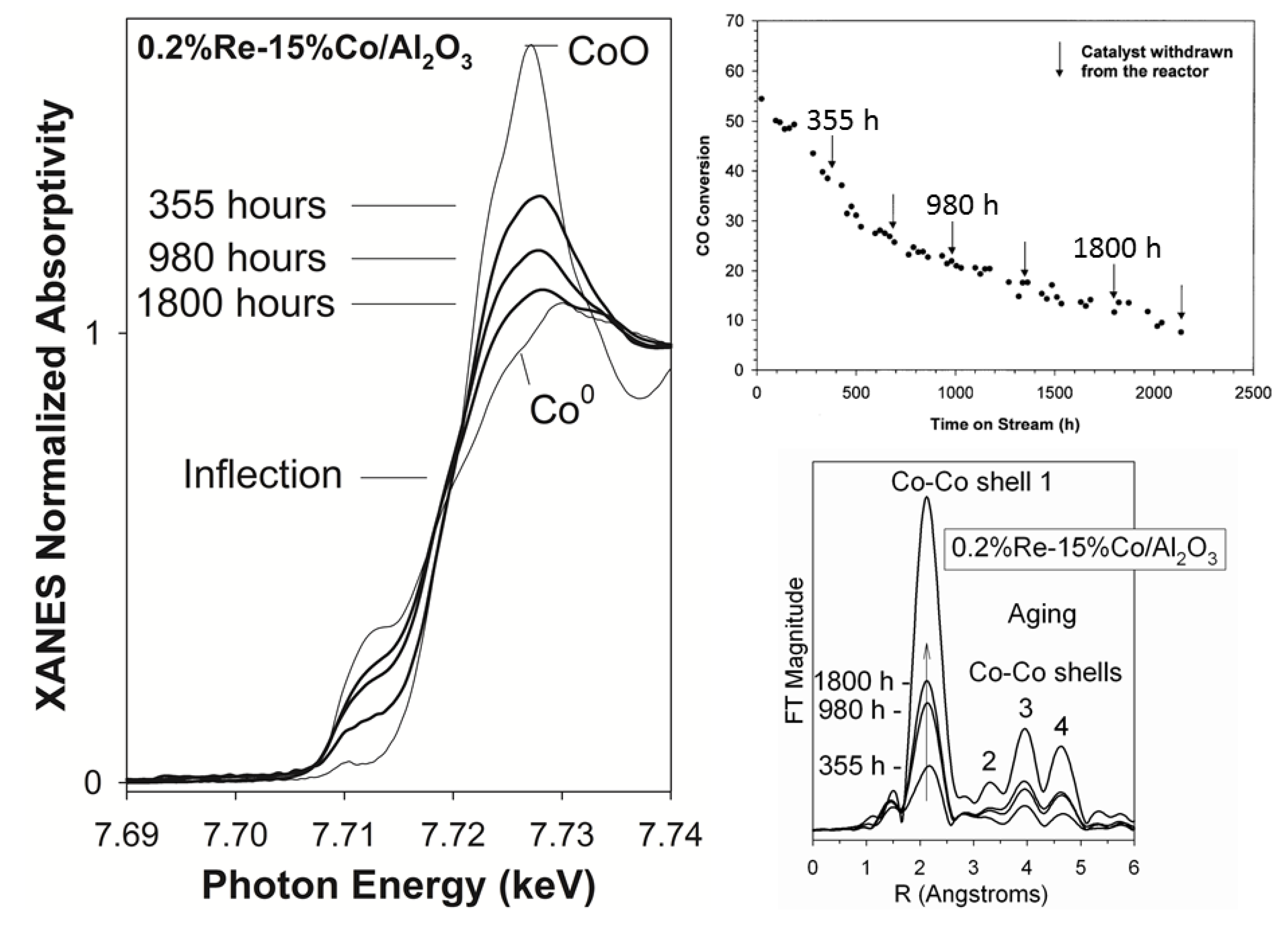{kind=link}
{kind=link}
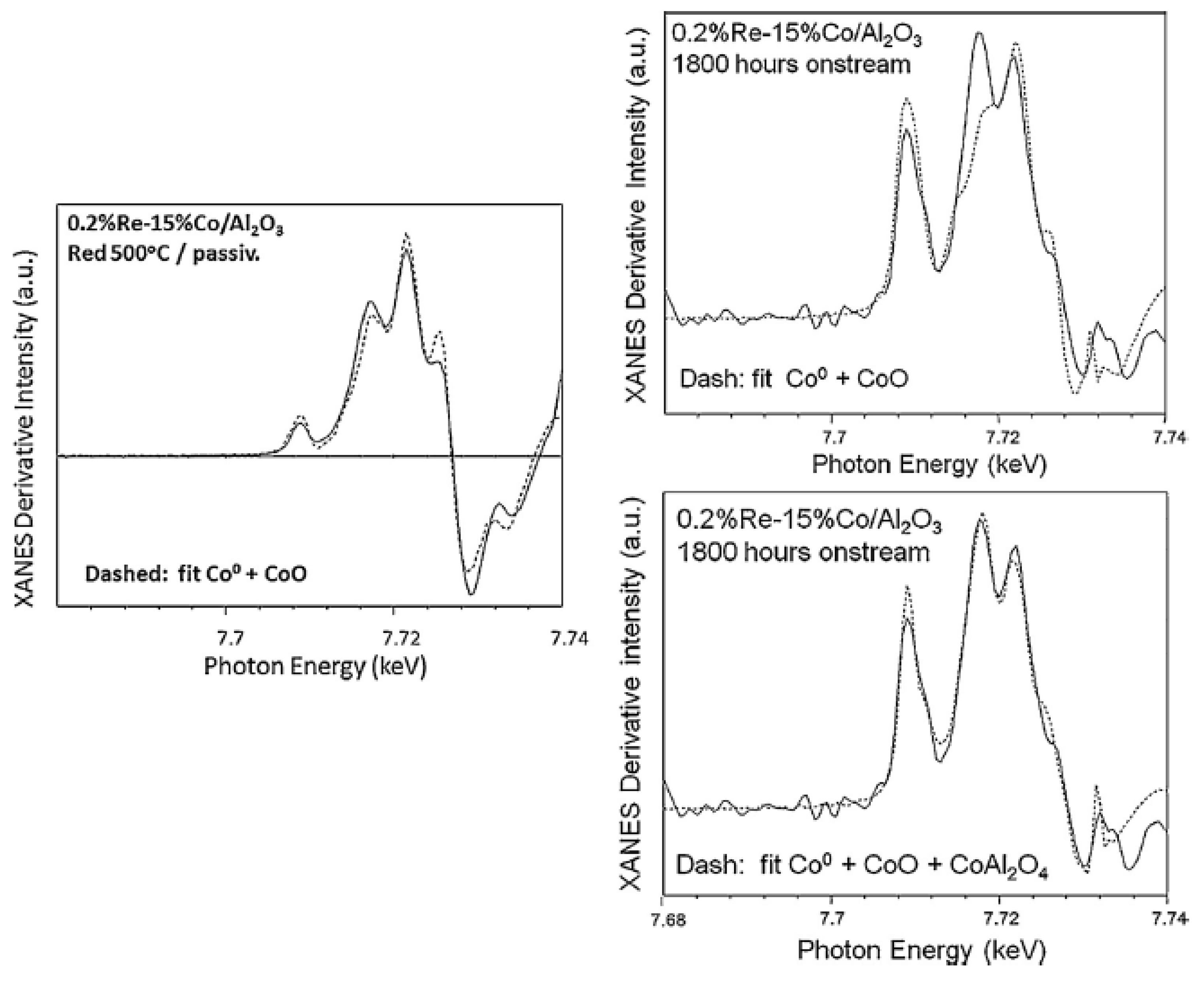{kind=link}
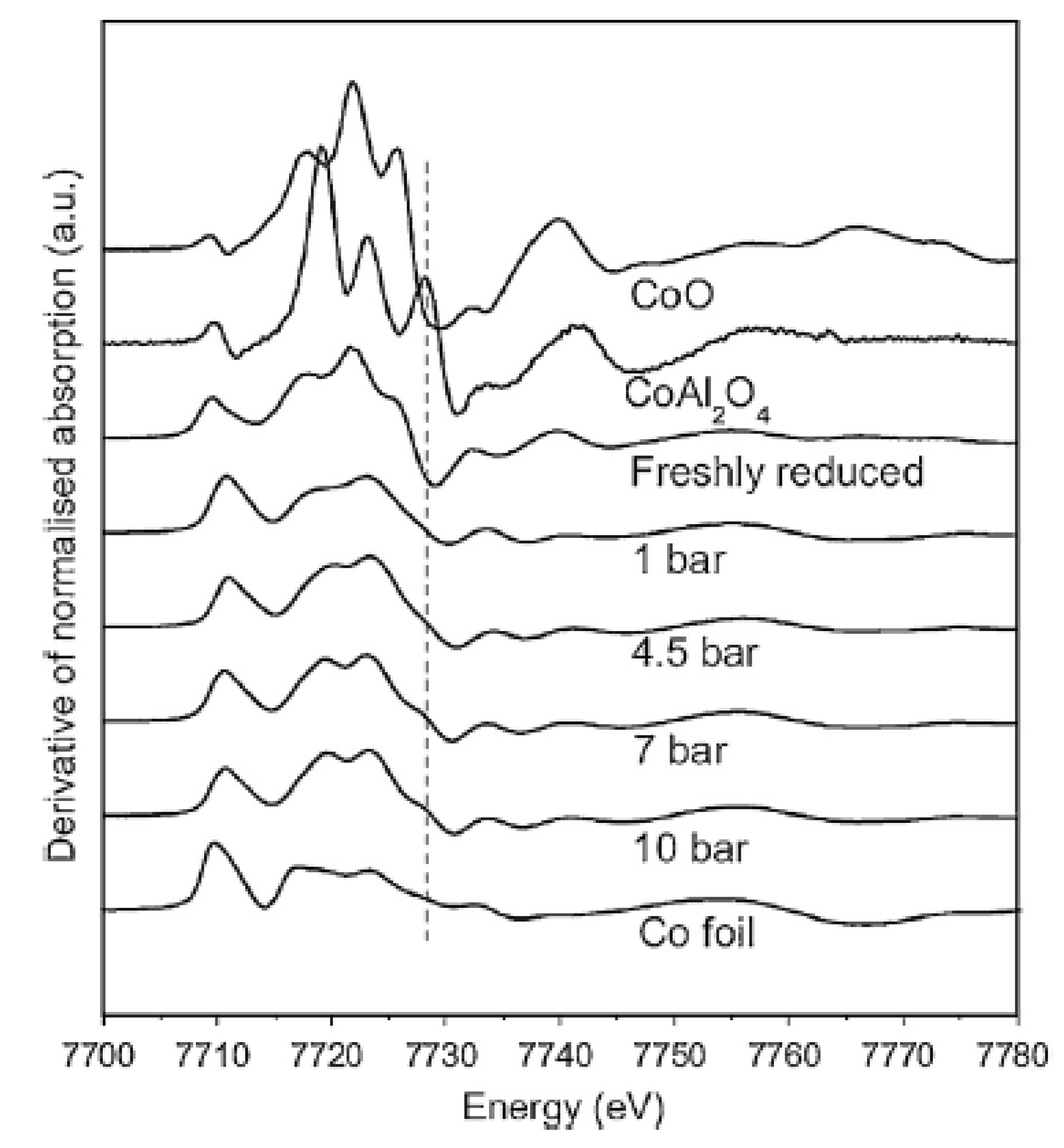{kind=link}
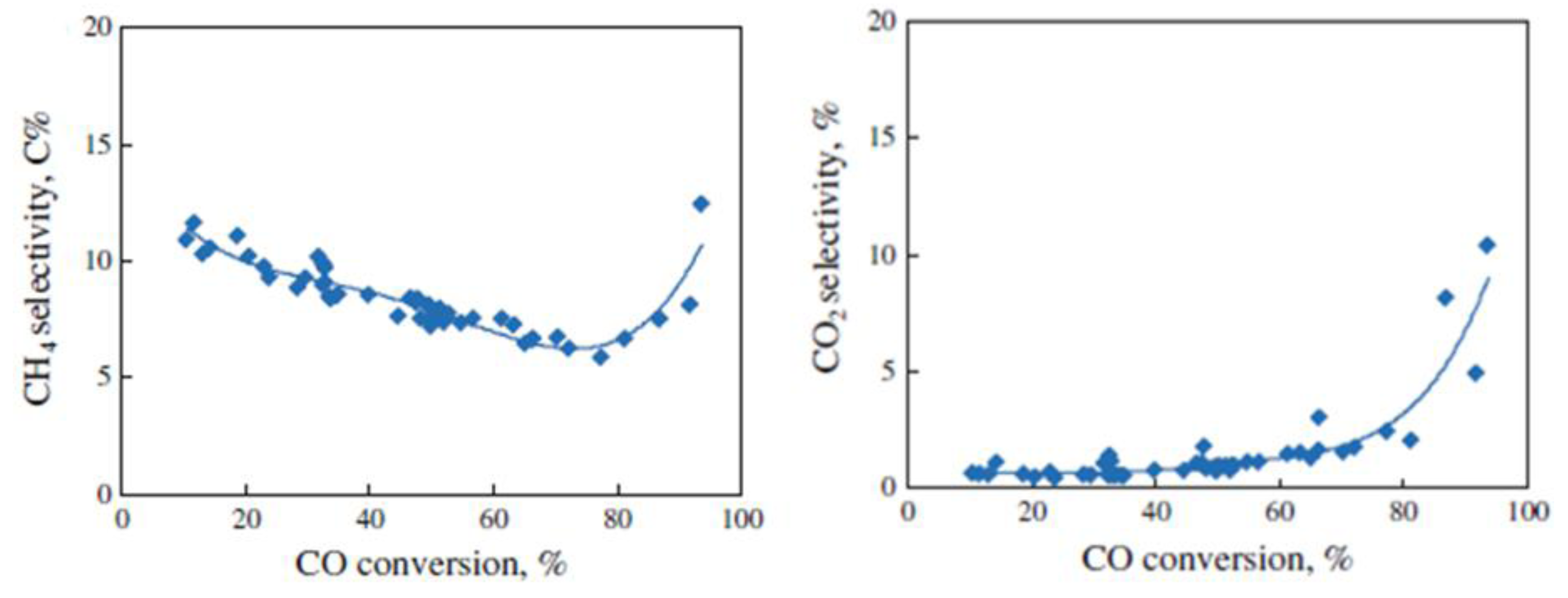{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | TOS (h) | XCO (%) | SV (NL/gcat/h) |
|---|---|---|---|
| 15%Co/Al2O3 | 26–98 | 28.7 | 4.2 |
| 0.49%Cu-15%Co/Al2O3 | 30–99 | 27.9 | 4.2 |
| 1.63%Cu-15%Co/Al2O3 | 25–104 | 14.2 | 4.2 |
| Catalyst | XCO (%) | SV (NL/gcat/h) | S(CH4) | S(C5+) | S(CO2) |
|---|---|---|---|---|---|
| 15%Co/Al2O3 | 47.8 | 2.0 | 8.9 | 80.6 | 0.82 |
| 0.49%Cu-15%Co/Al2O3 | 50.6 | 1.7 | 9.9 | 76.6 | 0.83 |
| 15%Co/Al2O3 | 28.7 | 4.2 | 9.2 | 81.6 | 0.67 |
| 1.63%Cu-15%Co/Al2O3 * | 29.9 | 1.0 | 21.6 | 47.7 | 1.51 |
2.1.2. Example #2—Silver and Gold
| Catalyst | TOS (h) | XCO (%) | SV (NL/gcat/h) |
|---|---|---|---|
| 15%Co/Al2O3 | 26–98 | 28.7 | 4.2 |
| 1.51%Au-15%Co/Al2O3 | 26–57 | 51.7 | 4.2 |
| 5.05%Au-15%Co/Al2O3 | 30–84 | 14.1 | 4.2 |
| 0.83%Ag-15%Co/Al2O3 | 20–47 | 50.4 | 4.2 |
| 2.76%Ag-15%Co/Al2O3 | 22–92 | 46.9 | 4.2 |
| Catalyst | XCO (%) | SV (NL/gcat/h) | S(CH4) | S(C5+) | S(CO2) |
|---|---|---|---|---|---|
| 15%Co/Al2O3 | 47.8 | 2.0 | 8.9 | 80.6 | 0.82 |
| 1.51%Au-15%Co/Al2O3 | 50.0 | 4.2 | 8.0 | 83.7 | 0.83 |
| 0.83%Ag-15%Co/Al2O3 | 50.4 | 4.2 | 7.7 | 83.6 | 0.94 |
| 2.76%Ag-15%Co/Al2O3 | 46.9 | 4.2 | 7.6 | 85.0 | 0.87 |
| 15%Co/Al2O3 | 28.7 | 4.2 | 9.2 | 81.6 | 0.67 |
| 5.05%Au-15%Co/Al2O3* | 27.1 | 1.0 | 18.0 | 60.1 | 1.68 |
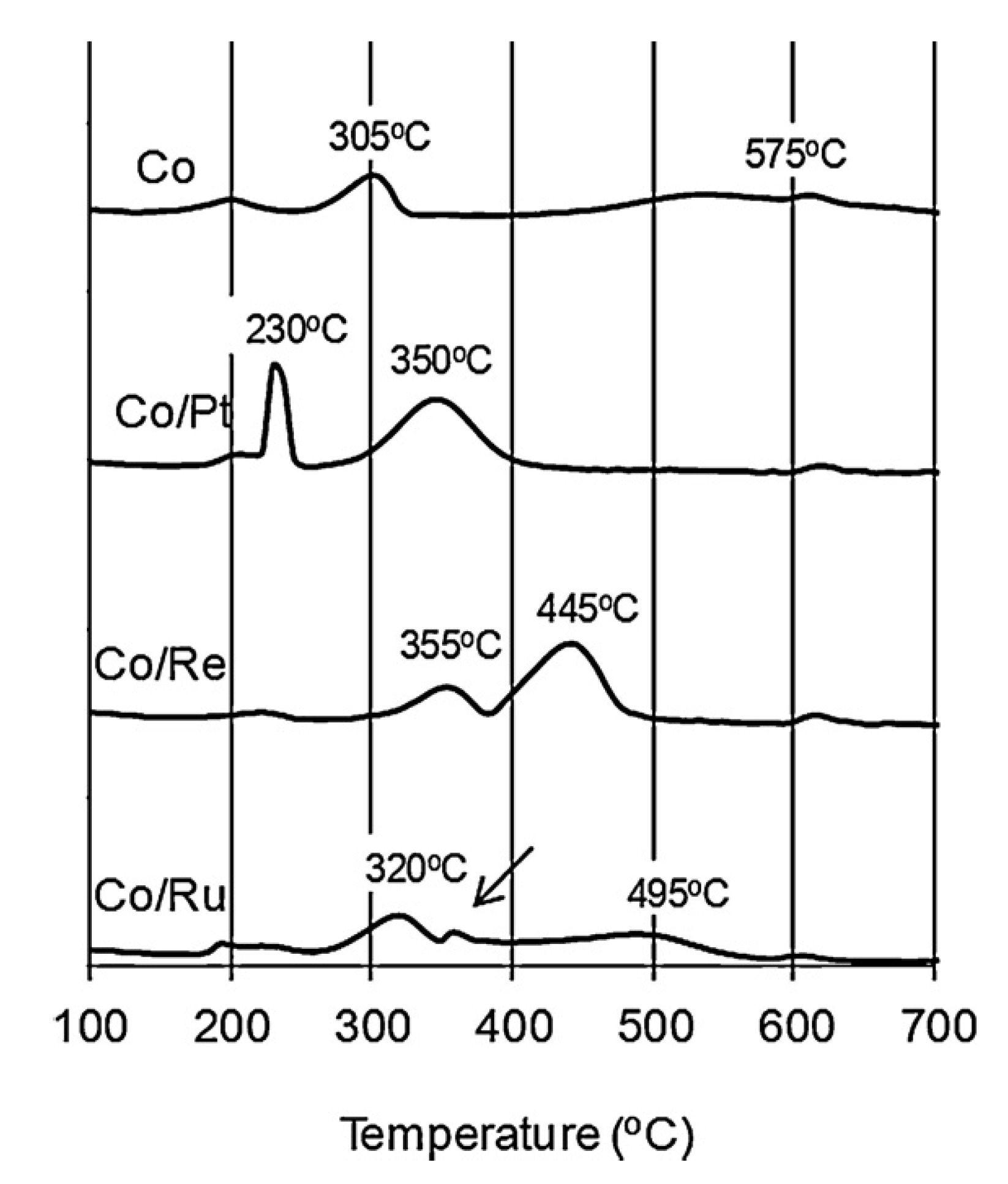
2.1.3. Example #3—Common Promoters (Pt, Re, Ru)
| Catalyst | XCO (%) | SV (NL/gcat/h) | S(CH4) | S(C5+) |
|---|---|---|---|---|
| 25%Co/Al2O3 | 49.4 | 4.3 | 7.9 | 83.4 |
| 0.26%Ru-25%Co/Al2O3 | 51.3 | 7.6 | 7.0 | 86.8 |
| 0.48%Re-25%Co/Al2O3 | 49.6 | 8.0 | 7.2 | 86.0 |
| 0.50%Pt-25%Co/Al2O3 | 48.0 | 5.6 | 8.3 | 83.0 |
| 0.27%Pd-25%Co/Al2O3 | 50.3 | 4.9 | 11.5 | 75.9 |
| Catalyst | Hydrocarbon selectivity (%) | ||
| C1 | C2-C4 | C5+ | |
| Co/NPA | 9.0 | 9.9 | 81.1 |
| CoRe/NPA | 8.8 | 9.5 | 81.7 |
| Co/MPA | 8.6 | 8.7 | 82.8 |
| CoRe/MPA | 8.4 | 8.3 | 83.4 |
| Co/WPA | 8.0 | 7.5 | 84.5 |
| CoRe/WPA | 8.0 | 7.2 | 84.9 |
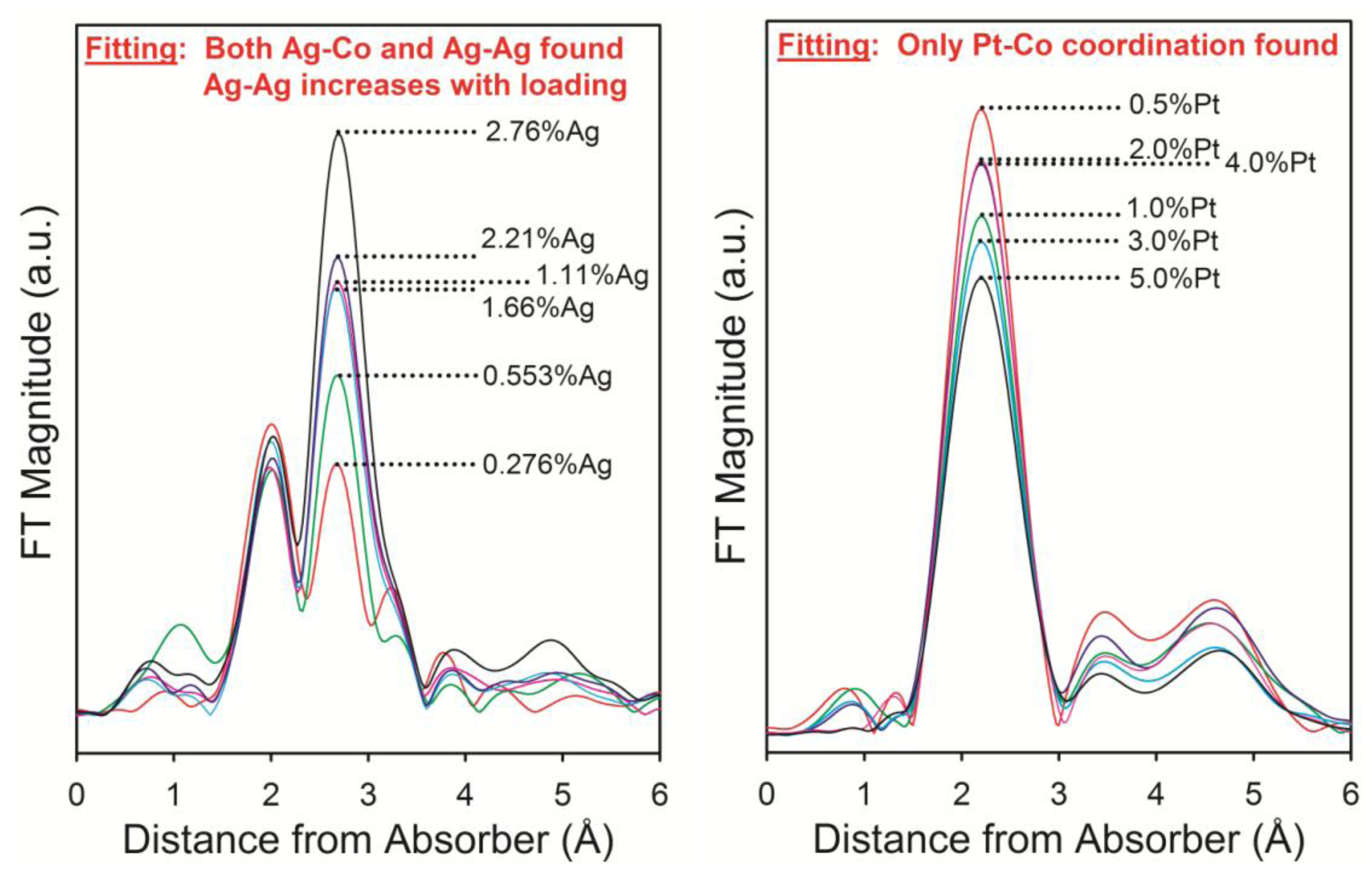
2.1.4. Example #4—Impact of Loading for Pt and Ag Promoted Catalysts
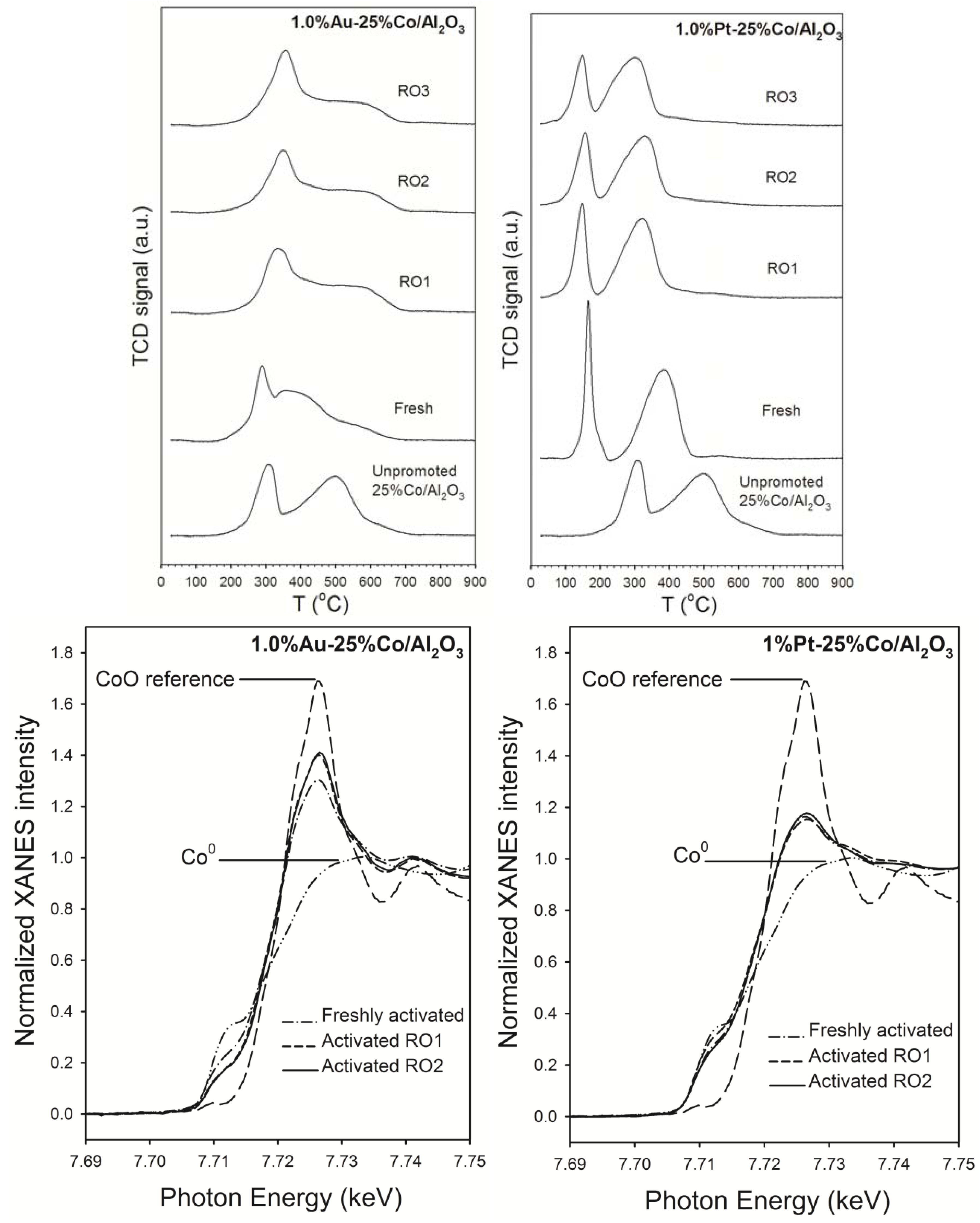
2.2. Influence of Promoter Addition on Oxidation and Complex Sintering of Cobalt
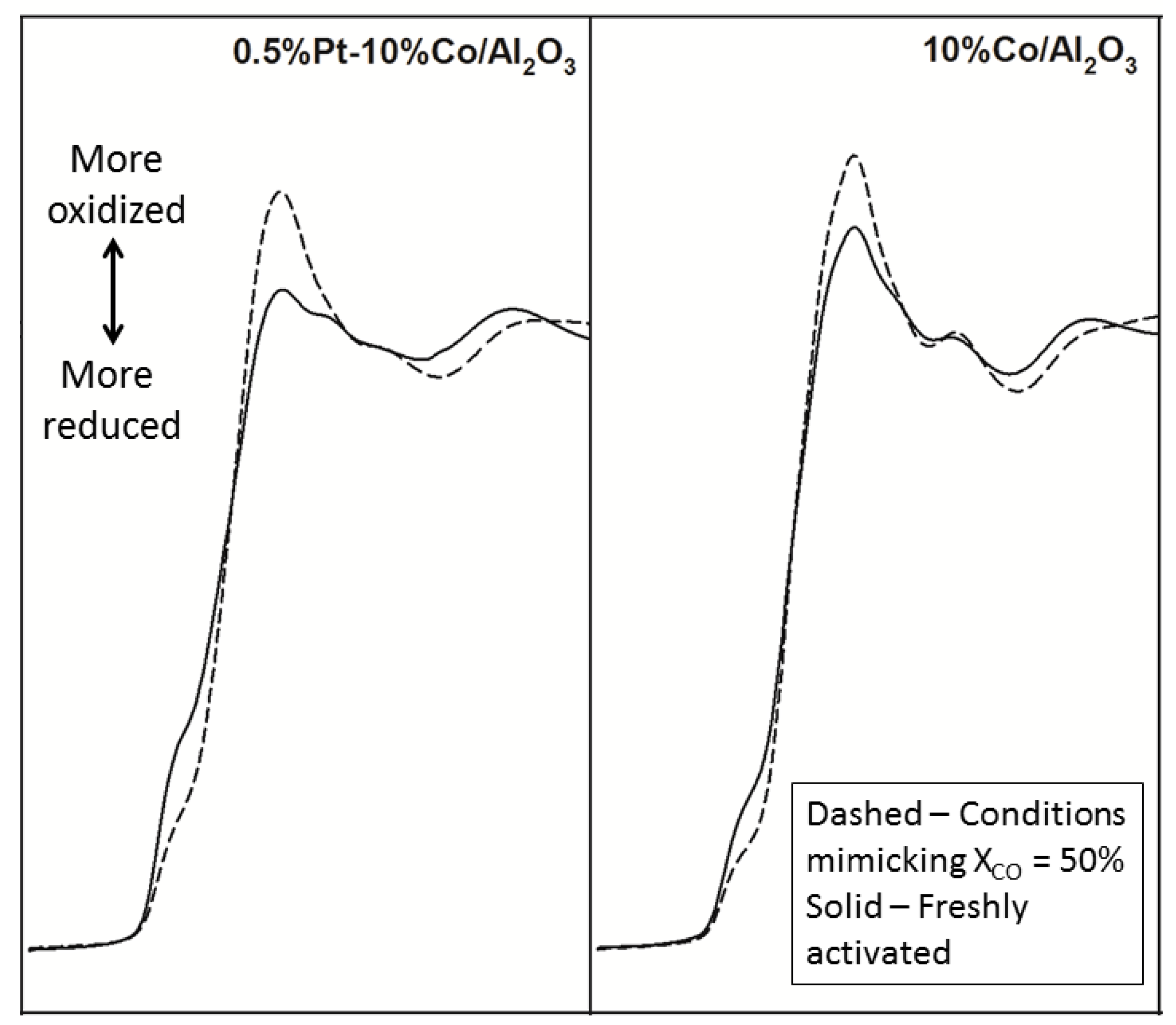
2.2.1. Reoxidation of Small Cobalt Crystallites at the Onset of FTS at Realistic Conversions

| Catalyst | XCO (%) | SV (NL/gcat/h) | S(CH4) | S(C5+) | S(CO2) |
|---|---|---|---|---|---|
| 25%Co/Al2O3 | 51.0 | 3.4–4.2 | 8.3 | 82.5 | 0.8 |
| 0.5%Pt-25%Co/Al2O3 | 52.0 | 1.7–12 | 9.1 | 81.2 | 1.1 |
| 2.0%Pt-25%Co/Al2O3 | 45.0 | 9.0–12 | 9.1 | 81.9 | 1.1 |
| 5.0%Pt-25%Co/Al2O3 | 52.5 | 10–16 | 9.5 | 80.7 | 3.2 |
| 0.276%Ag-25%Co/Al2O3 | 46.4 | 8.8–12 | 7.4 | 84.1 | 0.4 |
| 1.11 %Ag-25%Co/Al2O3 | 48.1 | 9.3–12 | 7.3 | 83.7 | 0.4 |
| 2.76%Ag-25%Co/Al2O3 | 44.5 | 7.0–12 | 7.6 | 84.1 | 0.6 |

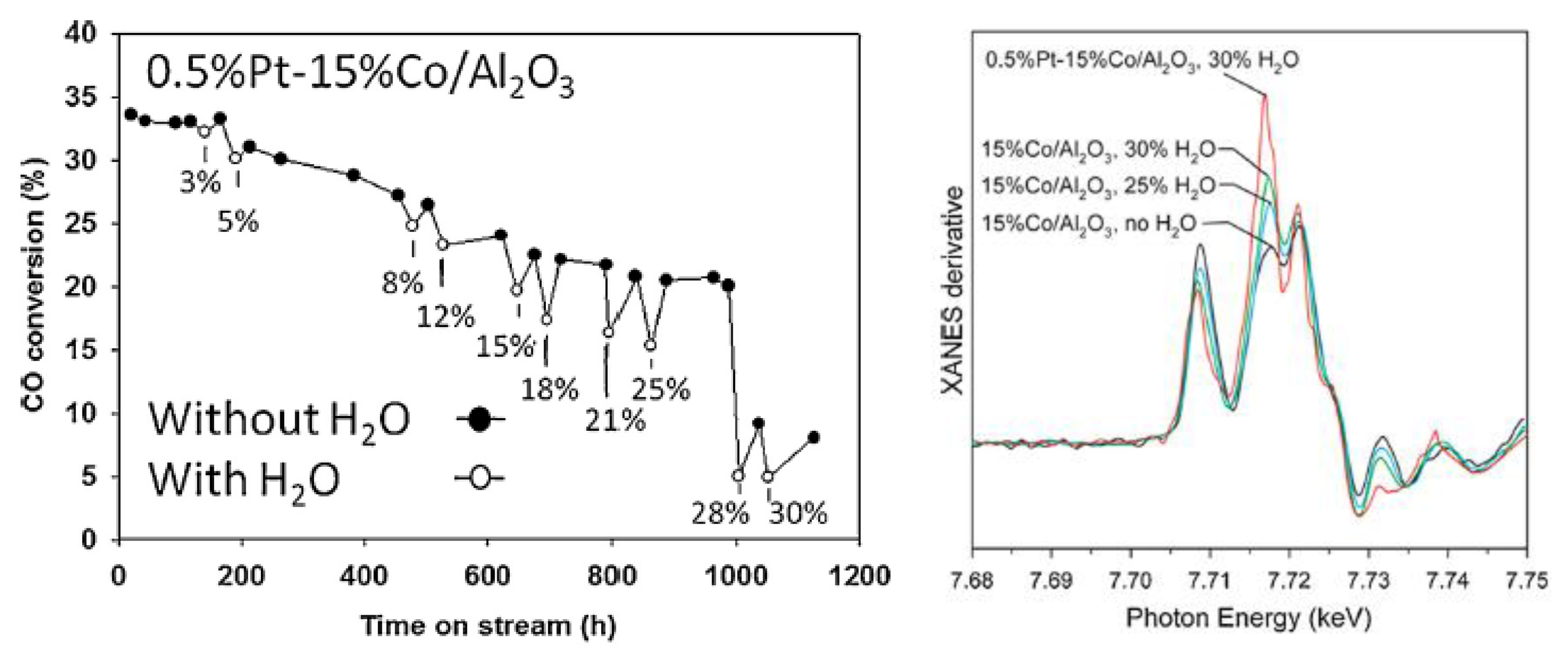
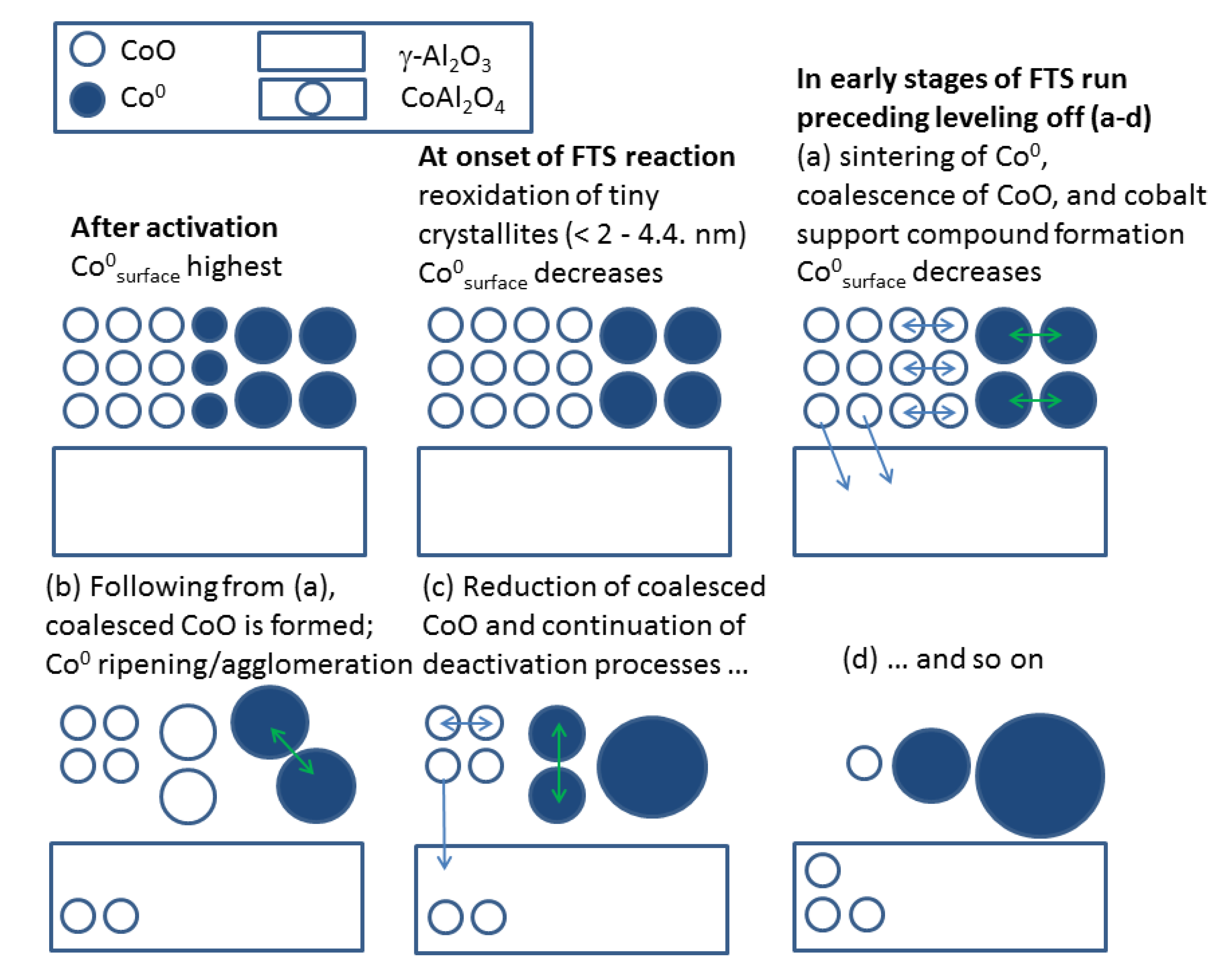
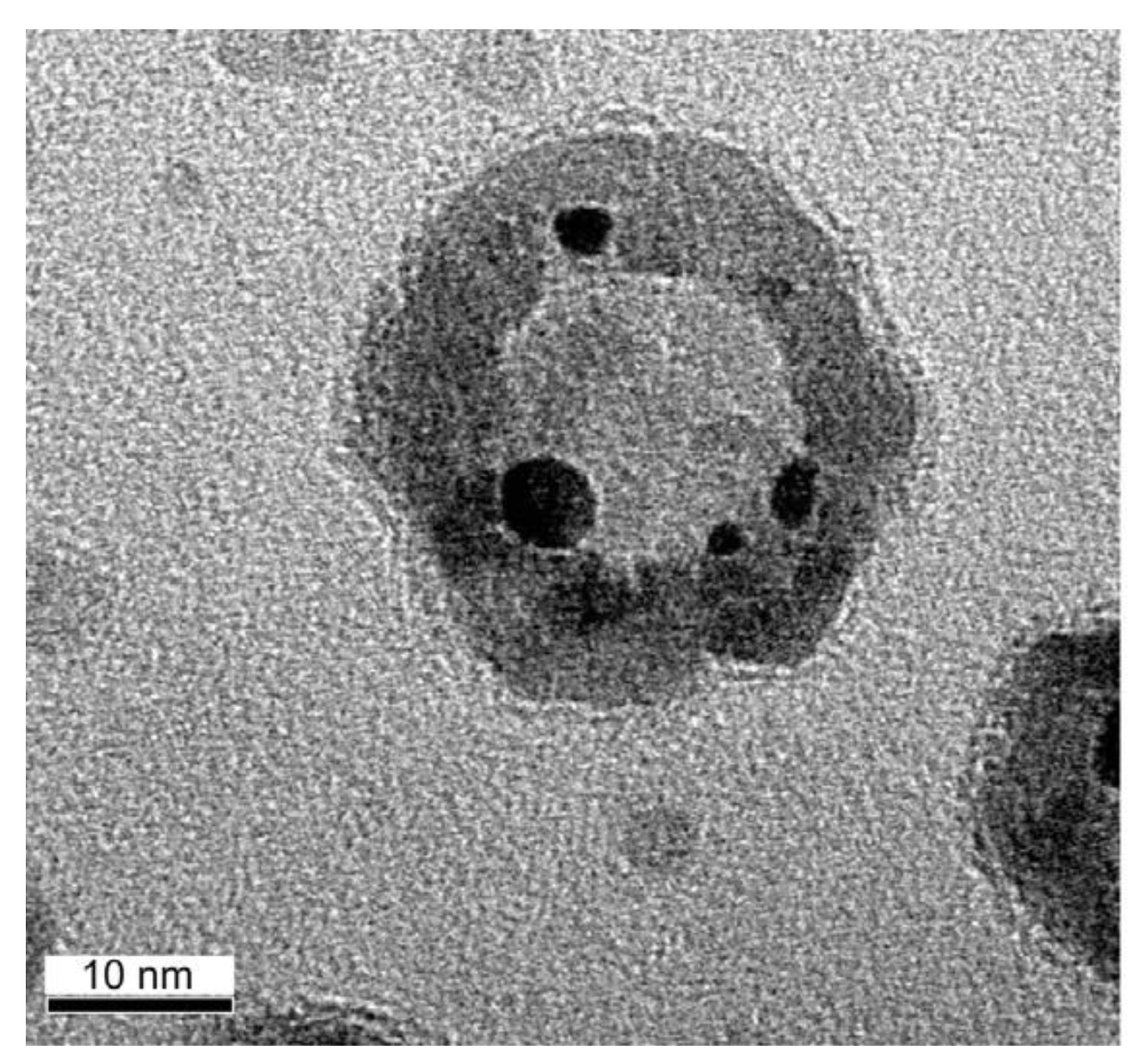
2.2.2. Sintering and Co Support Compound Formation during Initial Deactivation Period Prior to Leveling-off Period







2.3. Regeneration


2.4. Modeling
2.4.1. Modeling of Site Suppression and Deactivation
2.4.2. Computational Methods Based on First Principles
3. Experimental Section
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Espinoza, R.L.; Visagie, J.L.; van Berge, P.J.; Bolder, F.H. Fischer-Tropsch catalysts containing iron and cobalt. US Patent No. 5,733,839, March 1998. [Google Scholar]
- Van Berge, P.J.; Barradas, S.; van de Loosdrecht, J.; Visagie, J.L. Advances in the cobalt catalyzed Fischer-Tropsch synthesis. Erdöl Erdgas Kohle 2001, 117, 138–142. [Google Scholar]
- Wang, W.-J.; Chen, Y.-W. Influence of metal loading on the reducibility and hydrogenation activity of cobalt/alumina catalysts. Appl. Catal. 1991, 77, 223–233. [Google Scholar]
- Jacobs, G.; Ji, Y.; Davis, B.H.; Cronauer, D.C.; Kropf, A.J.; Marshall, C.L. Fischer-Tropsch synthesis: Temperature programmed EXAFS/XANES investigation of the influence of support type, cobalt loading, and noble metal promoter addition to the reduction behavior of cobalt oxide particles. Appl. Catal. A 2007, 333, 177–191. [Google Scholar] [CrossRef]
- Reuel, R.C.; Bartholomew, C.H. The stoichiometries of H2 and CO adsorptions on cobalt: Effects of support and preparation. J. Catal. 1984, 85, 63–77. [Google Scholar] [CrossRef]
- Vada, S.; Hoff, A.; Adnanes, E.; Schanke, D.; Holmen, A. Fischer-Tropsch synthesis on supported cobalt catalysts promoted by platinum and rhenium. Top. Catal. 1995, 2, 155–162. [Google Scholar] [CrossRef]
- Jacobs, G.; Das, T.K.; Zhang, Y.-Q.; Li, J.; Racoillet, G.; Davis, B.H. Fischer-Tropsch synthesis: support, loading, and promoter effects on the reducibility of cobalt catalysts. Appl. Catal. A 2002, 233, 263–281. [Google Scholar] [CrossRef]
- Farrauto, R.J.; Bartholomew, C.H. Fundamentals of Industrial Catalytic Processes; John Wiley & Sons: Chichester, UK, 2003; p. 700. [Google Scholar]
- Cook, K.M.; Poudyal, S.; Miller, J.T.; Bartholomew, C.H.; Hecker, W.C. Reducibility of alumina-supported cobalt Fischer-Tropsch catalysts: Effects of noble metal type, distribution, retention, chemical state, bonding, and influence on cobalt crystallite size. Appl. Catal. A 2012, 449, 69–80. [Google Scholar] [CrossRef]
- Jacobs, G.; Ribeiro, M.C.; Ma, W.; Ji, Y.; Khalid, S.; Sumodjo, P.T.A.; Davis, B.H. Group 11 (Cu, Ag, Au) promotion of 15%Co/Al2O3 Fischer-Tropsch synthesis catalysts. Appl. Catal. A 2009, 361, 137–151. [Google Scholar] [CrossRef]
- Aldossary, M.A.; Sharma, P.; Ojeda, M.; Gupta, M.; Fierro, J.L.; Spivey, J.J. Effect of different Cu loading on Fe-Mg catalyst for Fischer-Tropsch synthesis. In Proceedings of ACS National Meeting & Exposition, Philadelphia, PA, USA, August 19–23, 2012; Curran Associates, Inc.: Red Hook, NY, USA, 2012; Volume 57, p. 944. [Google Scholar]
- Li, S.; Li, A.; Krishnamoorthy, S.; Iglesia, E. Effects of Zn, Cu, and K promoters on the structure and on the reduction, carburization, and catalytic behavior of iron-based Fischer-Tropsch synthesis catalysts. Catal. Lett. 2001, 77, 197–205. [Google Scholar] [CrossRef]
- Cesar, D.V.; Perez, C.A. Quantitative XPS analysis of bimetallic Cu-Co catalysts. Phys. Status Solidi A 2001, 187, 321–326. [Google Scholar] [CrossRef]
- Todic, B.; Bhatelia, T.; Froment, G.F.; Ma, W.; Jacobs, G.; Davis, B.H.; Bukur, D.B. Kinetic model of Fischer-Tropsch synthesis in a slurry reactor on Co/Re/Al2O3 catalyst. Ind. Eng. Chem. Res. 2013, 52, 669–679. [Google Scholar] [CrossRef]
- Jacobs, G.; Ma, W.; Gao, P.; Todic, B.; Bhatelia, T.; Bukur, D.B.; Khalid, S.; Davis, B.H. Fischer-Tropsch synthesis: differences observed in local atomic structure and selectivity with Pd compared to typical promoters (Pt, Re, Ru) of Co/Al2O3 catalysts. (Special Issue in honor of the late Prof. Laszlo Guczi 1932–2012). Top. Catal. 2012, 55, 811–817. [Google Scholar] [CrossRef]
- Jacobs, G.; Sarkar, A.; Ji, Y.; Luo, M.-S.; Dozier, A.; Davis, B.H. Fischer-Tropsch synthesis: assessment of the ripening of cobalt clusters and mixing between Co and Ru promoter via oxidation-reduction cycles over lower Co-loaded Ru-Co/Al2O3 catalysts. Ind. Eng. Chem. Res. 2008, 47, 672–680. [Google Scholar] [CrossRef]
- Borg, O.; Hammer, N.; Eri, S.; Lindvag, O.A.; Myrstad, R.; Blekkan, E.A.; Ronning, M.; Rytter, E.; Holmen, A. Fischer-Tropsch synthesis over un-promoted and Re-promoted gamma-Al2O3 supported cobalt catalysts with different pore sizes. Catal. Today 2009, 142, 70–77. [Google Scholar] [CrossRef]
- Bazin, D.; Borko, L.; Koppany, Zs.; Kovacs, I.; Stefler, G.; Sajo, L.I.; Schay, Z.; Guczi, L. Re-Co/NaY and Re-Co/Al2O3 bimetallic catalysts: In situ EXAFS and catalytic activity. Catal. Lett. 2002, 84, 169–182. [Google Scholar] [CrossRef]
- Jacobs, G.; Chaney, J.A.; Patterson, P.M.; Das, T.K.; Davis, B.H. Fischer-Tropsch synthesis: Study of the promotion of Re on the reduction property of Co/Al2O3 catalysts by in situ EXAFS/XANES of Co K and Re LIII edges and XPS. Appl. Catal. A 2004, 264, 203–212. [Google Scholar] [CrossRef]
- Ronning, M.; Nicholson, D.G.; Holmen, A. In situ EXAFS study of the bimetallic interaction in a rhenium-promoted alumina-supported cobalt Fischer-Tropsch catalyst. Catal. Lett. 2001, 72, 141–146. [Google Scholar] [CrossRef]
- Ma, W.; Jacobs, G.; Ji, Y.; Bhatelia, T.; Bukur, D.B.; Khalid, S.; Davis, B.H. Fischer-Tropsch synthesis: Influence of CO conversion on selectivities, H2/CO usage ratios, and catalyst stability for a Ru promoted Co/Al2O3 catalyst using a slurry phase reactor. Top. Catal. 2011, 54, 757–767. [Google Scholar] [CrossRef]
- Guczi, L.; Bazin, D.; Kovacs, I.; Borko, L.; Schay, Z.; Lynch, J.; Parent, P.; Lafon, C.; Stefler, G.; Koppany, Z.; Sajo, I. Structure of Pt-Co/Al2O3 and Pt-Co/NaY bimetallic catalysts: Characterization by in situ EXAFS, TPR, XPS and by activity in CO (Carbon Monoxide) Hydrogenation. Top. Catal. 2002, 20, 129–139. [Google Scholar] [CrossRef]
- Jacobs, G.; Chaney, J.A.; Patterson, P.M.; Das, T.K.; Maillot, J.C.; Davis, B.H. Fischer-Tropsch synthesis: Study of the promotion of Pt on the reduction property of Co/Al2O3 catalysts by in situ EXAFS of Co K and Pt LIII edges and XPS. J. Synch. Rad. 2004, 11, 414–422. [Google Scholar] [CrossRef]
- Sadeqzadeh, M.; Karaca, H.; Safonova, O.V.; Fongarland, P.; Chambrey, S.; Roussel, P.; Griboval-Constant, A.; Lacroix, M.; Curulla-Ferré, D.; Luck, F.; et al. Identification of the active species in the working alumina-supported cobalt catalyst under various conditions of Fischer–Tropsch synthesis. Catal. Today 2011, 164, 62–67. [Google Scholar] [CrossRef]
- Iglesia, E.; Soled, S.L.; Fiato, R.A.; Via, G.H. Bimetallic synergy in cobalt-ruthenium Fischer-Tropsch synthesis catalysts. J. Catal. 1993, 143, 345–368. [Google Scholar] [CrossRef]
- Chonco, Z.H.; Nabaho, D.; Claeys, M.; van Steen, E. The role of reduction promoters in Fischer-Tropsch catalysts for the production of liquid fuels. In Proceedings of 23rd Meeting of the North American Catalysis Society, 2–7 June 2013, Louisville, KY, USA.
- Ionkina, O.; Subramanian, M.A.; Chao, W.; Makar, K.M.; Manzer, L.E. Fischer-Tropsch processes and catalysts with promoters. US Patent 20020010221A1, January 2002. [Google Scholar]
- Jermwongratanachai, T.; Jacobs, G.; Ma, W.; Shafer, W.D.; Gnanamani, M.K.; Gao, P.; Kitiyanan, B.; Davis, B.H.; Klettlinger, J.L.S.; Yen, C.H.; et al. Fischer-Tropsch synthesis: Comparisons between Pt and Ag promoted Co/Al2O3 catalysts for reducibility, local atomic structure, catalytic activity, and oxidation-reduction (OR) cycles. Appl. Catal. 2013, 464–465, 165–180. [Google Scholar]
- Redjala, T.; Remita, H.; Apostolescu, G.; Mostafavi, M.; Thomazeau, C.; Uzio, D. Bimetallic Au-Pd and Ag-Pd clusters synthesized by gamma or electron beam radiolysis and study of the reactivity/structure relationships in the selective hydrogenation of but-1,3-diene. Oil Gas Sci. Technol. 2006, 61, 789–797. [Google Scholar] [CrossRef]
- Van Steen, E.; Claeys, M.; Dry, M.E.; van de Loosdrecht, J.; Viljoen, E.L.; Visagie, J.L. Stability of nanocrystals: thermodynamic analysis of oxidation and re-reduction of cobalt in water/hydrogen mixtures. J. Phys. Chem. B 2005, 109, 3575–3577. [Google Scholar]
- Saib, A.M.; Moodley, D.J.; Ciobica, I.M.; Hauman, M.M.; Sigwebela, B.H.; Weststrate, C.J.; Niemantsverdriet, J.W.; van de Loosdrecht, J. Fundamental understanding of deactivation and regeneration of cobalt Fischer-Tropsch synthesis catalysts. Catal. Today 2010, 154, 271–282. [Google Scholar] [CrossRef]
- Saib, A.M.; Borgna, A.; van de Loosdrecht, J.; van Berge, P.J.; Niemantsverdriet, J.W. XANES study of the susceptibility of nano-sized cobalt crystallites to oxidation during realistic Fischer-Tropsch synthesis. Appl. Catal. 2006, 312, 12–19. [Google Scholar] [CrossRef]
- Bezemer, G.L; Bitter, J.H.; Kuipers, H.P.C.E.; Oosterbeek, H.; Holewijn, J.E.; Xu, X.D.; Kapteijn, F.; van Dillen, A.J.; de Jong, K.P. Cobalt particle size effects in the Fischer-Tropsch reaction studied with carbon nanofiber supported catalysts. J. Am. Chem. Soc. 2006, 128, 3956–3964. [Google Scholar] [CrossRef]
- Borg, Ø.; Dietzel, P.D.C.; Spjelkavik, A.I.; Tveten, E.Z.; Walmsley, J.C.; Diplas, S.; Eri, S.; Holmen, A.; Rytter, E. Fischer-Tropsch synthesis: cobalt particle size and support effects on intrinsic activity and product distribution. J. Catal. 2008, 259, 161–164. [Google Scholar] [CrossRef]
- Azzam, K.; Jacobs, G.; Ma, W.; Davis, B.H. Effect of cobalt particle size on the catalyst intrinsic activity for Fischer-Tropsch synthesis. Catal. Lett. 2014, 144, 389–394. [Google Scholar] [CrossRef]
- Jermwongratanachai, T.; Jacobs, G.; Shafer, W.D.; Ma, W.; Pendyala, V.R.R.; Davis, B.H.; Kitiyanan, B.; Khalid, S.; Cronauer, D.C.; Kropf, A.J.; Marshall, C.L. Fischer-Tropsch synthesis: Oxidation of a fraction of cobalt crystallites in research catalysts at the onset of FT at partial pressures mimicking 50% CO conversion. Top. Catal. in press.
- Fischer, N.; Clapham, B.; Feltes, T.E.; van Steen, E.; Claeys, M. The reoxidation of cobalt Fischer-Tropsch catalysts. In Proceedings of Syngas 2012 Convention, Cape Town, South Africa, 1–4 April 2012.
- Bertole, C.J.; Mims, C.A.; Kiss, G. The effect of water on the cobalt-catalyzed Fischer-Tropsch synthesis. J. Catal. 2002, 210, 84–96. [Google Scholar] [CrossRef]
- Sadeqzadeh, M.; Hong, J.; Fongarland, P.; Curulla-Ferre, D.; Luck, F.; Bousquet, J.; Schweich, D.; Khodakov, A.Y. Mechanistic modeling of cobalt based catalyst sintering in a fixed bed reactor under different conditions of Fischer-Tropsch synthesis. Ind. Eng. Chem. Res. 2012, 51, 11955–11964. [Google Scholar] [CrossRef]
- Jacobs, G.; Das, T.K.; Patterson, P.M.; Luo, M.; Conner, W.A.; Davis, B.H. Fischer-Tropsch synthesis: Effect of water on Co/Al2O3 catalysts and XAFS characterization of reoxidation phenomena. Appl. Catal. A 2004, 270, 65–76. [Google Scholar]
- Soled, S.; Kliewer, C.; Kiss, G.; Baumgartner, J. Reversible and irreversible changes in Co Fischer-Tropsch catalysts during synthesis. In Proceedings of 21st Meeting of the North American Catalysis Society, San Francisco, CA, USA, 7–12 June 2009.
- Jacobs, G.; Ma, W.; Gao, P.; Todic, B.; Bhatelia, T.; Bukur, D.B.; Davis, B.H. The application of synchrotron methods in characterizing iron and cobalt Fischer-Tropsch synthesis catalysts. Catal. Today 2013, 214, 100–139. [Google Scholar] [CrossRef]
- Moodley, D.J.; Saib, A.M.; van de Loosdrecht, J.; Welker-Nieuwoudt, C.A.; Sigwebela, B.H.; Niemantsverdriet, J.W. The impact of cobalt aluminate formation on the deactivation of cobalt-based Fischer-Tropsch synthesis catalysts. Catal. Today 2011, 171, 192–200. [Google Scholar] [CrossRef]
- Das, T.K.; Jacobs, G.; Patterson, P.M.; Conner, W.A.; Davis, B.H. Fischer-Tropsch synthesis: Characterization and catalytic properties of rhenium promoted cobalt alumina catalysts. Fuel 2003, 82, 805–815. [Google Scholar] [CrossRef]
- Jacobs, G.; Sarkar, A.; Ji, Y.; Patterson, P.M.; Das, T.K.; Luo, M.; Davis, B.H. Fischer-Tropsch synthesis: characterization of interactions between reduction promoters and Co for Co/Al2O3–based GTL catalysts. In Proceedings of AIChE Annual Meeting, San Francisco, CA, USA, 12–17 November 2006.
- Karaca, H.; Hong, J.; Fongarland, P.; Roussel, P.; Griboval-Constant, A.; Lacroix, M.; Hortmann, K.; Safonova, O.V.; Khodakov, A.Y. In situ XRD investigation of the evolution of alumina-supported cobalt catalysts under realistic conditions of Fischer-Tropsch synthesis. Chem. Commun. 2010, 46, 788–790. [Google Scholar]
- Jacobs, G.; Patterson, P.M.; Zhang, Y.-Q.; Das, T.K.; Li, J.; Davis, B.H. Fischer-Tropsch synthesis: Deactivation of noble metal-promoted Co/Al2O3 catalysts. Appl. Catal. A 2002, 233, 215–226. [Google Scholar]
- Kim, C.J. Water addition for increased CO/H2 hydrocarbon activity over catalysts comprising * e There is virtually no difference. carried out for thousands of hours, the time ranges specified are very close to each othecobalt, ruthenium, and mixtures thereof which may include a promoter metal. U.S. Patent 5,227,407, July 1993. [Google Scholar]
- Li, J.; Zhan, X.; Zhang, Y.-Q.; Jacobs, G.; Das, T.K.; Davis, B.H. Fischer-Tropsch synthesis: Effect of water on the deactivation of Pt promoted Co/Al2O3 catalysts. Appl. Catal. A 2002, 228, 203–212. [Google Scholar]
- Jacobs, G.; Das, T.K.; Patterson, P.M.; Li, J.; Sanchez, L.; Davis, B.H. Fischer-Tropsch synthesis: XAFS studies of the effect of water on a Pt-promoted Co/Al2O3 catalyst. Appl. Catal. A 2003, 247, 335–343. [Google Scholar]
- Weststrate, C.J.; Saib, A.M.; Niemantsverdriet, J.W. Promoter segregation in Pt and Ru promoted cobalt model catalysts during oxidation-reduction treatments. Catal. Today 2013, 215, 2–7. [Google Scholar] [CrossRef]
- Jermwongratanachai, T.; Jacobs, G.; Shafer, W.D.; Pendyala, V.R.R.; Ma, W.; Gnanamani, M.K.; Hopps, S.; Thomas, G.A.; Kitiyanan, B.; Khalid, S.; et al. Fischer-Tropsch synthesis: TPR and XANES analysis of the impact of oxidation-reduction (OR) cycles on the reducibility of Co/alumina catalysts with different promoters (Pt, Ru, Re, Ag, Au, Rh, Ir). Catal. Today 2014, in press. [Google Scholar]
- Li, J.; Jacobs, G.; Das, T.K.; Zhang, Y.-Q.; Davis, B.H. Fischer-Tropsch synthesis: Effect of water on the catalytic properties of a Co/SiO2 catalyst. Appl. Catal. A 2002, 236, 67–76. [Google Scholar]
- Dalai, A.K.; Das, T.K.; Chaudhari, K.V.; Jacobs, G.; Davis, B.H. Fischer-Tropsch synthesis: Water effects on Co supported on wide and narrow-pore silica. Appl. Catal. A 2005, 289, 135–142. [Google Scholar]
- Ma, W.; Jacobs, G.; Sparks, D.E.; Spicer, R.L.; Davis, B.H.; Klettlinger, J.L.S.; Yen, C.H. Fischer-Tropsch synthesis: Kinetics and water effect study over 25%Co/Al2O3 catalysts. Catal. Today 2014, in press. [Google Scholar]
- Logdberg, S.; Boutonnet, M.; Walmsley, J.C.; Jaras, S.; Holmen, A.; Blekkan, E.A. Effect of water on the space-time yield of different supported cobalt catalysts during Fischer-Tropsch synthesis. Appl. Catal. A 2011, 393, 109–121. [Google Scholar]
- Ma, W.; Jacobs, G.; Sparks, D.E.; Gnanamani, M.K.; Pendyala, V.R.R.; Yen, C.H.; Klettlinger, J.L.S.; Tomsik, T.M.; Davis, B.H. Fischer-Tropsch synthesis: support and cobalt cluster size effects on kinetics over Co/Al2O3 and Co/SiO2 catalysts. Fuel 2011, 90, 756–765. [Google Scholar] [CrossRef]
- Studies in Surface Science and Catalysis. Catalyst Deactivation 1991: Proceedings of the 5th International Symposium; Bartholomew, C.H., Butt, J.B., Eds.; Elsevier: Amsterdam, The Netherlands, 1991; Volume 68. [Google Scholar]
- Bartholomew, C.H. Mechanisms of catalyst deactivation. Appl. Catal. A 2001, 212, 17–60. [Google Scholar] [CrossRef]
- Eggenhuisen, T.M.; Munnik, P.; Talsma, H.; de Jongh, P.E.; de Jong, K.P. Freeze-drying for controlled nanoparticle distribution in Co/SiO2 Fischer-Tropsch catalyst. J. Catal. 2013, 297, 306–313. [Google Scholar] [CrossRef]
- Graham, U.M.; Jacobs, G.; Gnanamani, M.; Lipka, S.; Shafer, W.D.; Swartz, C.; Jermwongratanachai, T.; Chen, R.; Rogers, F.; Davis, B.H. Fischer Tropsch Synthesis: High Oxygenate-Selectivity of Cobalt Catalysts supported on Hydrothermal Carbons. In ACS Catal.; submitted for publication; 2014. [Google Scholar]
- van de Loosdrecht, J.; Datt, M.; Visagie, J.L. Carbon coated supports for cobalt based Fischer-Tropsch catalysts. Top. Catal. 2014, in press. [Google Scholar]
- Rane, S.; Borg, O.; Yang, J.; Rytter, E.; Holmen, A. Effect of alumina phases on hydrocarbon selectivity in Fischer-Tropsch synthesis. Appl. Catal. A 2010, 388, 160–167. [Google Scholar]
- Sietsma, J.R.A.; den Breejen, J.P.; de Jongh, P.E.; van Dillen, J.; Bitter, J.H.; de Jong, K.P. Highly active cobalt-on-silica catalysts for the Fischer-Tropsch synthesis obtained via a novel calcination. Stud. Surf. Sci. Catal. 2007, 167, 85–90. [Google Scholar] [CrossRef]
- Lu, J.; Elam, J.W.; Stair, P.C. Synthesis and stabilization of supported metal catalysts by atomic layer deposition. Accts. Chem. Res. 2013, 46, 1806–1815. [Google Scholar] [CrossRef]
- Soled, S.L.; Kiss, G.; Kliewer, C.; Baumgartner, J.; El-Malki, E.-M. Learnings from Co Fischer-Tropsch catalyst studies. In Abstracts of Papers, ENFL-412, 245th ACS National Meeting & Exposition, New Orleans, LA, USA, 7–11 April 2013.
- Argyle, M.D.; Frost, T.S.; Bartholomew, C.H. Cobalt Fischer Tropsch catalyst deactivation modeled using generalized power law expressions. Top. Catal. 2014, in press. [Google Scholar]
- Bakken, V.; Bergene, E.; Rytter, E.; Swang, O. Bimetallic cobalt/rhenium systems: Preferred position of rhenium through an interdisciplinary approach. Catal. Lett. 2010, 135, 21–25. [Google Scholar] [CrossRef]
- Tan, K.F.; Xu, J.; Chang, J.; Borgna, A.; Saeys, M. Carbon deposition on Co catalysts during Fischer-Tropsch synthesis: A computational and experimental study. J. Catal. 2010, 274, 121–129. [Google Scholar] [CrossRef]
- Swart, J.C.W.; Ciobica, I.M.; van Santen, R.A.; van Steen, E. Intermediates in the formation of graphitic carbon on a flat FCC-Co(111) surface. J. Phys. Chem. C 2008, 112, 12899–12904. [Google Scholar]
- Burghgraef, H. A quantum chemical study of CH and CC bond activation on transition metals. PhD Thesis, Eindhoven University of Technology, Eindhoven, The Netherlands, June 1995. [Google Scholar]
- Ciobica, I.M.; van Santen, R.A.; van Berge, P.J.; van de Loosdrecht, J. Adsorbate induced reconstruction of cobalt surfaces. Surf. Sci. 2008, 602, 17–27. [Google Scholar] [CrossRef]
- Wilson, J.; de Groot, C. Atomic-scale restructuring in high-pressure catalysis. J. Phys. Chem. 1995, 99, 7860–7866. [Google Scholar] [CrossRef]
- Schulz, H.; Nie, Z.; Ousmanov, F. Construction of the Fischer–Tropsch regime with cobalt catalysts. Catal. Today 2001, 71, 351–360. [Google Scholar] [CrossRef]
- Balakrishnan, N.; Joseph, B.; Bhethanabotla, V.R. Effect of Pt and Ru promoters on deactivation of Co catalysts by C deposition during Fischer-Tropsch synthesis: A DFT study. Appl. Catal. A 2013, 462–463, 107–115. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jacobs, G.; Ma, W.; Davis, B.H. Influence of Reduction Promoters on Stability of Cobalt/g-Alumina Fischer-Tropsch Synthesis Catalysts. Catalysts 2014, 4, 49-76. https://doi.org/10.3390/catal4010049
Jacobs G, Ma W, Davis BH. Influence of Reduction Promoters on Stability of Cobalt/g-Alumina Fischer-Tropsch Synthesis Catalysts. Catalysts. 2014; 4(1):49-76. https://doi.org/10.3390/catal4010049
Chicago/Turabian StyleJacobs, Gary, Wenping Ma, and Burtron H. Davis. 2014. "Influence of Reduction Promoters on Stability of Cobalt/g-Alumina Fischer-Tropsch Synthesis Catalysts" Catalysts 4, no. 1: 49-76. https://doi.org/10.3390/catal4010049
APA StyleJacobs, G., Ma, W., & Davis, B. H. (2014). Influence of Reduction Promoters on Stability of Cobalt/g-Alumina Fischer-Tropsch Synthesis Catalysts. Catalysts, 4(1), 49-76. https://doi.org/10.3390/catal4010049