
Recent Advances on Electro-Oxidation of Ethanol on Pt- and Pd-Based Catalysts: From Reaction Mechanisms to Catalytic Materials



Abstract
:
{kind=link}
{kind=link}
{kind=link}
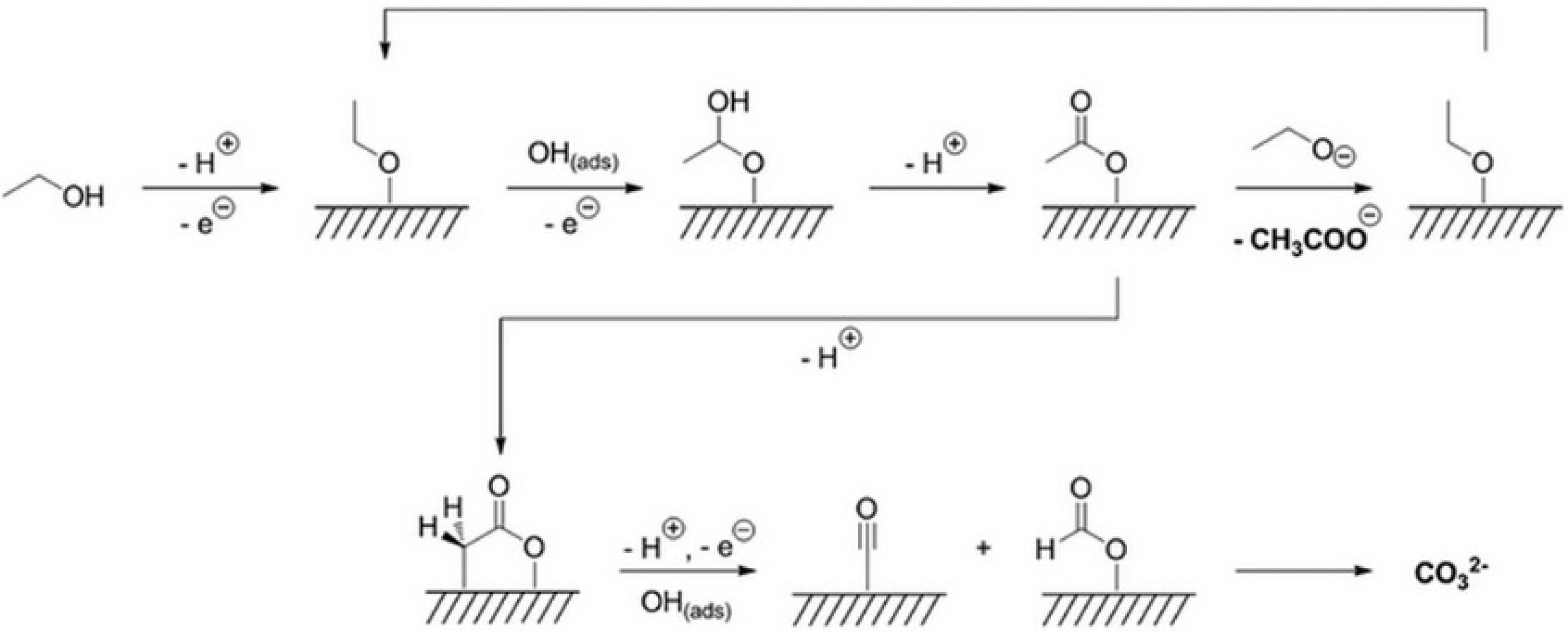{kind=link}
{kind=link}
{kind=link}
{kind=link}
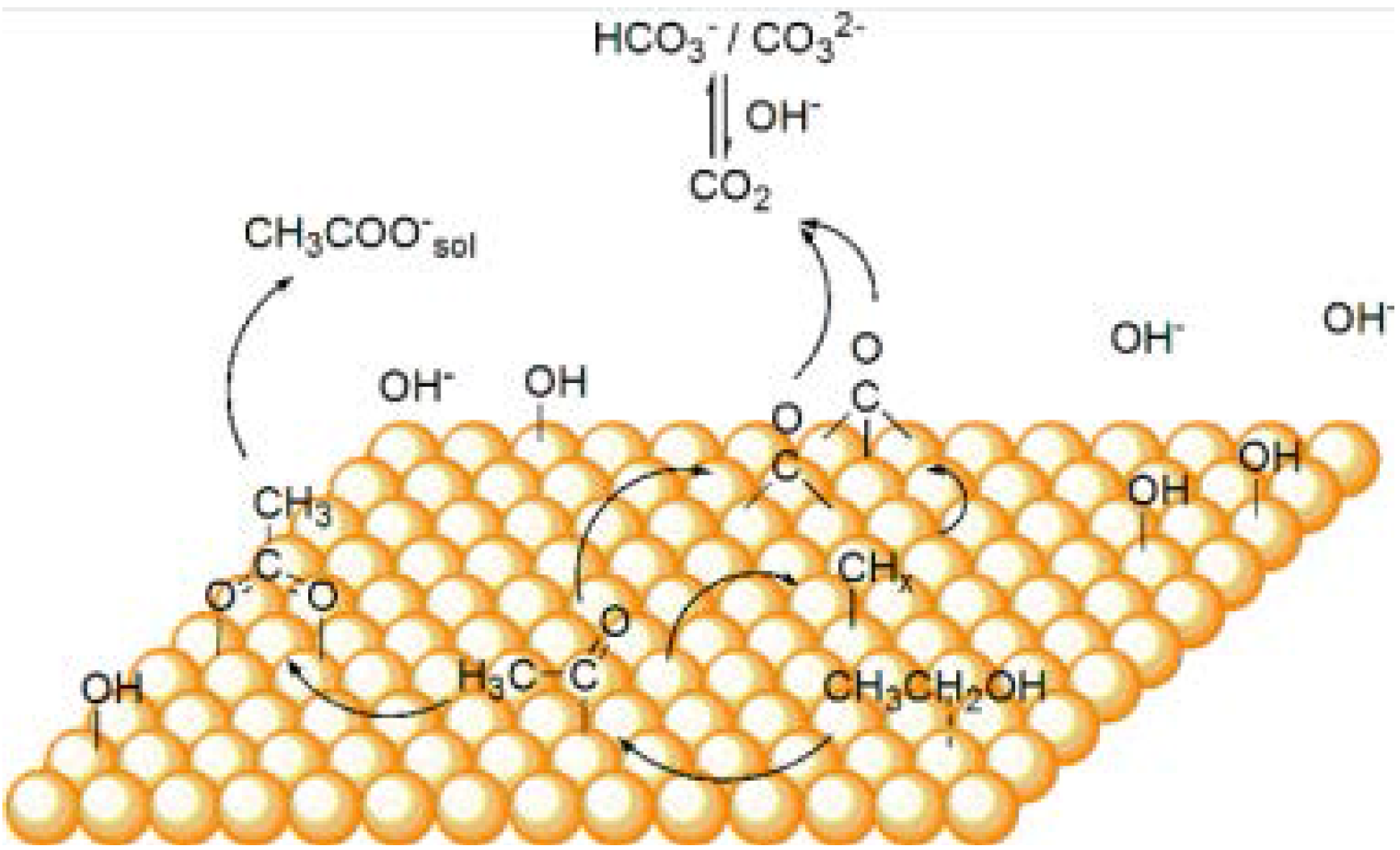
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
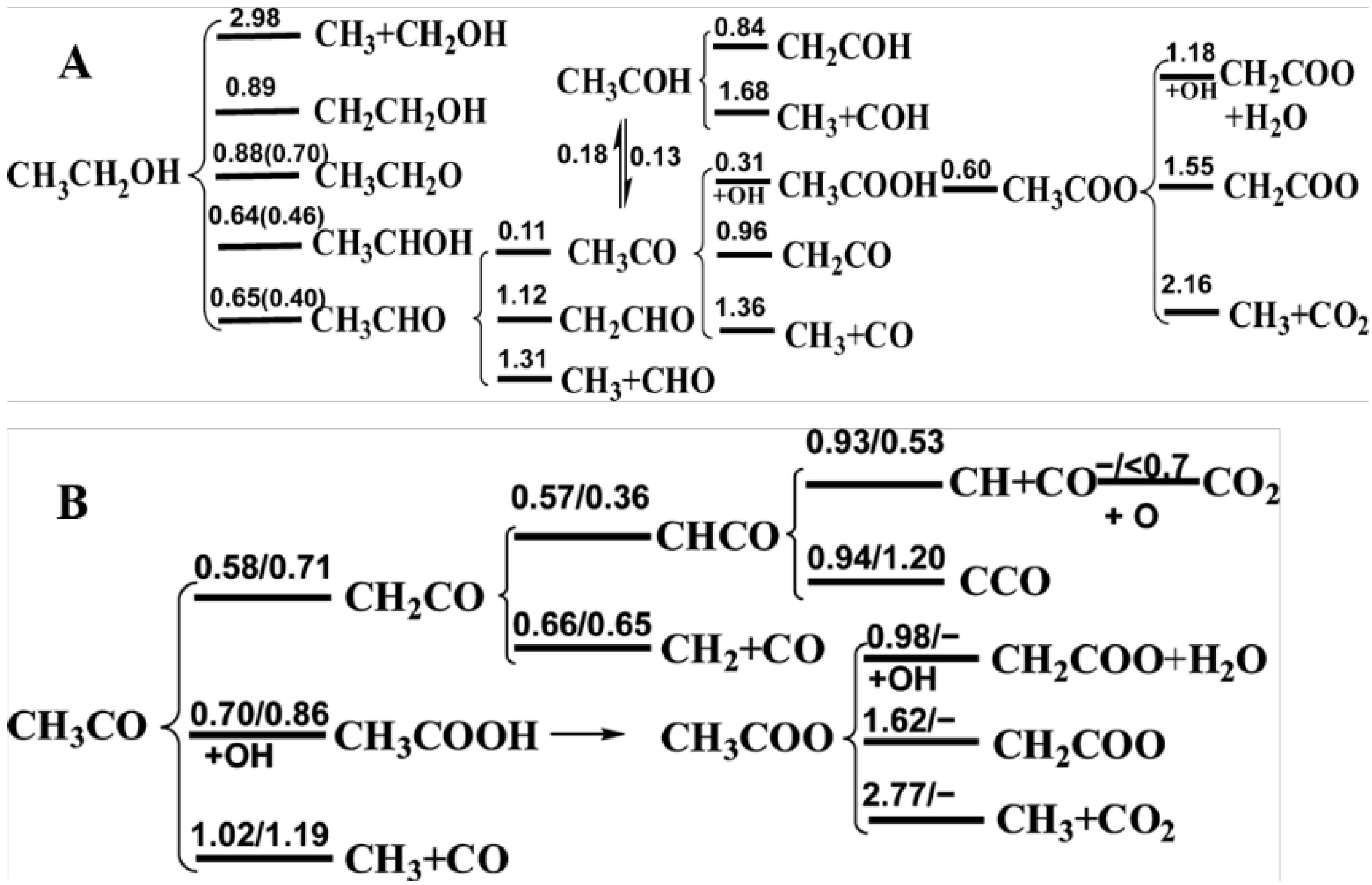
{kind=link}
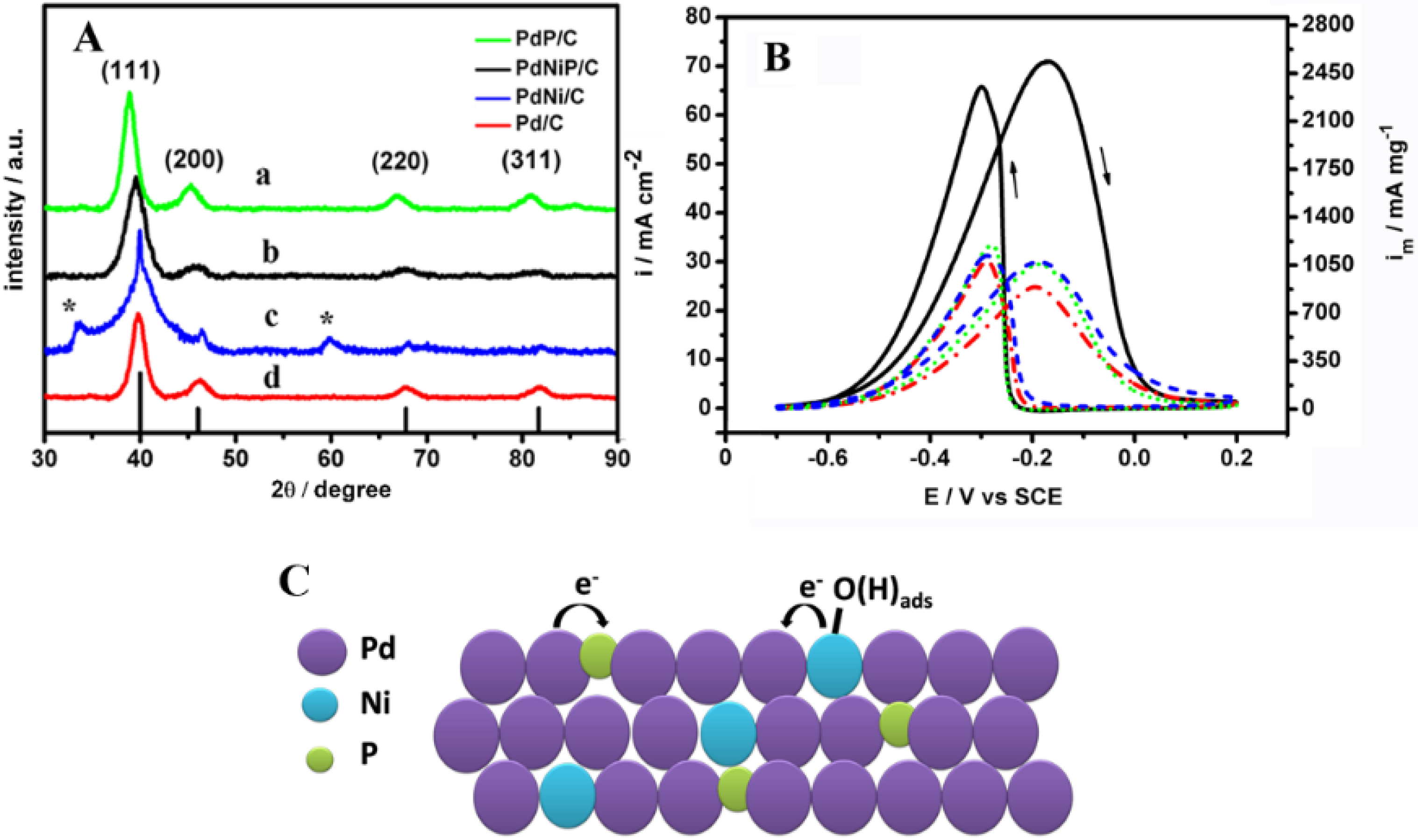
{kind=link}
1. Introduction
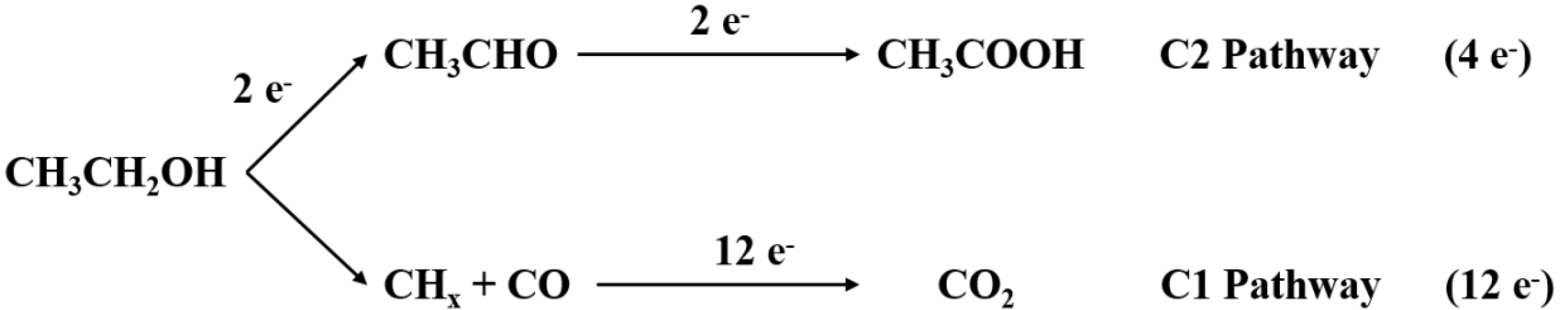
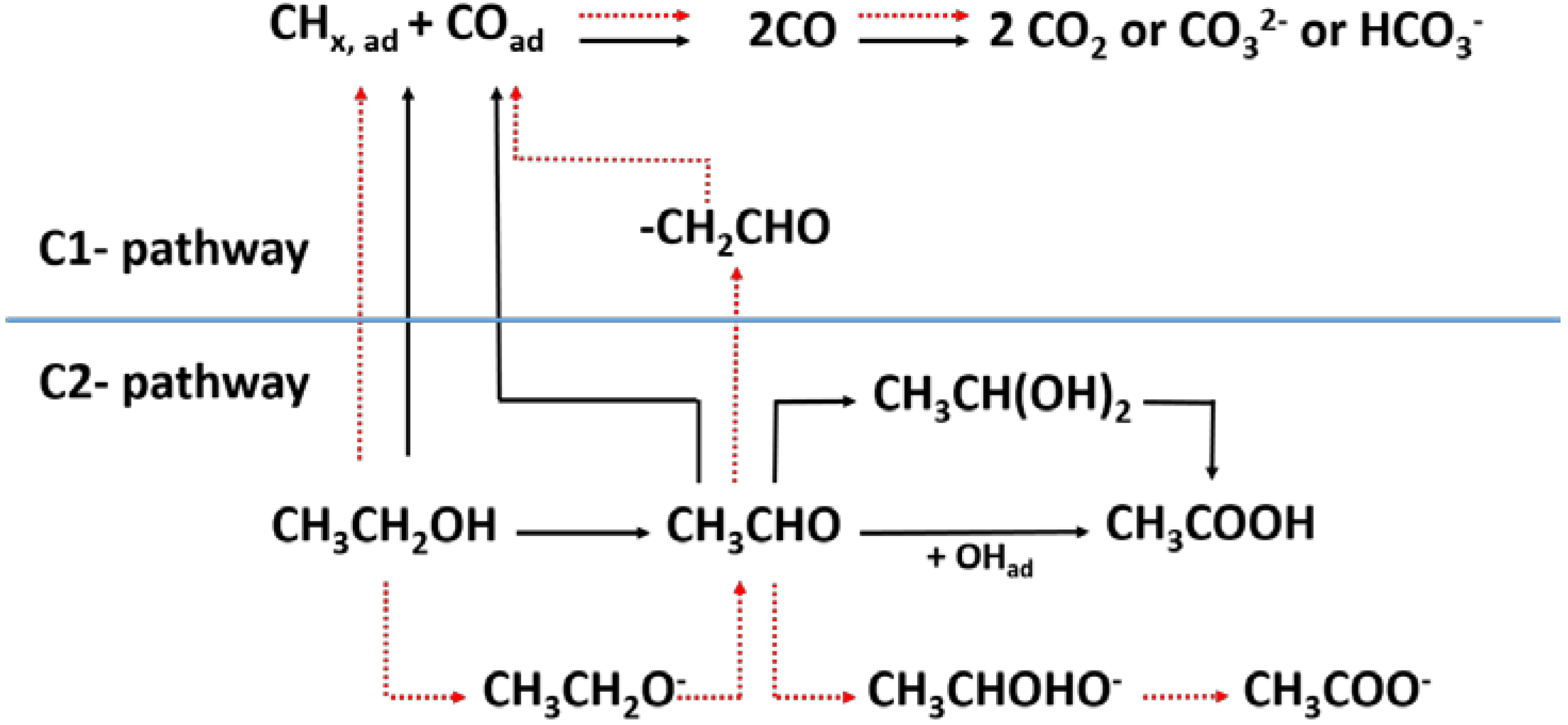
2. Reaction Mechanism of EOR

- C1 pathway:CH3–CH2OH + 3H2O→2CO2 + 12H+ + 12e−CH3–CH2OH + 5H2O→2HCO3− + 14H+ + 12e−CH3–CH2OH + 5H2O→2CO32− + 16H+ + 12e−
- C2 pathway:CH3–CH2OH + H2O→CH3–COOH + 4H+ + 4e−CH3–CH2OH→CH3–CHO + 2H+ + 2e−
2.1. Experimental Detection and Quantification of Reaction Intermediates and Products



2.2. Theoretical Studies



3. Catalytic Role of the Electrode Materials
3.1. Principles in Rational Design
3.2. Pt and Pd Based Electrocatalysts
3.2.1. Pt Based Catalysts



3.2.2. Pd Based Catalysts



4. Conclusions and Outlook
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Antolini, E.; Gonzalez, E.R. Alkaline direct alcohol fuel cells. J. Power Sources 2010, 195, 3431–3450. [Google Scholar] [CrossRef]
- Kamarudin, M.Z.F.; Kamarudin, S.K.; Masdar, M.S.; Daud, W.R.W. Review: Direct ethanol fuel cells. Int. J. Hydrogen Energy 2013, 38, 9438–9453. [Google Scholar] [CrossRef]
- Teng, X. Anodic Catalyst Design for the Ethanol Oxidation Fuel Cell. Available online: http://www.formatex.info/energymaterialsbook/book/473-484.pdf (accessed on 17 August 2015).
- Yao, L.X.; Chang, Y.H. Shaping china’s energy security: The impact of domestic reforms. Energy Policy 2015, 77, 131–139. [Google Scholar] [CrossRef]
- Xuan, J.; Leung, M.K.; Leung, D.Y.; Ni, M. A review of biomass-derived fuel processors for fuel cell systems. Renew. Sustain. Energy Rev. 2009, 13, 1301–1313. [Google Scholar] [CrossRef]
- Wee, J.-H. Applications of proton exchange membrane fuel cell systems. Renew. Sustain. Energy Rev. 2007, 11, 1720–1738. [Google Scholar] [CrossRef]
- Antolini, E. Catalysts for direct ethanol fuel cells. J. Power Sources 2007, 170, 1–12. [Google Scholar] [CrossRef]
- Rao, L.; Jiang, Y.; Zhang, B.; You, L.; Li, Z.; Sun, S. Electrocatalytic oxidation of ethanol. Prog. Chem. 2014, 26, 727–736. [Google Scholar]
- Li, M.; Kowal, A.; Sasaki, K.; Marinkovic, N.; Su, D.; Korach, E.; Liu, P.; Adzic, R.R. Ethanol oxidation on the ternary Pt–Rh–SnO2/C electrocatalysts with varied Pt:Rh:Sn ratios. Electrochim. Acta 2010, 55, 4331–4338. [Google Scholar] [CrossRef]
- Demirci, U.B. Theoretical means for searching bimetallic alloys as anode electrocatalysts for direct liquid-feed fuel cells. J. Power Sources 2007, 173, 11–18. [Google Scholar] [CrossRef]
- Bianchini, C.; Shen, P.K. Palladium-based electrocatalysts for alcohol oxidation in half cells and in direct alcohol fuel cells. Chem. Rev. 2009, 109, 4183–4206. [Google Scholar] [CrossRef] [PubMed]
- Antolini, E. Palladium in fuel cell catalysis. Energy Environ. Sci. 2009, 2, 915–931. [Google Scholar] [CrossRef]
- Chang, S.C.; Leung, L.W.H.; Weaver, M.J. Metal crystallinity effects in electrocatalysis as probed by real-time FTIR spectroscopy: Electrooxidation of formic acid, methanol, and ethanol on ordered low-index platinum surfaces. J. Phys. Chem. 1990, 94, 6013–6021. [Google Scholar] [CrossRef]
- Hitmi, H.; Belgsir, E.; Léger, J.-M.; Lamy, C.; Lezna, R. A kinetic analysis of the electro-oxidation of ethanol at a platinum electrode in acid medium. Electrochim. Acta 1994, 39, 407–415. [Google Scholar] [CrossRef]
- Vigier, F.; Coutanceau, C.; Hahn, F.; Belgsir, E.; Lamy, C. On the mechanism of ethanol electro-oxidation on Pt and PtSn catalysts: Electrochemical and in situ IR reflectance spectroscopy studies. J. Electroanal. Chem. 2004, 563, 81–89. [Google Scholar] [CrossRef]
- Raskó, J.; Dömök, M.; Baán, K.; Erdőhelyi, A. FTIR and mass spectrometric study of the interaction of ethanol and ethanol-water with oxide-supported platinum catalysts. Appl. Catal. A 2006, 299, 202–211. [Google Scholar] [CrossRef]
- Fang, X.; Wang, L.; Shen, P.K.; Cui, G.; Bianchini, C. An in situ fourier transform infrared spectroelectrochemical study on ethanol electrooxidation on Pd in alkaline solution. J. Power Sources 2010, 195, 1375–1378. [Google Scholar] [CrossRef]
- Lai, S.C.S.; Kleijn, S.E.F.; Ozturk, F.T.Z.; Vellinga, V.C.V.; Koning, J.; Rodriguez, P.; Koper, M.T.M. Effects of electrolyte pH and composition on the ethanol electro-oxidation reaction. Catal. Today 2010, 154, 92–104. [Google Scholar] [CrossRef]
- Zhou, Z.Y.; Wang, Q.A.; Lin, J.L.; Tian, N.; Sun, S.G. In situ FTIR spectroscopic studies of electrooxidation of ethanol on Pd electrode in alkaline media. Electrochim. Acta 2010, 55, 7995–7999. [Google Scholar] [CrossRef]
- Christensen, P.A.; Jones, S.W.M.; Hamnett, A. In situ FTIR studies of ethanol oxidation at polycrystalline Pt in alkaline solution. J. Phys. Chem. C 2012, 116, 26109–26109. [Google Scholar] [CrossRef]
- Christensen, P.A.; Jones, S.W.; Hamnett, A. An in situ FTIR spectroscopic study of the electrochemical oxidation of ethanol at a Pb-modified polycrystalline Pt electrode immersed in aqueous KOH. Phys. Chem. Chem. Phys. 2013, 15, 17268–17276. [Google Scholar] [CrossRef] [PubMed]
- Anjos, D.M.; Hahn, F.; Leger, J.M.; Kokoh, K.B.; Tremiliosi, G. In situ FTIRS studies of the electrocatalytic oxidation of ethanol on Pt alloy electrodes. J. Solid State Electrochem. 2007, 11, 1567–1573. [Google Scholar] [CrossRef]
- Yang, Y.-Y.; Ren, J.; Li, Q.-X.; Zhou, Z.-Y.; Sun, S.-G.; Cai, W.-B. Electrocatalysis of ethanol on a Pd electrode in alkaline media: An in situ attenuated total reflection surface-enhanced infrared absorption spectroscopy study. ACS Catal. 2014, 4, 798–803. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, K.; Cai, W.-B. Enhanced electrocatalysis of ethanol on dealloyed Pd–Ni–P film in alkaline media: An infrared spectroelectrochemical investigation. Electrochim. Acta 2015, 162, 100–107. [Google Scholar] [CrossRef]
- Leung, L.W.H.; Chang, S.C.; Weaver, M.J. Real-time FTIR spectroscopy as an electrochemical mechanistic probe—Electrooxidation of ethanol and related species on well-defined Pt(111) surfaces. J. Electroanal. Chem. 1989, 266, 317–336. [Google Scholar] [CrossRef]
- Shao, M.H.; Adzic, R.R. Electrooxidation of ethanol on a Pt electrode in acid solutions: In situ ATR-SEIRAS study. Electrochim. Acta 2005, 50, 2415–2422. [Google Scholar] [CrossRef]
- Willsau, J.; Heitbaum, J. Elementary steps of ethanol oxidation on Pt in sulfuric acid as evidenced by isotope labelling. J. Electroanal. Chem. Interface 1985, 194, 27–35. [Google Scholar] [CrossRef]
- Iwasita, T.; Pastor, E. A DEMS and FTIR spectroscopic investigation of adsorbed ethanol on polycrystalline platinum. Electrochim. Acta 1994, 39, 531–537. [Google Scholar] [CrossRef]
- Wang, J.; Wasmus, S.; Savinell, R. Evaluation of ethanol, 1-propanol, and 2-propanol in a direct oxidation polymer-electrolyte fuel cell a real-time mass spectrometry study. J. Electrochem. Soc. 1995, 142, 4218–4224. [Google Scholar] [CrossRef]
- Wang, H.; Jusys, Z.; Behm, R.J. Ethanol electrooxidation on a carbon-supported Pt catalyst: Reaction kinetics and product yields. J. Phys. Chem. B 2004, 108, 19413–19424. [Google Scholar] [CrossRef]
- Wang, H.; Jusys, Z.; Behm, R.J. Ethanol and acetaldehyde adsorption on a carbon-supported Pt catalyst: A comparative DEMS study. Fuel Cells 2004, 4, 113–125. [Google Scholar] [CrossRef]
- Colmenares, L.; Wang, H.; Jusys, Z.; Jiang, L.; Yan, S.; Sun, G.Q.; Behm, R.J. Ethanol oxidation on novel, carbon supported Pt alloy catalysts-model studies under defined diffusion conditions. Electrochim. Acta 2006, 52, 221–233. [Google Scholar] [CrossRef]
- Wang, H.; Jusys, Z.; Behm, R. Ethanol electro-oxidation on carbon-supported Pt, PtRu and Pt3Sn catalysts: A quantitative DEMS study. J. Power Sources 2006, 154, 351–359. [Google Scholar] [CrossRef]
- Rao, V.; Cremers, C.; Stimming, U. Investigation of the ethanol electro-oxidation in alkaline membrane electrode assembly by differential electrochemical mass spectrometry. Fuel Cells 2007, 7, 417–423. [Google Scholar] [CrossRef]
- Wang, Q.; Sun, G.Q.; Jiang, L.H.; Xin, Q.; Sun, S.G.; Jiang, Y.X.; Chen, S.P.; Jusys, Z.; Behm, R.J. Adsorption and oxidation of ethanol on colloid-based Pt/C, PtRu/C and Pt3Sn/C catalysts: In situ FTIR spectroscopy and on-line DEMS studies. Phys. Chem. Chem. Phys. 2007, 9, 2686–2696. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, M.C.; Aran-Ais, R.M.; Feliu, J.M.; Kontturi, K.; Kallio, T. Pt catalysts modified with Bi: Enhancement of the catalytic activity for alcohol oxidation in alkaline media. J. Catal. 2014, 312, 78–86. [Google Scholar] [CrossRef]
- Sun, S.; Halseid, M.C.; Heinen, M.; Jusys, Z.; Behm, R.J. Ethanol electrooxidation on a carbon-supported Pt catalyst at elevated temperature and pressure: A high-temperature/high-pressure DEMS study. J. Power Sources 2009, 190, 2–13. [Google Scholar] [CrossRef]
- Kavanagh, R.; Cao, X.M.; Lin, W.F.; Hardacre, C.; Hu, P. Origin of low CO2 selectivity on platinum in the direct ethanol fuel cell. Angew. Chem. Int. Ed. Engl. 2012, 51, 1572–1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, T.; Lin, W.F.; Hardacre, C.; Hu, P. Role of water and adsorbed hydroxyls on ethanol electrochemistry on Pd: New mechanism, active centers, and energetics for direct ethanol fuel cell running in alkaline medium. J. Phys. Chem. C 2014, 118, 5762–5772. [Google Scholar] [CrossRef]
- Sheng, T.; Lin, W.-F.; Hardacre, C.; Hu, P. Significance of β-dehydrogenation in ethanol electro-oxidation on platinum doped with Ru, Rh, Pd, Os and Ir. Phys. Chem. Chem. Phys. 2014, 16, 13248–13254. [Google Scholar] [CrossRef] [PubMed]
- Neurock, M. First-Principles Modeling for the Electro-Oxidation of Small Molecules. In Handbook of Fuel Cells; Vielstich, W.G., Lamm, A., Yokokawa, H., Eds.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2010. [Google Scholar]
- Cui, G.F.; Song, S.Q.; Shen, P.K.; Kowal, A.; Bianchini, C. First-principles considerations on catalytic activity of Pd toward ethanol oxidation. J. Phys. Chem. C 2009, 113, 15639–15642. [Google Scholar] [CrossRef]
- Wang, H.F.; Liu, Z.P. Comprehensive mechanism and structure-sensitivity of ethanol oxidation on platinum: New transition-state searching method for resolving the complex reaction network. J. Am. Chem. Soc. 2008, 130, 10996–11004. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.F.; Liu, Z.P. Selectivity of direct ethanol fuel cell dictated by a unique partial oxidation channel. J. Phys. Chem. C 2007, 111, 12157–12160. [Google Scholar] [CrossRef]
- Asiri, H.A.; Anderson, A.B. Mechanisms for ethanol electrooxidation on Pt(111) and adsorption bond strengths defining an ideal catalyst. J. Electrochem. Soc. 2015, 162, F115–F122. [Google Scholar] [CrossRef]
- Zhiani, M.; Majidi, S.; Rostami, H.; Taghiabadi, M.M. Comparative study of aliphatic alcohols electrooxidation on zero-valent palladium complex for direct alcohol fuel cells. Int. J. Hydrogen Energy 2015, 40, 568–576. [Google Scholar] [CrossRef]
- Liang, Z.X.; Zhao, T.S.; Xu, J.B.; Zhu, L.D. Mechanism study of the ethanol oxidation reaction on palladium in alkaline media. Electrochim. Acta 2009, 54, 2203–2208. [Google Scholar] [CrossRef]
- Buso-Rogero, C.; Herrero, E.; Feliu, J.M. Ethanol oxidation on Pt single-crystal electrodes: Surface-structure effects in alkaline medium. Chemphyschem 2014, 15, 2019–2028. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.J.; Li, M.; Zhang, L.; Chan, S.H. Supported PtAu catalysts with different nano-structures for ethanol electrooxidation. Electrochim. Acta 2014, 123, 233–239. [Google Scholar] [CrossRef]
- Hammer, B.; Nørskov, J.K. Theoretical Surface Science and Catalysis-Calculations and Concepts. Adv. Catal. 2000, 45, 71–129. [Google Scholar]
- Srinivasan, S.; Dave, B.B.; Murugesamoorthi, K.A.; Parthasarathy, A.; Appleby, A.J. Overview of Fuel Cell Technology. In Fuel cell Systems; Plenum Press: New York, NY, USA, 1993. [Google Scholar]
- Lamy, C.; Belgsir, E.; Leger, J. Electrocatalytic oxidation of aliphatic alcohols: Application to the direct alcohol fuel cell (DAFC). J. Appl. Electrochem. 2001, 31, 799–809. [Google Scholar] [CrossRef]
- Zhou, W.; Zhou, Z.; Song, S.; Li, W.; Sun, G.; Tsiakaras, P.; Xin, Q. Pt based anode catalysts for direct ethanol fuel cells. Appl. Catal. B 2003, 46, 273–285. [Google Scholar] [CrossRef]
- Lai, S.C.S.; Koper, M.T.M. Ethanol electro-oxidation on platinum in alkaline media. Phys. Chem. Chem. Phys. 2009, 11, 10446–10456. [Google Scholar] [CrossRef] [PubMed]
- Camara, G.A.; Iwasita, T. Parallel pathways of ethanol oxidation: The effect of ethanol concentration. J. Electroanal. Chem. 2005, 578, 315–321. [Google Scholar] [CrossRef]
- Belgsir, E.M.; Bouhier, E.; Yei, H.E.; Kokoh, K.B.; Beden, B.; Huser, H.; Leger, J.M.; Lamy, C. Electrosynthesis in aqueous medium: A kinetic study of the electrocatalytic oxidation of oxygenated organic molecules. Electrochim. Acta 1991, 36, 1157–1164. [Google Scholar] [CrossRef]
- Tarnowski, D.J.; Korzeniewski, C. Effects of surface step density on the electrochemical oxidation of ethanol to acetic acid. J. Phys. Chem. B 1997, 101, 253–258. [Google Scholar] [CrossRef]
- Kutz, R.B.; Braunschweig, B.; Mukherjee, P.; Dlott, D.D.; Wieckowski, A. Study of ethanol electrooxidation in alkaline electrolytes with isotope labels and sum-frequency generation. J. Phys. Chem. Lett. 2011, 2, 2236–2240. [Google Scholar] [CrossRef]
- Ke, X.; Deng, L.L.; Shen, P.K.; Cu, G.F. Electrochemical quartz crystal microbalance (EQCM) characterization of electrodeposition and catalytic activity of Pd-based electrocatalysts for ethanol oxidation. Chem. Res. Chin. Univ. 2010, 26, 443–448. [Google Scholar]
- Huang, L.; Sorte, E.; Sun, S.-G.; Tong, Y.Y.J. A straightforward implementation of in situ solution electrochemical 13C NMR spectroscopy for studying reactions on commercial electrocatalysts: Ethanol oxidation. Chem. Commun. 2015, 51, 8086–8088. [Google Scholar] [CrossRef] [PubMed]
- Leung, L.W.H.; Weaver, M.J. Real-time FTIR spectroscopy as a quantitative kinetic probe of competing electrooxidation pathways of small organic molecules. J. Phys. Chem. 1988, 92, 4019–4022. [Google Scholar] [CrossRef]
- Gao, P.; Chang, S.C.; Zhou, Z.H.; Weaver, M.J. Electrooxidation pathways of simple alcohols at platinum in pure nonaqueous and concentrated aqueous environments as studied by real-time FTIR spectroscopy. J. Electroanal. Chem. 1989, 272, 161–178. [Google Scholar] [CrossRef]
- Buso-Rogero, C.; Grozovski, V.; Vidal-Iglesias, F.J.; Solla-Gullon, J.; Herrero, E.; Feliu, J.M. Surface structure and anion effects in the oxidation of ethanol on platinum nanoparticles. J. Mater. Chem. A 2013, 1, 7068–7076. [Google Scholar] [CrossRef]
- Paulino, M.E.; Nunes, L.M.; Gonzalez, E.R.; Tremiliosi-Filho, G. In situ FTIR spectroscopic study of ethanol oxidation on Pt (111)/Rh/Sn surface: The anion effect. Electrochem. Commun. 2015, 52, 85–88. [Google Scholar] [CrossRef]
- Lai, S.C.; Koper, M.T. Electro-oxidation of ethanol and acetaldehyde on platinum single-crystal electrodes. Faraday Discuss. 2009, 140, 399–416. [Google Scholar] [CrossRef]
- Ma, L.; Chu, D.; Chen, R. Comparison of ethanol electro-oxidation on Pt/C and Pd/C catalysts in alkaline media. Int. J. Hydrogen Energy 2012, 37, 11185–11194. [Google Scholar] [CrossRef]
- Zhou, Z.-Y.; Chen, D.-J.; Li, H.; Wang, Q.; Sun, S.-G. Electrooxidation of dimethoxymethane on a platinum electrode in acidic solutions studied by in situ FTIR spectroscopy. J. Phys. Chem. C 2008, 112, 19012–19017. [Google Scholar] [CrossRef]
- Osawa, M. Surface-Enhanced Infrared Absorption Spectroscopy. In Handbook of Vibrational Spectroscopy; Osawa, M.C., Griffiths, P.R., Eds.; Wiley: New York, NY, USA; Chichester, UK, 2002; Volume 1, pp. 785–799. [Google Scholar]
- Yang, Y.Y.; Zhang, H.X.; Cai, W.B. Recent experimental progresses on electrochemical ATR-SEIRAS. J. Electrochem. 2013, 19, 6–16. [Google Scholar]
- Heinen, M.; Jusys, Z.; Behm, R.J. Ethanol, acetaldehyde and acetic acid adsorption/electrooxidation on a Pt thin film electrode under continuous electrolyte flow: An in situ ATR-FTIRs flow cell study. J. Phys. Chem. C 2010, 114, 9850–9864. [Google Scholar] [CrossRef]
- Bittins-Cattaneo, B.; Wilhelm, S.; Cattaneo, E.; Buschmann, H.W.; Vielstich, W. Intermediates and products of ethanol oxidation on platinum in acid solution. Ber. Bunsenges. Phys. Chem. 1988, 92, 1210–1218. [Google Scholar] [CrossRef]
- Lai, S.C.; Koper, M.T. The influence of surface structure on selectivity in the ethanol electro-oxidation reaction on platinum. J. Phys. Chem. Lett. 2010, 1, 1122–1125. [Google Scholar] [CrossRef]
- Norskov, J.K.; Bligaard, T.; Rossmeisl, J.; Christensen, C.H. Towards the computational design of solid catalysts. Nat. Chem. 2009, 1, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, S.G.; Silva, J.C.M.; Buzzo, G.S.; de Souza, R.F.B.; Spinace, E.V.; Neto, A.O.; Assumpcao, M.H.M.T. Electrochemical and fuel cell evaluation of PtAu/C electrocatalysts for ethanol electro-oxidation in alkaline media. Int. J. Hydrogen Energy 2014, 39, 10121–10127. [Google Scholar]
- Climent, V.; Gómez, R.; Orts, J.M.; Feliu, J.M. Thermodynamic analysis of the temperature dependence of oh adsorption on Pt(111) and Pt(100) electrodes in acidic media in the absence of specific anion adsorption. J. Phys. Chem. B 2006, 110, 11344–11351. [Google Scholar] [CrossRef] [PubMed]
- Kowal, A.; Li, M.; Shao, M.; Sasaki, K.; Vukmirovic, M.; Zhang, J.; Marinkovic, N.; Liu, P.; Frenkel, A.; Adzic, R. Ternary Pt/Rh/SnO2 electrocatalysts for oxidizing ethanol to CO2. Nat. Mater. 2009, 8, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Assumpção, M.; Nandenha, J.; Buzzo, G.; Silva, J.; Spinacé, E.; Neto, A.; de Souza, R. The effect of ethanol concentration on the direct ethanol fuel cell performance and products distribution: A study using a single fuel cell/attenuated total reflectance-fourier transform infrared spectroscopy. J. Power Sources 2014, 253, 392–396. [Google Scholar] [CrossRef]
- Greeley, J.; Norskov, J.K.; Mavrikakis, M. Electronic structure and catalysis on metal surfaces. Annu. Rev. Phys. Chem. 2002, 53, 319–348. [Google Scholar] [CrossRef] [PubMed]
- Rabis, A.; Rodriguez, P.; Schmidt, T.J. Electrocatalysis for polymer electrolyte fuel cells: Recent achievements and future challenges. ACS Catal. 2012, 2, 864–890. [Google Scholar] [CrossRef]
- Du, W.X.; Mackenzie, K.E.; Milano, D.F.; Deskins, N.A.; Su, D.; Teng, X.W. Palladium-Tin alloyed catalysts for the ethanol oxidation reaction in an alkaline medium. ACS Catal. 2012, 2, 287–297. [Google Scholar] [CrossRef]
- Zhu, W.; Ke, J.; Wang, S.-B.; Ren, J.; Wang, H.-H.; Zhou, Z.-Y.; Si, R.; Zhang, Y.-W.; Yan, C.-H. Shaping single-crystalline trimetallic Pt–Pd–Rh nanocrystals toward high-efficiency C–C splitting of ethanol in conversion to CO2. ACS Catal. 2015, 5, 1995–2008. [Google Scholar] [CrossRef]
- Camara, G.A.; de Lima, R.B.; Iwasita, T. Catalysis of ethanol electrooxidation by PtRu: The influence of catalyst composition. Electrochem. Commun. 2004, 6, 812–815. [Google Scholar] [CrossRef]
- Neto, A.O.; Giz, M.J.; Perez, J.; Ticianelli, E.A.; Gonzalez, E.R. The electro-oxidation of ethanol on Pt–Ru and Pt–Mo particles supported on high-surface-area carbon. J. Electrochem. Soc. 2002, 149, A272–A279. [Google Scholar] [CrossRef]
- Lamy, C.; Rousseau, S.; Belgsir, E.M.; Coutanceau, C.; Leger, J.M. Recent progress in the direct ethanol fuel cell: Development of new platinum-tin electrocatalysts. Electrochim. Acta 2004, 49, 3901–3908. [Google Scholar] [CrossRef]
- Zhou, W.J.; Song, S.Q.; Li, W.Z.; Zhou, Z.H.; Sun, G.Q.; Xin, Q.; Douvartzides, S.; Tsiakaras, P. Direct ethanol fuel cells based on PtSn anodes: The effect of Sn content on the fuel cell performance. J. Power Sources 2005, 140, 50–58. [Google Scholar] [CrossRef]
- Tayal, J.; Rawat, B.; Basu, S. Effect of addition of rhenium to Pt-based anode catalysts in electro-oxidation of ethanol in direct ethanol PEM fuel cell. Int. J. Hydrogen Energy 2012, 37, 4597–4605. [Google Scholar] [CrossRef]
- He, Q.G.; Shyam, B.; Macounova, K.; Krtil, P.; Ramaker, D.; Mukerjee, S. Dramatically enhanced cleavage of the C–C bond using an electrocatalytically coupled reaction. J. Am. Chem. Soc. 2012, 134, 8655–8661. [Google Scholar] [CrossRef] [PubMed]
- Suffredini, H.B.; Salazar-Banda, G.R.; Avaca, L.A. Enhanced ethanol oxidation on PbOx-containing electrode materials for fuel cell applications. J. Power Sources 2007, 171, 355–362. [Google Scholar] [CrossRef]
- Huang, Y.; Cai, J.; Liu, M.; Guo, Y. Fabrication of a novel PtPbBi/C catalyst for ethanol electro-oxidation in alkaline medium. Electrochim. Acta 2012, 83, 1–6. [Google Scholar] [CrossRef]
- Matsumoto, F. Ethanol and methanol oxidation activity of PtPb, PtBi, and PtBi2 intermetallic compounds in alkaline media. Electrochemistry 2012, 80, 132–138. [Google Scholar] [CrossRef]
- Gunji, T.; Tanabe, T.; Jeevagan, A.J.; Usui, S.; Tsuda, T.; Kaneko, S.; Saravanan, G.; Abe, H.; Matsumoto, F. Facile route for the preparation of ordered intermetallic Pt3Pb–PtPb core-shell nanoparticles and its enhanced activity for alkaline methanol and ethanol oxidation. J. Power Sources 2015, 273, 990–998. [Google Scholar] [CrossRef]
- Tusi, M.M.; Polanco, N.S.; da Silva, S.G.; Spinacé, E.V.; Neto, A.O. The high activity of PtBi/C electrocatalysts for ethanol electro-oxidation in alkaline medium. Electrochem. Commun. 2011, 13, 143–146. [Google Scholar] [CrossRef]
- Figueiredo, M.C.; Santasalo-Aarnio, A.; Vidal-Iglesias, F.J.; Solla-Gullon, J.; Feliu, J.M.; Kontturi, K.; Kallio, T. Tailoring properties of platinum supported catalysts by irreversible adsorbed adatoms toward ethanol oxidation for direct ethanol fuel cells. Appl Catal. B 2013, 140, 378–385. [Google Scholar] [CrossRef]
- Li, M.; Cullen, D.A.; Sasaki, K.; Marinkovic, N.S.; More, K.; Adzic, R.R. Ternary electrocatalysts for oxidizing ethanol to carbon dioxide: Making Ir capable of splitting C–C bond. J. Am. Chem. Soc. 2013, 135, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.; Ouyang, J.Y. Ternary niaupt nanoparticles on reduced graphene oxide as catalysts toward the electrochemical oxidation reaction of ethanol. ACS Catal. 2015, 5, 1371–1380. [Google Scholar] [CrossRef]
- Jacob, J.M.; Corradini, P.G.; Antolini, E.; Santos, N.A.; Perez, J. Electro-oxidation of ethanol on ternary Pt–Sn–Ce/C catalysts. Appl. Catal. B 2015, 165, 176–184. [Google Scholar] [CrossRef]
- Zhao, Y.H.; Wang, R.Y.; Han, Z.X.; Li, C.Y.; Wang, Y.S.; Chi, B.; Li, J.Q.; Wang, X.J. Electrooxidation of methanol and ethanol in acidic medium using a platinum electrode modified with lanthanum-doped tantalum oxide film. Electrochim. Acta 2015, 151, 544–551. [Google Scholar] [CrossRef]
- Datta, J.; Dutta, A.; Mukherjee, S. The beneficial role of the cometals Pd and Au in the carbon-supported PtPdAu catalyst toward promoting ethanol oxidation kinetics in alkaline fuel cells: Temperature effect and reaction mechanism. J. Phys. Chem. C 2011, 115, 15324–15334. [Google Scholar] [CrossRef]
- Yang, X.; Yang, Q.; Xu, J.; Lee, C.-S. Bimetallic PtPd nanoparticles on nafion-graphene film as catalyst for ethanol electro-oxidation. J. Mater. Chem. 2012, 22, 8057–8062. [Google Scholar] [CrossRef]
- Liao, F.L.; Lo, T.W.B.; Sexton, D.; Qu, J.; Wu, C.T.; Tsang, S.C.E. PdFe nanoparticles as selective catalysts for C–C cleavage in hydrogenolysis of vicinal diol units in biomass-derived chemicals. Catal. Sci. Technol. 2015, 5, 887–896. [Google Scholar] [CrossRef]
- Almeida, T.S.; van Wassen, A.R.; van Dover, R.B.; de Andrade, A.R.; Abruña, H.D. Combinatorial PtSnM (M = Fe, Ni, Ru and Pd) nanoparticle catalyst library toward ethanol electrooxidation. J. Power Sources 2015, 284, 623–630. [Google Scholar] [CrossRef]
- Carmo, M.; Sekol, R.C.; Ding, S.Y.; Kumar, G.; Schroers, J.; Taylor, A.D. Bulk metallic glass nanowire architecture for electrochemical applications. ACS Nano 2011, 5, 2979–2983. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yuan, X.X.; Xia, X.Y.; Du, J.; Ma, Z.; Ma, Z.F. Effects of Mo doping on properties of Pt/C as catalyst towards electro-oxidation of ethanol. J. Inorg. Mater. 2014, 29, 1044–1048. [Google Scholar]
- Jiang, L.; Colmenares, L.; Jusys, Z.; Sun, G.Q.; Behm, R.J. Ethanol electrooxidation on novel carbon supported Pt/SnOx/C catalysts with varied Pt:Sn ratio. Electrochim. Acta 2007, 53, 377–389. [Google Scholar] [CrossRef]
- Li, M.; Liu, P.; Adzic, R.R. Platinum monolayer electrocatalysts for anodic oxidation of alcohols. J. Phys. Chem. Lett. 2012, 3, 3480–3485. [Google Scholar] [CrossRef]
- Cheng, F.L.; Dai, X.C.; Wang, H.; Jiang, S.P.; Zhang, M.; Xu, C.W. Synergistic effect of Pd-Au bimetallic surfaces in Au-covered Pd nanowires studied for ethanol oxidation. Electrochim. Acta 2010, 55, 2295–2298. [Google Scholar] [CrossRef]
- Mourdikoudis, S.; Chirea, M.; Zanaga, D.; Altantzis, T.; Mitrakas, M.; Bals, S.; Liz-Marzán, L.M.; Perez-Juste, J.; Pastoriza-Santos, I. Governing the morphology of Pt–Au heteronanocrystals with improved electrocatalytic performance. Nanoscale 2015, 7, 8739–8747. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, M.C.; Solla-Gullón, J.; Vidal-Iglesias, F.J.; Nisula, M.; Feliu, J.M.; Kallio, T. Carbon-supported shape-controlled Pt nanoparticle electrocatalysts for direct alcohol fuel cells. Electrochem. Commun. 2015, 55, 47–50. [Google Scholar] [CrossRef]
- Tian, N.; Zhou, Z.-Y.; Sun, S.-G.; Ding, Y.; Wang, Z.L. Synthesis of tetrahexahedral platinum nanocrystals with high-index facets and high electro-oxidation activity. Science 2007, 316, 732–735. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, A.; Silva, E.; Moares, L.; Antonini, L.; Abellah, M.Y.; Malfatti, C. Pd-based catalysts for ethanol oxidation in alkaline electrolyte. Am. J. Min. Metal. 2014, 2, 64–69. [Google Scholar]
- Kumar, K.S.; Haridoss, P.; Seshadri, S.K. Synthesis and characterization of electrodeposited Ni–Pd alloy electrodes for methanol oxidation. Surf. Coat. Technol. 2008, 202, 1764–1770. [Google Scholar] [CrossRef]
- Hosseini-Sarvari, M.; Khanivar, A.; Moeini, F. Magnetically recoverable nano Pd/Fe3O4/ZnO catalyst: Preparation, characterization, and application for the synthesis of 2-oxazolines and benzoxazoles. J. Mater. Sci. 2015, 50, 3065–3074. [Google Scholar] [CrossRef]
- Li, G.; Jiang, L.; Jiang, Q.; Wang, S.; Sun, G. Preparation and characterization of PdxAgy/C electrocatalysts for ethanol electrooxidation reaction in alkaline media. Electrochim. Acta 2011, 56, 7703–7711. [Google Scholar] [CrossRef]
- Zhu, L.D.; Zhao, T.S.; Xu, J.B.; Liang, Z.X. Preparation and characterization of carbon-supported sub-monolayer palladium decorated gold nanoparticles for the electro-oxidation of ethanol in alkaline media. J. Power Sources 2009, 187, 80–84. [Google Scholar] [CrossRef]
- Wang, H.; Xu, C.W.; Cheng, F.L.; Jiang, S.P. Pd nanowire arrays as electrocatalysts for ethanol electrooxidation. Electrochem. Commun. 2007, 9, 1212–1216. [Google Scholar] [CrossRef]
- Dutta, A.; Datta, J. Outstanding catalyst performance of pdauni nanoparticles for the anodic reaction in an alkaline direct ethanol (with anion-exchange membrane) fuel cell. J. Phys. Chem. C 2012, 116, 25677–25688. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, F.F.; Yang, Y.Y.; Cai, W.B. Carbon supported Pd–Ni–P nanoalloy as an efficient catalyst for ethanol electro-oxidation in alkaline media. J. Power Sources 2013, 243, 369–373. [Google Scholar] [CrossRef]
- Bahemmat, S.; Ghassemzadeh, M.; Afsharpour, M.; Harms, K. Synthesis, characterization and crystal structure of a Pd(ii) complex containing a new bis-1,2,4-triazole ligand: A new precursor for the preparation of Pd(0) nanoparticles. Polyhedron 2015, 89, 196–202. [Google Scholar] [CrossRef]
- Da Silva, S.G.; Assumpcao, M.H.M.T.; Silva, J.C.M.; de Souza, R.F.B.; Spinace, E.V.; Neto, A.O.; Buzzo, G.S. PdSn/C electrocatalysts with different atomic ratios for ethanol electro-oxidation in alkaline media. Int. J. Electrochem. Sci. 2014, 9, 5416–5424. [Google Scholar]
- Mao, H.M.; Wang, L.L.; Zhu, P.P.; Xu, Q.J.; Li, Q.X. Carbon-supported PdSn SnO2 catalyst for ethanol electro-oxidation in alkaline media. Int. J. Hydrogen Energy 2014, 39, 17583–17588. [Google Scholar] [CrossRef]
- Bambagioni, V.; Bianchini, C.; Filippi, J.; Oberhauser, W.; Marchionni, A.; Vizza, F.; Psaro, R.; Sordelli, L.; Foresti, M.L.; Innocenti, M. Ethanol oxidation on electrocatalysts obtained by spontaneous deposition of palladium onto nickel-zinc materials. ChemSusChem 2009, 2, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Takeguchi, T.; Anzai, Y.; Kikuchi, R.; Eguchi, K.; Ueda, W. Preparation and characterization of CO-tolerant Pt and Pd anodes modified with SnO2 nanoparticles for PEFC. J. Electrochem. Soc. 2007, 154, B1132–B1137. [Google Scholar] [CrossRef]
- Mao, H.; Huang, T.; Yu, A.-S. Facile synthesis of trimetallic Cu1Au0.15Pd1.5/C catalyst for ethanol oxidation with superior activity and stability. J. Mater. Chem. A 2014, 2, 16378–16380. [Google Scholar] [CrossRef]
- Shen, P.K.; Xu, C. Alcohol oxidation on nanocrystalline oxide Pd/C promoted electrocatalysts. Electrochem. Commun. 2006, 8, 184–188. [Google Scholar] [CrossRef]
- Wang, L.; Lavacchi, A.; Bevilacqua, M.; Bellini, M.; Fornasiero, P.; Filippi, J.; Innocenti, M.; Marchionni, A.; Miller, H.A.; Vizza, F. Energy efficiency of alkaline direct ethanol fuel cells employing nanostructured palladium electrocatalysts. Chemcatchem 2015, 7, 2214–2221. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.M.; Wang, Y.Z.; Li, J.P. Acid-treatment-assisted synthesis of Pt–Sn/graphene catalysts and their enhanced ethanol electro-catalytic activity. Int. J. Hydrogen Energy 2015, 40, 990–997. [Google Scholar] [CrossRef]
- Xu, C.; Cheng, L.; Shen, P.; Liu, Y. Methanol and ethanol electrooxidation on Pt and Pd supported on carbon microspheres in alkaline media. Electrochem. Commun. 2007, 9, 997–1001. [Google Scholar] [CrossRef]
- Wang, A.L.; He, X.J.; Lu, X.F.; Xu, H.; Tong, Y.X.; Li, G.R. Palladium-Cobalt nanotube arrays supported on carbon fiber cloth as high-performance flexible electrocatalysts for ethanol oxidation. Angew. Chem. Int. Ed. 2015, 54, 3669–3673. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Abe, K.; Yamaura, S.-I.; Yamamoto, Y.; Asao, N. Fabrication of Pd–Ni–P metallic glass nanoparticles and their application as highly durable catalysts in methanol electro-oxidation. Chem. Mater. 2014, 26, 1056–1061. [Google Scholar] [CrossRef]
- Jiang, R.Z.; Tran, D.T.; McClure, J.P.; Chu, D. A class of (Pd–Ni–P) electrocatalysts for the ethanol oxidation reaction in alkaline media. ACS Catal. 2014, 4, 2577–2586. [Google Scholar] [CrossRef]
- Shao, A.F.; Wang, Z.B.; Chu, Y.Y.; Jiang, Z.Z.; Yin, G.P.; Liu, Y. Evaluation of the performance of carbon supported Pt–Ru–Ni–P as anode catalyst for methanol electrooxidation. Fuel Cells 2010, 10, 472–477. [Google Scholar] [CrossRef]
- Lo, Y.L.; Hwang, B.J. Kinetics of ethanol oxidation on electroless Ni–P/SnO2/Ti electrodes in KOH solutions. J. Electrochem. Soc. 1995, 142, 445–450. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Zou, S.; Cai, W.-B. Recent Advances on Electro-Oxidation of Ethanol on Pt- and Pd-Based Catalysts: From Reaction Mechanisms to Catalytic Materials. Catalysts 2015, 5, 1507-1534. https://doi.org/10.3390/catal5031507
Wang Y, Zou S, Cai W-B. Recent Advances on Electro-Oxidation of Ethanol on Pt- and Pd-Based Catalysts: From Reaction Mechanisms to Catalytic Materials. Catalysts. 2015; 5(3):1507-1534. https://doi.org/10.3390/catal5031507
Chicago/Turabian StyleWang, Ye, Shouzhong Zou, and Wen-Bin Cai. 2015. "Recent Advances on Electro-Oxidation of Ethanol on Pt- and Pd-Based Catalysts: From Reaction Mechanisms to Catalytic Materials" Catalysts 5, no. 3: 1507-1534. https://doi.org/10.3390/catal5031507
APA StyleWang, Y., Zou, S., & Cai, W. -B. (2015). Recent Advances on Electro-Oxidation of Ethanol on Pt- and Pd-Based Catalysts: From Reaction Mechanisms to Catalytic Materials. Catalysts, 5(3), 1507-1534. https://doi.org/10.3390/catal5031507