1. Introduction
The major part of the presently consumed global energy is dependent on fossil fuels including oil, coal and natural gas. According to a British Petroleum p.l.c. statistical review of world energy, 86% of the energy consumed worldwide in 2014 comes from these sources [
1]. To reduce the dependence on fossil fuel, also considering environmental concerns, renewable and sustainable energy is needed [
2,
3]. With its availability, renewability and CO
2 neutrality, biomass appears to be an alternative and sustainable resource for transportation fuels. The use of first-generation biofuels such as bioethanol and biodiesel (FAME—Fatty Acid Methyl Ester) has been established around the world for blending of conventional gasoline or diesel fuel. However, the use of edible oils and arable land for biofuels competes with the food supply chain and affects availability and prices. Nevertheless, the governments of many countries set ambitious goals to promote the use of renewable sources, e.g., the U.S. Department of Energy has set a target to produce 20% of transportation fuel from biomass [
4].
Several technologies for production of second-generation fuels (utilizing complete biomass) like gasification (and subsequent Fischer-Tropsch process), hydrothermal upgrading and pyrolysis are developing. Bio-oil, e.g., obtained from fast pyrolysis of biomass, is considered a promising feedstock for liquid fuel production; however, its quality is far away from conventional fuels due to its high water and oxygenates content. This necessitates additional treatment to remove oxygen in general. Among available technologies, hydrodeoxygenation (HDO), which is performed at high pressure of external hydrogen (50–300 bar) at moderate and high temperature (200–450 °C) in the presence of a heterogeneous catalyst, has evolved quickly into a major technology for bio-oil upgrading. The commercial NExBTL (Neste Oil) leading to HVO (hydrogenated vegetable oil) and the similar Ecofining (UOP/Eni) processes are two prime HDO examples.
Nonetheless, analysis of bio-oil and evaluation of conversion processes are still challenging due to the complex mixture with a high number of functionalities. As a result, particularly at bench scale, more attention has been put on studies of individual model compounds to get more insight into the chemistry before switching to bio-oil. Therefore, the rather less reactive phenolic compounds (phenol, catechol, anisole, guaiacol) have attracted significantly more attention compared to other groups like furanes, acids, aldehydes, ketones and alcohols [
5,
6].
To take advantage of available hydroprocessing technology of the conventional refinery, the same catalysts and conditions were applied for bio-oil HDO in the first known studies. However, the hydrodesulfurization (HDS) catalysts (sulfides of CoMo or NiMo/γ-Al
2O
3) deactivated quickly due to loss of sulfur and coke deposition and the support is unstable against the abundant amount of water in bio-oil [
7,
8]. Meanwhile, commercial hydrotreatment catalysts (e.g., supported noble metal catalysts with Pt, Pd, Ru, Rh) show much higher activity and stability than HDS catalysts [
9,
10,
11,
12,
13,
14,
15,
16,
17,
18,
19,
20]. However, such precious metal catalysts are much more expensive than HDS catalysts, thus further progress is needed to realize viable economic alternative formulations.
Various supported non-noble monometallic catalysts (such as Fe/SiO
2, Co/SiO
2, copper chromite, and Ni-based catalysts) have been already used in HDO studies [
21,
22,
23,
24,
25,
26]. Although Ni-based catalysts showed the highest activity, coke deposition and deactivation due to Ni agglomeration and leaching into aqueous phase are still a problem [
21,
23]. Few groups also reported on bimetallic catalysts for HDO of model compounds and bio-oil due to their properties being different compared to the corresponding monometallic catalysts [
27]. It was reported that bimetallic Ni-Cu/δ-Al
2O
3 catalysts improve product properties of HDO oil [
28]. Bimetallic Ni-Pt/γ-Al
2O
3 and Co-Pt/γ-Al
2O
3 catalysts show high conversion and change the product selectivity in HDO of m-cresol compared to monometallic Pt/γ-Al
2O
3 catalysts [
29]. Other groups reported on the HDO of model compounds (e.g., m-cresol, guaiacol) or bio-oil over bimetallic catalysts such as Pt-Fe/C, Ni-Fe/SiO
2, Ni-Fe/Al
2O
3 [
30,
31,
32,
33,
34]. For instance, vapor phase HDO of guaiacol using Pd-Fe/C catalyst significantly improved HDO yield for toluene/benzene/trimethylbenzene (80%) compared to Fe/C and Pd/C monometallic catalysts [
30]. Besides the fact that the precious metal catalysts rule out the application, the hydrothermal stability of some supports (e.g., Al
2O
3) was limited. More insights into the HDO of model compounds and bio-oil have been given in relevant reviews [
35,
36,
37,
38].
As stated above, phenols are refractory at the desired reaction conditions and therefore suited as probe compounds to understand the fundamental chemistry of HDO reaction. Depending on types of catalysts and reaction conditions (temperature, pressure, solvent properties, etc.), there are two main reaction pathways to eliminate oxygen from phenol, either the direct deoxygenation to benzene (DDO route) or the hydrogenation of the aromatic ring followed by dehydration for oxygen removal (HYD route) [
10,
21,
39]. This suggests that multiple chemical transformations are needed which require different sites to activate hydrogen and to cleave C–O bonds. Depending on the catalyst nature, several possible active sites for phenol HDO reaction have been proposed. In bifunctional catalysts, metal sites and acid sites (e.g., oxides of oxophilic metals like MoO
3, Al
2O
3, WO
3, ZrO
2 or zeolites) are present at the same time and are involved in the steps along the reaction pathways [
8,
22,
39,
40]. In extension, few kinetic studies on HDO of phenol on different catalysts, e.g., Co-MoS
2/Al
2O
3 [
39], Pd/C, Ru/C, Rh/C or Ni catalysts combined with several aqueous acidic solutions (e.g., H
3PO
4, CH
3COOH and Nafion
® solution), or solid acids (HZSM-5, Nafion
®/SiO
2, ZrO
2) are known [
19,
22,
23]. Rate-determining steps in the reaction sequence for phenol HDO strongly depend on the nature of catalyst (metal and acid sites) and reaction conditions.
Most of the mentioned model studies were carried out either in batch autoclaves with short run times [
16,
19,
20,
21,
22,
23,
41] or under gas phase reaction conditions [
30,
31,
32,
33,
42]. Recent HDO studies reported application of continuous flow reactors using organic solvents (e.g., 1-octanol, 1-octane, 1-hexane) [
43,
44,
45] or supercritical water [
46]. Those studies often have not put the focus on the spent catalysts after long-term reaction. However, understanding of catalyst structure and stability is crucial for development of an effective catalyst. Compared to a batch reactor, the use of a continuous reactor is favorable because it allows long-term studies to not be limited by chemical equilibrium. In addition, it provides better kinetic control of reaction steps which helps to elucidate complex reaction networks. The mechanisms are different in gas phase and aqueous phase reactions due to solvent effects, different concentration profiles on the catalyst surface and mass transfer limitations [
47]. From a technological point of view, continuous and heterogeneously catalyzed operation is often preferred, e.g., as catalyst separation from products is no issue.
In recent batch studies, we have developed supported bimetallic Ni-Co catalysts (homogeneous Ni-Co alloy), which were successfully tested in aqueous phase HDO of phenol, to replace noble metal catalysts and to overcome known drawbacks (deactivation, coke formation, costs) [
48,
49]. Batch runs (250 °C, 2 h) revealed that bimetallic catalysts with an equimolar Ni:Co ratio (10 wt % each on HZSM-5) outperformed monometallic Co, Cu and Ni, bimetallic Ni-Cu and Ni-Co catalysts with other metal ratio in terms of conversion and yield toward saturated hydrocarbons. Moreover, the bimetallic Ni-Co catalysts formed significantly less coke deposits. However, similar to previous studies, these short-time batch tests did not give satisfactory information about catalyst stability.
Thus, various bimetallic Ni-Co catalysts with different acidic supports and a corresponding monometallic catalyst as a benchmark were tested in both a batch autoclave and a continuous fixed bed reactor setup. Long-term experiments together with characterization of fresh and spent catalysts using various techniques (N2 physisorption, XRD, XPS, TEM, IR) indicated crucial features and gave more insight into reaction network and catalyst stability.
3. Materials and Methods
3.1. Catalyst Preparation
All monometallic and bimetallic catalysts were prepared by the wet (co-)impregnation method. Ni(NO3)2·6H2O (Merck) and Co(NO3)2·6H2O (Fluka) were used as precursors for Ni and Co, respectively. Common zeolites HZSM-5 (PZ-2/25H, SiO2/Al2O3 = 34, Zeochem AG), HBeta (PB/H, SiO2/Al2O3 = 25, Zeochem AG) and NH4Y (CBV712, SiO2/Al2O3 = 12, Zeolyst) together with ZrO2 (Saint Gobain) were used as supports. Prior to impregnation, NH4Y was calcined (550 °C, 4 h) to get the HY form. The other supports were calcined at 450 °C for 2 h to remove physisorbed water. The calculated amount of precursor was dissolved in a known amount of deionized water and the solution was then impregnated onto supports under stirring at room temperature (RT) for 16 h. Water was removed by a rotary evaporator and the samples were ground, dried overnight and calcined (550 °C, 4 h). These samples are denoted as “fresh”. The calcined catalyst was reduced in a tubular quartz reactor in pure H2 flow (100 mL/min) with a heating rate of 10 K/min to 650 °C and kept for 4 h. These samples are denoted as “pre-reduced”.
3.2. Catalyst Characterization
The metal (Ni, Co, Al) and Si contents of the samples were determined by inductively coupled plasma-optical emission spectroscopy (ICP-OES) using a 715-ES device (Varian, Inc., Palo Alto, CA, USA) and atomic adsorption spectroscopy (AAS) using an Analyst 300 apparatus (PerkinElmer, Inc., Waltham, MA, USA), respectively. The coke deposition on spent catalyst was analyzed using a Truespace CHNS analyzer (LECO Corporation, Saint Joseph, MI, USA).
Nitrogen physisorption method served for calculating the specific surface area according to the BET theory. The measurements were performed on a Micromeritics ASAP 2010 apparatus (Micromeritics GmbH, Aachen, Germany) at −196 °C. Prior to analysis, the calcined catalysts were degassed at 200 °C in vacuum for 4 h.
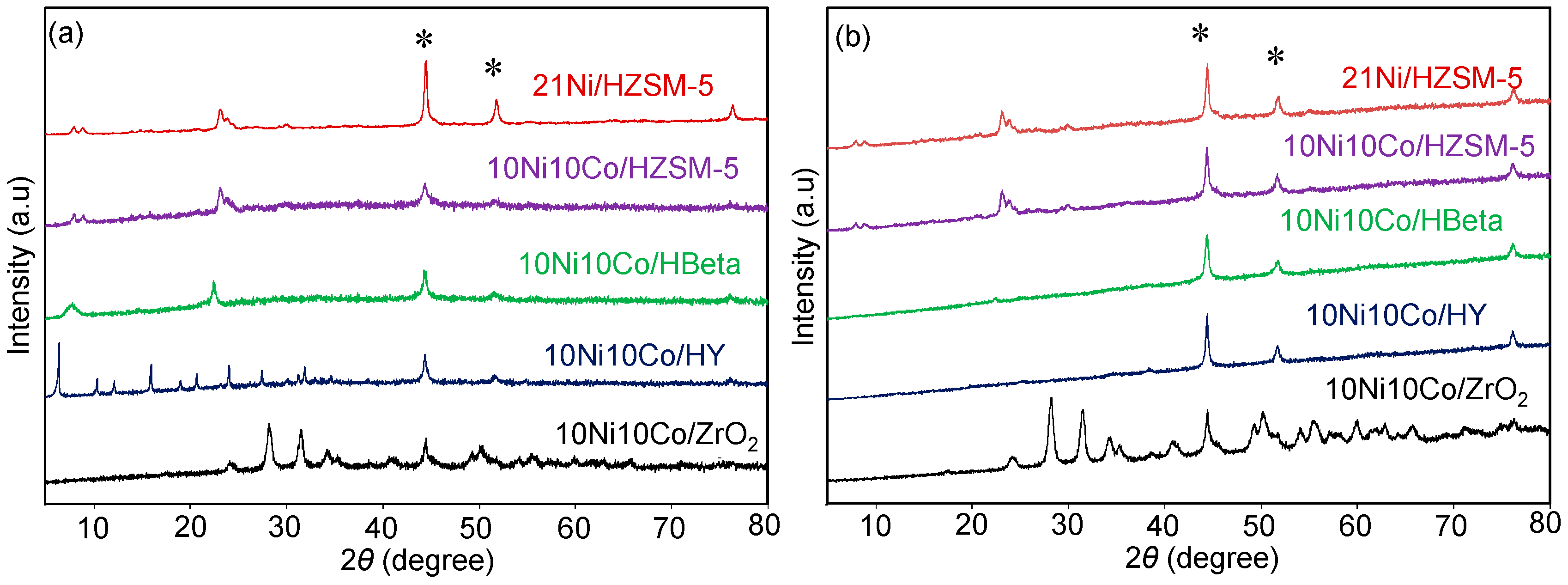
X-ray powder diffraction (XRD) measurements were carried out by a X’Pert Pro theta/theta diffractometer (Panalytical B.V., Almelo, Netherlands) with CuKα radiation (λ = 1.5418 Å, 40 kV, 40 mA) and an X’Celerator RTMS Detector (Panalytical B.V., Almelo, Netherlands). The samples were scanned in the 2θ range of 5°–80° and the Scherrer equation was used for crystallite size calculation.
X-ray photoelectron spectroscopy (XPS) measurements were carried out with a VG ESCALAB 220iXL instrument (Thermo Fisher Scientific, Inc., Waltham, MA, USA) with monochromatic AlKα radiation (E = 1486.6 eV). The peak areas were determined after background subtraction and fitting with Gaussian-Lorentzian curves. The amount of each component in the near-surface region can be calculated based on these peak areas with division by the element-specific Scofield factor and the transmission function of the spectrometer.
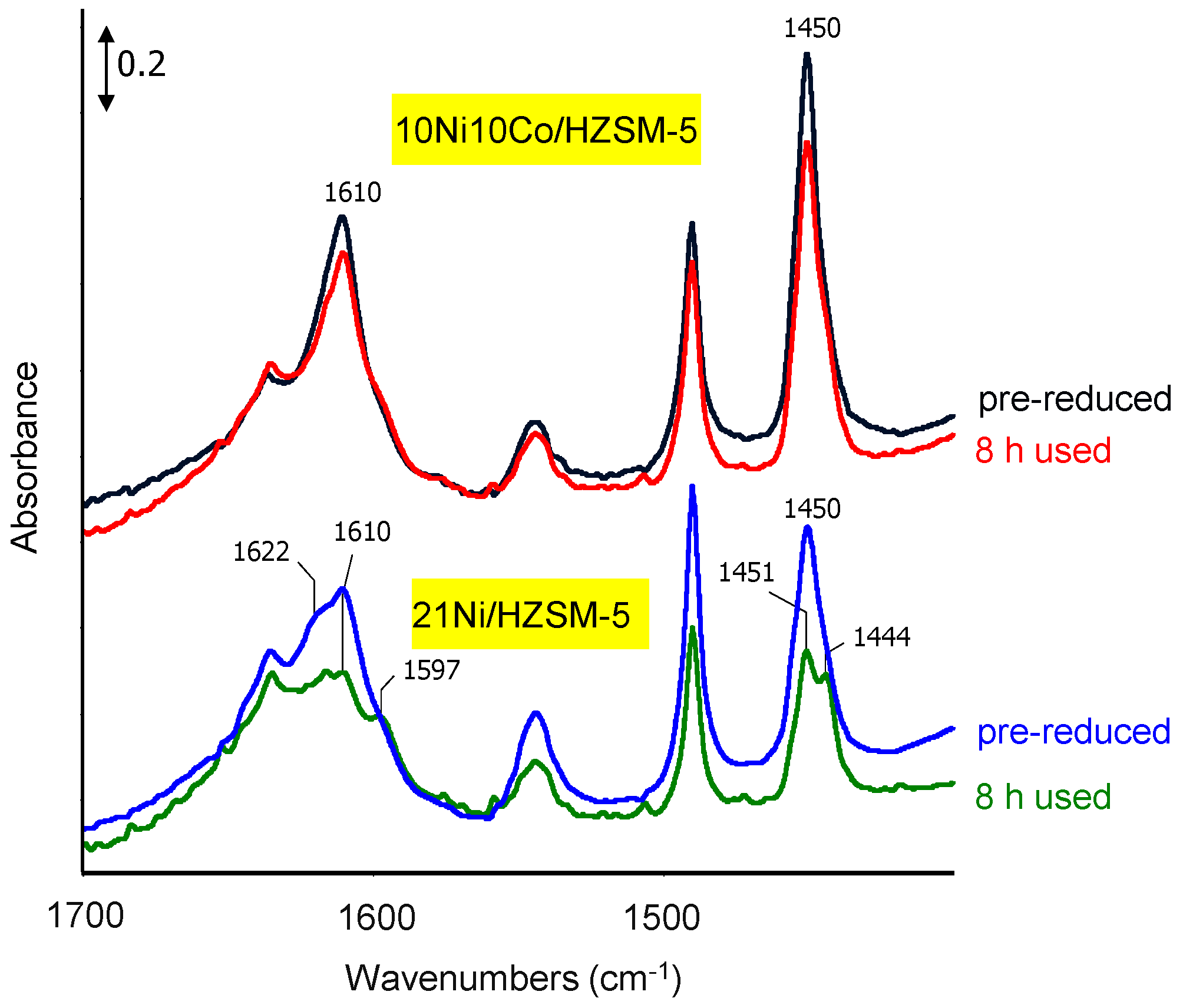
Infrared spectroscopic measurements of adsorbed pyridine (pyr-IR) were used for acidity characterization and were carried out on a Tensor 27 spectrometer (Bruker Optik GmbH, Ettlingen, Germany) equipped with a homemade reaction cell with CaF2 windows. The pre-reduced catalysts were pretreated in 5% H2/He at 400 °C for 10 min. After cooling to room temperature (RT) and evacuation, the pyridine adsorption was performed until saturation. Then, the reaction cell was evacuated to remove physisorbed pyridine and finally the desorption of pyridine was followed by heating the sample in vacuum to 300 °C and recording spectra every 50 K.
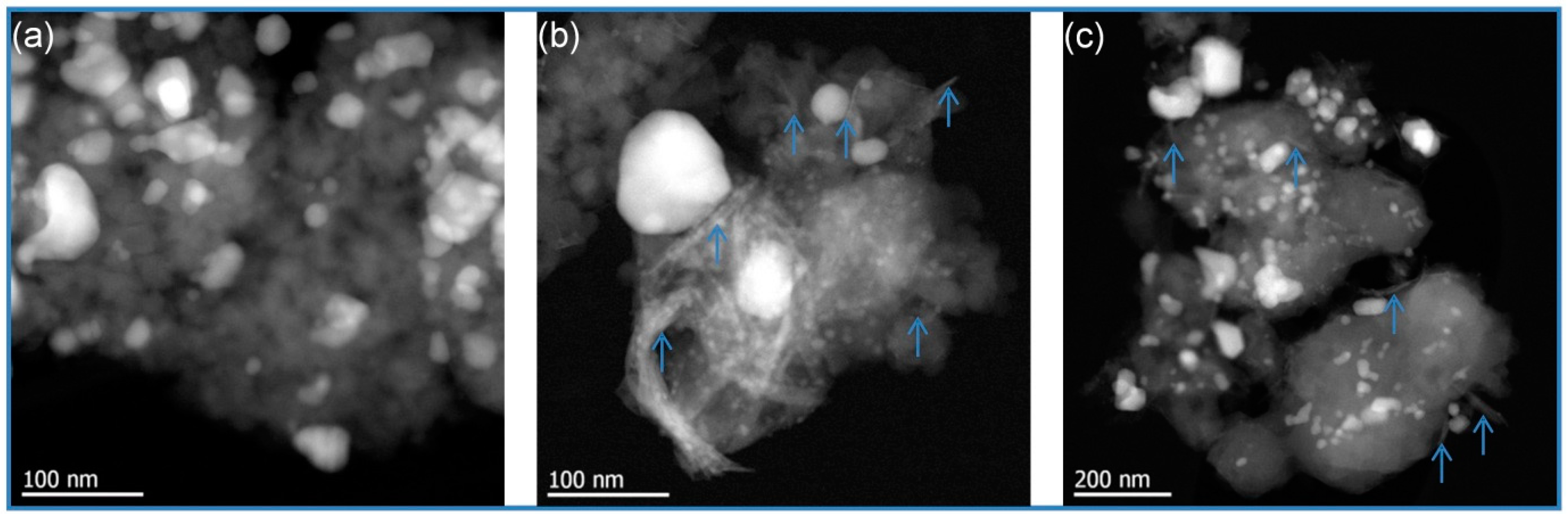
Transmission electron microscopy (TEM) was employed to gain more information on metal sites, their dispersion and particle size on the catalysts. The measurements were performed at 200 kV on a JEM-ARM200F (JEOL (Germany) GmbH, Freising, Germany) which is aberration-corrected by a CESCOR (CEOS) for the scanning transmission (STEM) applications. The microscope is equipped with a JED-2300 (JEOL) energy dispersive X-ray spectrometer (EDXS) for chemical analysis. High-angle annular dark field (HAADF) combined with EDXS imaging was operated with a spot size of 0.16 nm and a 40 µm condenser aperture. For the bright field STEM images, annular bright field (BF) with beam stopper and a 3 mm BF aperture was used. The sample was ground and deposited on a holey carbon-supported Cu grid and transferred to the microscope.
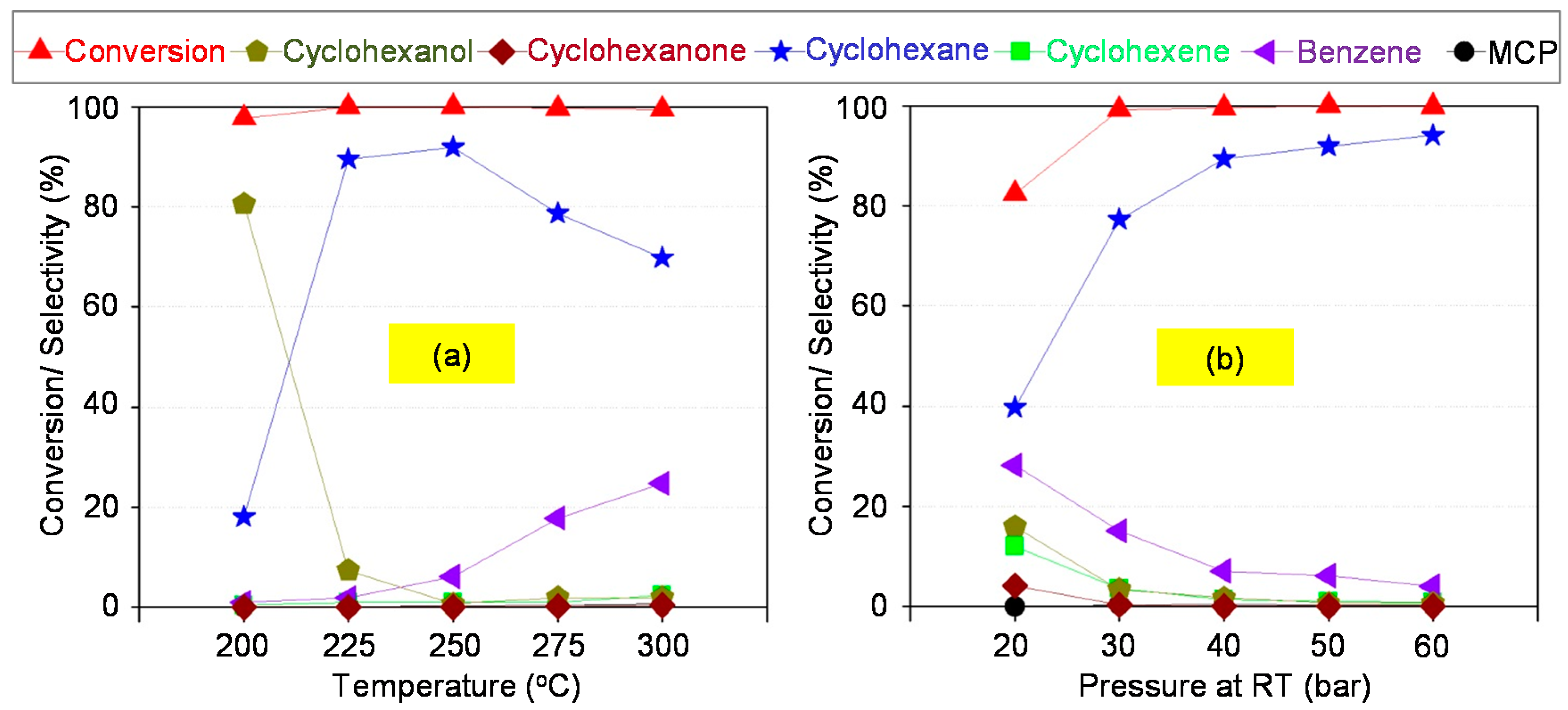
3.3. HDO of Phenol in Batch Reactor
In a typical test, 0.5 g (5.3 mmol) phenol (Merck), 10 g H2O, and 0.025 g of pre-reduced catalyst (in powder form) were loaded into an autoclave (25 mL volume; Parr Instruments, Moline, IL, USA). The reactor was flushed with Ar and then with H2 to remove air, pressurized with H2 to 50 bar at RT and heated to 250 °C. The start time was recorded when the reaction temperature was reached and then stirring speed was set to 650 rpm. After reaction, the autoclave was cooled to RT and the gas was analyzed by gas chromatography using a HP 5890 device (Hewlett-Packard, Inc., Palo Alto, CA, USA) online from autoclave via a transfer line. This gas chromatograph (GC) was equipped with both flame ionization detector (FID) (Poraplot Q column, 25 m × 0.53 mm × 0.20 µm) and thermal conductivity detector (TCD) (HP PLOT Molesieve column, 25 m × 0.53 mm) both using Ar as carrier gas. Liquid products (organic and aqueous phase) were analyzed by a Shimadzu GC 17A apparatus (Shimadzu Deutschland GmbH, Duisburg, Germany) with autosampler and FID (CP-FFAP column, 25 m × 0.32 mm) using He as carrier gas. Mesitylene and 1,4-dioxane were used as the internal standards for quantification of organic and aqueous phase, respectively. Pure components were used for peak identification and calibration.
Conversion and selectivity (product distribution) were calculated basing on the number of carbon moles defined as follows:
Carbon balances were calculated from detected products in gas and liquid phase and reached more than 90% in this work. Missing carbon was due to deposits on the surface of catalysts, some unknown minor peaks in chromatograms and losses during work-up.
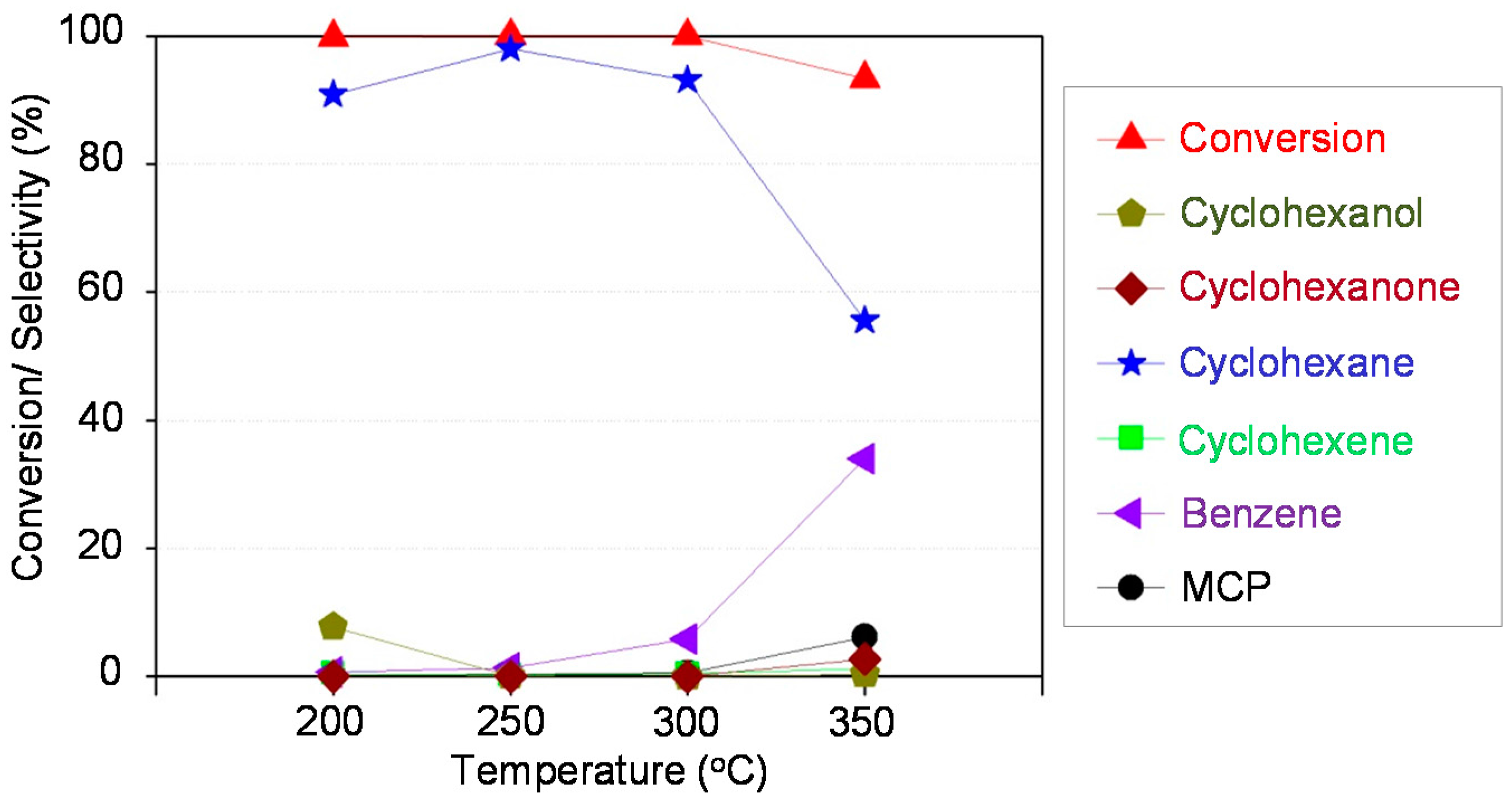
3.4. HDO of Phenol in a Continuous Fixed Bed Reactor
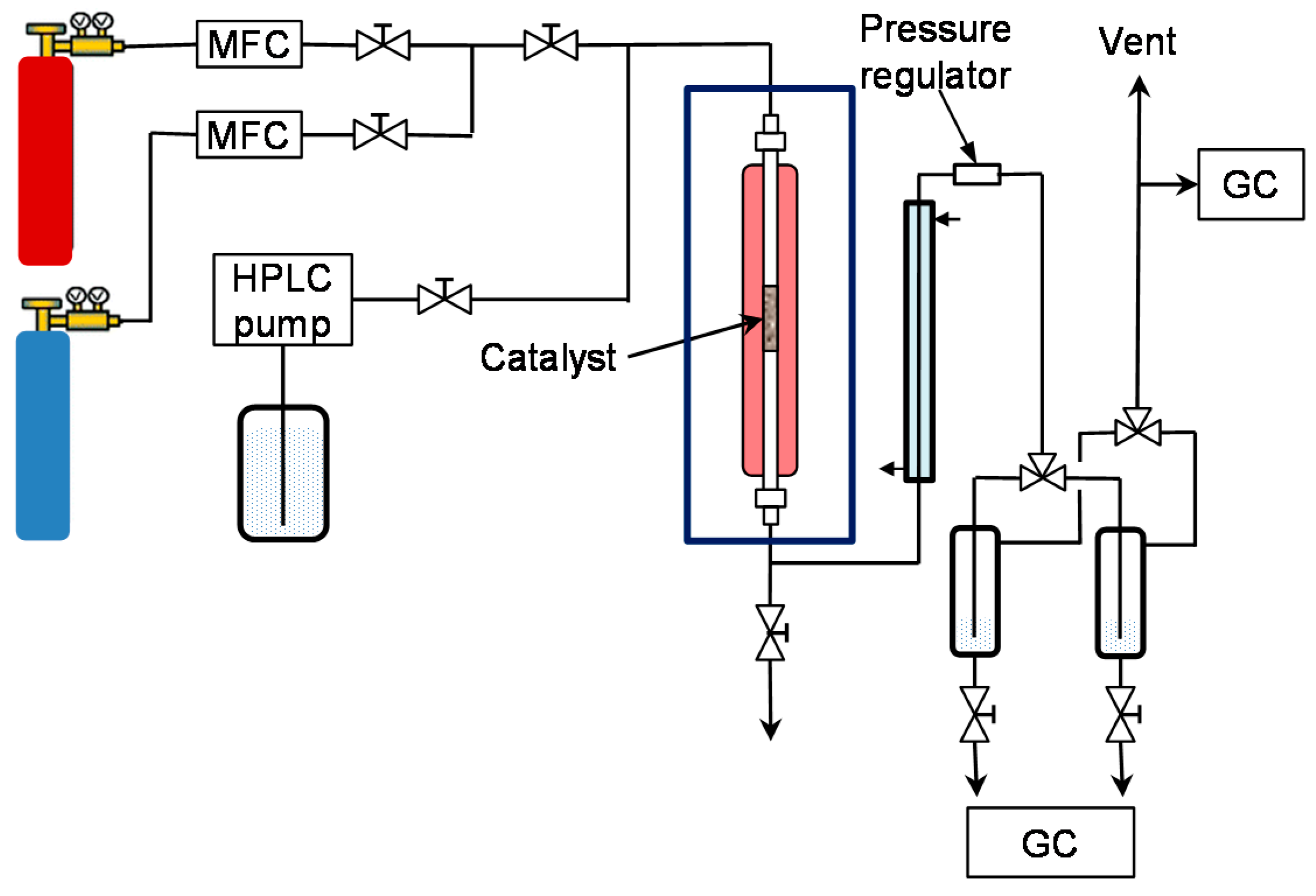
The setup consisted of a stainless steel tubular reactor (i.d. = 5 mm, l = 310 mm, V = 7 mL, material: SS316) that was heated by an electric heating jacket. Reactor temperature was controlled by using a thermocouple placed inside the heating jacket. In addition, a movable thermocouple was placed in a guiding tube (1/16”) inside the reactor to measure the temperature of the catalyst bed. The reactor was placed in a heating box (set to 150 °C) to avoid undesired downstream condensation.
Liquid feed (3 wt % phenol in water was kept constant in this study) was dosed with a flowrate of 0.5 mL/min by an HPLC pump (Gilson, Middleton, WI, USA). The declared space velocity (WHSV) refers to
x g catalyst (
x = 0.3, 0.4 and 0.5 g, respectively) and 0.9 g phenol/h metered as aqueous solution per hour. N
2 (as auxiliary gas) and H
2 were fed from commercial cylinders (Air Liquide, 99.999%) downward into the reactor. The outlet of the reactor was connected to a countercurrent water cooler. Backpressure regulation (Swagelok, Solon, OH, USA) was used to release the reaction pressure to ambient and then the gas and liquid were separated in a system of two parallel separators (switchable with a three-way valve) that was placed in iced water to avoid any vaporization and escape of products. The scheme of the continuous setup is given in
Figure 9.
The catalyst for continuous runs was shaped into pellets, then crushed to a particle size of 180–250 µm (0.3–0.5 g). The catalyst was diluted with silica (100–160 µm, Supelco) to get a volume of reaction zone of 2 mL that was placed between two silica layers and quartz wool plugs at each reactor end. Prior to each run, the externally pre-reduced catalyst was treated again at 450 °C in H2 flow of 100 mL/min for 30 min and then cooled to RT in N2 at 50 mL/min. Then, the feed solution was pumped to the reactor up to the desired pressure, then H2 was fed into the reactor and finally the catalyst bed was heated to reaction temperature. The system was run at least for 2 h to achieve steady-state operation. Thus, the standard test program for each catalyst consisted of 3 h for stabilization and subsequently 8 h or more on-stream at the desired temperature. The liquid effluent was collected over 15 min in the separator in which toluene was preloaded as absorbent. The aqueous and toluene phases were analyzed by the above described GCs. Only small traces of toluene and cyclohexane (together with non-converted H2) were found in the gas phase, thus only the liquid phase was taken into account for calculation of conversion and selectivity.
Conversion and selectivity (product distribution) were calculated from the number of carbon moles in inlet and outlet stream with the following equations:
The carbon balances of all these experiments were >93%.
4. Conclusions
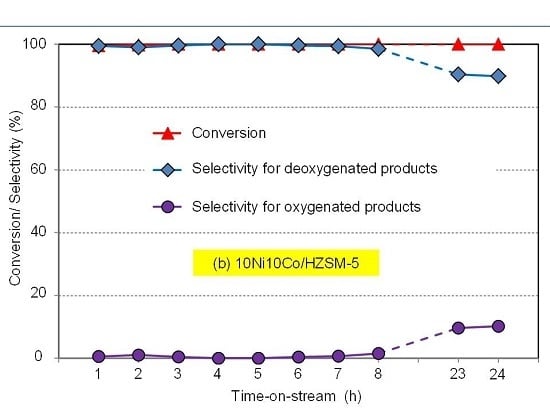
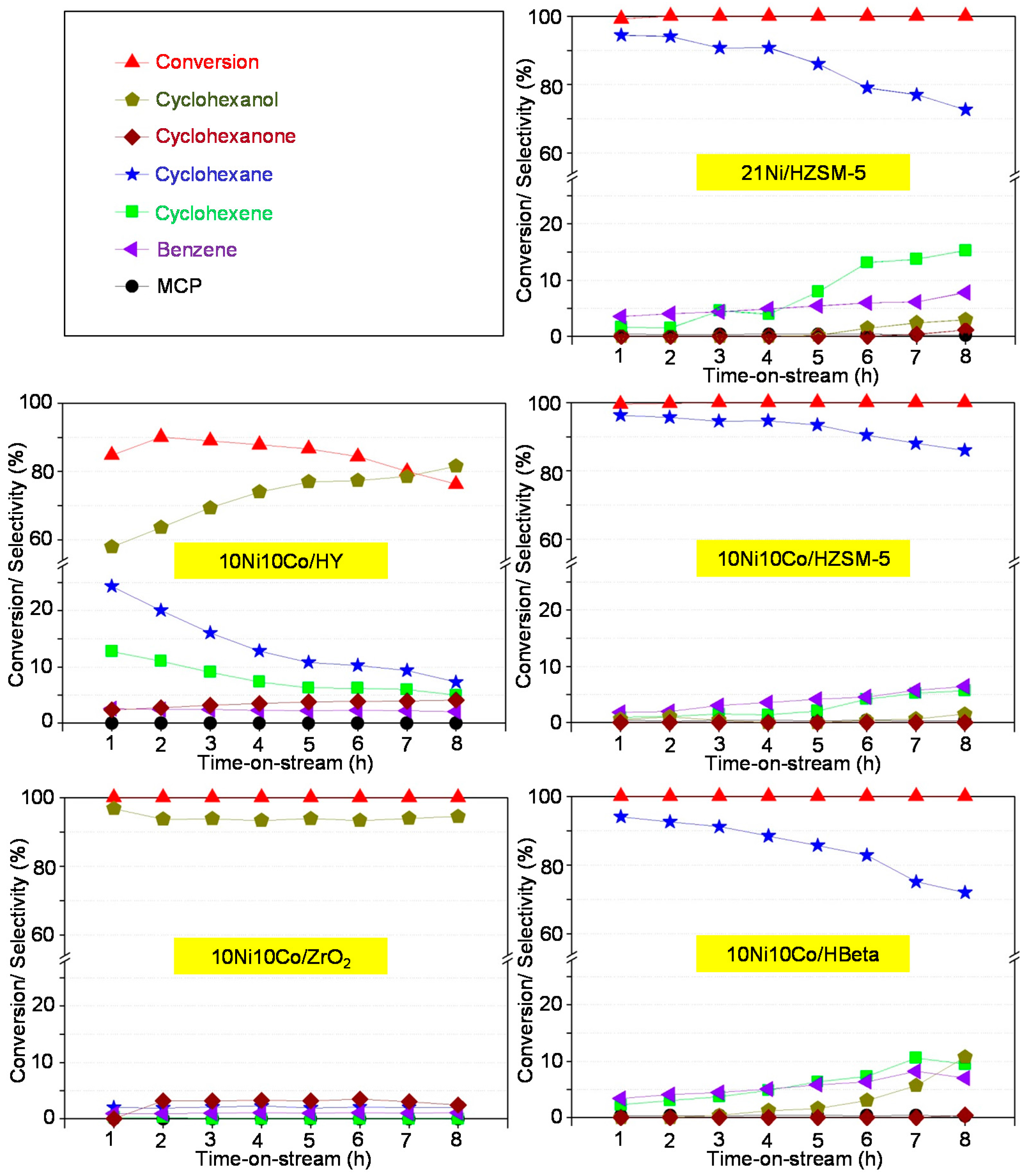
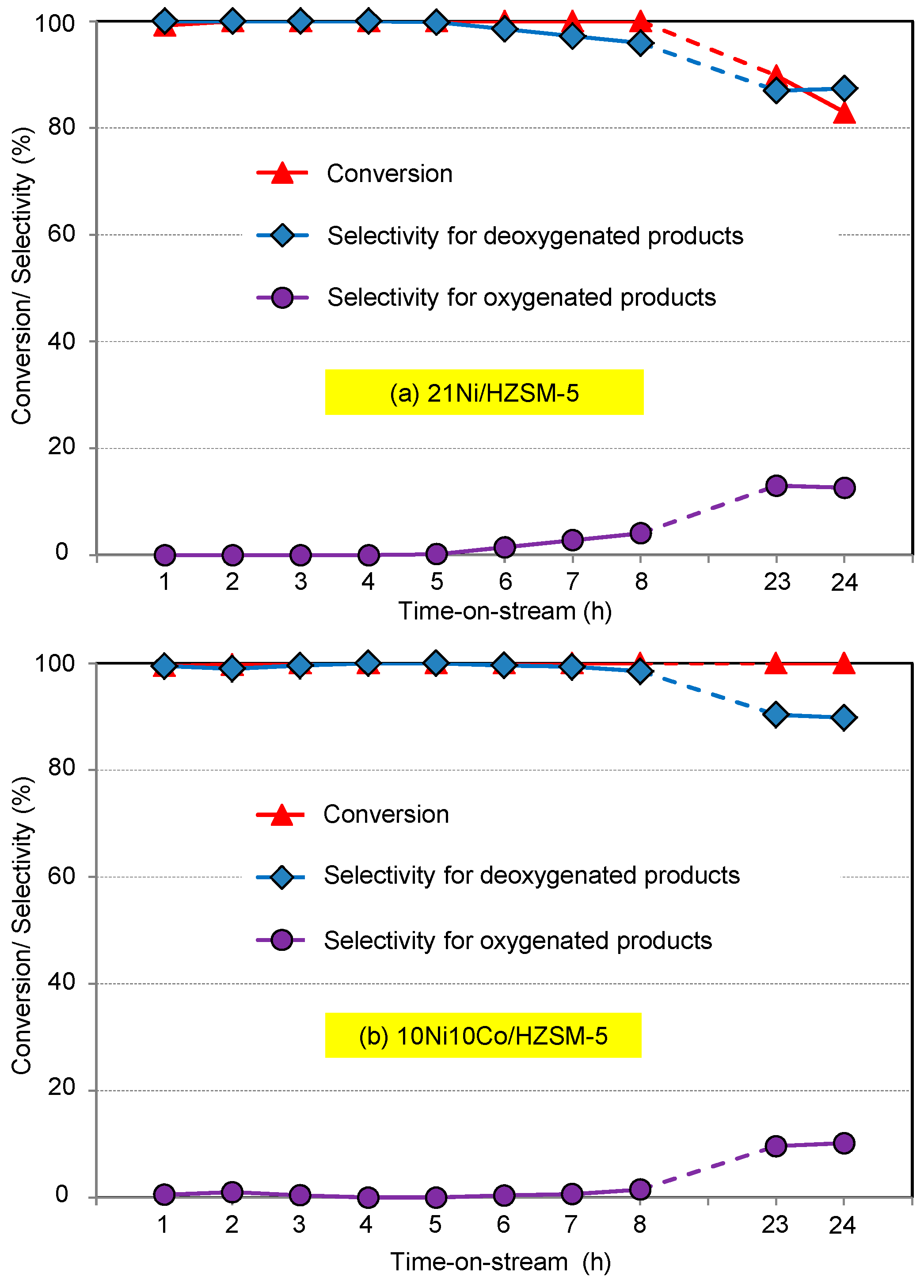
Aqueous phase hydrodeoxygenation (HDO) of phenol with Ni-Co-based catalysts in both batch and continuous flow mode reveals the best performance using HZSM-5 as support compared to HBeta, HY and ZrO2. This is connected first to its high acid site density and strength, because such sites promote oxygen-removing steps like dehydration. The second crucial feature is the high hydrothermal stability. Therefore, the 10Ni10Co/HZSM-5 catalyst sample is the most stable catalyst and revealed the best catalytic performance. However, some deactivation with respect to particle growth, surface area decrease and acidity drop may be seen.
Ni-Co alloyed particles, as evidenced by TEM, seem to be better suited than the corresponding monometallic nickel catalysts, because Co stabilizes small Ni species, even in grown particles. The strong temperature dependence of conversion and particularly selectivity is based on two major effects: First, hydrogenation on Ni-Co metal sites starts at quite low temperatures around 200 °C, whereas the activation energy for C-O cleavage is higher and thus dehydration steps need temperatures above 225 °C. Second, at temperatures above 250 °C, undesired dehydrogenation towards benzene and isomerization come to the foreground, and chemical equilibrium rules the product distribution.
The initial catalyst screening with a batch autoclave was useful for catalyst discrimination and evaluating the effect of some reaction parameters (e.g., H2 pressure, temperature). However, after transferring the reaction to continuous mode, some crucial features of the catalyst performance—specifically related to deactivation such as surface blocking, metal particle agglomeration, and loss of active metal and acidity—could be observed. Interestingly, some of these effects seem to affect the deoxygenation selectivity rather than the phenol conversion.


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}