1. Introduction
Over the past few decades, the need for economically and environmentally compatible chemical processes has increased exponentially, along with global environmental concerns. As a result, the concept of green chemistry has emerged, which endorses the use of mild chemical reaction conditions, such as the use of non-polluting reactants, visible light as main energy source, and water as solvent, which, in addition, serves as O atom and electron sources [
1]. In this context, oxidation reactions such as the conversion of alkanes into alcohols—one of first steps in the synthesis of products issued from the petrochemical industry—would benefit from significant improvements. Current chemical processes take place in wasteful organic solvents at high temperature, in addition to lacking in efficiency and/or selectivity [
2,
3]. Although promising metal catalysts have been developed and show encouraging implementation potential [
4], several aspects such as efficacy, regio- and stereoselectivity, compound stability, or environmental toxicity remain to be addressed. In addition, these reactions often resort to oxidant reactants that might have a direct or indirect impact on the environment [
2,
3].
Natural enzymes provide us with astonishing examples of extremely efficient, selective, and stable catalysts. Oxidases constitute a widely investigated class of enzymes, most of which contain one or several metal ions at the heart of their redox center. For example, a non-heme iron enzyme such as phenylalanine hydroxylase (PAH) [
5] catalyzes the hydroxylation of the aromatic side-chain of phenylalanine to generate tyrosine, whereas di-iron enzymes such as methane monooxygenases (MMO) allow the transformation of methane into methanol [
6,
7]. Cytochrome P450 enzymes constitute a large family of hemoproteins that can perform the oxidation of various substrates at high catalytic rates using molecular oxygen as the sole source of oxidant through a reductive activation mechanism [
8]. During the catalytic cycle, two electrons are provided by redox mediators, Nicotinamide Adenine Dinucleotide (NADH), Nicotinamide Adenine Dinucleotide phosphate (NADPH), etc., allowing the formation of a very reactive high-valent iron-oxo intermediate responsible for substrate oxidation.
Over the years, P450 enzymes have been largely studied and utilized for catalytic purposes. However, electron delivery to the iron center remains the principal hurdle to high catalytic efficiency. On the one hand, natural redox cofactors are expensive, requiring the need to recycle them or to avoid their use. On the other hand, the electrons are not directly transferred to the heme iron, but instead are delivered through a P450 reductase that involves two flavin redox cofactors, i.e., Flavin Adenine Dinucleotide (FAD) and Flavin Mononucleotide (FMN). Two electrons are initially transferred from NADPH to FAD to yield FADH
2, which then transfers them to FMN. The obtained FMNH
2, then delivers sequentially, one by one, these two electrons to the iron atom at precise steps of the catalytic cycle [
9]. Such a complex process, which is also driven by protein conformational changes, is then far from reproducible, and numerous approaches have been developed to mimic this catalytic reaction. For instance, approaches like the use of alternative oxygen atom donors (PhIO, ROOH, H
2O
2, KHSO
5, etc.) [
10], chemical or electrochemical reductions [
11], and, more recently, reductase proteins that were substituted with ruthenium-based photosensitizers capable of gathering electrons upon light irradiation and transferring them to cytochrome (P450) enzymes [
12,
13].
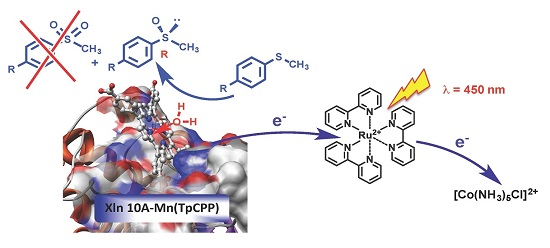
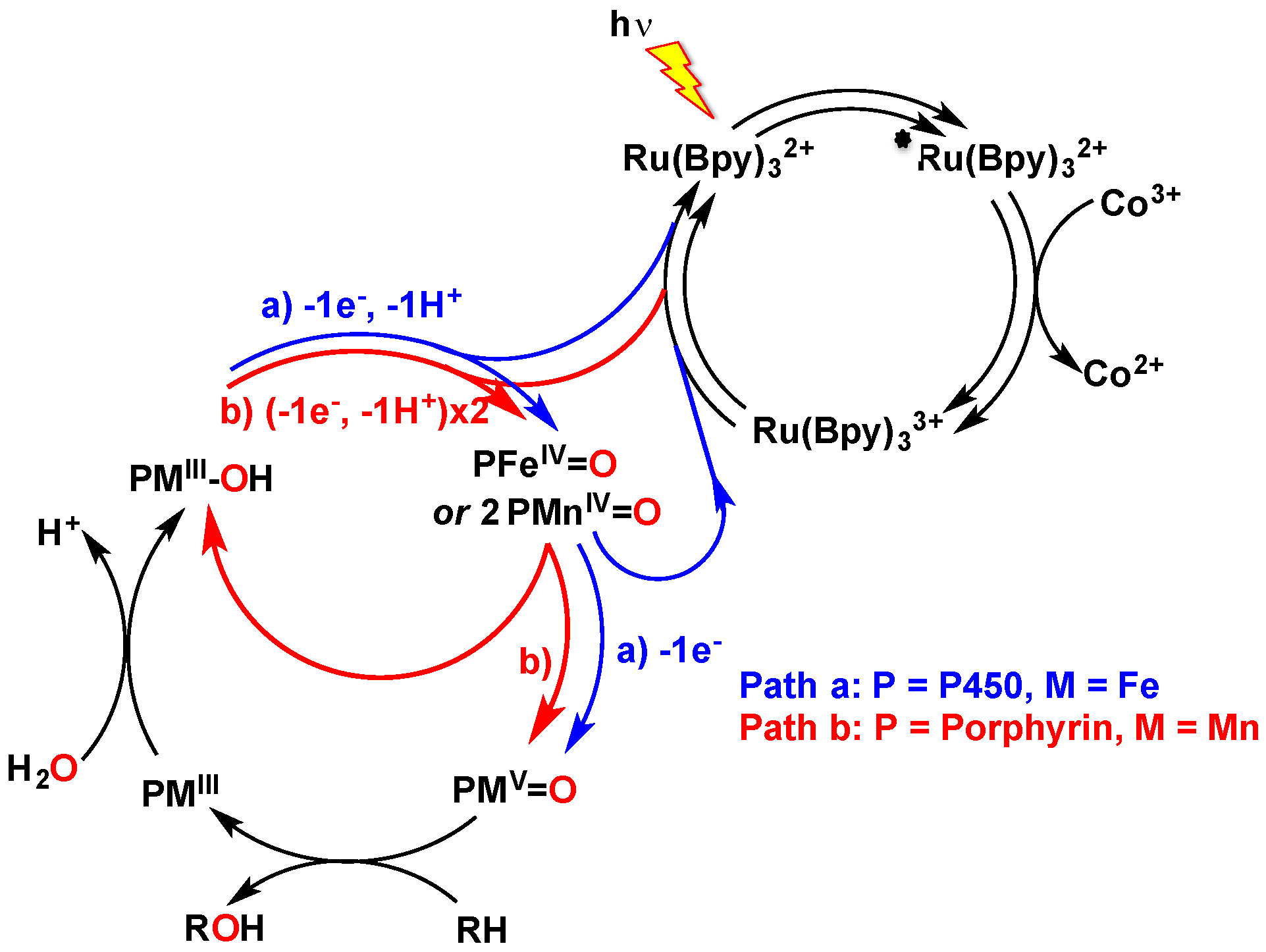
This latter field of research is appealing because it utilizes visible light as the sole energy input in the chemical reaction, which can be used either under reductive or oxidative experimental conditions. The former pathway relies on the photo-reduction by diethyldithiocarbamate (DTC) of a Ru
II-chromophore that is covalently attached to the apo-P450, which then catalyzes the reductive activation of oxygen leading, in the presence of H
+, to a high-valent P450Fe
V-oxo species that performs the oxidation of fatty acids [
12,
13]. Conversely, the oxidative pathway relies on the ability to quench the excited state of a [Ru
II(bpy)
3]
2+ chromophore with an irreversible electron acceptor such as [Co
III(NH
3)
5Cl]
2+, in order to form the highly oxidative [Ru
III(bpy)
3]
3+, which then catalyzes the two-electron oxidation of an iron-bound water molecule with formation of the high-valent iron-oxo species (
Scheme 1, Path a) [
14].
The oxidative pathway was also used with biomimetic systems involving manganese porphyrins as catalysts and water as an oxygen source, which were reported originally by Calvin’s group [
15], and more recently by Fukuzumi et al. [
16]. In these systems, the authors showed that the photogenerated highly oxidative [Ru
III(bpy)
3]
3+ can oxidize the Mn
III-OH complex into an Mn
IV-oxo species. In this case, the Mn
V-oxo species was not directly formed by a second one-electron oxidation, but rather arose from the disproportionation of the Mn
IV-oxo species into Mn
III and Mn
V=O (
Scheme 1, Path b) [
16]. Such a system could catalyze the oxidation of water-soluble substrates, including alkenes, alkanes, and sulfides [
16]. Previous work from our group also showed that the moderately enantioselective light-induced oxidation of thioanisole by water as the oxygen atom source could occur in the presence of a system that associated a protein that induced chirality, BSA (Bovine Serum Albumin), a manganese corrole as oxygen atom transfer catalyst, and [Ru
II(bpy)
3]
2+ as photosensitisizer [
17].
In parallel, other work also allowed us to show the strong affinity of manganese sodium tetra-
para-carboxylatophenyl-porphyrin (Mn-TCPP) for xylanase 10A (Xln10A) from
Streptomyces lividans, a glycoside hydrolase that catalyzes the hydrolysis of (1-4)-β-
d-xylosidic bonds in xylan biopolymers to release xylose [
18]. Xylanase 10A is an abundant and resistant protein. It is easily purified and its sequence and three-dimensional structure are known, which makes it a good candidate as a protein scaffold for the production of biohybrid catalysts resulting from the combination of a natural protein and an artificial metal complex. We expected this combination of enzymatic and molecular catalysis to yield hybrid catalysts showing improved efficacy, selectivity, and stability. Studies on the reactivity of the Mn-TCPP-Xln10A hybrid showed that it possessed good mono-oxygenase activity for the epoxidation of styrene-type compounds, in addition to yielding important enantiomeric excesses, in particular for the epoxidation of
p-methoxystyrene by oxone. This selectivity was further illustrated by molecular modeling studies that showed the existence of hydrogen bonds between the methoxy substituent of the substrate and a tyrosine residue of the protein [
18].
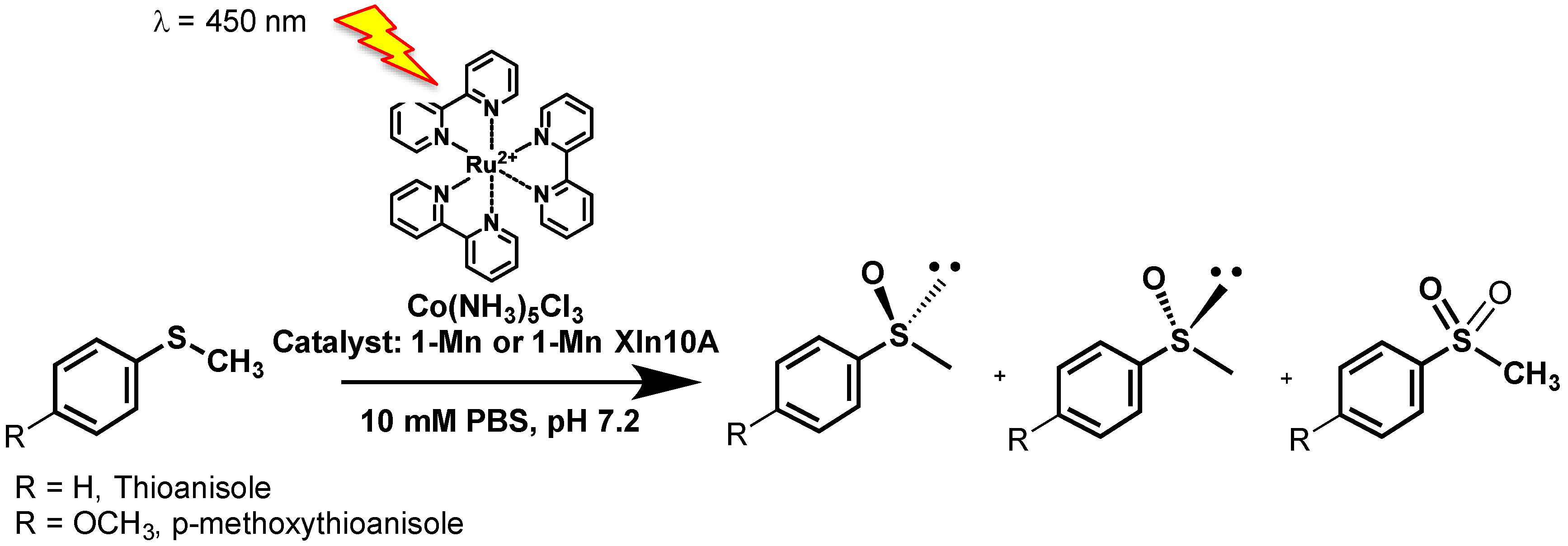
We thus decided to use our Mn-TCPP-Xln10A biohybrid as a catalyst for the selective photo-induced oxidation of organic substrates in the presence of [RuII(bpy)3]2+ as a photosensitizer and [CoIII(NH3)5Cl]2+ as a sacrificial oxidant, with water as an oxygen atom source. We report that both Mn-TCPP and its complex with xylanase 10A can catalyze the photoinduced sulfoxidation of thioanisole and 4-methoxythioanisole with slight enantiomeric excesses, using water as an oxygen atom source.
3. Discussion
The aforementioned results show that both the manganese(III)-meso-tetrakis-
para-carboxyphenylporphyrin
1-Mn and its complex with xylanase 10A, the
1-Mn-Xln 10A hemozyme, catalyze the photo-assisted oxidation of thioanisole derivatives with formation of the sulfoxide as major product (
Scheme 2,
Table 1). With both thioanisole and
p-methoxy-thioanisole, the reaction is highly chemoselective and leads to the corresponding sulfoxide as a major product with greater yields with
1-Mn alone as catalyst (125 ± 5 TON and 100 ± 4 TON, respectively) than with
1-Mn-Xln10A as catalyst (25 ± 2 TON and 10 ± 1 TON, respectively). This could indicate that the insertion of
1-Mn into the hydrophobic pocket of Xln10A [
18] hinders the electron transfer from the manganese center to the photo-activated ruthenium complex. The results observed in the presence of
1-Mn-Xln10A are, however, comparable to those obtained under the same conditions for the oxidation of thioanisole catalyzed by an Mn-corrole complex and Mn-corrole-BSA artificial metalloenzyme, which respectively lead to the chemoselective formation of the corresponding sulfoxide with 32 ± 3 and 21 ± 7 TON (
Table 1) [
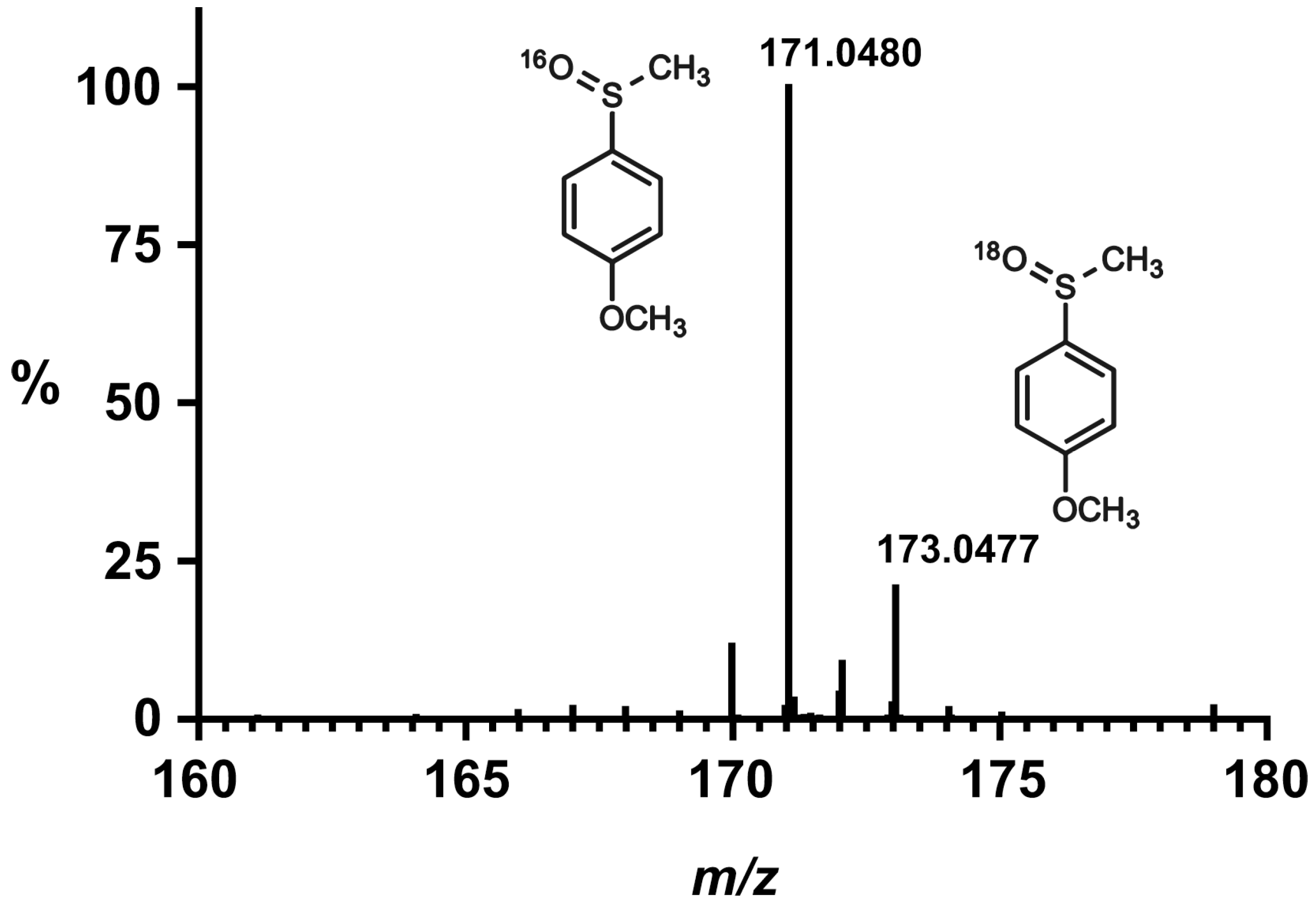
17]. In addition, as studies in the presence of H
218O show the incorporation of
18O in the obtained sulfoxide, and no reaction occurs in the absence of the [Ru
II(bpy)
3]
2+ photoactivator, it is reasonable to propose that the reaction follows a mechanism similar to that depicted in
Scheme 1. Water would thus act as the oxygen source, leading to an Mn
III-OH that would be oxidized into an Mn
IV-oxo species by the photogenerated highly oxidative [Ru
III(bpy)
3]
3+ species. For the
1-Mn homogenous catalyst, the final Mn
V-oxo oxidizing species would likely arise from the disproportionation of the Mn
IV-oxo species into Mn
III and Mn
V=O (
Scheme 1, Path b) as reported for other manganese porphyrin complexes [
15,
16] whereas it would be formed upon oxidation of the Mn
IV-oxo by the photogenerated [Ru
III(bpy)
3]
3+ in the
1-Mn-Xln10A system (
Scheme 1, pathway a) [
14] because disproportionation between complexes embedded in Xln10A is unlikely.
Since the oxidation of 2-(CH
3-thio)ethanol catalyzed by the Mn(III)-5,10,15,20-tetrakis (2,4,6-trimethyl-3-sulfonatophenyl)porphyrin (Mn-TMPS) under the same conditions was also reported to lead to the formation of the corresponding sulfoxide with 30 TON (
Table 1) [
16],
1-Mn-Xln10A is an equally interesting artificial metalloenzyme for the chemoselective photoinduced oxidation of sulfides in water. It is noteworthy, however, that these results illustrate lower yields relative to those obtained in the presence of supramolecular assemblies in which a ruthenium-tris-bipyridyl photoactivable moiety (Ru
Phot) is covalently attached to another Ru(tpy)(bpy)(H
2O) catalytic moiety (Ru
cat) that photoinduces the sulfoxidation of thioanisole derivatives, such as thioanisole,
p-Methoxy-, and
p-bromo-thioanisole using water as an oxygen source, in the presence of [Co
III(NH
3)
5Cl]
2+ as sacrificial reductant with 200–745 TON (
Table 1) [
22,
23].
As expected, no enantioselectivity was observed for the oxidation of thioanisole and
p-methoxy thioanisole with that of
1-Mn alone as catalyst (
Table 1). Surprisingly, no enantioselectivity could be observed for the oxidation of thioanisole in the presence of Xln10A, which contrasted with the 12%–16% ee observed for the same reaction in the presence of Mn-Corrole-BSA as catalyst [
17] (
Table 1). The oxidation of
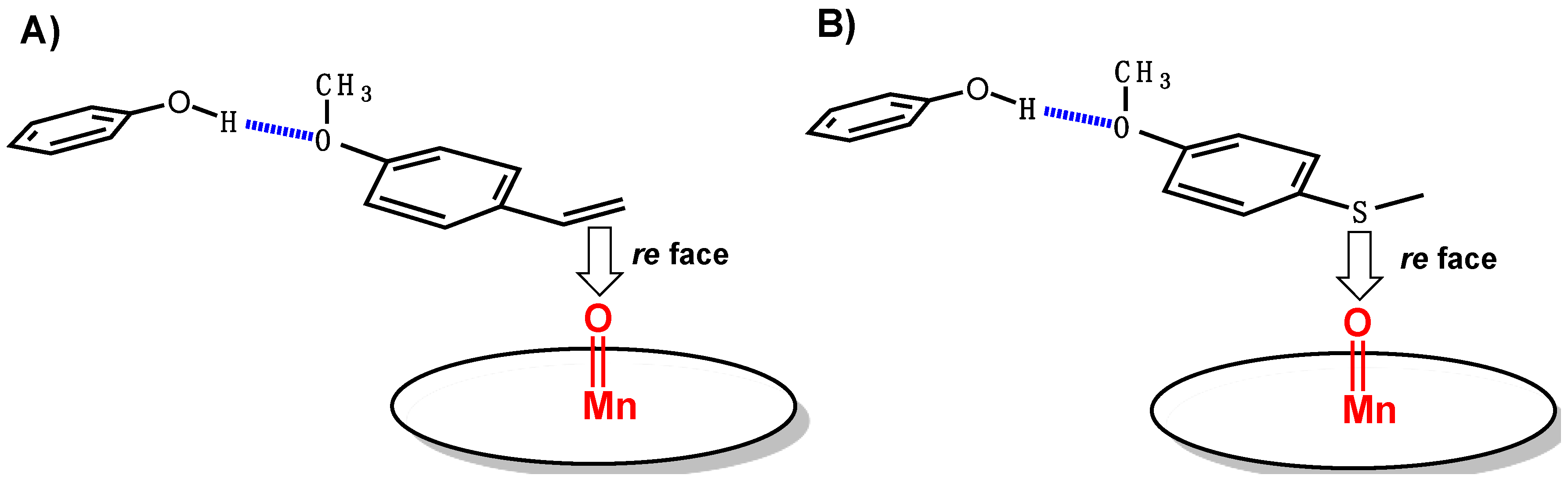
p-methoxy-thioanisole, which was chosen in the hope of a selective pro-R positioning of the sulfide similar to that previously reported for
p-methoxy-styrene [
18] (
Scheme 3) was only slightly enantioselective, though it led to the R sulfoxide as the major enantiomer, as was expected. However, the ee value, 7% ± 1%, was far from that obtained for the epoxidation of
p-methoxy-styrene by oxone catalyzed by
1-Mn-Xln10A, (80% ee in favor of the R epoxide). This could be due to a denaturation of the protein during catalysis. Indeed, when the Xln10A/
1-Mn ratio increases, the ee remains constant, whereas the TON decreases, in agreement with a radical polymerization initiated by its direct reaction with the [Ru
II(bpy)
3]
3+ intermediate.
4. Materials and Methods
4.1. Physical Measurements
ESI-HRMS was determined on a micrOTOF-Q II 10027 apparatus (Bruker Daltonics, Billerica, MA, USA), UV/Vis spectra were recorded on an UVIKON-XL spectrophotometer (BioTek Instruments, Inc., Winooski, VE, USA) fluorescence spectra were recorded on a CARY Eclipse fluorometer (Agilent Technologies, Santa Clara, CA, USA). Gas chromatographs (Shimadzu Scientific Instruments, Inc., Columbia, MA, USA) were obtained with a Shimazu GC 2010 plus apparatus equipped with a Zebron ZB-SemiVolatiles Gas Chromatography (GC) column (30 m × 0.25 mm × 0.25 µm). HPLC (Agilent Technologies, Santa Clara, CA, USA) was performed on an Agilent Infinity 1260 apparatus equipped with a chiral column (Chiralcel OD-H, Daicel Co., Osaka, Japan, 250 mm × Φ4.6 mm).
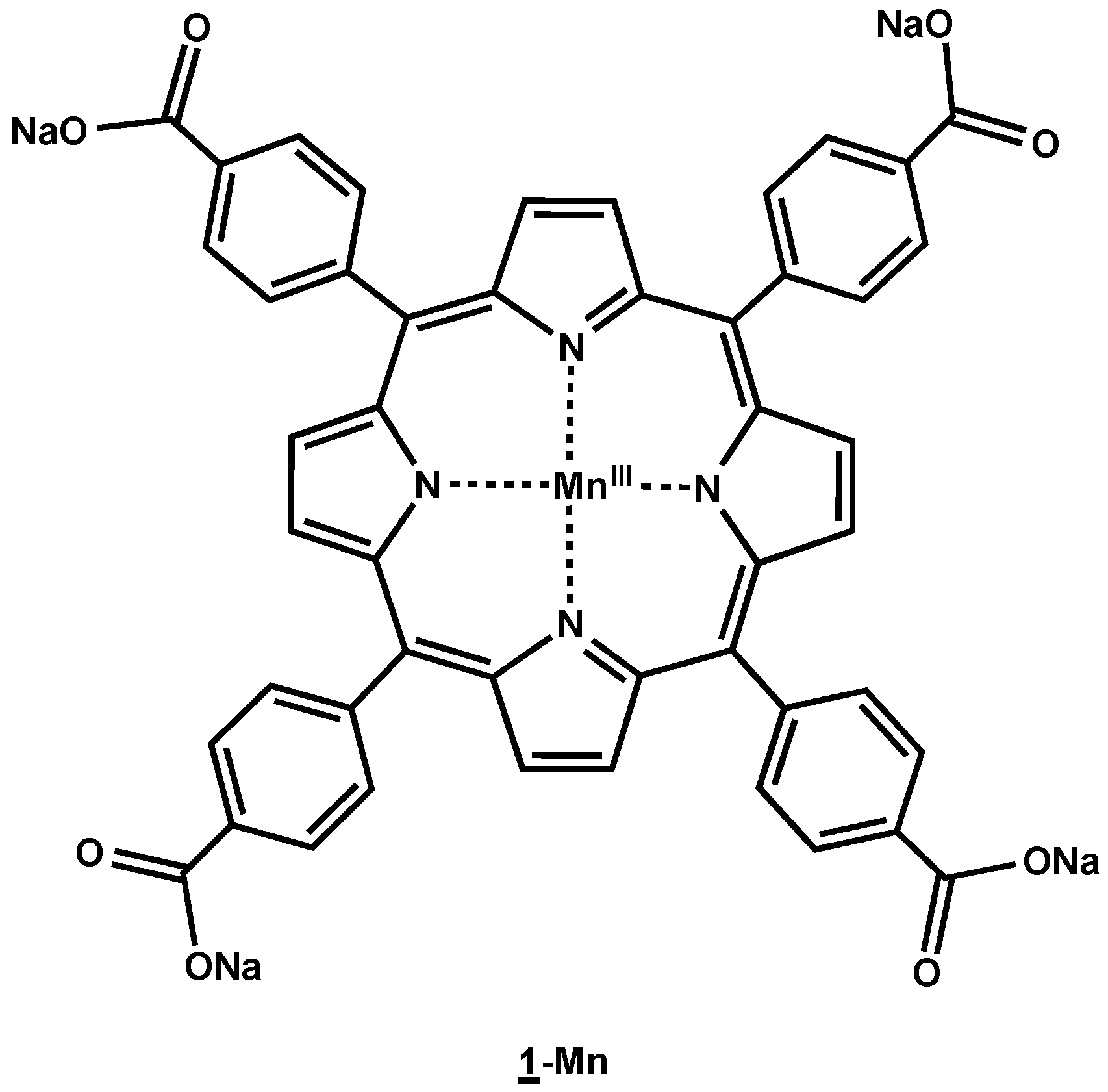
4.2. Synthesis of MnIII-meso-tetrakis(para-carboxyphenyl)porphyrin (1-Mn)
meso-Tetrakis(
para-carboxyphenyl)porphyrin (
1) was prepared in 10.5% yield by condensation of p-carboxybenzaldehyde with pyrrole in propionic acid according to the procedure of Adler et al. [
19,
20], and its molecular properties were found to be identical to those already reported [
19]:
1H-NMR (
d6-DMSO) 8.85 (8H, s), 8.38 (8H, d,
J = 8 Hz), 8.32 (8H, d,
J = 8 Hz), −2.95 (2H, s, NH); Matrix-assisted Laser Desorption-Time Of Flight (MALDI-TOF)-MS:
m/
z = 791.21 (M + H
+). The insertion of manganese was performed by treatment of
1 (2.5 mM) in AcONa buffer (pH 4.0, 50 mM) with Mn(OAc)
2, 4H
2O (11 equiv.) at 100 °C for 4 h. The progress of the reaction was monitored by UV/Vis spectroscopy. After 2 h under reflux, the Mn metalation was quantitative. The excess of manganese salt was then removed by successive purifications on P6DG exclusion gel and then on Chelex. After lyophylisation,
1-Mn was obtained as a purple solid in a quantitative yield. Its properties were found to be identical to those already reported [
18]: UV/Vis (pH 7.0, sodium phosphate buffer, 50 mM): λ
max (ε): 468 (93 × 10
3 M
−1·cm
−1), 565, 600 nm; MALDI HR-MS (ESI):
m/
z = 843.12396, calculated for C
48H
28MnN
4O
8, and
m/
z = 843.12132 (
∆m = 5.04 ppm).
4.3. Preparation and Characterization of the Artificial Metalloenzyme 1-Mn-Xln10A
Xylanase 10A was first purified from the supernatant of
S. lividans culture as reported earlier [
19]. The various
1-Mn-Xln10A hemozyme samples were then prepared by incubation for 30 min at room temperature (RT) of Xln10A in sodium phosphate buffer (50 mM, pH 7.0) with various amounts of
1-Mn.
1-Mn-Xln10A was then characterized by UV/Vis and fluorescence spectroscopy studies as already reported [
18].
4.4. UV/Vis Spectroscopy Experiments
1-Mn (5 μM) was incubated with Xln10A (1.5 equiv.) in 50 mM sodium phosphate buffer (pH 7.0) at room temperature for 30 min, and the UV/Vis spectrum was recorded between 280 nm and 700 nm. The spectrum obtained was found identical to that already reported [
18]: UV/Vis (pH 7.0, sodium phosphate buffer, 50 mM): λ
max (ε): 468 (92.3 × 10
3 M
−1·cm
−1), 565,600 nm.
4.5. Determination of the KD Values for 1-Mn-Xln10A Complexes
The KD value for 1-Mn-Xln10A complexes as well as the 1-Mn/Xln10A stoichiometry were determined as follows: the fluorescence spectrum of Xln10A, 5 μM in 50 mM sodium phosphate buffer, pH 7.2, at 25 °C was first recorded between 300 and 400 nm after excitation of the sample at 290 nm. The quenching of fluorescence was then followed by progressive addition of a 500 μM solution of 1-Mn in 50 mM phosphate buffer pH 7.0. A double reciprocal plot of the residual fluorescence intensity at 340 nm as a function of the 1-Mn final concentration afforded a KD value of 1.5 μM for the 1-Mn-Xln10A complex and a 1/1 1-Mn/Xln10A stoichiometry.
4.6. Photoassisted Oxidation of Sulfides
For a typical photo-oxidation reaction, 150 µM of pure xylanase 10A, 37.5 µM porphyrin, 130 µL of 3.04 mM [RuII(bpy)3]2+ in phosphate buffer saline PBS, and either 11 µL thioanisole or 30 µL 4-methoxy thioanisole were added to a 800 µL degassed 50 mM phosphate buffer solution (pH 7.0) containing 14 mM [CoIII(NH3)5Cl]2+. The solution was degassed by three freeze/thaw cycles, and the samples were illuminated with a 450 nm diode and stirred for 10 minutes at RT. The final concentrations of each component were 12 mM [CoIII(NH3)5Cl]2+ (390 eq.), 130 µM xylanase 10A (4.2 eq.), 32.5 µM Mn-porphyrin (1 eq.), 0.42 mM [RuII(bpy)3]2+ (14 eq.), and 84 mM 4-methoxy thioanisole (2700 eq.), or 99 mM thioanisole (3200 eq.). Control experiments without protein were performed maintaining the above concentrations. Control experiments without chromophore were performed by substituting the [RuII(bpy)3]2+ solution by the equivalent PBS solution.
4.7. Extraction and Analysis of the Oxidation Products
After reaction, the content of the vial was eluted through a short (3 cm) silica plug to which 100 µL 10
−2 M acetophenone solution acting as internal standard had been previously added. The column was washed with 2 mL ethyl acetate in order to elute all the components. The aqueous phase of the resulting filtrate was removed and the organic phase was dried using NaSO
4 prior to GC analysis. For detection of thioanisole and its oxidation products, the applied temperature gradient was as follows: from 100 °C to 130 °C at 5 °C/min. and then 130 °C to 300 °C at 50 °C/min. which was further held constant for 3 min. The injector and flame injector detection (FID) temperature were set at 300 °C. Elution retention times were (min.): acetophenone (4.03), thioanisole (4.32), thioanisole sulfoxide (7.56), and thioanisole sulfone (8.04). To study the enantiomeric excess of the obtained sulfoxide, the solvent was evaporated and the residue was re-dissolved in the minimum amount of isopropanol prior to HPLC analysis. The enantiomeric excesses were then determined by HPLC analysis using a chiral column (Chiralcel OD-H, Daicel Co., IlKirch, France, 250 mm × Φ4.6 mm). For the oxidation of thioanisole, samples were eluted with hexane/isopropanol (95:5,
v/
v, 1.0 mL/min.), and, for the oxidation of
p-methoxy-thioanisole, elution was done with hexane/isopropanol (97:3,
v/
v, 1.2 mL/min). All products were detected at 254 nm. Authentic samples of each sulfoxide enantiomer were prepared by the method previously described by Li et al. [
24], and their retention time under the above described conditions are, respectively, 17 min and 21 min. for the
R and
S thioanisole sulfoxides, and 32 min and 36 min. for the
R and
S p-methoxy-thioanisole sulfoxides.



 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}