
P-Stereogenic Phosphines for the Stabilisation of Metal Nanoparticles. A Surface State Study

 , ,
, , 
Abstract
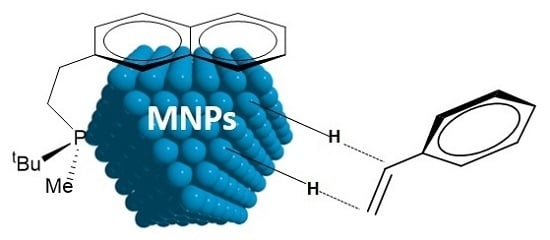
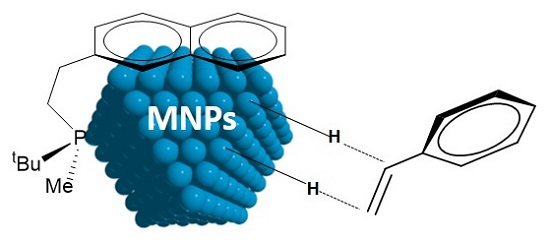
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
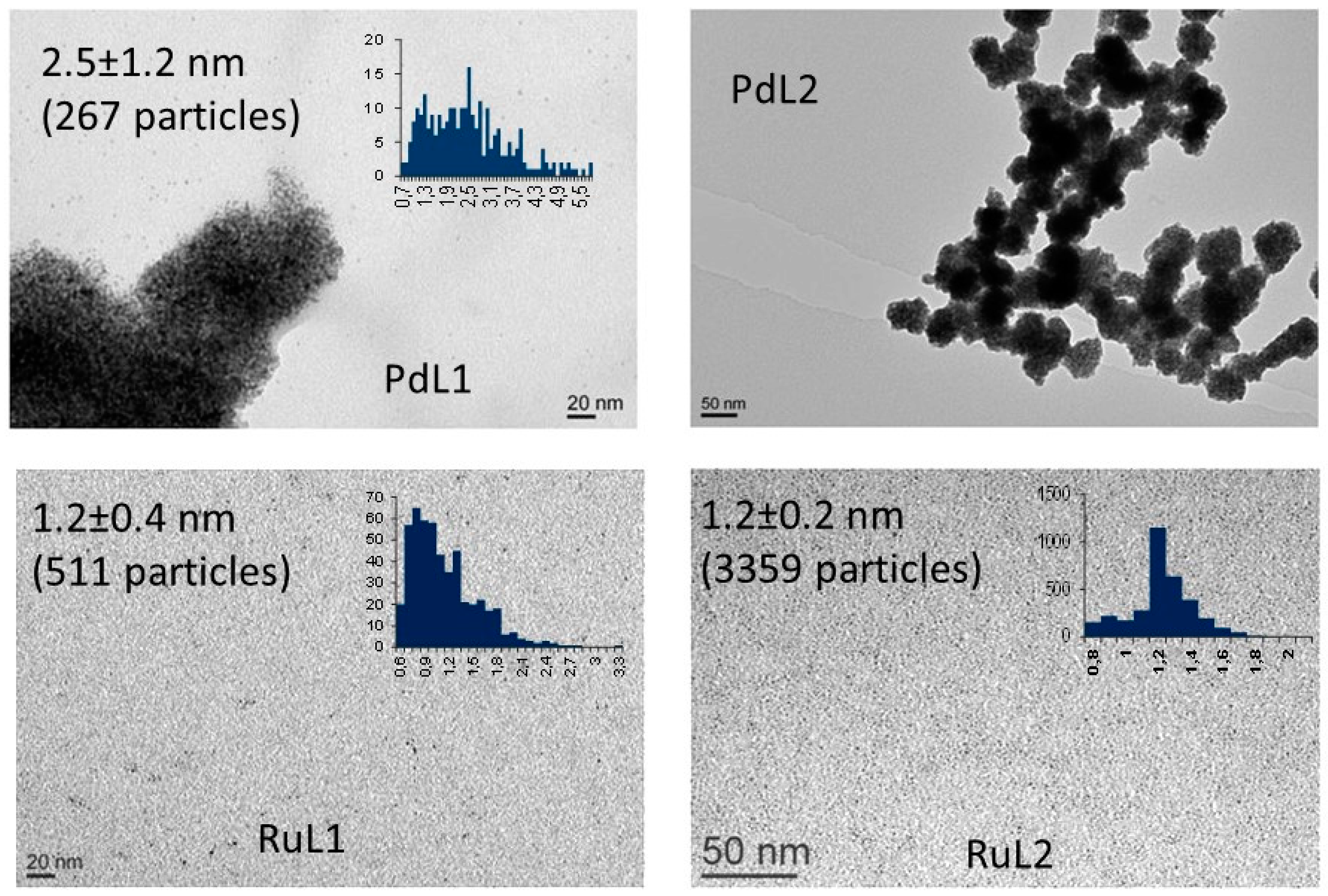
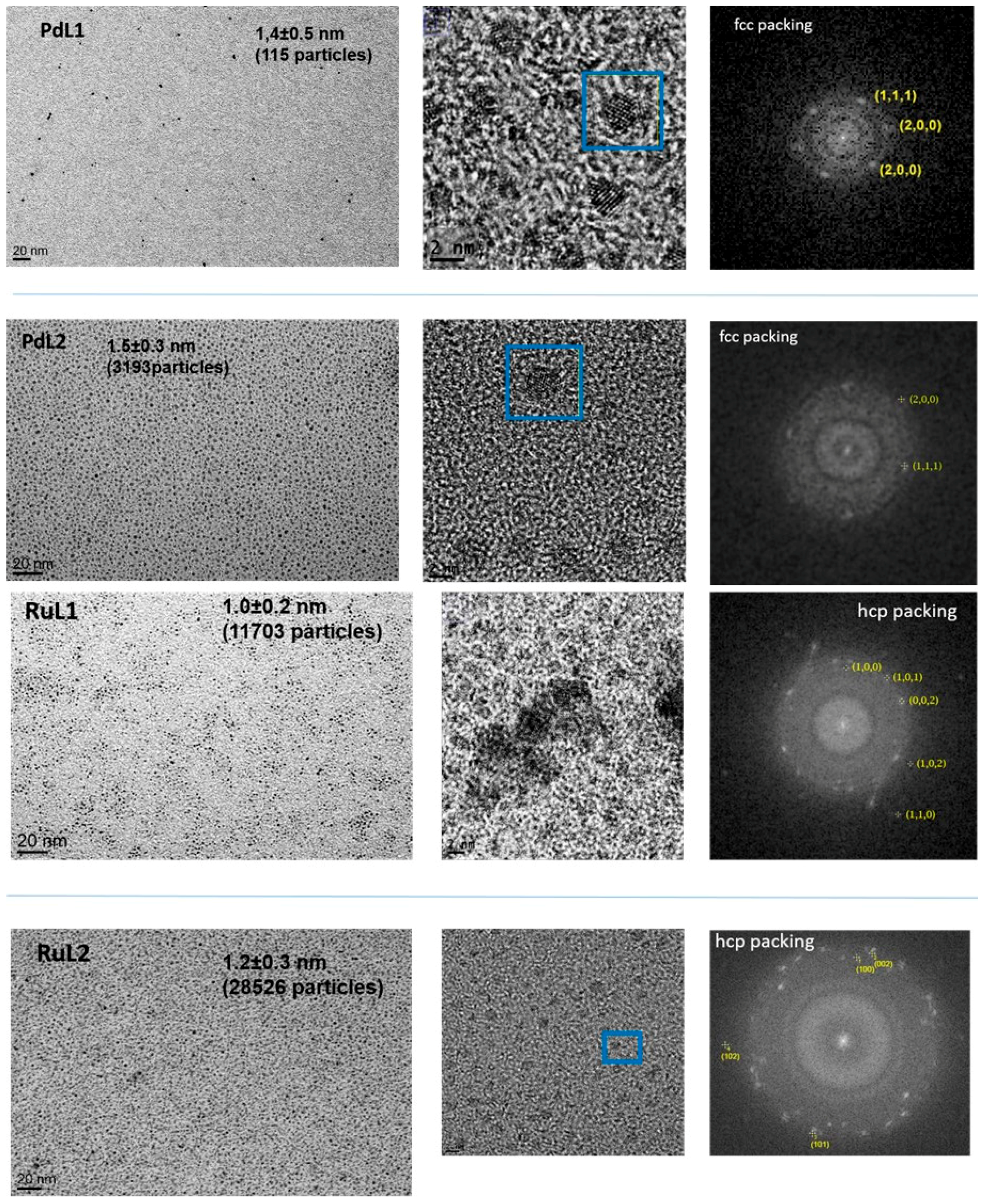
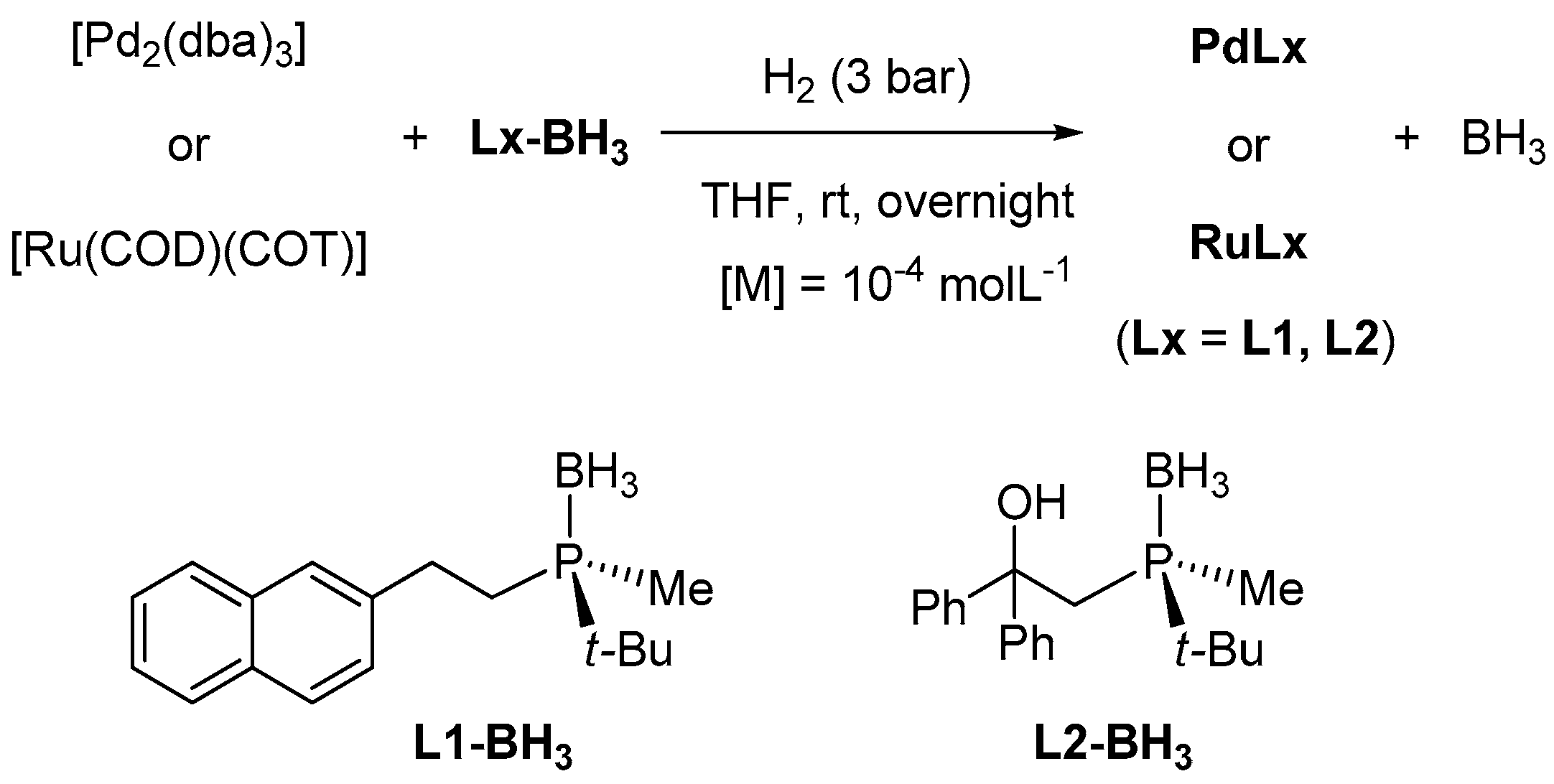
2.1. Synthesis and Characterisation of PdNPs and RuNPs
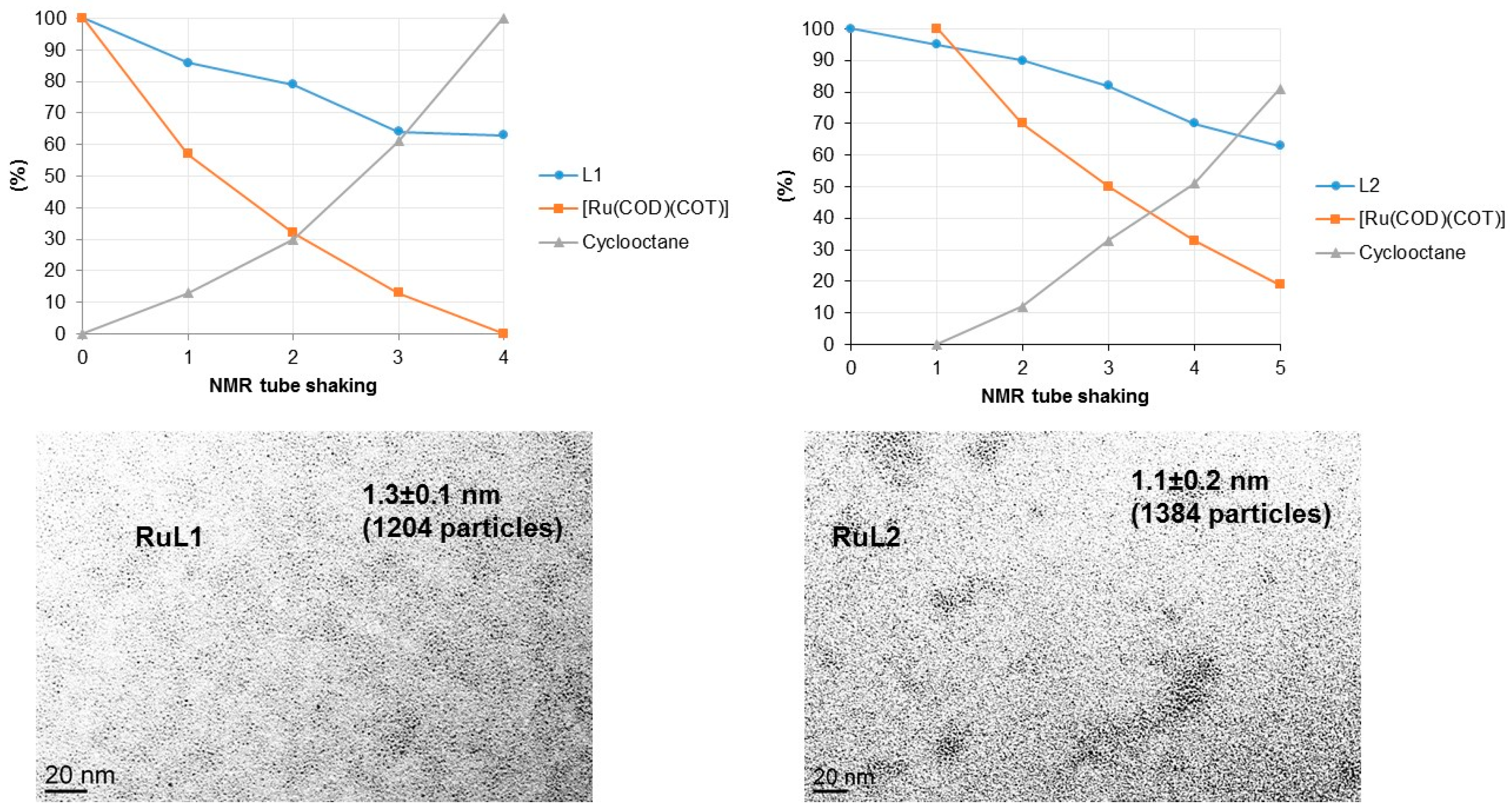
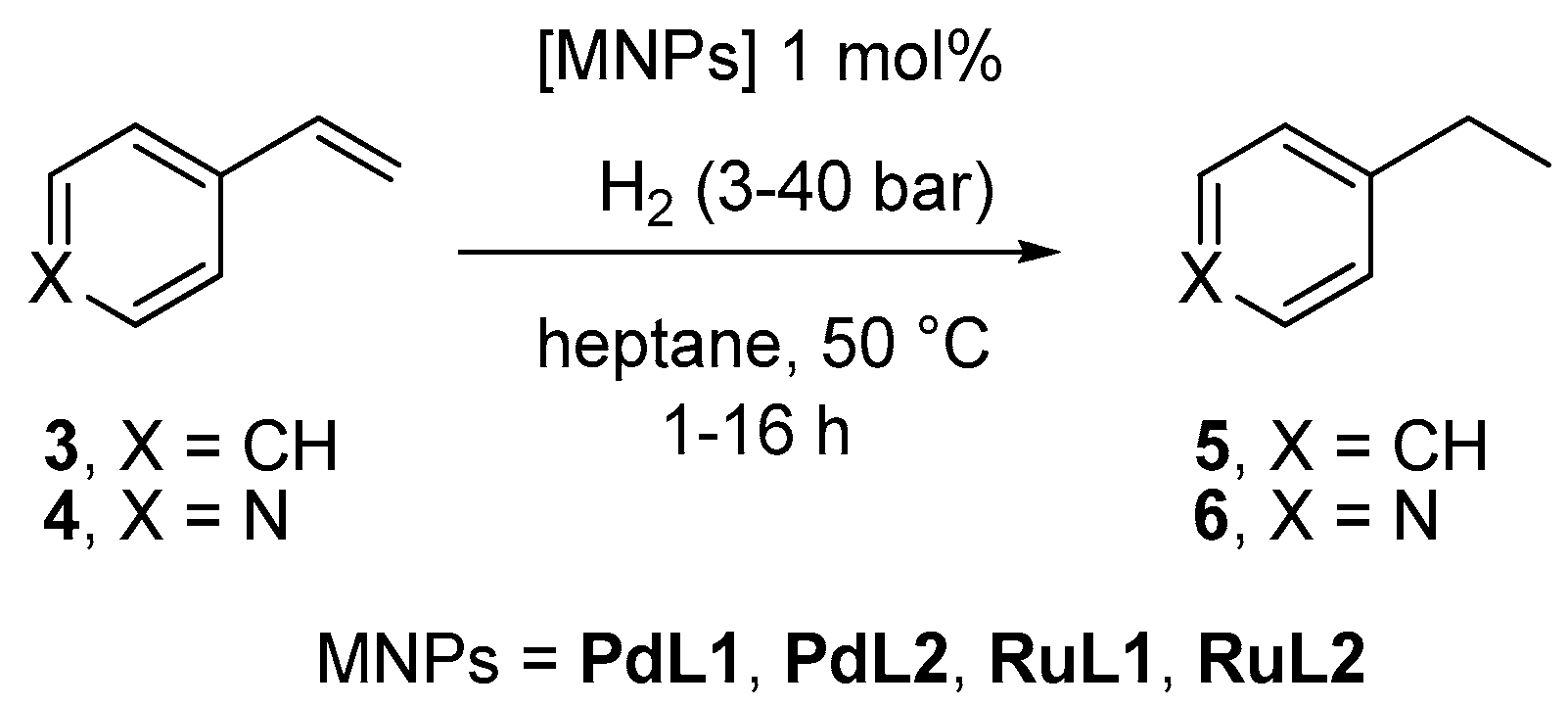
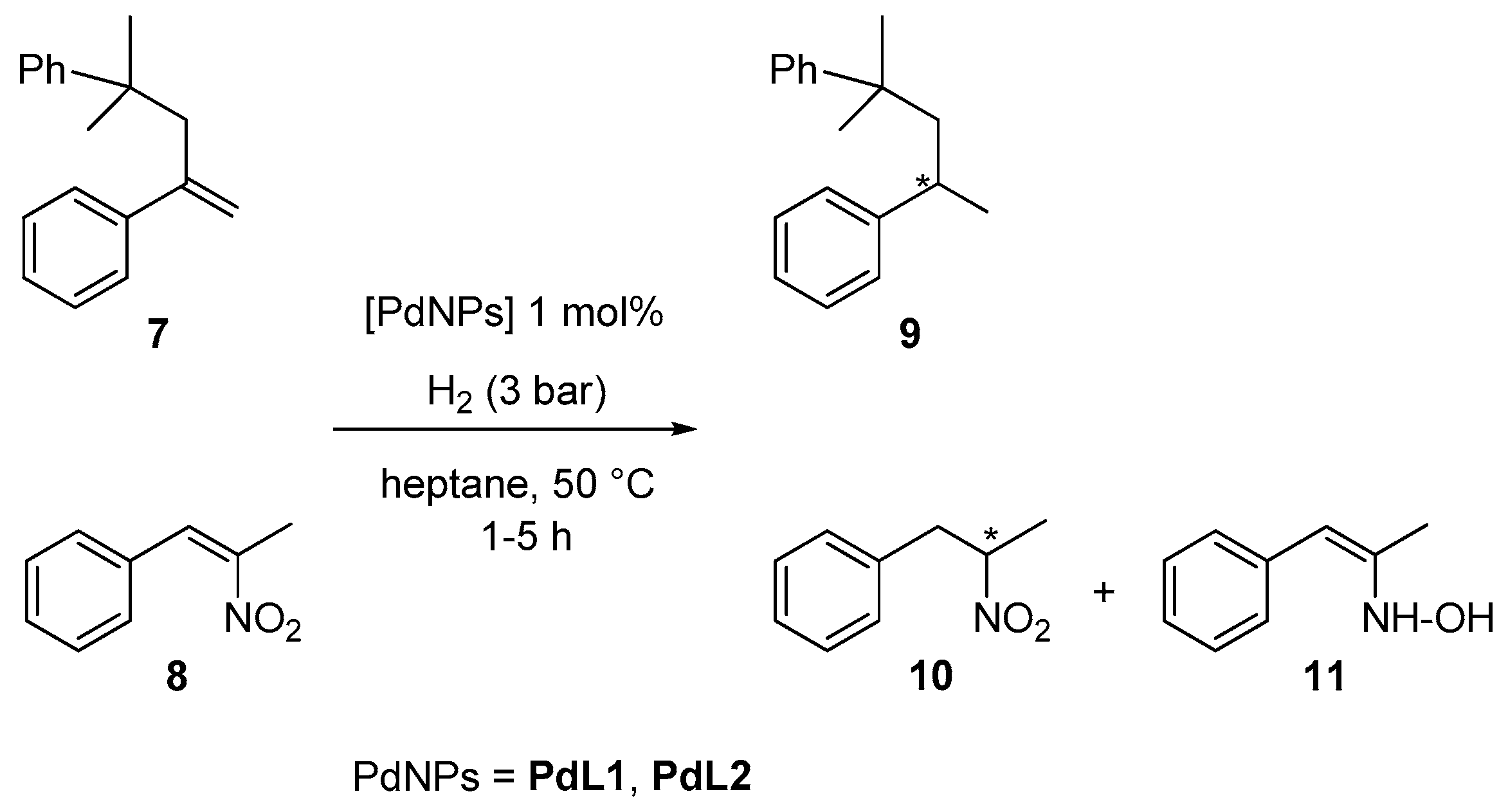
2.2. Hydrogenation Reactivity
3. Materials and Methods
3.1. General
3.2. Synthesis of PdNPs and RuNPs Stabilised by Optically Pure Borane-Protected Phosphines, L1-BH3 and L2-BH3
3.3. Hydrogenation Reactions Catalysed by PdNPs and RuNPs
3.4. Hydrogenation of L1
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bönnemann, H.; Richards, R.M. Nanoscopic Metal Particles—Synthetic Methods and Potential Applications. Eur. J. Inorg. Chem. 2001, 2001, 2455–2480. [Google Scholar] [CrossRef]
- Reetz, M.T.; Helbig, W.; Quaiser, S.A.; Stimming, U.; Breuer, N.; Vogel, R. Visualization of Surfactants on Nanostructured Clusters by a Combination of STM and High-Resolution TEM. Science 1995, 267, 367–369. [Google Scholar] [CrossRef] [PubMed]
- Trapp, O.; Weber, S.K.; Bauch, S.; Hofstadt, W. High-Throughput Screening of Catalysts by Combining Reaction and Analysis. Angew. Chem. Int. Ed. 2007, 46, 7307–7310. [Google Scholar] [CrossRef] [PubMed]
- Trapp, O.; Weber, S.K.; Bauch, S.; Bäcker, T.; Hofstadt, W.; Spliethoff, B. High Throughput Kinetic Study of Hydrogenations over Palladium Nanoparticles—Combination of Reaction and Analysis. Chem. Eur. J. 2008, 14, 4657–4666. [Google Scholar] [CrossRef] [PubMed]
- Gmeiner, J.; Seibicke, M.; Behrens, S.; Spliethoff, B.; Trapp, O. Investigation of the Hydrogenation of 5-Methylfurfural by Noble Metal Nanoparticles in a Microcapillary Reactor. ChemSusChem 2016, 6, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Gmeiner, J.; Behrens, S.; Spliethoff, B.; Trapp, O. Ruthenium Nanoparticles in High-Throughput Studies of Chemoselective Carbonyl Hydrogenation Reactions. ChemCatChem 2016, 8, 571–576. [Google Scholar] [CrossRef]
- Yasukawa, T; Miyamura, H.; Kobayashi, S. Chiral metal nanoparticle-catalyzed asymmetric C–C bond formation reactions. Chem. Soc. Rev. 2014, 43, 1450–1461. [Google Scholar] [CrossRef] [PubMed]
- Barbaro, P.; Dal Santo, V.; Liguori, F. Emerging strategies in sustainable fine-chemical synthesis: Asymmetric catalysis by metal nanoparticles. Dalton Trans. 2010, 39, 8391–8402. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Pericàs, M.A. Functionalized nanoparticles as catalysts for enantioselective processes. Org. Biomol. Chem. 2009, 7, 2669–2677. [Google Scholar] [CrossRef] [PubMed]
- Favier, I.; Madec, D.; Teuma, E.; Gómez, M. Palladium nanoparticles applied in organic synthesis as catalytic precursors. Curr. Org. Chem. 2011, 15, 3127–3174. [Google Scholar] [CrossRef]
- Gellman, A.J.; Tysoe, W.T.; Zaera, F. Surface Chemistry for Enantioselective Catalysis. Catal. Lett. 2015, 145, 220–232. [Google Scholar] [CrossRef]
- Mallat, T.; Orglmeister, E.; Baiker, A. Asymmetric Catalysis at Chiral Metal Surfaces. Chem. Rev. 2007, 107, 4863–4890. [Google Scholar] [CrossRef] [PubMed]
- Gual, A.; Godard, C.; Castillón, S.; Claver, C. Soluble transition-metal nanoparticles-catalysed hydrogenation of arenes. Dalton Trans. 2010, 39, 11499–11512. [Google Scholar] [CrossRef] [PubMed]
- Studer, M.; Blaser, H.-U.; Exner, C. Enantioselective Hydrogenation Using Heterogeneous Modified Catalysts: An Update. Adv. Synth. Catal. 2003, 345, 45–65. [Google Scholar] [CrossRef]
- Yasukawa, T.; Suzuki, A.; Miyamura, H.; Nishino, K.; Kobayashi, S. Chiral Metal Nanoparticle Systems as Heterogeneous Catalysts beyond Homogeneous Metal Complex Catalysts for Asymmetric Addition of Arylboronic Acids to α,β-Unsaturated Carbonyl Compounds. J. Am. Chem. Soc. 2015, 137, 6616–6623. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Lee, I.; Zaera, F. Correlated bifunctionality in heterogeneous catalysts: Selective tethering of cinchonidine next to supported Pt nanoparticles. Catal. Sci. Technol. 2015, 5, 680–689. [Google Scholar] [CrossRef]
- Kirby, F.; Moreno-Marrodan, C.; Baán, Z.; Bleeker, B.F.; Barbaro, P.; Berben, P.H.; Witte, P.T. NanoSelect Precious Metal Catalysts and their Use in Asymmetric Heterogeneous Catalysis. ChemCatChem 2014, 6, 2904–2909. [Google Scholar] [CrossRef]
- Schmidt, E.; Vargas, A.; Mallat, T.; Baiker, A. Shape-Selective Enantioselective Hydrogenation on Pt Nanoparticles. J. Am. Chem. Soc. 2009, 131, 12358–12367. [Google Scholar] [CrossRef] [PubMed]
- Gross, E.; Dean Toste, F.; Somorjai, G.A. Polymer-Encapsulated Metallic Nanoparticles as a Bridge between Homogeneous and Heterogeneous Catalysis. Catal. Lett. 2015, 145, 126–138. [Google Scholar] [CrossRef]
- Lili, L.; Xin, Z.; Shumin, R.; Ying, Y.; Xiaoping, D.; Jinsen, G.; Chunming, X.; Jing, H. Catalysis by metal–organic frameworks: Proline and gold functionalized MOFs for the aldol and three-component coupling reactions. RSC Adv. 2014, 4, 13093–13107. [Google Scholar] [CrossRef]
- Khiar, N.; Navas, N.; Elhalem, E.; Valdivia, V.; Fernandez, I. Proline-coated gold nanoparticles as a highly efficient nanocatalyst for the enantioselective direct aldol reaction in water. RSC Adv. 2013, 3, 3861–3864. [Google Scholar] [CrossRef]
- Jansat, S.; Gómez, M.; Philippot, K.; Muller, G.; Guiu, E.; Claver, C.; Castillón, S.; Chaudret, B. A Case for Enantioselective Allylic Alkylation Catalyzed by Palladium Nanoparticles. J. Am. Chem. Soc. 2004, 126, 1592–1593. [Google Scholar] [CrossRef] [PubMed]
- Favier, I.; Balanta Castillo, A.; Godard, C.; Castillón, S.; Claver, C.; Gómez, M.; Teuma, E. Efficient recycling of a chiral palladium catalytic system for asymmetric allylic substitutions in ionic liquid. Chem. Commun. 2011, 47, 7869–7871. [Google Scholar] [CrossRef] [PubMed]
- Dieguez, M.; Pamies, O.; Mata, Y.; Teuma, E.; Gómez, M.; Ribaudo, F.; van Leeuwen, P.W.N.M. Palladium nanoparticles in allylic alkylations and Heck reactions: The molecular nature of the catalyst studied in a membrane reactor. Adv. Synth. Catal. 2008, 350, 2583–2598. [Google Scholar] [CrossRef]
- Gual, A.; Axet, M.R.; Philippot, K.; Chaudret, B.; Denicourt-Nowicki, A.; Roucoux, A.; Castillón, S.; Claver, C. Diphosphite ligands derived from carbohydrates as stabilizers for ruthenium nanoparticles: Promising catalytic systems in arene hydrogenation. Chem. Commun. 2008, 2759–2761. [Google Scholar] [CrossRef] [PubMed]
- Mella, C.; Ávila, M.; Sánchez, A.; Marzialetti, T.; Reyes, P.; Ruiz, D. Chiral Rh/SiO2 catalysts for enantioselective hydrogenation reactions. The role of (S,S)-DIPAMP as chiral modifier and stabilizer on metallic nanoparticles synthesis. J. Chil. Chem. Soc. 2013, 58, 2125–2130. [Google Scholar] [CrossRef]
- Ruiz, D.; Mella, C.; Fierro, J.L.; Reyes, P. Silica supported rhodium metal nanoparticles stabilized with (-)-DIOP. Effect of ligand concentration and metal loading on enantioselective hydrogenation of ketones. J. Chil. Chem. Soc. 2012, 57, 1394–1399. [Google Scholar] [CrossRef]
- Gonzalez-Galvez, D.; Nolis, P.; Philippot, K.; Chaudret, B.; van Leeuwen, P.W.N.M. Phosphine-Stabilized Ruthenium Nanoparticles: The Effect of the Nature of the Ligand in Catalysis. ACS Catal. 2012, 2, 317–321. [Google Scholar] [CrossRef]
- Tamura, M.; Fujihara, H. Chiral Bisphosphine BINAP-Stabilized Gold and Palladium Nanoparticles with Small Size and Their Palladium Nanoparticle-Catalyzed Asymmetric Reaction. J. Am. Chem. Soc. 2003, 125, 15742–15743. [Google Scholar] [CrossRef] [PubMed]
- Sawai, K.; Tatumi, R.; Nakahodo, T.; Fujihara, H. Asymmetric Suzuki–Miyaura Coupling Reactions Catalyzed by Chiral Palladium Nanoparticles at Room Temperature. Angew. Chem. Int. Ed. 2008, 47, 6917–6919. [Google Scholar] [CrossRef] [PubMed]
- Cano, I.; Tschan, M.J.-L.; Martínez-Prieto, L.M.; Philippot, K.; Chaudret, B.; van Leeuwen, P.W.N.M. Enantioselective hydrogenation of ketones by iridium nanoparticles ligated with chiral secondary phosphine oxides. Catal. Sci. Technol. 2016, 6, 3758–3766. [Google Scholar] [CrossRef]
- Kamer, P.C.J.; van Leeuwen, P.W.N.M. Phosphorous (III) Ligands in Homogeneous Catalysis: Design and Synthesis; Wiley & Sons Inc.: Chichester, UK, 2012; p. 566. [Google Scholar]
- Grabulosa, A. P-Stereogenic Ligands in Enantioselective Catalysis; RSC Catalysis Series No. 7; Spivey, J.J., Ed.; RSC: Cambridge, UK, 2010; p. 520. [Google Scholar]
- Börner, A. Phosphorus Ligands in Asymmetric Catalysis: Synthesis and Applications; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008; Volumes 1–3, p. 1546. [Google Scholar]
- Aznar, R.; Muller, G.; Sainz, D.; Font-Bardia, M.; Solans, X. Synthesis and Reactivity of P-Chiral Tethered (η1:η6-phosphinoarene)ruthenium Complexes. Organometallics 2008, 27, 1967–1969. [Google Scholar] [CrossRef]
- Johansson, M.J.; Schwartz, L.; Amedjkouh, M.; Kann, N. New chiral amine ligands in the desymmetrization of prochiral phosphine boranes. Tetrahedron Asymmetry 2004, 15, 3531–3538. [Google Scholar] [CrossRef]
- Lara, P.; Philippot, K.; Chaudret, B. Organometallic Ruthenium Nanoparticles: A Comparative Study of the Influence of the Stabilizer on their Characteristics and Reactivity. ChemCatChem 2013, 5, 28–45. [Google Scholar] [CrossRef]
- Philippot, K.; Chaudret, B. Organometallic approach to the synthesis and surface reactivity of noble metal nanoparticles. C. R. Chim. 2003, 6, 1019–1034. [Google Scholar] [CrossRef]
- Favier, I.; Gómez, M.; Teuma, E. Palladium and ruthenium nanoparticles: Reactivity and coordination at the metallic surface. C. R. Chim. 2009, 12, 533–545. [Google Scholar] [CrossRef]
- López-Vinasco, A.M.; Favier, I.; Pradel, C.; Huerta, L.; Guerrero-Ríos, I.; Teuma, E.; Gómez, M.; Martin, E. Unexpected bond activations promoted by palladium nanoparticles. Dalton Trans. 2014, 43, 9038–9044. [Google Scholar] [CrossRef] [PubMed]
- Favier, I.; Massou, S.; Teuma, E.; Philippot, K.; Chaudret, B.; Gómez, M. A new and specific mode of stabilization of metallic nanoparticles. Chem. Commun. 2008, 3296–3298. [Google Scholar] [CrossRef] [PubMed]
- Favier, I.; Lavedan, P.; Massou, S.; Teuma, E.; Philippot, K.; Chaudret, B.; Gómez, M. Hydrogenation Processes at the Surface of Ruthenium Nanoparticles: A NMR Study. Top. Catal. 2013, 56, 1253–1261. [Google Scholar] [CrossRef]
- Chahdoura, F.; Pradel, C.; Gómez, M. Palladium Nanoparticles in Glycerol: A Versatile Catalytic System for C-X Bond Formation and Hydrogenation Processes. Adv. Synth. Catal. 2013, 355, 3648–3660. [Google Scholar] [CrossRef]
- Tschan, M.J.-L.; Diebolt, O.; van Leeuwen, P.W.N.M. Ruthenium Metal Nanoparticles in Hydrogenation: Influence of Phosphorus-Ligands. Top. Catal. 2014, 57, 1054–1065. [Google Scholar] [CrossRef]
- Escárcega-Bobadilla, M.V.; Tortosa, C.; Teuma, E.; Pradel, C.; Orejón, A.; Gómez, M.; Masdeu-Bultó, A.M. Ruthenium and rhodium nanoparticles as catalytic precursors in supercritical carbon dioxide. Catal. Today 2009, 148, 398–404. [Google Scholar] [CrossRef]
- Rafter, E.; Gutmann, T.; Löw, F.; Buntkowsky, G.; Philippot, K.; Chaudret, B.; van Leeuwen, P.W.N.M. Secondary phosphine oxides as pre-ligands for nanoparticle stabilization. Catal. Sci. Technol. 2013, 3, 595–599. [Google Scholar] [CrossRef]
- Jiang, H.-Y.; Zhen, X.-X. Tuning the chemoselective hydrogenation of aromatic ketones, aromatic aldehydes and quinolines catalyzed by phosphine functionalized ionic liquid stabilized ruthenium nanoparticles. Catal. Sci. Technol. 2015, 5, 3728–3734. [Google Scholar] [CrossRef]
- Wu, Z.; Jiang, H. Efficient palladium and ruthenium nanocatalysts stabilized by phosphine functionalized ionic liquid for selective hydrogenation. RSC Adv. 2015, 5, 34622–34629. [Google Scholar] [CrossRef]
- Delbecq, F.; Loffreda, D.; Sautet, P. Heterogeneous Catalytic Hydrogenation: Is Double Bond/Surface Coordination Necessary? J. Phys. Chem. Lett. 2010, 1, 323–326. [Google Scholar] [CrossRef]
- Bera, T.; Thybaut, J.W.; Marin, G.B. Extension of the Single-Event Microkinetic Model to Alkyl Substituted Monoaromatics Hydrogenation on a Pt Catalyst. ACS Catal. 2012, 2, 1305–1318. [Google Scholar] [CrossRef] [Green Version]







© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raluy, E.; Grabulosa, A.; Lavedan, P.; Pradel, C.; Muller, G.; Favier, I.; Gómez, M. P-Stereogenic Phosphines for the Stabilisation of Metal Nanoparticles. A Surface State Study. Catalysts 2016, 6, 213. https://doi.org/10.3390/catal6120213
Raluy E, Grabulosa A, Lavedan P, Pradel C, Muller G, Favier I, Gómez M. P-Stereogenic Phosphines for the Stabilisation of Metal Nanoparticles. A Surface State Study. Catalysts. 2016; 6(12):213. https://doi.org/10.3390/catal6120213
Chicago/Turabian StyleRaluy, Eva, Arnald Grabulosa, Pierre Lavedan, Christian Pradel, Guillermo Muller, Isabelle Favier, and Montserrat Gómez. 2016. "P-Stereogenic Phosphines for the Stabilisation of Metal Nanoparticles. A Surface State Study" Catalysts 6, no. 12: 213. https://doi.org/10.3390/catal6120213
APA StyleRaluy, E., Grabulosa, A., Lavedan, P., Pradel, C., Muller, G., Favier, I., & Gómez, M. (2016). P-Stereogenic Phosphines for the Stabilisation of Metal Nanoparticles. A Surface State Study. Catalysts, 6(12), 213. https://doi.org/10.3390/catal6120213