1. Introduction
Noble metal heterogeneous catalysts have found various industrial applications and, thus, play a major role in the world economy. Recently, there has been a great deal of interest in bimetallic catalysts research due to their novel chemical (catalytic) and physical (especially magnetic and electronic) properties. CoPd bimetallic catalysts have potentially high activity and selectivity for many reactions, including Fuel cells [
1,
2], Fischer-Tropsch [
3], NO reduction [
4], CO hydrogenation [
5], formic acid electrooxidation [
6], methanol oxidation reaction [
7], ethanol synthesis from syngas [
8], and hydrogenolysis [
9,
10,
11]. Small particle size (~10–100 Å) and high dispersion lends catalysts cost effective, efficient and economical. Among various known catalyst synthetic methods [
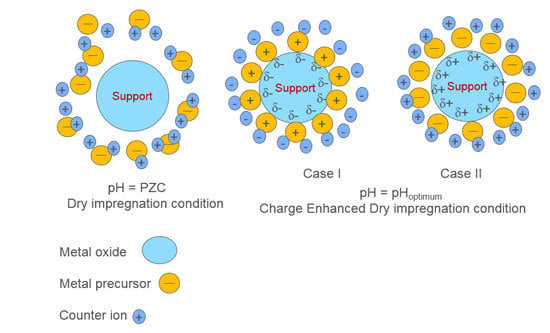
12], ‘Strong Electrostatic Adsorption' (SEA) has proven to be one such preparation method, which can simply and effectively fulfill the above-mentioned catalysts needs. The principle consideration of SEA is that ions adsorb on oxide surface only when it is charged. If the oxide surface is positively charged then it can adsorb anions and
vice versa. Moreover, ions cannot adsorb at point of zero charge (PZC) of metal oxides, as the surface is neutral due to unaltered surface hydroxyl groups. PZC can be determined by pH titrations. When the oxide is in contact with a solution of which the pH is less than PZC, the hydroxyl groups protonate and acquire a positive charge and show affinity towards anions like [PtCl
6]
2−. Similarly, when it is contacted with higher pH solution, the oxide surface shows affinity towards cations like [Pt(NH
3)
4]
2+. In either case, at a particular pH, the affinity attains a maximum and that particular pH is known as the point of strong electrostatic attraction. A catalyst prepared by contacting a metal precursor and oxide at this particular pH usually leads to mono dispersed metal particles with <15% standard deviation [
13]. The SEA preparation is carried out by contacting support material for 1 h with an aqueous solution of metal complex solution at a particular pH, at which point attraction between the support and metal complex is at the maximum, followed by filtering, drying and pretreatment.
Dry impregnation (DI) is the most commonly employed method in academia, as well as in industry. In DI protocols, a metal precursor salt/s is/are usually dissolved in a pore volume equivalent of water, and the pores of metal oxide support are filled in, which is usually followed by drying at room temperature, high-temperature drying, and pretreatment. The pretreatment steps consist of heating the catalyst sample at a high temperature in the presence of a controlled atmosphere or air. The DI technique is very simple to employ, and minimizes metal salt waste, but may or may not result in small metal particle sizes.
In the present work, a Charge Enhanced Dry Impregnation (CEDI) [
14], which is a combination of SEA and DI, is employed to synthesize monometallic and bimetallic heterogeneous catalysts. CEDI can be defined as SEA at DI conditions. The technique simply consists of adding sufficient acid or base to the DI solution (not in trivial amounts; but which can be pre-estimated [
14], thereby charging the system for metal ion adsorption).
2. Results
Table 1 gives the metal content, support and surface area, support point of zero charge, metal precursor used, reduction temperature, metal uptake, surface area weighted, and number weighted average particle size of Pd, Co, and CoPd, synthesized using SEA, CEDI and DI.
Table 1 depicts that nearly 20 wt. % and 5.3 wt. % Pd was adsorbed from the solution phase on Black Pearls 2000 (BP) and Vulcan XC-72 (VXC) carbons in three consecutive adsorption procedures using SEA. The particle sizes obtained were 33 Å in the case of 20 wt. % Pd/BP, and 29 Å in the case of 5.3 wt. % Pd/VXC. The adsorption of cation [Pd(NH
3)
4]
2+ on BPo
x and VXCo
x from the solution phase via SEA was nearly 24 wt. % Pd and 6 wt. % Pd, respectively. The particle sizes obtained were 30 Å in the case of 24 wt. % Pd/BPo
x, and 28 Å in the case of 6 wt. % Pd/VXCo
x. Repetition of the adsorption procedure more than three times did not increase the metal uptake to a considerable level. The particle size remained almost the same, between 28–33 Å, in all the above cases.
The CEDI procedure for 20 wt. % Pd using BP and VXC with H2PdCl4 yielded particles with a small particle size of about 25 and 33 Å, with a very narrow size distribution. However, the CEDI procedure for BPox and VXCox with [Pd(NH3)4]2+ resulted in slightly bigger particle sizes of about 63 and 73 Å. As weight loadings near 20% are currently favored for many fuel cell applications, the current results are directly applicable, especially for catalysts prepared with the Cl− free metal precursor [Pd(NH3)4](NO3)2 as it overcomes the catalyst poisoning effect caused by Cl− ions.
Catalysts with varying particle sizes can be prepared by varying the reduction temperature. As an example, by increasing the reduction temperature from 200 to 850 °C, particle sizes ranging from 17 to 241 Å were obtained for 10 wt. % Pd/BP (
Table 1). The Co/carbon catalysts were prepared by mixing the Co(H
2O)
6∙(NO
3)
2 in HNO
3 with the support. Interestingly, at 400 °C, reduced Co/C catalyst did not show any well-formed particles, but catalyst reduced at above 600 °C showed well-formed particles. The 5 wt. % Co on BP2000 and VXC yielded 39 Å and 55 Å particles, respectively, with a narrow size distribution.
Bimetallic alloy particles with varying Co/Pd ratios were prepared by first mixing the Co(H2O)6∙(NO3)2 and [Pd(NH3)4](NO3)2 together in 1 M HNO3, and then mixing with the support. The final pH of the slurry was between pH 1.5–2.2. All three ratios of CoPd on VXC and BP2000 showed well-formed particles after reducing at 400 °C and 600 °C.
CoPd bimetallic catalysts supported on BP and VXC were also synthesized using the CEDI route. In the case of different ratios of Co and Pd being loaded on the BP2000 support, 400-°C- and 600-°C-reduced catalysts showed almost same particle sizes, between 25 Å to 28 Å, except in the case of 5 wt. % Co + 5 wt. % Pd reduced at 600 °C, which showed a particle size of 35 Å. It should be noted that standard deviation in all bimetallic catalysts was below ±8 Å. Increasing the loading to 10 wt. % Co + 5 wt. % Pd and 10 wt. % Co + 2.5 wt. % Pd resulted in slightly bigger particles, i.e., 32 Å and 33 Å.
In the case of catalysts (5 wt. % Co + 5 wt. % Pd), (5 wt. % Co + 2.5 wt. % Pd), (5 wt. % Co + 1.25 wt. % Pd), and 5 wt. % Co on VXC, synthesized using CEDI and reduced at 600 °C, small particles of 36 Å, 30 Å, 33 Å, and 55 Å, respectively, were obtained, with near monodispersion,
i.e., ±15 Å.
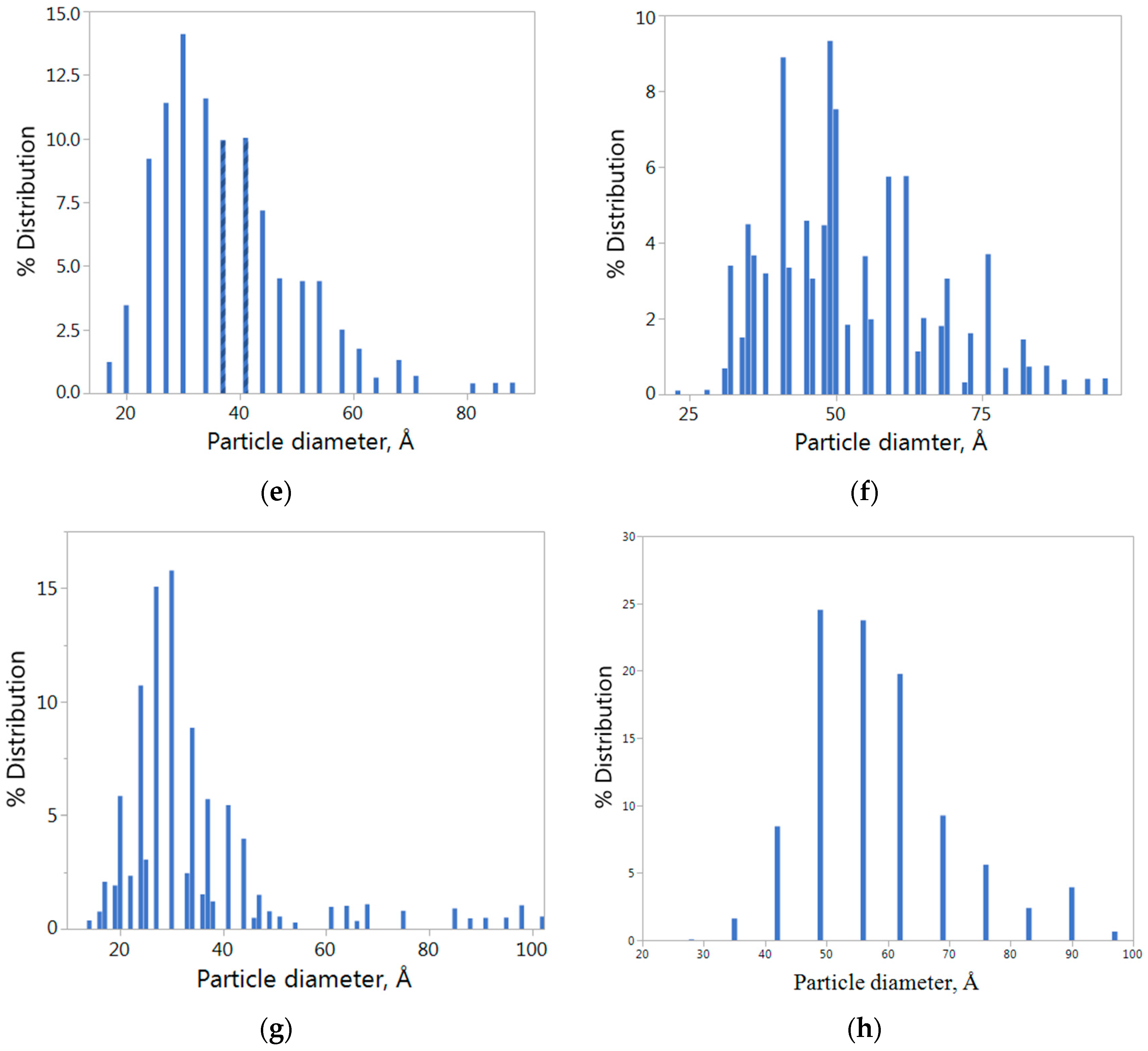
Figure 1a–d gives the STEM images of all three different CoPd and Co catalysts supported on VXC.
Figure 1e–h are particle size distribution graphs corresponding to
Figure 1a–d. The particles are not symmetrically distributed, but, rather, are skewed to the right in almost all cases. It was intriguing that overall bimetallic particles are smaller and more spherical in shape than their individual metals are after reducing at the same temperature.
As a control experiment, catalysts were prepared by using deionized water via regular DI (5 wt. % Co + 5 wt. % Pd)/VXC, (5 wt. % Co + 2.5 wt. % Pd)/VXC and (5 wt. % Co + 5 wt. % Pd)/BP2000 were prepared and pretreated at 600 °C. The pH of the slurry containing metal ions and carbon was around 6.9. As expected, particles obtained were very large and standard deviations were very broad,
viz. 126 ± 81 Å, 124 ± 85 Å, and 134 ± 90 Å in the case of (5 wt. % Co + 5 wt. % Pd)/VXC, (5 wt. % Co + 2.5 wt. % Pd)/VXC and (5 wt. % Co + 5 wt. % Pd)/BP2000, respectively, which confirms that the DI is unable to charge the system for an effective interaction between the metal ions and support.
Figure 2 shows the STEM images of 5 wt. % Co + 5 wt. % Pd/VXC, (5 wt. % Co + 2.5 wt. % Pd)/VXC and (5 wt. % Co + 5 wt. % Pd)/BP2000 prepared by DI. STEM analysis also showed that more Pd metal aggregated than Co metal during pretreatment.
In our earlier work, monometallic Co and Pd were synthesized using the DI method, where 20 wt. % Co/VXC synthesized using Co(NO
3)
2 resulted in aggregated lumps without a proper size or shape. On the other hand, 1 wt. % Co/VXC synthesized using Co(NH
3)
6)
3+ resulted in big particles and a broader size distribution,
Xn = 187 ± 133 Å [
15]. Similarly, 1.9 wt. % Pd/VXC synthesized via DI resulted in particles of 29 ± 17 Å [
16]. On the other hand, 20 wt. % Pd/VXC synthesized via CEDI also resulted in the same range, which, if prepared via DI, would have resulted in very big particles. This, once again, demonstrates the advantage of CEDI over the commonly-practiced DI method.
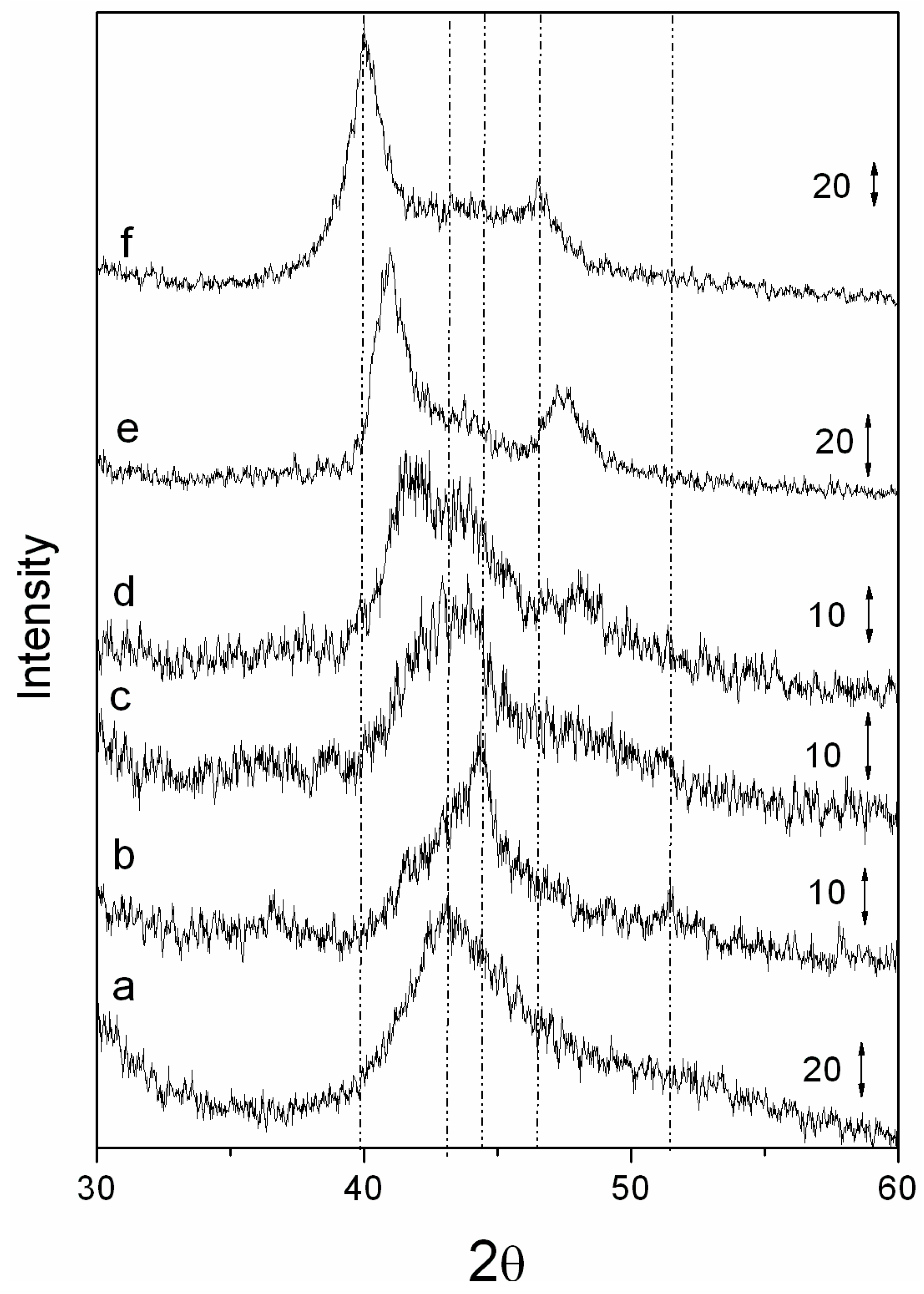
Figure 3 gives the diffraction pattern of Pd/carbon reduced catalysts. Diffraction patterns showed that palladium was in the metallic state. The diffraction patterns of the bimetallic catalysts confirmed that particles possessed a well-formed alloy phase. Particles reduced at 400 °C and 500 °C showed the presence of a small amount (5%–10%) of spinel phase, in addition to the metallic alloy phase, inferring that 400 and 500 °C are not enough to fully reduce the catalyst to its metallic phase. Moreover, these results also show that Co
2+ first transforms into a spinel phase,
i.e., (Co
1−xPd
x)
3O
4 and then converts to a Co
1−xPd
x metallic phase. Alloy catalysts reduced at 600 °C showed no spinel phase, but rather a metallic fcc phase. Juszczyle
et al. [
11] have used co-impregnation of PdCl
2 and CoCl
2 on a SiO
2 support to synthesized bimetallic alloy catalysts, and observed that the metallic precursor can reduce at 380 °C. The lower reduction temperature is possibly due to different precursors and supports. The formation of an alloy can be seen in the diffraction pattern shown in
Figure 3.
Figure 3 also gives the diffraction patterns for pure VXC, as well as monometallic Co/VXC and Pd/VXC. With the increase in Pd content, the (111) and (200) diffraction peak position shifted towards that of a pure Pd peak position, indicating very good CoPd alloy formation and not a physical mixture. The diffraction peak position for different catalysts is given in
Table 2. One archived bimetallic system involving cobalt and palladium is CoPd2, PDF# 50-1437. The peak position of (111) and (200) for this system occurs at 2θ = 40.77 and 47.439, which are very close to the peak position of 5 wt. % Co + 5 wt. % Pd/VXC system. These data confirm that well-alloyed nanoparticles have formed on the carbon support.
In view of electrostatic adsorption, it is understandable that [PdCl
4]
2− adsorbs at low pH on protonated, positively charged surfaces and [Pd(NH
3)
4]
2+ and [Co(H
2O)
6]
2+ at high pH on deprotonated, negatively charged surfaces, but it is surprising to note that both [Pd(NH
3)
4]
2+ and [Co(H
2O)
6]
2+ adsorb well, even at low pH < 2.5, and result in well-dispersed metallic particles upon reduction. To understand the actual mechanism of adsorption, control experiments were done by dissolving [Pd(NH
3)
4](NO
3)
2 and Co(H
2O)
6∙(NO
3)
2 into HNO
3 and observed the reaction by using a UV-visible spectrometer. In 2–3 hours, the yellowish color of [Pd(NH
3)
4](NO
3)
2 slowly changed to reddish brown, indicating the transformation of the above-mentioned complex to [Pd(NO
3)
4]
2−. UV-visible spectrum of [Pd(NH
3)
4]
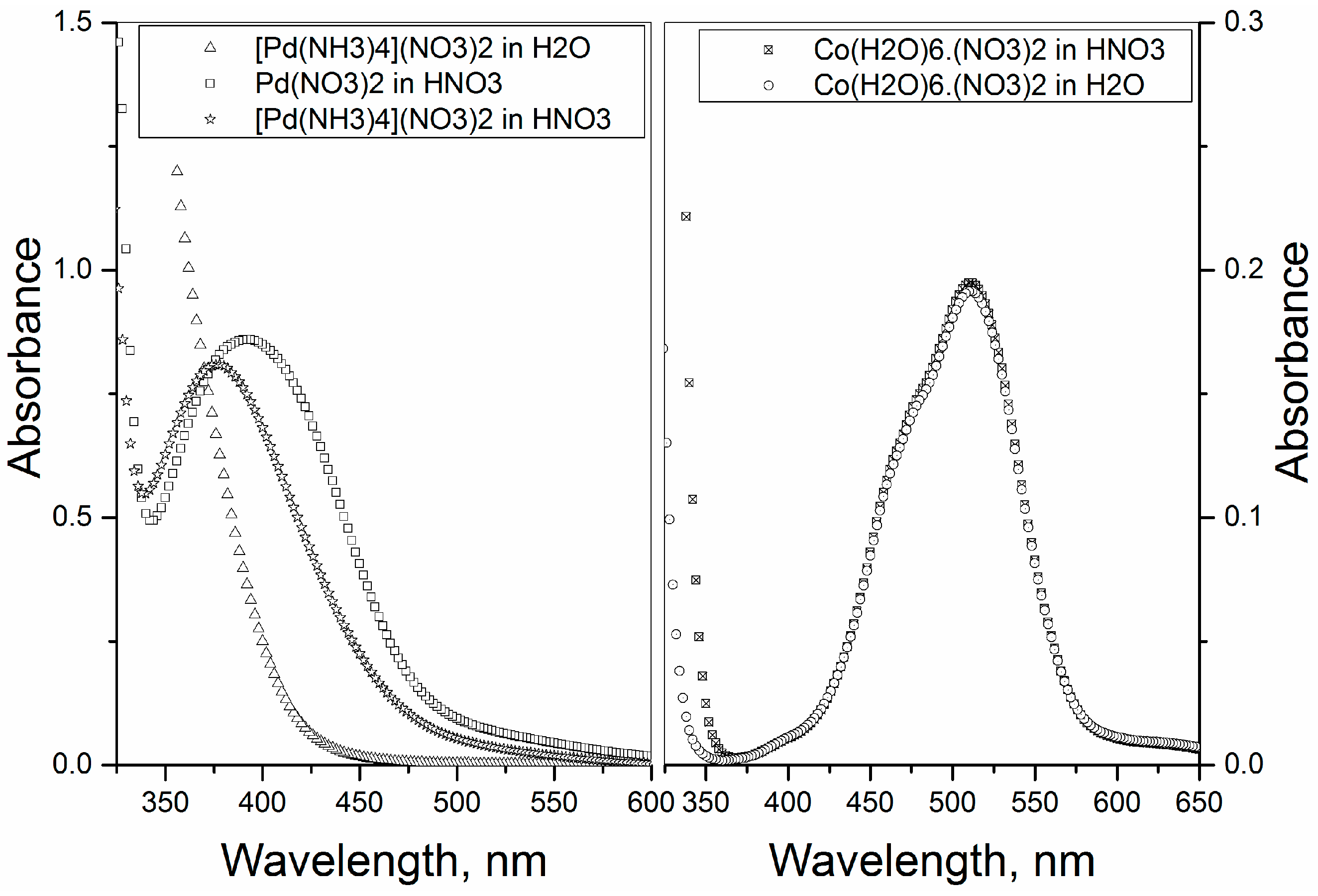
2+ does not have a characteristic λmax, but absorption increases steadily from about 450 nm towards UV region (
Figure 4 (left)).
The spectra of the transformed compound shows a peak at λ
max = 375 nm (
Figure 4 (left)) and of which absorption spectrum is very similar to that of pure Pd(NO
3)
2 salt dissolved in 1 M HNO
3, which shows a λ
max at 392 nm (
Figure 4 (left)). These spectra are in good agreement with the literature [
17]. The slight difference in λ
max between two systems indicates a difference in complexation [
17]. The absorption spectra of Co(H
2O)
6∙(NO
3)
2 dissolved in H
2O and 1 M HNO
3 is shown in
Figure 4 (right). The present assumption is that Co(H
2O)
6∙(NO
3)
2 dissolved in HNO
3 transforms into [Co(NO
3)
4]
2−, but the similar absorption spectra in H
2O and HNO
3 makes it difficult to make conclusions. Thus, even though the starting precursors were [Pd(NH
3)
4]
2+ and [Co(H
2O)
6]
2+, their transformed species are anions, namely [Pd(NO
3)
4]
2− and [Co(NO
3)
4]
2−; and these anionic species adsorbs strongly on high PZC materials to yield highly-dispersed metal particles.
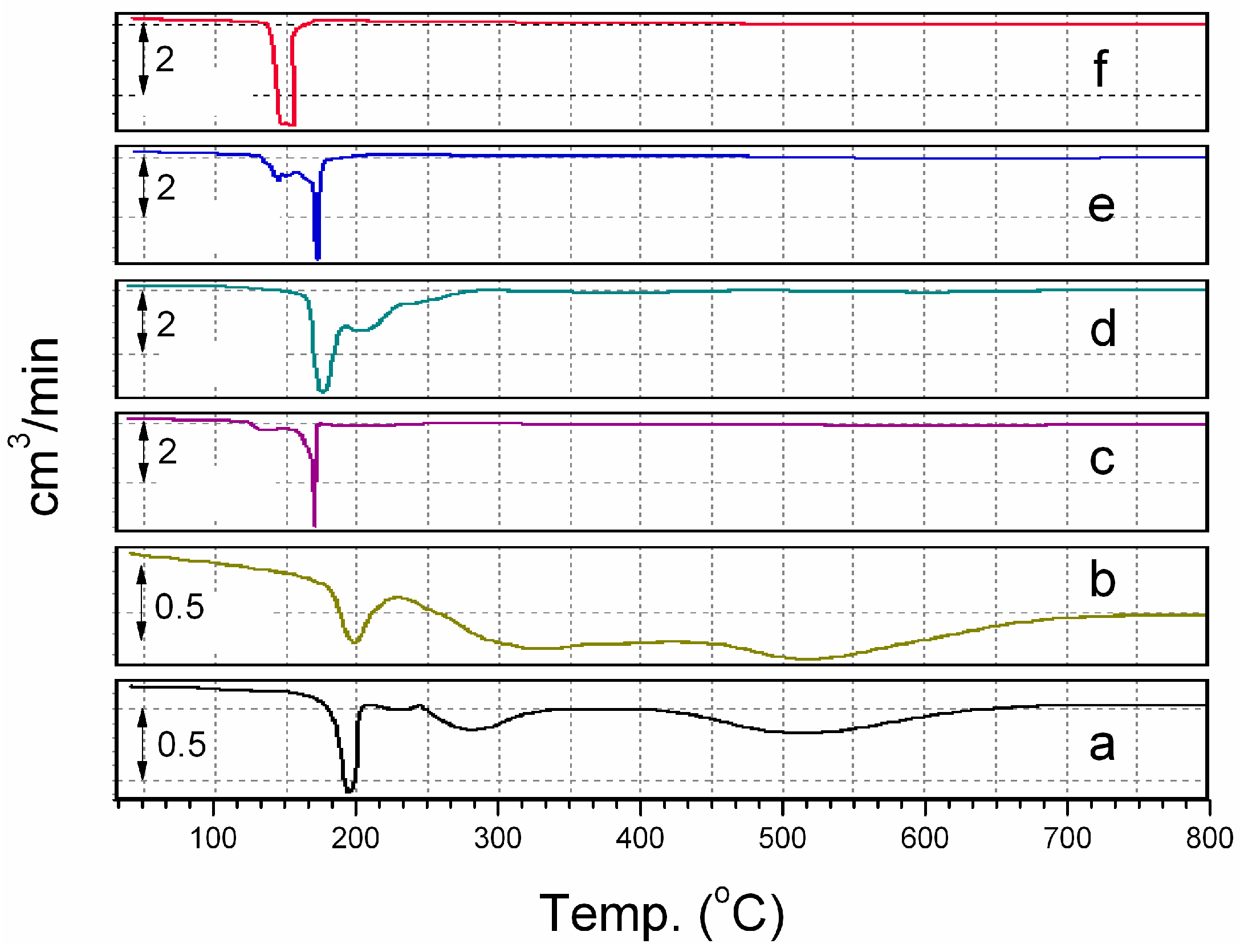
Figure 5 gives the temperature programmed reduction profiles for (a) a physical mixture of Co(H
2O)
6∙(NO
3)
2 and VXC, (b) 5 wt. % Co/VXC, (c) 5 wt. % Co + 1.25 wt. % Pd/VXC, (d) 5 wt. % Co + 2.5 wt. % Pd/VXC, (e) 5 wt. % Co + 5 wt. % Pd/VXC and (f) 5 wt. % Pd/VXC. From (a) it can be seen that the Co(H
2O)
6∙(NO
3)
2 TPR profile contains three major peaks. The first peak at 195 °C can be considered as the decomposition of Co(H
2O)
6∙(NO
3)
2 to Co
3O
4, the second peak, at 280 °C, the reduction of Co
3O
4 to CoO, and the third peak, at 510 °C, the reduction of CoO to Co. A similar trend is observed in (b). Pd/VXC (f) shows two peaks merged together, the decomposition and reduction of Pd complex occurs almost together at around 150 °C. In the case of CoPd/VXC systems, the Pd complex decomposition peak and reduction peak are separate from each other. The Pd reduction peak and Co complex decomposition peak come together and almost merge in the case of 5 wt. % Co + 5 wt. % Pd (e) at 172 °C. It can be inferred that, at this temperature, formation of (Co
1−xPd
x)
3O
4 takes place, which can be reduced to CoPd in two stages at around 330° and 600 °C.
3. Materials and Methods
Two types of carbons, namely Vulcan XC-72 (VXC) and Black Pearls 2000 (BP) were used to prepare the catalysts. The point of zero charge of these carbons is 8.9 and 9.5, respectively. These carbon samples were oxidized by boiling in concentrated 15.9 M HNO
3 for 5 h, which brought down the PZC to 2.5 and 2.65 for BPo
x and VXCo
x, respectively, due to surface oxidation and formation of acidic functional groups [
18]. Co(NO
3)
2∙6H
2O, PdCl
2 and [Pd(NH
3)
4](NO
3)
2 were used as precursors. During the preparation of catalysts using the CEDI method, using VXC and BP supports; Co(H
2O)
6(NO
3)
2 and [Pd(NH
3)
4](NO
3)
2 precursors are dissolved in 1 M HNO
3, and when using activated carbons, VXCo
x and BPo
x precursors are dissolved in 1 M NH
4OH instead of H
2O. PdCl
2 was dissolved in concentrated HCl, which results in speciation of Pd salt to H
2PdCl
4. The amount of precursor solution is equal to the pore volume of the support. The pore volume of the VXC, BP, VXCo
x and BPo
x were 2.5, 7, 2, and 1.6 mL per gram of the solid, respectively, as determined by titration with distilled water. When these precursor solutions are mixed with carbon, the final pH of the slurry arrives at the pH at which strong electrostatic attraction between support and precursor is maximal, and results in strong adsorption of metal precursors onto the support. Drying the slurry at room temperature, followed by drying at 110 °C and high-temperature reduction in flowing pure H
2 results in highly dispersed monometallic/bimetallic nanoparticles supported on carbons. In the case of usual dry impregnation (DI), metal precursors were dissolved in a pore volume equivalent of distilled water instead of HNO
3 or NH
4OH.
The equilibrium adsorption uptake values of H
2PdCl
4 and/or [Pd(NH
3)
4](NO
3)
2 onto carbon was obtained using published data [
16]; namely, pH 1–2 for [PdCl
4]
2− and 11–12 for [Pd(NH
3)
4]
2+. To overcome the oxide buffering effect [
17], the adsorption step was performed in the presence of excel liquid. The surface loading is termed as the amount of support surface area per liter of impregnation solution. For example, to obtain a surface loading of 500 m
2/L, a 0.1 g of 250 m
2/g carbon needs to be dispersed in 50 mL of solution. Two hundred and fifty milliliter polypropylene bottles, containing about 200 mL of 200 ppm H
2PdCl
4 or [Pd(NH
3)
4](NO
3)
2 solution, was taken in a 250-mL polypropylene bottle and shaken for 1 h for adsorption of the metal precursor on the support. About 3–4 mL of solution was filtered and analyzed using Inductively Coupled Plasma spectroscopy (ICP) (Supplied by Perkin-Elmer Optima 2000, American Fork, UT, USA) to quantify metal adsorption. Amounts of 0.1–1 N HCl, HNO
3, NH
4OH and NaOH were employed to adjust solution pH. The remaining solution was filtered and metal adsorbed oxide material was collected and dried at room temperature, and then at 110 °C for 12 h, followed by reduction at 200 °C for 1 h. The adsorption to reduction procedure was repeated three times to increase the metal content of the catalysts as SEA is limited to one monolayer of metal complex ion.
Microscopy measurements (STEM) were done using an electron microscope supplied by JEOL, Model JEM-2010F FasTEM FEI (JEOL USA, Inc., Peabody, MA, USA), operated at 200 kV and with an extracting voltage of 4500 V. Particle distribution, number weighted mean particles size (), and standard deviation and were calculated by considering about 1000 particles, where x is the diameter of the particle, XN is number weighted mean particle size, and f is frequency of occurrence.
The surface area weighted mean particle size () and standard deviation associated with that were also calculated using the same data. Surface area weighted mean particle size (XA) and standard deviation have been used in most places, unless otherwise stated. The closeness in XA and XN values shows the homogeneity of the metal dispersion.
An AutoChem II instrument (Micromeritics Instrument Corporation, Norcross, GA, USA), supplied by Micromeritics, was used to perform temperature programmed reduction (TPR) experiments. A 50-mg sample, 50 mL/min 10% H2 in Ar and 10 °C/min temperature ramp was considered for all TPR experiments.
A Siemens D5000 diffractometer was employed in X-ray diffraction analyses using Cu Kα radiation (λ = 1.5406 Å) operating at 30 kV and 40 mA, and operated in Bragg–Brentano geometry. A step size of 0.01° and 2.5 s exposure at each point was used. The scan was performed in the 2θ range of 20°–70° in a “Locked-coupled” mode. Powder diffraction pattern (PDF) data provided by JCPDS—International Centre for Diffraction Data—was used to interpret X-ray diffractograms.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}