
A Short Review on the Catalytic Activity of Hydrotalcite-Derived Materials for Dry Reforming of Methane


 ,
, 
Abstract
:
1. Introduction
- (i)
- Employing an appropriate preparation method in order to control Ni crystal size and thus inhibit coke growth.
- (ii)
- Using metal oxides with strong Lewis basicity as supports or promoters, since basic sites enhance CO2 adsorption. Metal oxides can promote the oxidation of carbon deposits (i.e. via the reverse Boudouard reaction), but, on the other hand, the supports exhibiting Lewis acidity enhance formation of coke deposits.
- (iii)
- Addition of a second metal, i.e., a noble metal, which may enhance the transport of hydrogen and/or oxygen between active sites and support by spillover, and can influence the mechanism of coke formation. Addition of promoters, such as Ce, Zr or La, in the aim of modifying the selectivity of the DRM process and/or enhancing the gasification of the carbon deposits.
- (iv)
- Sulphur passivation of Ni catalysts, which blocks the step edge sites where coke build-up is initiated.
- (v)
- Changing reaction conditions by the addition of oxidizing agents, such as water or oxygen, which can help oxidize carbon deposits.
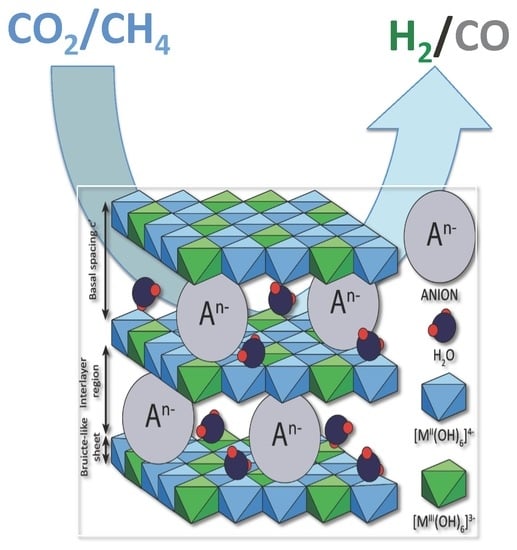
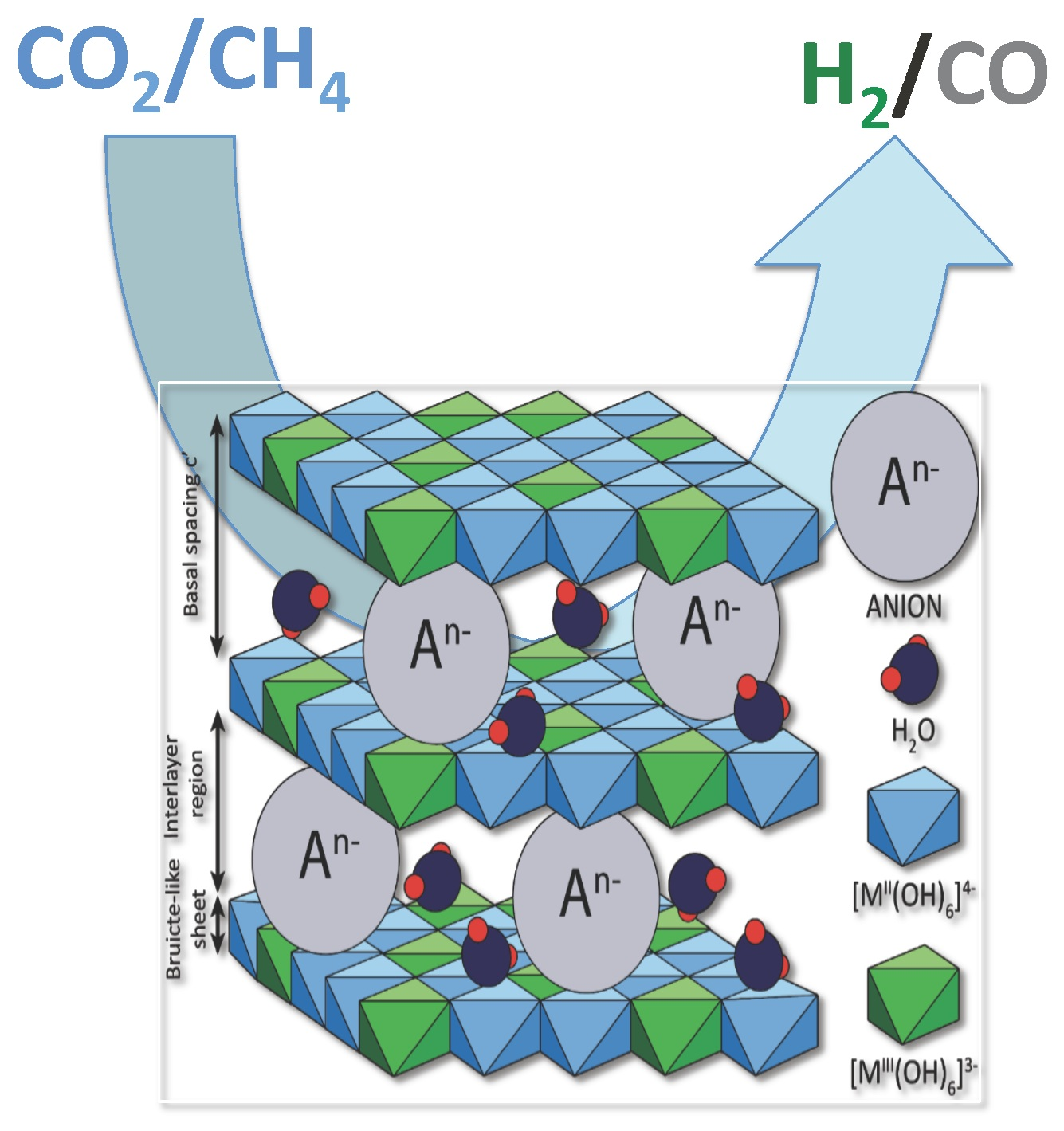
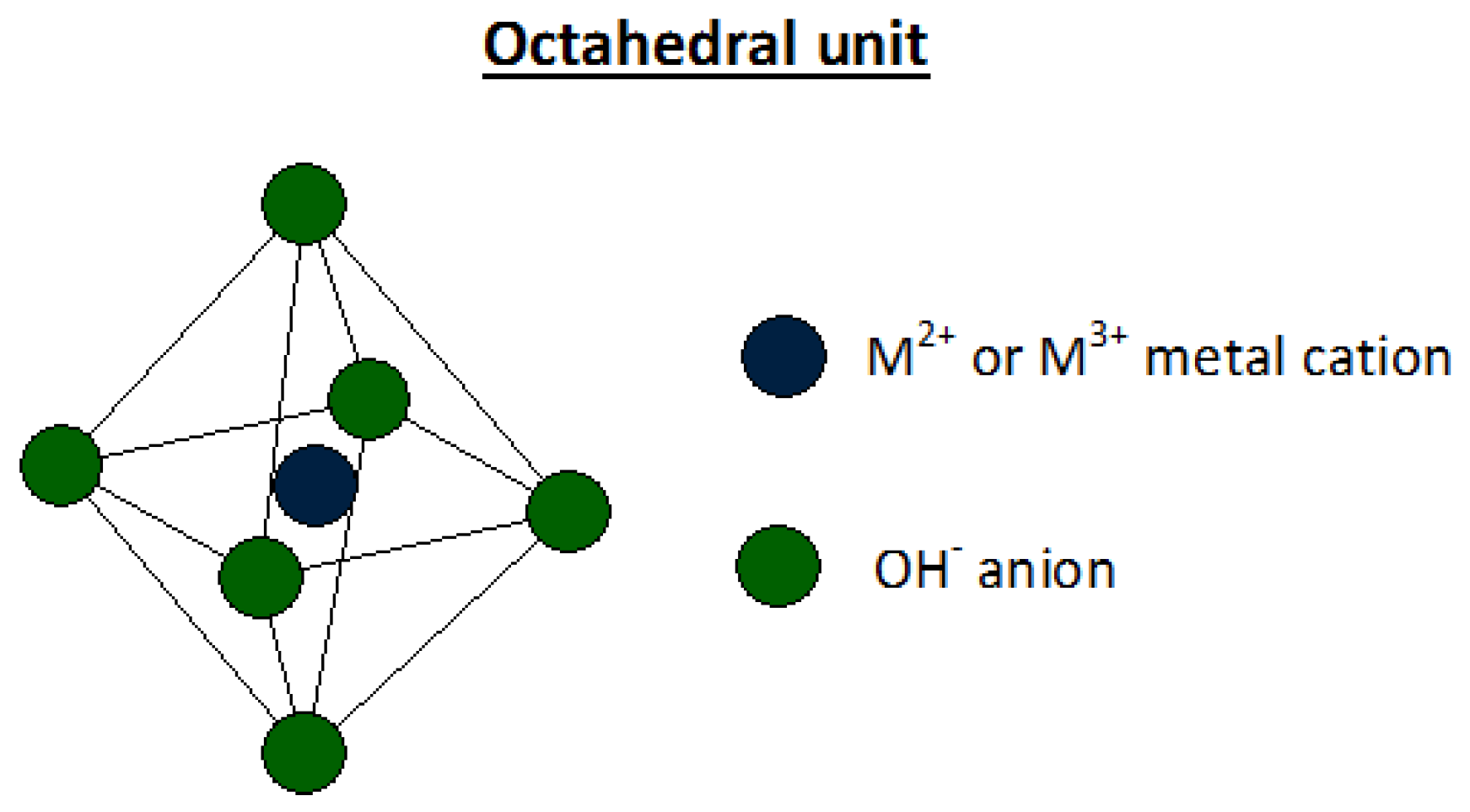
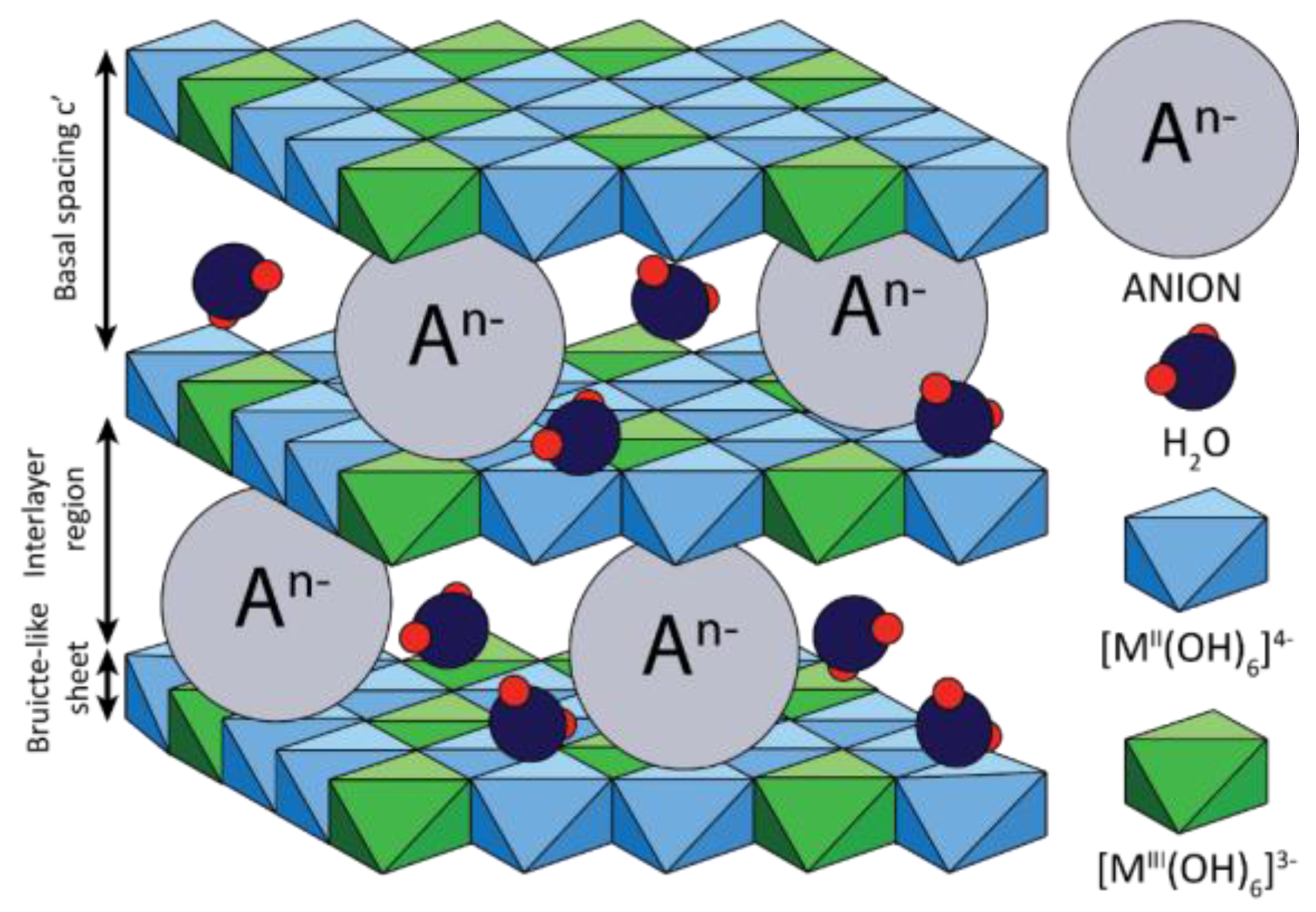
2. Hydrotalcites
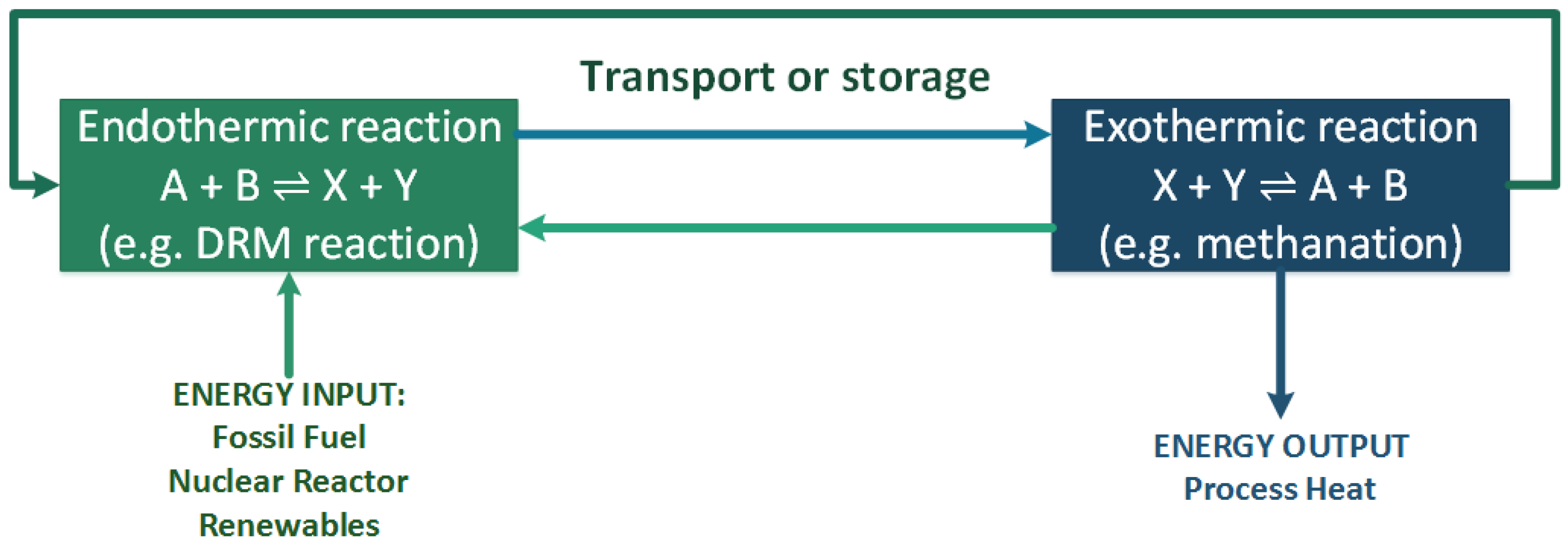
3. Catalytic Activity of Hydrotalcite-Derived Materials in Dry Reforming of Methane (DRM)
3.1. Ni/Mg/Al and Ni/Al Hydrotalcite-Derived Catalysts
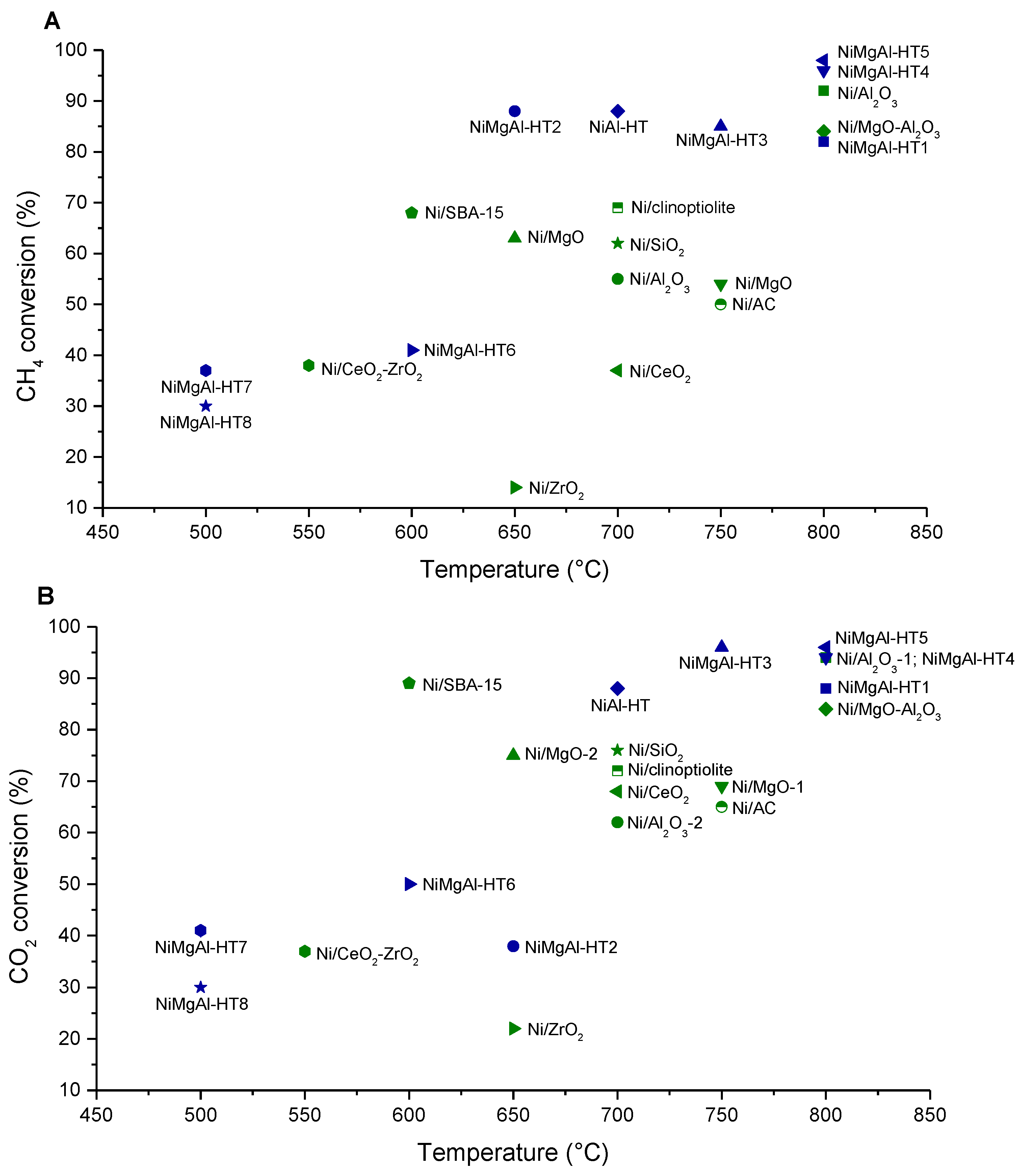
3.1.1. Effect of Mg/Al, Ni/Mg and Ni/Al Molar Ratios
3.1.2. Effect of the Method of Ni Introduction into Hydrotalcite (HT) Structure
3.1.3. Influence of the Air-Calcination Temperature
3.2. Effect of the Addition of Different Promoters
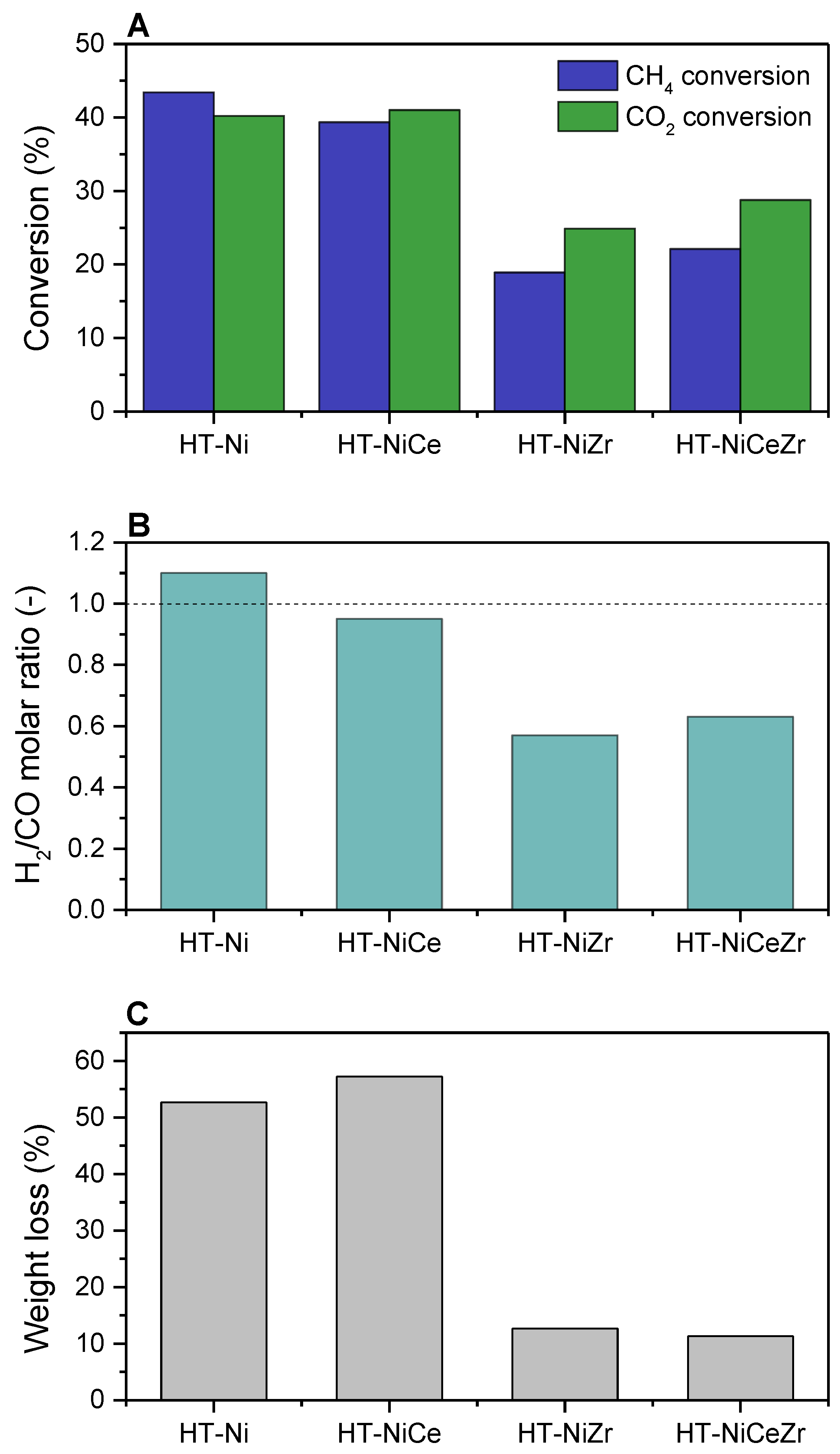
3.2.1. Ce Promotion
3.2.2. Other Promoters
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- BP. Energy charting tool. British Petroleum BP plc. Available online: http://tools.bp.com/energy-charting-tool (accessed on 2 January 2016).
- Lavoie, J.-M. Review on dry reforming of methane, a potentially more environmentally-friendly approach to the increasing natural gas exploitation. Front. Chem. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- U.S. Energy Information Administration. International Energy Statistics. Available online: http://www.eia.gov/cfapps/ipdbproject/IEDIndex3.cfm (accessed on 6 January 2016).
- United Nations Framework Convention on Climate Change. Kyoto Protocol. Available online: http://unfccc.int/kyoto_protocol/items/2830.php (accessed on 1 September 2016).
- United Nations Conference on Climate Change (COP21). Available online: http://www.cop21.gouv.fr/en/ (accessed on 1 September 2016).
- COM(2014) 15 Final, Communication From The Commission To The European Parliament, The Council, The European Economic and Social Committee and The Committee of the Regions, A Policy Framework for Climate and Energy in the Period from 2020 to 2030; European Commission: Brussels, Belgium, 2014; pp. 1–18.
- European Commission. Energy Roadmap 2050; Publications Office of the Eueopean Union: Luxembourg, Belgium, 2012. [Google Scholar]
- Bradford, M.C.J.; Vannice, M.A. CO2 Reforming of CH4. Catal. Rev. 1999, 41, 1–42. [Google Scholar] [CrossRef]
- Suhartanto, T.; York, A.P.E.; Hanif, A.; Al-Megren, H.; Green, M.L.H. Potential utilisation of Indonesia's Natuna natural gas field via methane dry reforming to synthesis gas. Catal. Lett. 2001, 71, 49–54. [Google Scholar] [CrossRef]
- Richardson, J.T.; Paripatyadar, S.A. Carbon dioxide reforming of methane with supported rhodium. Appl. Catal. 1990, 61, 293–309. [Google Scholar] [CrossRef]
- McCrary, J.H.; McCrary, G.E.; Chubb, T.A.; Nemecek, J.J.; Simmons, D.E. An experimental study of the CO2 CH4 reforming-methanation cycle as a mechanism for converting and transporting solar energy. Sol. Energy 1982, 29, 141–151. [Google Scholar] [CrossRef]
- Chubb, T.A. Characteristics of CO2-CH4 reforming-methanation cycle relevant to the solchem thermochemical power system. Solar Energy 1980, 24, 341–345. [Google Scholar] [CrossRef]
- Mortensen, P.M.; Dybkjær, I. Industrial scale experience on steam reforming of CO2-rich gas. Appl. Catal. A Gen. 2015, 495, 141–151. [Google Scholar] [CrossRef]
- Teuner, C.; Neumann, P.; Von Linde, F. The Calcor Standard and Calcor Economy Processes. Oil Gas Eur. Mag. 2001, 3, 44–46. [Google Scholar]
- Reitmeier, R.E.; Atwood, K.; Bennett, H.; Baugh, H. Production of Synthetic Gas–Reaction of Light Hydrocarbons with Steam and Carbon Dioxide. Ind. Eng. Chem. 1948, 40, 620–626. [Google Scholar] [CrossRef]
- Więcław-Solny, L.; Łabojko, G.; Babiński, P. Możliwości przemysłowego wykorzystania ditlenku węgla-badania nad zastosowaniem CO2 w procesie otrzymywania gazu syntezowego. Polit. Energ. 2009, 12, 633–642. [Google Scholar]
- Rostrup-Nielsen, J. 40 years in catalysis. Catal. Today 2006, 111, 4–11. [Google Scholar]
- Yun Hang, H. Advances in Catalysts for CO2 Reforming of Methane. In Advances in CO2 Conversion and Utilization; American Chemical Society: Washington, DC, USA, 2010; pp. 155–174. [Google Scholar]
- Hei, M.J.; Chen, H.B.; Yi, J.; Lin, Y.J.; Lin, Y.Z.; Wei, G.; Liao, D.W. CO2-reforming of methane on transition metal surfaces. Surf. Sci. 1998, 417, 82–96. [Google Scholar] [CrossRef]
- Kroll, V.C.H.; Swaan, H.M.; Mirodatos, C. Methane Reforming Reaction with Carbon Dioxide Over Ni/SiO2 Catalyst: I. Deactivation Studies. J. Catal. 1996, 161, 409–422. [Google Scholar] [CrossRef]
- Hu, Y.H.; Ruckenstein, E. Catalytic Conversion of Methane to Synthesis Gas by Partial Oxidation and CO2 Reforming. Advances in Catalysis; Academic Press: Washington, DC, USA, 2004; pp. 297–345. [Google Scholar]
- Ay, H.; Üner, D. Dry reforming of methane over CeO2 supported Ni, Co and Ni–Co catalysts. Appl. Catal. B Environ. 2015, 179, 128–138. [Google Scholar] [CrossRef]
- Becerra, A.; Dimitrijewits, M.; Arciprete, C.; Castro Luna, A. Stable Ni/Al2O3 catalysts for methane dry reforming. Granul. Matter 2001, 3, 79–81. [Google Scholar] [CrossRef]
- Kim, J.-H.; Suh, D.J.; Park, T.-J.; Kim, K.-L. Effect of metal particle size on coking during CO2 reforming of CH4 over Ni–alumina aerogel catalysts. Appl. Catal. A Gen. 2000, 197, 191–200. [Google Scholar] [CrossRef]
- Li, L.; Zhang, L.-M.; Zhang, Y.-H.; Li, J.-L. Effect of Ni loadings on the catalytic properties of Ni/MgO(111) catalyst for the reforming of methane with carbon dioxide. J. Fuel Chem. Technol. 2015, 43, 315–322. [Google Scholar] [CrossRef]
- Jafarbegloo, M.; Tarlani, A.; Mesbah, A.W.; Sahebdelfar, S. One-pot synthesis of NiO–MgO nanocatalysts for CO2 reforming of methane: The influence of active metal content on catalytic performance. J. Nat. Gas Sci. Eng. 2015, 27, 1165–1173. [Google Scholar] [CrossRef]
- Odedairo, T.; Chen, J.; Zhu, Z. Metal–support interface of a novel Ni–CeO2 catalyst for dry reforming of methane. Catal. Commun. 2013, 31, 25–31. [Google Scholar] [CrossRef]
- Pompeo, F.; Nichio, N.N.; Ferretti, O.A.; Resasco, D. Study of Ni catalysts on different supports to obtain synthesis gas. Int. J. Hydrog. Energ. 2005, 30, 1399–1405. [Google Scholar] [CrossRef]
- Kroll, V.C.H.; Swaan, H.M.; Lacombe, S.; Mirodatos, C. Methane Reforming Reaction with Carbon Dioxide over Ni/SiO2 Catalyst: II. A Mechanistic Study. J. Catal. 1996, 164, 387–398. [Google Scholar] [CrossRef]
- Gálvez, M.E.; Albarazi, A.; Da Costa, P. Enhanced catalytic stability through non-conventional synthesis of Ni/SBA-15 for methane dry reforming at low temperatures. Appl. Catal. A Gen. 2015, 504, 143–150. [Google Scholar] [CrossRef]
- Zhang, Z.; Verykios, X.E. Carbon dioxide reforming of methane to synthesis gas over Ni/La2O3 catalysts. Appl. Catal. A Gen. 1996, 138, 109–133. [Google Scholar] [CrossRef]
- Kim, S.S.; Lee, S.M.; Won, J.M.; Yang, H.J.; Hong, S.C. Effect of Ce/Ti ratio on the catalytic activity and stability of Ni/CeO2–TiO2 catalyst for dry reforming of methane. Chem. Eng. J. 2015, 280, 433–440. [Google Scholar] [CrossRef]
- Min, J.-E.; Lee, Y.-J.; Park, H.-G.; Zhang, C.; Jun, K.-W. Carbon dioxide reforming of methane on Ni–MgO–Al2O3 catalysts prepared by sol–gel method: Effects of Mg/Al ratios. J. Ind. Eng. Chem. 2015, 26, 375–383. [Google Scholar] [CrossRef]
- Alipour, Z.; Rezaei, M.; Meshkani, F. Effect of Ni loadings on the activity and coke formation of MgO-modified Ni/Al2O3 nanocatalyst in dry reforming of methane. J. Energy Chem. 2014, 23, 633–638. [Google Scholar] [CrossRef]
- Xu, L.; Song, H.; Chou, L. Ordered mesoporous MgO–Al2O3 composite oxides supported Ni based catalysts for CO2 reforming of CH4, Effects of basic modifier and mesopore structure. Int. J. Hydrog. Energ. 2013, 38, 7307–7325. [Google Scholar] [CrossRef]
- Radlik, M.; Adamowska-Teyssier, M.; Krztoń, A.; Kozieł, K.; Krajewski, W.; Turek, W.; Da Costa, P. Dry reforming of methane over Ni/Ce0.62Zr0.38O2 catalysts: Effect of Ni loading on the catalytic activity and on H2/CO production. Comptes Rendus Chim. 2015, 18, 1242–1249. [Google Scholar] [CrossRef]
- Luisetto, I.; Tuti, S.; Battocchio, C.; Lo Mastro, S.; Sodo, A. Ni/CeO2–Al2O3 catalysts for the dry reforming of methane: The effect of CeAlO3 content and nickel crystallite size on catalytic activity and coke resistance. Appl. Catal. A Gen. 2015, 500, 12–22. [Google Scholar] [CrossRef]
- Luengnaruemitchai, A.; Kaengsilalai, A. Activity of different zeolite-supported Ni catalysts for methane reforming with carbon dioxide. Chem. Eng. J. 2008, 144, 96–102. [Google Scholar] [CrossRef]
- Nimwattanakul, W.; Luengnaruemitchai, A.; Jitkarnka, S. Potential of Ni supported on clinoptilolite catalysts for carbon dioxide reforming of methane. Int. J. Hydrog. Energ. 2006, 31, 93–100. [Google Scholar] [CrossRef]
- Jabbour, K.; El Hassan, N.; Davidson, A.; Massiani, P.; Casale, S. Characterizations and performances of Ni/diatomite catalysts for dry reforming of methane. Chem. Eng. J. 2015, 264, 351–358. [Google Scholar] [CrossRef]
- Liu, Y.; He, Z.; Zhou, L.; Hou, Z.; Eli, W. Simultaneous oxidative conversion and CO2 reforming of methane to syngas over Ni/vermiculite catalysts. Catal. Commun. 2013, 42, 40–44. [Google Scholar] [CrossRef]
- Daza, C.E.; Kiennemann, A.; Moreno, S.; Molina, R. Dry reforming of methane using Ni–Ce catalysts supported on a modified mineral clay. Appl. Catal. A Gen. 2009, 364, 65–74. [Google Scholar] [CrossRef]
- Ma, Q.; Wang, D.; Wu, M.; Zhao, T.; Yoneyama, Y.; Tsubaki, N. Effect of catalytic site position: Nickel nanocatalyst selectively loaded inside or outside carbon nanotubes for methane dry reforming. Fuel 2013, 108, 430–438. [Google Scholar] [CrossRef]
- Chen, Y.-G.; Ren, J. Conversion of methane and carbon dioxide into synthesis gas over alumina-supported nickel catalysts. Effect of Ni-Al2O3 interactions. Catal. Lett. 1994, 29, 39–48. [Google Scholar] [CrossRef]
- Bhattacharyya, A.; Chang, V.W. CO2 Reforming of Methane to Syngas: Deactivation Behavior of Nickel Aluminate Spinel Catalysts. In Studies in Surface Science and Catalysis; Delmon, B., Froment, G.F., Eds.; Elsevier: Ostend, Belgium, 1994; pp. 207–213. [Google Scholar]
- Baktash, E.; Littlewood, P.; Schomäcker, R.; Thomas, A.; Stair, P.C. Alumina coated nickel nanoparticles as a highly active catalyst for dry reforming of methane. Appl. Catal. B Environ. 2015, 179, 122–127. [Google Scholar] [CrossRef]
- Zanganeh, R.; Rezaei, M.; Zamaniyan, A. Dry reforming of methane to synthesis gas on NiO–MgO nanocrystalline solid solution catalysts. Int. J. Hydrog. Energ. 2013, 38, 3012–3018. [Google Scholar] [CrossRef]
- Zanganeh, R.; Rezaei, M.; Zamaniyan, A. Preparation of nanocrystalline NiO–MgO solid solution powders as catalyst for methane reforming with carbon dioxide: Effect of preparation conditions. Adv. Powder Technol. 2014, 25, 1111–1117. [Google Scholar] [CrossRef]
- Cavani, F.; Trifirò, F.; Vaccari, A. Hydrotalcite-type anionic clays: Preparation, properties and applications. Catal. Today 1991, 11, 173–301. [Google Scholar] [CrossRef]
- Forano, C.; Costantino, U.; Prévot, V.; Gueho, C.T. Chapter 14.1—Layered Double Hydroxides (LDH). In Developments in Clay Science; Faïza, B., Gerhard, L., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 745–782. [Google Scholar]
- Evans, D.G.; Slade, R.C.T. Structural Aspects of Layered Double Hydroxides. In Layered Double Hydroxides; Duan, X., Evans, D.G., Eds.; Springer: Berlin, Germany, 2006; pp. 1–87. [Google Scholar]
- Rives, V.; Carriazo, D.; Martín, C. Heterogeneous Catalysis by Polyoxometalate-Intercalated Layered Double Hydroxides. In Pillared Clays and Related Catalysts; Gil, A., Korili, A.S., Trujillano, R., Vicente, A.M., Eds.; Springer: New York, NY, USA, 2010; pp. 319–397. [Google Scholar]
- Zhu, Y.; Zhang, S.; Chen, B.; Zhang, Z.; Shi, C. Effect of Mg/Al ratio of NiMgAl mixed oxide catalyst derived from hydrotalcite for carbon dioxide reforming of methane. Catal. Today 2016, 264, 163–170. [Google Scholar] [CrossRef]
- Perez-Lopez, O.W.; Senger, A.; Marcilio, N.R.; Lansarin, M.A. Effect of composition and thermal pretreatment on properties of Ni–Mg–Al catalysts for CO2 reforming of methane. Appl. Catal. A Gen. 2006, 303, 234–244. [Google Scholar] [CrossRef]
- Guo, J.; Lou, H.; Zhao, H.; Chai, D.; Zheng, X. Dry reforming of methane over nickel catalysts supported on magnesium aluminate spinels. Appl. Catal. A Gen. 2004, 273, 75–82. [Google Scholar] [CrossRef]
- Gonzalez, A.R.; Asencios, Y.J.O.; Assaf, E.M.; Assaf, J.M. Dry reforming of methane on Ni-Mg-Al nano-spheroid oxide catalysts prepared by the sol-gel method from hydrotalcite-like precursors. Appl. Surf. Sci. 2013, 280, 876–887. [Google Scholar] [CrossRef]
- Tsyganok, A.I.; Tsunoda, T.; Hamakawa, S.; Suzuki, K.; Takehira, K.; Hayakawa, T. Dry reforming of methane over catalysts derived from nickel-containing Mg-Al layered double hydroxides. J. Catal. 2003, 213, 191–203. [Google Scholar] [CrossRef]
- Lin, X.; Li, R.; Lu, M.; Chen, C.; Li, D.; Zhan, Y.; Jiang, L. Carbon dioxide reforming of methane over Ni catalysts prepared from Ni–Mg–Al layered double hydroxides: Influence of Ni loadings. Fuel 2015, 162, 271–280. [Google Scholar] [CrossRef]
- Daza, C.E.; Moreno, S.; Molina, R. Co-precipitated Ni–Mg–Al catalysts containing Ce for CO2 reforming of methane. Int. J. Hydrog. Energ. 2011, 36, 3886–3894. [Google Scholar] [CrossRef]
- Daza, C.E.; Cabrera, C.R.; Moreno, S.; Molina, R. Syngas production from CO2 reforming of methane using Ce-doped Ni-catalysts obtained from hydrotalcites by reconstruction method. Appl. Catal. A Gen. 2010, 378, 125–133. [Google Scholar] [CrossRef]
- Touahra, F.; Sehailia, M.; Ketir, W.; Bachari, K.; Chebout, R.; Trari, M.; Cherifi, O.; Halliche, D. Effect of the Ni/Al ratio of hydrotalcite-type catalysts on their performance in the methane dry reforming process. Appl. Petrochem. Res. 2016, 6, 1–13. [Google Scholar] [CrossRef]
- Bhattacharyya, A.; Chang, V.W.; Schumacher, D.J. CO2 reforming of methane to syngas: I: Evaluation of hydrotalcite clay-derived catalysts. Appl. Clay Sci. 1998, 13, 317–328. [Google Scholar] [CrossRef]
- Basile, F.; Basini, L.; Amore, M.D.; Fornasari, G.; Guarinoni, A.; Matteuzzi, D.; Pieroc, G.D.; Trifiròa, F.; Vaccaria, A. Ni/Mg/Al Anionic Clay Derived Catalysts for the Catalytic Partial Oxidation of Methane: Residence Time Dependence of the Reactivity Features. J. Catal. 1998, 173, 247–256. [Google Scholar] [CrossRef]
- Dębek, R.; Motak, M.; Duraczyska, D.; Launay, F.; Galvez, M.E.; Grzybek, T.; Da Costa, P. Methane dry reforming over hydrotalcite-derived Ni-Mg-Al mixed oxides: The influence of Ni content on catalytic activity, selectivity and stability. Catal. Sci. Technol. 2016, 6, 6705–6715. [Google Scholar] [CrossRef]
- Djebarri, B.; Gonzalez-Delacruz, V.M.; Halliche, D.; Bachari, K.; Saadi, A.; Caballero, A.; Holgado, J.; Cherifi, O. Promoting effect of Ce and Mg cations in Ni/Al catalysts prepared from hydrotalcites for the dry reforming of methane. React. Kinet. Mech. Catal. 2013, 111, 259–275. [Google Scholar] [CrossRef]
- Dębek, R.; Zubek, K.; Motak, M.; Galvez, M.E.; Da Costa, P.; Grzybek, T. Ni–Al hydrotalcite-like material as the catalyst precursors for the dry reforming of methane at low temperature. Comptes Rendus Chim. 2015, 18, 1205–1210. [Google Scholar] [CrossRef]
- Mette, K.; Kühl, S.; Düdder, H.; Kähler, K.; Tarasov, A.; Muhler, M.; Behrens, M. Stable Performance of Ni Catalysts in the Dry Reforming of Methane at High Temperatures for the Efficient Conversion of CO2 into Syngas. Chemcatchem 2014, 6, 100–104. [Google Scholar] [CrossRef]
- Mette, K.; Kühl, S.; Tarasov, A.; Düdder, H.; Kähler, K.; Muhler, M.; Schloegl, R.; Behrens, M. Redox dynamics of Ni catalysts in CO2 reforming of methane. Catal. Today 2015, 242, 101–110. [Google Scholar] [CrossRef]
- Abdelsadek, Z.; Sehailia, M.; Halliche, D.; Gonzalez-Delacruz, V.M.; Holgado, J.P.; Bachari, K.; Caballero, A.; Cherifi, O. In-situ hydrogasification/regeneration of NiAl-hydrotalcite derived catalyst in the reaction of CO2 reforming of methane: A versatile approach to catalyst recycling. J. CO2 Util. 2016, 14, 98–105. [Google Scholar] [CrossRef]
- Dudder, H.; Kahler, K.; Krause, B.; Mette, K.; Kuhl, S.; Behrens, M.; Scherer, V.; Muhler, M. The role of carbonaceous deposits in the activity and stability of Ni-based catalysts applied in the dry reforming of methane. Catal. Sci. Technol. 2014, 4, 3317–3328. [Google Scholar] [CrossRef]
- Shishido, T.; Sukenobu, M.; Morioka, H.; Furukawa, R.; Shirahase, H.; Takehira, K. CO2 reforming of CH4 over Ni/Mg–Al oxide catalysts prepared by solid phase crystallization method from Mg–Al hydrotalcite-like precursors. Catal. Lett. 2001, 73, 21–26. [Google Scholar] [CrossRef]
- Tan, P.; Gao, Z.; Shen, C.; Du, Y.; Li, X.; Huang, W. Ni-Mg-Al solid basic layered double oxide catalysts prepared using surfactant-assisted coprecipitation method for CO2 reforming of CH4. Chin. J. Catal. 2014, 35, 1955–1971. [Google Scholar] [CrossRef]
- Tsyganok, A.I.; Suzuki, K.; Hamakawa, S.; Takehira, K.; Hayakawa, T. Mg–Al Layered Double Hydroxide Intercalated with [Ni(EDTA)]2− Chelate as a Precursor for an Efficient Catalyst of Methane Reforming with Carbon Dioxide. Catal. Lett. 2001, 77, 75–86. [Google Scholar] [CrossRef]
- Dębek, R.; Zubek, K.; Motak, M.; Da Costa, P.; Grzybek, T. Effect of nickel incorporation into hydrotalcite-based catalyst systems for dry reforming of methane. Res. Chem. Intermed. 2015, 41, 9485–9495. [Google Scholar] [CrossRef]
- Hou, Z.; Yashima, T. Meso-porous Ni/Mg/Al catalysts for methane reforming with CO2. Appl. Catal. A Gen. 2004, 261, 205–209. [Google Scholar] [CrossRef]
- Li, N.; Shen, C.; Tan, P.; Zou, Z.; Huang, W. Effect of phase transformation on the stability of Ni-Mg-Al catlyst for dry reforming of methane. Indian J. Chem. 2015, 54A, 1198–1205. [Google Scholar]
- Rives, V. Characterisation of layered double hydroxides and their decomposition products. Mater. Chem. Phys. 2002, 75, 19–25. [Google Scholar] [CrossRef]
- Lwin, Y.; Yarmo, M.A.; Yaakob, Z.; Mohamad, A.B.; Ramli Wan Daud, W. Synthesis and characterization of Cu–Al layered double hydroxides. Mater. Res. Bull. 2001, 36, 193–198. [Google Scholar] [CrossRef]
- Kannan, S.; Dubey, A.; Knozinger, H. Synthesis and characterization of CuMgAl ternary hydrotalcites as catalysts for the hydroxylation of phenol. J. Catal. 2005, 231, 381–392. [Google Scholar] [CrossRef]
- Velu, S.; Suzuki, K.; Kapoor, M.P.; Tomura, S.; Ohashi, F.; Osaki, T. Effect of Sn Incorporation on the Thermal Transformation and Reducibility of M(II)Al-Layered Double Hydroxides [M(II) = Ni or Co]. Chem. Mater. 2000, 12, 719–730. [Google Scholar] [CrossRef]
- Yu, M.; Zhu, Y.-A.; Lu, Y.; Tong, G.; Zhu, K.; Zhou, X. The promoting role of Ag in Ni-CeO2 catalyzed CH4-CO2 dry reforming reaction. Appl. Catal. B Environ. 2015, 165, 43–56. [Google Scholar] [CrossRef]
- Daza, C.E.; Gallego, J.; Moreno, J.A.; Mondragón, F.; Moreno, S.; Molina, R. CO2 reforming of methane over Ni/Mg/Al/Ce mixed oxides. Catal. Today 2008, 133–135, 357–366. [Google Scholar] [CrossRef]
- Daza, C.E.; Gallego, J.; Mondragón, F.; Moreno, S.; Molina, R. High stability of Ce-promoted Ni/Mg–Al catalysts derived from hydrotalcites in dry reforming of methane. Fuel 2010, 89, 592–603. [Google Scholar] [CrossRef]
- Daza, C.E.; Moreno, S.; Molina, R. Ce-incorporation in mixed oxides obtained by the self-combustion method for the preparation of high performance catalysts for the CO2 reforming of methane. Catal. Commun. 2010, 12, 173–179. [Google Scholar] [CrossRef]
- Lino, A.V.P.; Assaf, E.M.; Assaf, J.M. Hydrotalcites derived catalysts for syngas production from biogas reforming: Effect of nickel and cerium load. Catal. Today. [CrossRef]
- Ren, H.-P.; Song, Y.-H.; Wang, W.; Chen, J.-G.; Cheng, J.; Jiang, J.; Liu, Z.T.; Hao, Z.; Lu, J. Insights into CeO2-modified Ni–Mg–Al oxides for pressurized carbon dioxide reforming of methane. Chem. Eng. J. 2015, 259, 581–593. [Google Scholar] [CrossRef]
- Dębek, R.; Radlik, M.; Motak, M.; Galvez, M.E.; Turek, W.; Da Costa, P.; Grzybek, T. Ni-containing Ce-promoted hydrotalcite derived materials as catalysts for methane reforming with carbon dioxide at low temperature—On the effect of basicity. Catal. Today 2015, 257, 59–65. [Google Scholar] [CrossRef]
- Dębek, R.; Gramatyka, A.; Motak, M.; Da Costa, P. Produkcja gazu syntezowego w reakcji suchego reformingu metanu na katalizatorach hydrotalkitowych, Syngas production by dry reforming of methane over hydrotalcite-derived catalysts. Przem. Chem. 2014, 93, 2026–2032. [Google Scholar]
- Dębek, R.; Galvez, M.E.; Launay, F.; Motak, M.; Grzybek, T.; Da Costa, P. Low temperature dry methane reforming over Ce, Zr and CeZr promoted Ni–Mg–Al hydrotalcite-derived catalysts. Int. J. Hydrog. Energ. 2016, 41, 11616–11623. [Google Scholar] [CrossRef]
- Yu, X.; Wang, N.; Chu, W.; Liu, M. Carbon dioxide reforming of methane for syngas production over La-promoted NiMgAl catalysts derived from hydrotalcites. Chem. Eng. J. 2012, 209, 623–632. [Google Scholar] [CrossRef]
- Serrano-Lotina, A.; Rodríguez, L.; Muñoz, G.; Martin, A.J.; Folgado, M.A.; Daza, L. Biogas reforming over La-NiMgAl catalysts derived from hydrotalcite-like structure: Influence of calcination temperature. Catal. Commun. 2011, 12, 961–967. [Google Scholar] [CrossRef]
- Serrano-Lotina, A.; Rodríguez, L.; Muñoz, G.; Daza, L. Biogas reforming on La-promoted NiMgAl catalysts derived from hydrotalcite-like precursors. J. Power Sources 2011, 196, 4404–4410. [Google Scholar] [CrossRef]
- Liu, H.; Wierzbicki, D.; Debek, R.; Motak, M.; Grzybek, T.; Da Costa, P.; Galvez, M. La-promoted Ni-hydrotalcite-derived catalysts for dry reforming of methane at low temperatures. Fuel 2016, 182, 8–16. [Google Scholar] [CrossRef]
- Lucrédio, A.F.; Assaf, J.M.; Assaf, E.M. Reforming of a model sulfur-free biogas on Ni catalysts supported on Mg(Al)O derived from hydrotalcite precursors: Effect of La and Rh addition. Biomass Bioenergy 2014, 60, 8–17. [Google Scholar] [CrossRef]
- Li, C.; Tan, P.J.; Li, X.D.; Du, Y.L.; Gao, Z.H.; Huang, W. Effect of the addition of Ce and Zr on the structure and performances of Ni-Mo/CeZr-MgAl(O) catalysts for CH4-CO2 reforming. Fuel Process. Technol. 2015, 140, 39–45. [Google Scholar] [CrossRef]
- Tsyganok, A.I.; Inaba, M.; Tsunoda, T.; Uchida, K.; Suzuki, K.; Takehira, K.; Hayakawa, T. Rational design of Mg–Al mixed oxide-supported bimetallic catalysts for dry reforming of methane. Appl. Catal. A Gen. 2005, 292, 328–343. [Google Scholar] [CrossRef]
- Long, H.; Xu, Y.; Zhang, X.; Hu, S.; Shang, S.; Yin, Y.; Dai, X. Ni-Co/Mg-Al catalyst derived from hydrotalcite-like compound prepared by plasma for dry reforming of methane. J. Energy Chem. 2013, 22, 733–739. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, H.; Dalai, A.K. Effects of metal content on activity and stability of Ni-Co bimetallic catalysts for CO2 reforming of CH4. Appl. Catal. A Gen. 2008, 339, 121–129. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Catalyst | Method of Hydrotalcite Synthesis | Cations in HTs Layers | Ni/Mg or Ni Loading 1 | M2+/M3+ | Calcination Conditions | Reaction Conditions | Conversion 2 | H2/CO (-) | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Temp. (°C) | CH4/CO2 | GHSV (h−1) | TOS (h) | CH4 (%) | CO2 (%) | ||||||||
| NiMgAl HTs | Co-precipitation at constant pH | Ni2+, Mg2+, Al3+ | 1, 0.2 | 2, 3 | nd | 815 | 1.25 | 720 | 250 | 70 | 52 | 1.0 | [62] |
| NiMgAl HT | Co-precipitation at constant pH | Ni2+, Mg2+, Al3+ | 1/6 | 2.45 | 700, 900 °C for 14 h | 750 | 1/1 | nd | nd | 29 | 50 | nd | [63] |
| NiMgAl HTs | Co-precipitation at constant pH | Ni2+, Mg2+, Al3+ | 3, 6, 9, 12, 15, 18 1 | 3 | 800 °C for 5 h | 600 | 1/1 3 | 60,000 | 25 | 41 | 50 | 0.7 | [58] |
| NiMgAl and NiAl HTs | Co-precipitation at constant pH | Ni2+, Mg2+, Al3+ | 3, 1, 0.33, 0.18, 0.06 | 3 | 550 °C for 4 h in air | 550 | 1/1 3 | 20,000 | 24 | 43 | 40 | 1.1 | [64] |
| NiAl and NiMgAl HTs | Co-precipitation at constant pH | Ni2+, Al3+ | - | 2 | 800 °C for 6 h in air | 750 | 1/1 3 | 3 × 105 | 8 | 68 | 90 | 0.7 | [65] |
| NiMgAl HTs | Co-precipitation at constant pH | Ni2+, Mg2+, Al3+ | 0.5, 1, 2, 5 | 0.4, 0.9, 2 | 400, 600, 800 °C for 6 h in air | 650 | ½ 3 | 45,000 4 | nd | 83 | 38 | 1.2 | [54] |
| NiAl HT | Co-precipitation at constant pH | Ni2+, Al3+ | 63 1 | 4 | 550 °C for 4 h in air | 550 | 2/13 | 20,000 | 4 | 48 | 54 | 2.6 | [66] |
| NiMgAl HT | Co-precipitation at constant pH | Ni2+, Mg2+, Al3+ | 2.94 | 2 | 350, 600, 800, 1000 °C | 900 | 32/40 3 | nd | 10 | 74 | nd | nd | [67,68] |
| NiMgAl HTs | Co-precipitation at constant pH | Ni2+, Mg2+, Al3+ | 1/5, 1/3, 1 | 6, 4, 2, 2/3, 2/5 | 500 °C for 10 h in air | 800 | 1/1 | 80,000 | 30 | 82 | 88 | nd | [53] |
| NiAl HTs | Co-precipitation at constant pH | Ni2+, Al3+ | - | 2, 3, 5, 8, 10 | 300, 400, 500, 600, 700, 800 °C for 6 h | 700 | 1/1 3 | nd | 10 | 88 | 88 | 1.0 | [61] |
| NiAl HT | Co-precipitation at constant pH | Ni2+, Al3+ | 44 1 | 2 | 450 °C for 4 h | 700 | 1/1 3 | nd | 30 | 94 | 94 | 0.9 | [69] |
| NiMgAl HTs | Co-precipitation at constant pH | Ni2+, Mg2+, Al3+ | 1, 5, 25, 50 4 | 2 | 600 °C for 3 h in air | 900 | 32/40 3 | nd | 10 | 67 | nd | 0.8 | [70] |
| Type of Catalyst | Method of Hydrotalcite Synthesis | Cations in HTs Layers | Ni/Mg or Ni Loading 1 | M2+/M3+ | Calcination Conditions | Reaction Conditions | Conversion 2 | H2/CO (-) | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Temp. (°C) | CH4/CO2 | GHSV (h−1) | TOS (h) | CH4 (%) | CO2 (%) | ||||||||
| Ni supported on MgAl HTs | Co-precipitation at constant pH | Mg2+, Al3+ | 1, 3, 5, 10, 15 1 | nd | 900°C for 5 h | 750 | 1/1 | 50,000 | 10 | 85 | 96 | 0.9 | [55] |
| NiMgAl HTs; Ni supported on MgAl HT | Co-precipitation at constant pH | Ni2+, Mg2+, Al3+ | 1/2 | 3 | 650 and 850 °C for 14 h in air | 800 | 1/1 3 | 54,000 | 6 | 94 | nd | nd | [71] |
| Impregnation of Ni2+ on MgAl HT | Mg2+, Al3+ | 25.1 1 | 3 | 92 | nd | nd | |||||||
| NiMgAl HTs | Surfactant assisted co-precipitation | Ni2+, Mg2+, Al3+ | 10 1 | 3 | 700 °C for 6 h | 800 | 1/1 | 60,000 | 35 | 47 | 62 | nd | [72] |
| Ni introduced into MgAl HTs | Co-precipitation in [Ni(EDTA)]2- | Mg2+, Al3+ | - | 3 | 500 °C for 16 h in air | 800 | 1/1 3 | nd | 150 | 98 | 95 | 1.0 | [73] |
| Ni introduced into MgAl HTs and NiMgAl HTs | Co-precipitation in [Ni(EDTA)]2– | Mg2+, Al3+ | 1/7 | 3.5 | 500 °C for 16 h in air | 800 | 1/1 3 | nd | 6 | 97 | 95 | 1.0 | [57] |
| Anion exchange | Mg2+, Al3+ | 1/11 | 3.3 | 97 | 95 | 1.0 | |||||||
| Reconstruction | Mg2+, Al3+ | 1/11 | 3.6 | 97 | 94 | 1.0 | |||||||
| Co-precipitation | Ni2+, Mg2+, Al3+ | 1/5 | 2.5 | 98 | 96 | 1.0 | |||||||
| NiMgAl HTs | Sol-gel method | Ni2+, Mg2+, Al3+ | 4, 15, 19 1 | nd | 500, 650 °C for 5 h | 800 | 1/1 3 | 2.94 × 10−5 | 8 | 96 | 94 | nd | [56] |
| NiMgAl HTs | Sol-gel method | Ni2+, Mg2+, Al3+ | 15 1 | 0.25–19 | 750 °C for 5 h in air | 800 | 1/1 3 | 36,000 | 40 | 84 | 89 | nd | [33] |
| Co-precipitation at constant pH | Ni2+, Mg2+, Al3+ | 2 | 84 | 89 | nd | ||||||||
| NiAl HT and Ni supported on MgAl HT | Co-precipitation at constant pH | Ni2+, Al3+ | 63 1 | 4 | 550 °C for 4 h in air | 550 °C | 2/1 | 20,000 | 1 | 48 | 57 | 2.7 | [74] |
| Adsorption of [Ni(EDTA)]2− | Mg2+, Al3+ | 0.8 1 | 3 | 25 | 38 | 1.6 | |||||||
| NiMgAl HTs | Co-precipitation at constant pH | Ni2+, Mg2+, Al3+ | 10 1 | 1.5–9 | 800 °C for 3 h | 800 | 1/1 | nd | 4 | 86 | 87 | nd | [75] |
| NiMgAl HTs | Co-precipitation at constant pH | Ni2+, Mg2+, Al3+ | 10 1 | 3 | 500–800 °C for 6 h | 800 | 1/1 | 8000 | 2000 | 92 | 95 | 0.9 | [76] |
| Type of Catalyst/Method 1 | Calcination Conditions 2 | Effect of Calcination Temperature | Ref. |
|---|---|---|---|
| NiAl HT/CP | 300, 400, 500, 600, 700 and 800 °C for 6 h | Calcination temperature <500 °C has minimal effect of activity; Increase in calcination temperature resulted in increased activity and stability | [61] |
| NiMgAl HT/CP | 400, 600, 800 °C for 6 h | Calcination temperature had a small influence on the activity and selectivity | [54] |
| NiMgAl HTsSG | 500, 650 °C for 5 h | Moderate calcination temperatures prevent formation of spinel phase. Higher calcination temperature resulted in the increased stability | [56] |
| NiMgAl HT/CP | 500, 600, 700, 800 °C for 6 h | No significant effect of calcination temperature on performance in DRM was observed | [76] |
| NiMgAl HT/CP | 350, 600, 800, 1000 °C | Calcination at 800 and 1000 °C resulted in formation of spinel phase. The optimal calcination temperature was selected to be 600 °C | [67,68] |
| Method of Hydrotalcite Synthesis | Method of Ce Introduction into HT Structure | Ni/Mg | Ce Content (wt %) | Calcination Conditions | Ref. |
|---|---|---|---|---|---|
| Co-precipitation in solution of Na2CO3, [Ce(EDTA)]− or [Ni(EDTA)]2− | At co-precipitation stage in form of Ce3+ cations or [Ce(EDTA)]− | 1/2 | 5 | 500 °C for 16 h in air | [82] |
| Co-precipitation at constant pH | Reconstruction method with solution of [Ce(EDTA)]− | 2 | 0, 1, 3, 5, 10 | 500 °C for 16 h in air | [83] |
| Co-precipitation at constant pH | Reconstruction method with solution of [Ce(EDTA)]− | 2 | 0, 1, 3, 10 | 500 °C for 16 h in air | [60] |
| Co-precipitation at constant pH | At co-precipiatation stage in form of Ce3+ cations | 2 | 24, 9, 4, 1,5 1 | 500 °C for 16 h in air | [59] |
| Co-precipitation at constant pH and self-combustion method | Reconstruction method with solution of [Ce(EDTA)]− | 2 | 3 | 500 °C for 16 h in air | [84] |
| Co-precipitation at constant pH | At co-precipitation stage in form of Ce3+ cations and by impregnation | 20 2 | 1–10 1 | 500 °C for 4 h | [86] |
| Co-precipitation at constant pH | Adsorption from the solution of [Ce(EDTA)]− | 0.6 2 | 1.15 | 550 °C for 4 h | [88] |
| Co-precipitation at constant pH | Adsorption from the solution of [Ce(EDTA)]− | 1/3 | 3.7 | 550 °C for 4 h | [87,89] |
| Co-precipitation at constant pH | At co-precipiatation stage in form of Ce3+ cations | 10, 25 2 | 5 | 650 °C for 5 h | [85] |
| Promoter | Catalyst | Promoter Loading (wt %)/Method 1 | Effect of Addition | Ref. |
|---|---|---|---|---|
| La | NiMgAl-HT | 0, 0.04, 0.11, 0.18 2/CP | Increased stability and activity | [90] |
| La | NiMgAl-HT | 1.1, 2/CP | Increased stability and decreased activity | [91,92] |
| La | NiMgAl-HT | 0, 1, 2, 4/CP | Increased activity, selectivity and stability | [93] |
| La | 10 wt % Ni impregnated on MgAl-HT | 10/IMP | Increased reducibility of Ni; promotes carbon formation | [94] |
| Rh | 1/IMP | |||
| CeZrO2 | NiMoMgAl-HT | 0, 5, 10, 15, 20/CP | Increased catalyst activity; promotes reducibility of Ni | [95] |
| Ru | 5 wt % Ni supported on MgAl-HT | 0.1/RE | Inhibits sintering; increased activity and stability | [96] |
| Co | NiMgAl and NiCoMgAl-HTs | 1, 4 3/CP | Increased activity and stability | [97] |
| Co | NiCoMgAl-HTs | 2.76–12.9/CP | nd 4 | [98] |
| Zr | NiMgAl-HT | 3/CP | Decreased reducibility; formation of small Ni crystallites; increased stability | [89] |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dębek, R.; Motak, M.; Grzybek, T.; Galvez, M.E.; Da Costa, P. A Short Review on the Catalytic Activity of Hydrotalcite-Derived Materials for Dry Reforming of Methane. Catalysts 2017, 7, 32. https://doi.org/10.3390/catal7010032
Dębek R, Motak M, Grzybek T, Galvez ME, Da Costa P. A Short Review on the Catalytic Activity of Hydrotalcite-Derived Materials for Dry Reforming of Methane. Catalysts. 2017; 7(1):32. https://doi.org/10.3390/catal7010032
Chicago/Turabian StyleDębek, Radosław, Monika Motak, Teresa Grzybek, Maria Elena Galvez, and Patrick Da Costa. 2017. "A Short Review on the Catalytic Activity of Hydrotalcite-Derived Materials for Dry Reforming of Methane" Catalysts 7, no. 1: 32. https://doi.org/10.3390/catal7010032
APA StyleDębek, R., Motak, M., Grzybek, T., Galvez, M. E., & Da Costa, P. (2017). A Short Review on the Catalytic Activity of Hydrotalcite-Derived Materials for Dry Reforming of Methane. Catalysts, 7(1), 32. https://doi.org/10.3390/catal7010032