The photocatalysts of the present study were doped with platinum using incipient impregnation, wet impregnation and a sol-gel method. Pt loadings were confirmed using XRF with deviations from nominal values not exceeding 10% in any case. The TiO2 semiconductors synthesized and doped with Pt exhibit a light grey colour for small Pt loadings (<1.00 wt %) and a dark grey colour for high Pt concentrations (>1.00 wt %). These photocatalysts were characterized using BET analysis, UV spectroscopy with diffuse reflectance, and X-ray diffraction (XRD). Furthermore, a near UV lamp was employed during the runs in the Photo-CREC-Water II Reactor. Various methodologies used for photocatalyst preparation as well as for hydrogen production experiments are described in the upcoming sections of this manuscript.
2.2. Photocatalytic Reactor
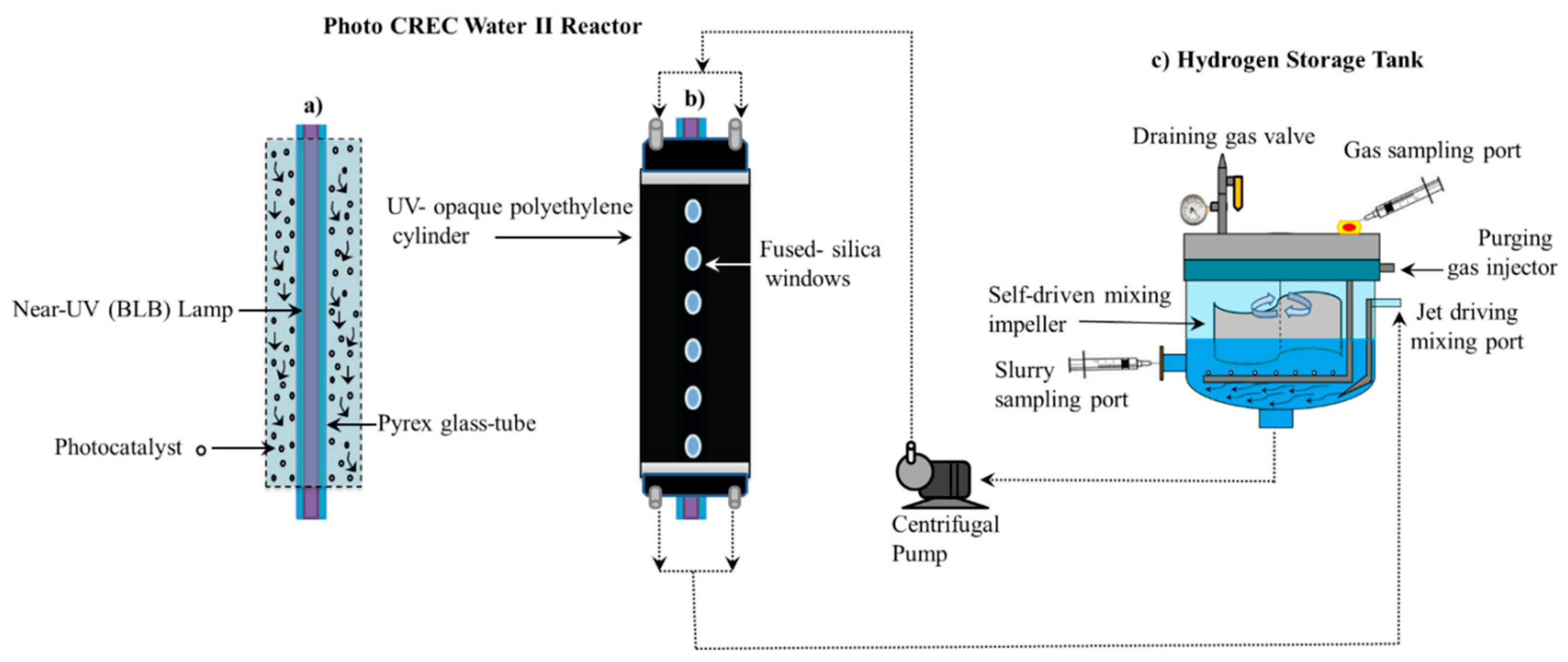
Figure 2 reports a schematic diagram of the Photo-CREC Water II Reactor and its accessories for hydrogen production used in the present study. This modified Photo-CREC Water II Reactor is a “well mixed” batch unit that produces hydrogen.
Figure 2 shows the overall unit configuration: a sealed stirred tank chamber connected in series with a tubular photocatalytic reactor. The Photo-CREC Water II Reactor includes the additional following components: (i) a BLB Lamp; (ii) a Pyrex glass tube; (iii) a UV-opaque polyethylene cylinder; (iv) fused-silica windows; (v) a centrifugal pump; (vi) a H
2 storing/mixing tank; (vii) a gas sampling port; (viii) a slurry sampling port; (ix) a purging gas injector; (x) a jet driving mixing port; (xi) a self-driven mixing impeller and (xii) a draining gas valve.
The experimental runs of the present study were performed by employing the following: (a) 6 L of distillate deionized water; (b) 0.90 g of photocatalyst and (c) 2 M of H
2SO
4 to adjust the solution pH to 4.00 ± 0.05. As well, the near UV lamps of the present study were characterized using a Solatell Spectroradiometer capable of measuring UV intensities in the 1 × 10
−7 to 2 × 10
−2 W/cm
2 range. This was used to determine the energy flux of the BLB lamp absorbed in the Photo-CREC Water II Reactor unit. Additional details regarding lamp characterization and macroscopic balances are reported in
Appendix B.
Table 1 reports the flux of absorbed photons for each photocatalyst using the macroscopic balance methodology described in Guayaquil-Sosa et al. [
8].
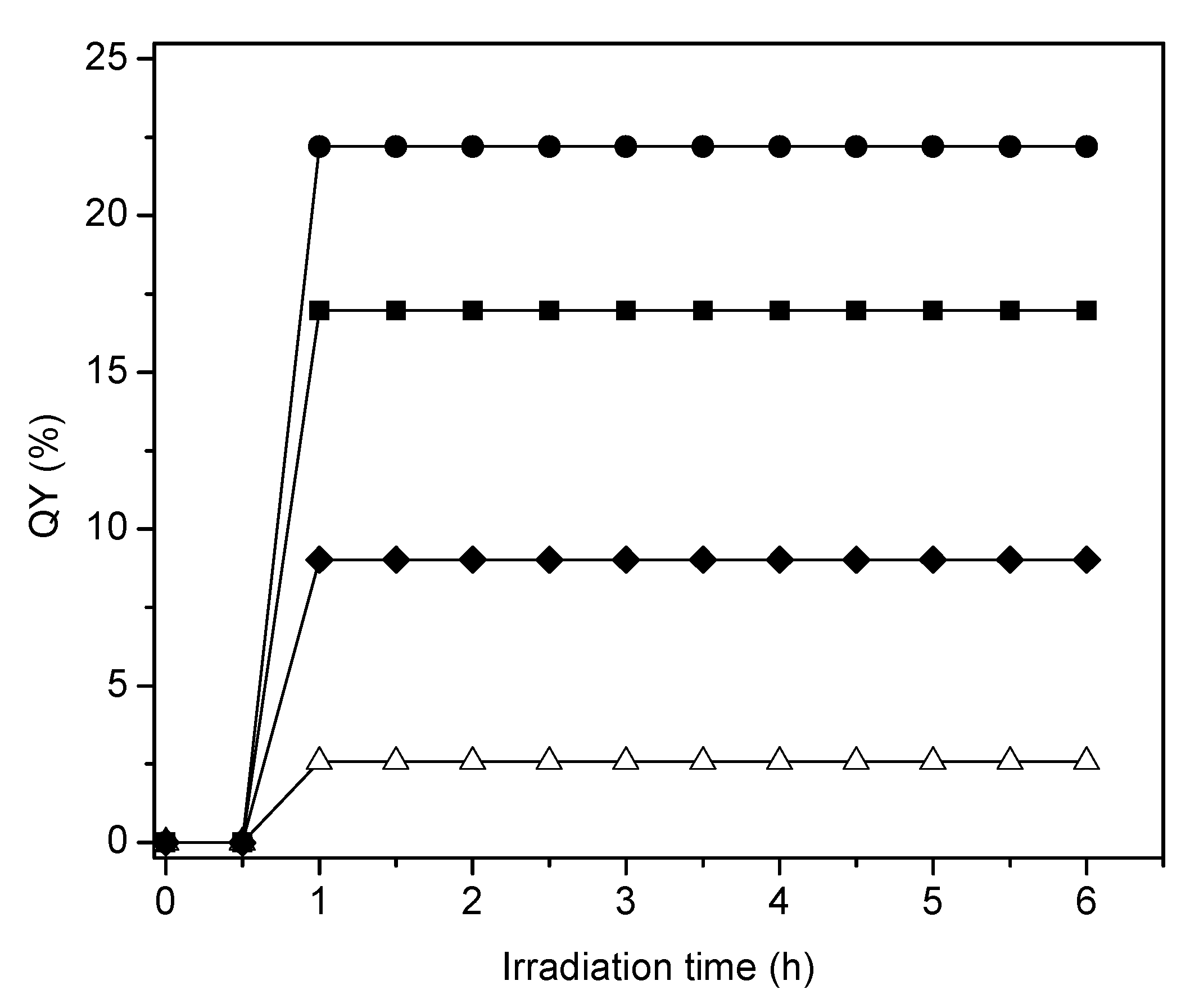
Table 1 shows that there is no significant difference between the photon absorption efficiencies of the various photocatalysts prepared either via incipient and wet impregnation. These absorption efficiencies remain in the 84–87% range. On the other hand, one can observe that the sol-gel synthesized mesoporous Pt photocatalysts displayed slightly higher efficiencies, with these small differences being attributed to the different surface morphology properties of the synthesized photocatalysts.
2.4. Photocatalyst Characterisation
Three photocatalyst characterisation techniques were employed in this study: (a) Specific surface area determined using a Micromeritics, ASAP 2010 unit, through nitrogen adsorption; (b) The band gap energy (Ebg) obtained from diffuse reflectance measurements (Cary 500 UV-Vis NIR Varian spectrophotometer) and (c) The X-ray diffraction (XRD) developed by employing a diffractometer RIGAKU Ultima IV and a multi-purpose diffractometer.
It is, in this respect, anticipated that semiconductors with larger specific surface areas doped with novel metals may lead to a higher density of metal active sites. Hence, this could enhance hydrogen evolution. Thus, specific surface areas were calculated using the Brunauer–Emmett–Teller (BET) method while the pore volume distributions were measured with the Barrett–Joyner–Halenda (BJH) model.
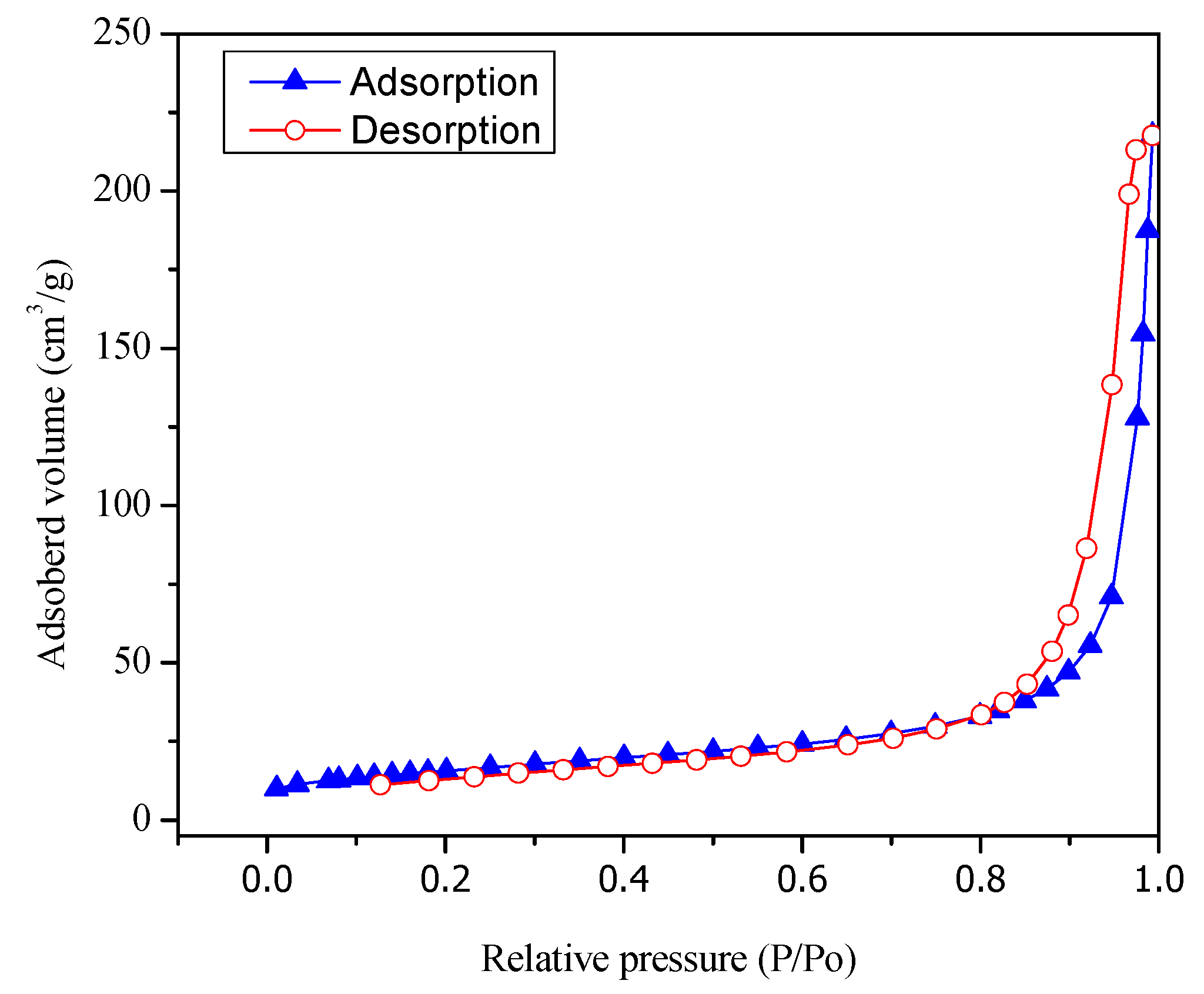
Figure 3 reports both adsorption and desorption isotherms type V for the Pt–TiO
2 prepared via wet impregnation with 1.70 wt % Pt loading.
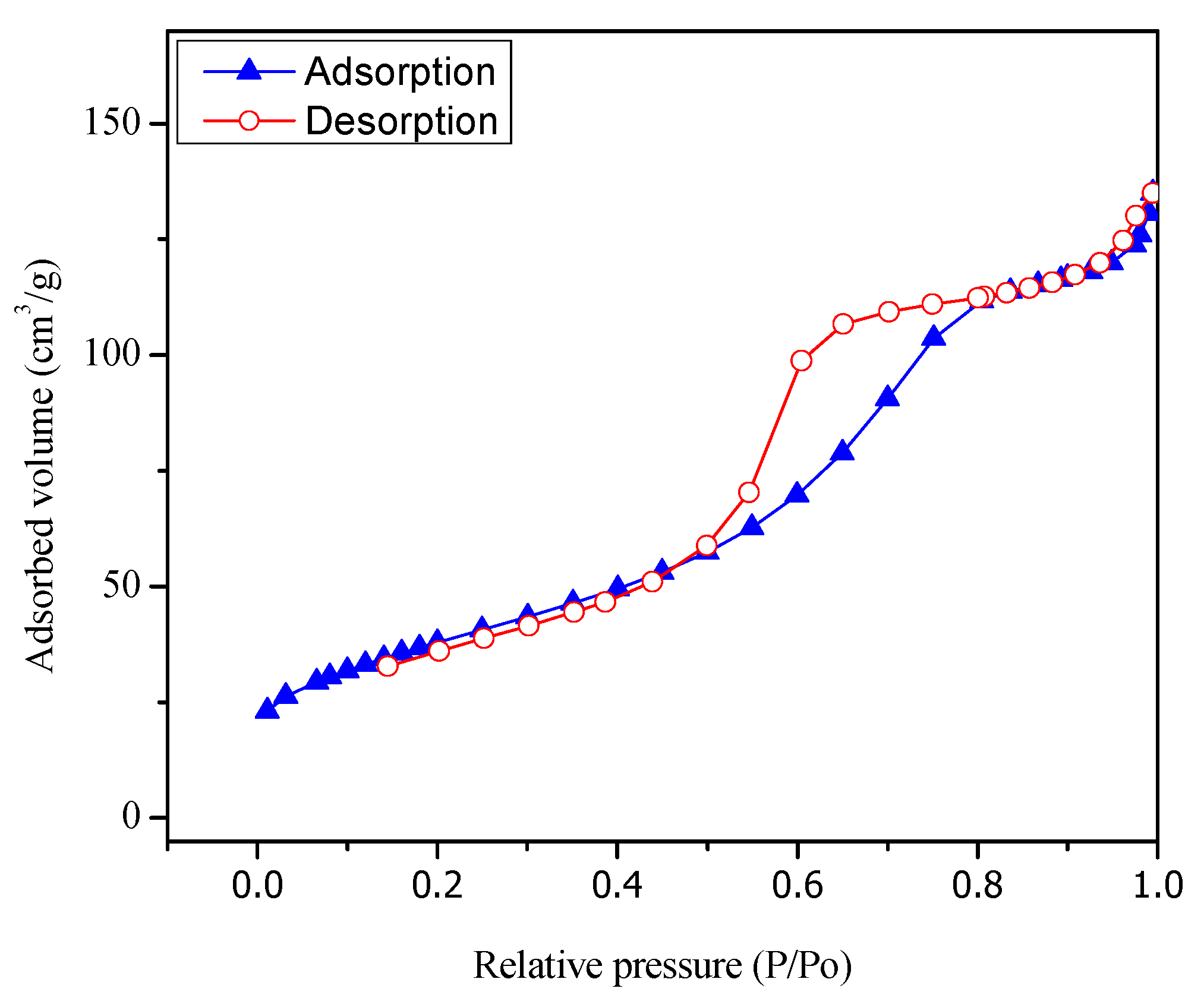
Figure 4 shows the adsorption isotherm for the sol-gel A synthesized TiO
2 with 1.70 wt % Pt. One can notice that the sol-gel A synthesized semiconductors display a type IV isotherm.
Table 2 reports the specific surface area of the photocatalysts synthesized by different methods.
One can also observe a 52 m2/g for the specific surface area of the Degussa P25 impregnated via incipient wetness with a Pt precursor. This specific surface area increases progressively to 116 and 150 m2/g for sol-gel A and sol-gel B, respectively. These findings point towards higher promoted specific surface areas for platinum-sol-gel prepared photocatalysts.
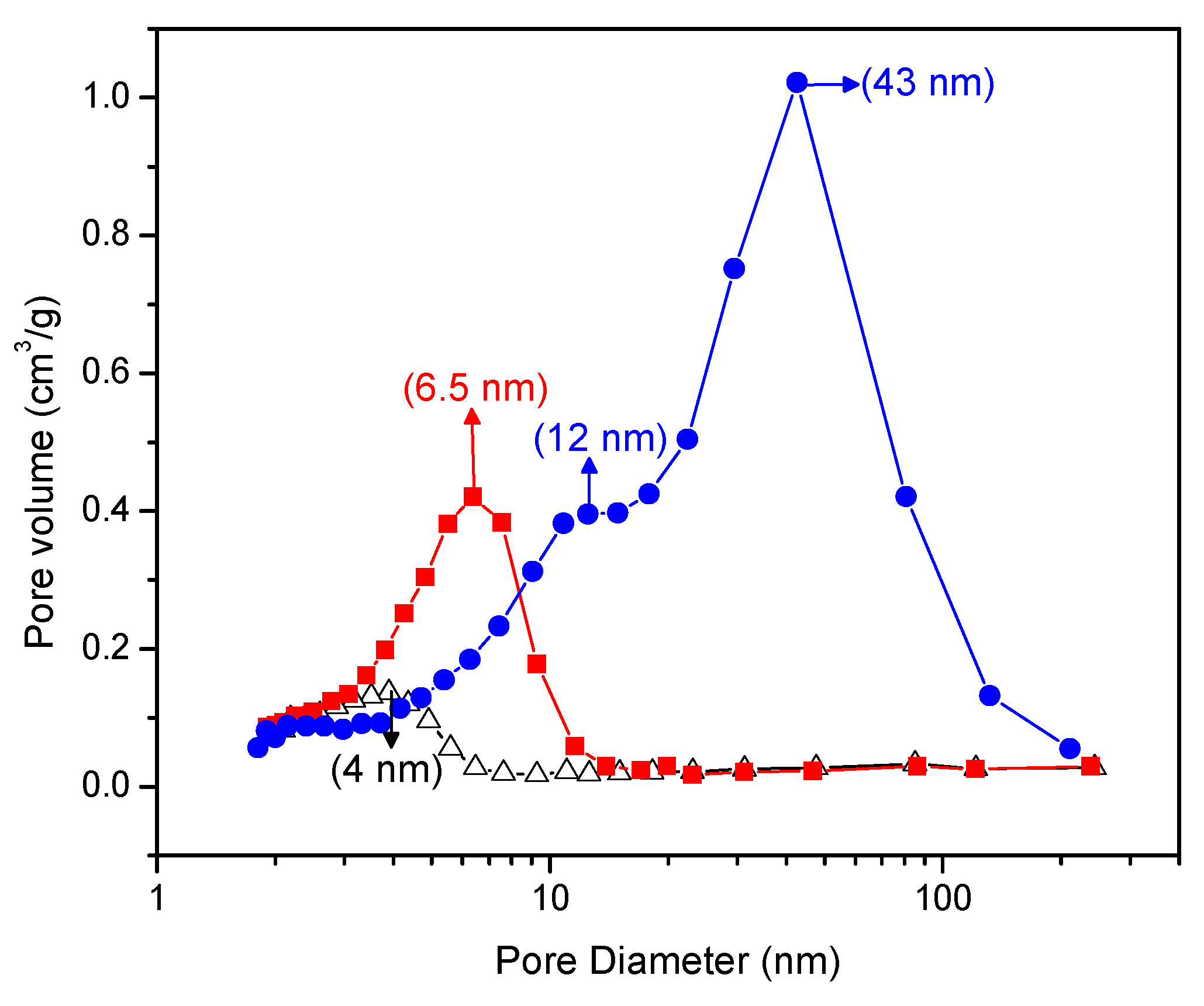
Figure 5 gives a Barrett–Joyner–Halenda plot that shows the pore size distribution of the Pt–TiO
2. One can clearly see in this figure that the doped Pt–TiO
2 obtained via incipient wet impregnation and the sol-gel A and sol-gel B TiO
2 doped with Pt differ in both pore size distribution and pore volume.
In this respect, and as shown in
Figure 5, the Pt–TiO
2 obtained via incipient impregnation shows a 4.0 nm average pore size, while the Pt added on sol-gel prepared via method A displays a 6.5 nm average pore size.
Furthermore, the Pt-doped photocatalyst synthesized using sol-gel method B shows a bimodal pore size distribution with dominant 12.0 nm and 43.0 nm pore sizes. As reported in
Figure 5, one can notice for the Pt on sol-gel prepared photocatalysts, and especially for the Pt on sol-gel prepared by method B, there is an increased volume fraction in the mesoporous pore range.
The band gap energy can be estimated using the Kubelka–Munk equation and the Tauc plot, as described in Guayaquil et al. [
8].
Figure 6 reports the Tauc plots for sol-gel materials with different platinum loadings.
Based on the red line slope extrapolation in
Figure 6, one can calculate the band gap energy (refer to the caption of
Figure 6) with the higher platinum loading photocatalysts yielding reduced band gaps. These band gaps range from 3.20 eV (Pristine TiO
2 via sol-gel A and B) to 2.70 eV (TiO
2-1.70 wt % Pt). Additionally, one can see that the Pt doped on TiO
2 via sol-gel yields semiconductors with the lowest band gap. This reduction of the band gap energy allows one to speculate about the enhanced activation of these semiconductors under visible light.
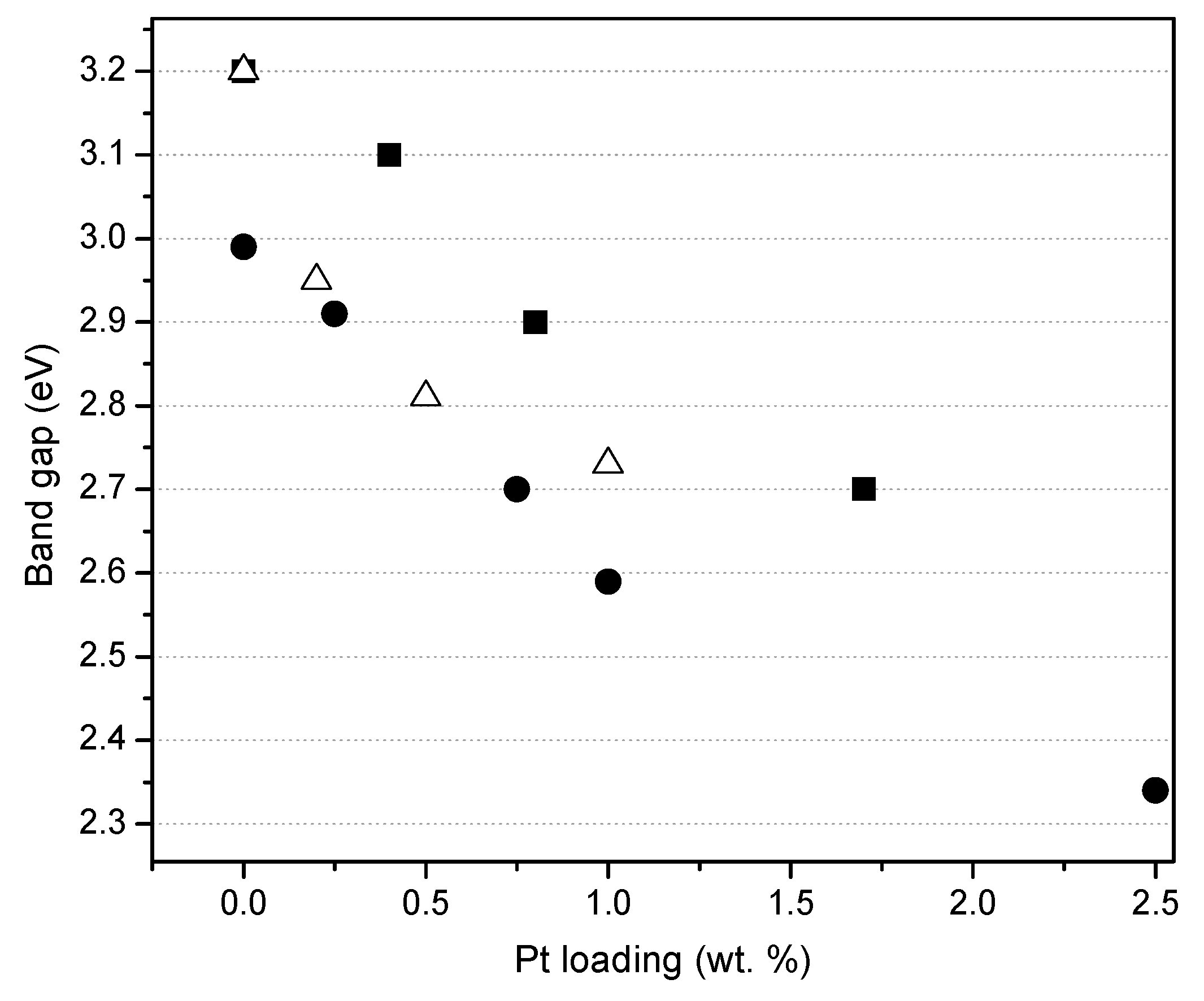
Figure 7 reports the effect of the platinum loading on the photocatalyst band gap, for the photocatalysts prepared by sol-gel. One can observe that the band gap for the Degussa P25 impregnated via incipient wetness with a Pt precursor, remains in the 3.10–3.20 eV range. Moreover, Pt–TiO
2 photocatalysts being prepared using both sol-gel methods leads to a significant reduction of the band gap from 3.20 to 2.70 eV as a function of the platinum loading.
Thus, it is confirmed, as reported in
Figure 7, that for the various platinum-loaded TiO
2 photocatalysts of the present study, Pt has a consistent beneficial effect, reducing the band gap energy.
Figure 8 reports a typical XRD diffractogram for TiO
2-based materials prepared by either wet or incipient impregnation. In addition, one can recognize the characteristic peaks of the two crystalline phases of titanium dioxide (anatase and rutile), with platinum also being included.
Figure 7 reports a change of the different XRD peaks for the wet and incipient platinum impregnated TiO
2–Degussa P25. One can notice that both photocatalysts display rutile (110) crystal plane and that this given the characteristic 27° Bragg angle band. One can also observe, as reported in
Table 3, that for the various sol-gel Pt–TiO
2, there is no recordable 27° Bragg angle. As a result, this shows that the various synthesized sol-gel Pt–TiO
2 are free of rutile (110) facet.
2.5. Photocatalytic Reaction Mechanism for Hydrogen Production
Experiments of hydrogen production using heterogeneous photocatalysis were carried out using the modified Photo-CREC Water II Reactor. There was a minimum of three repeats for each experimental condition considered. Hydrogen generation runs were performed at room temperature and pressure, under an inert argon atmosphere, with a 2.00% v/v ethanol concentration, and with a pH of 4.00 ± 0.05.
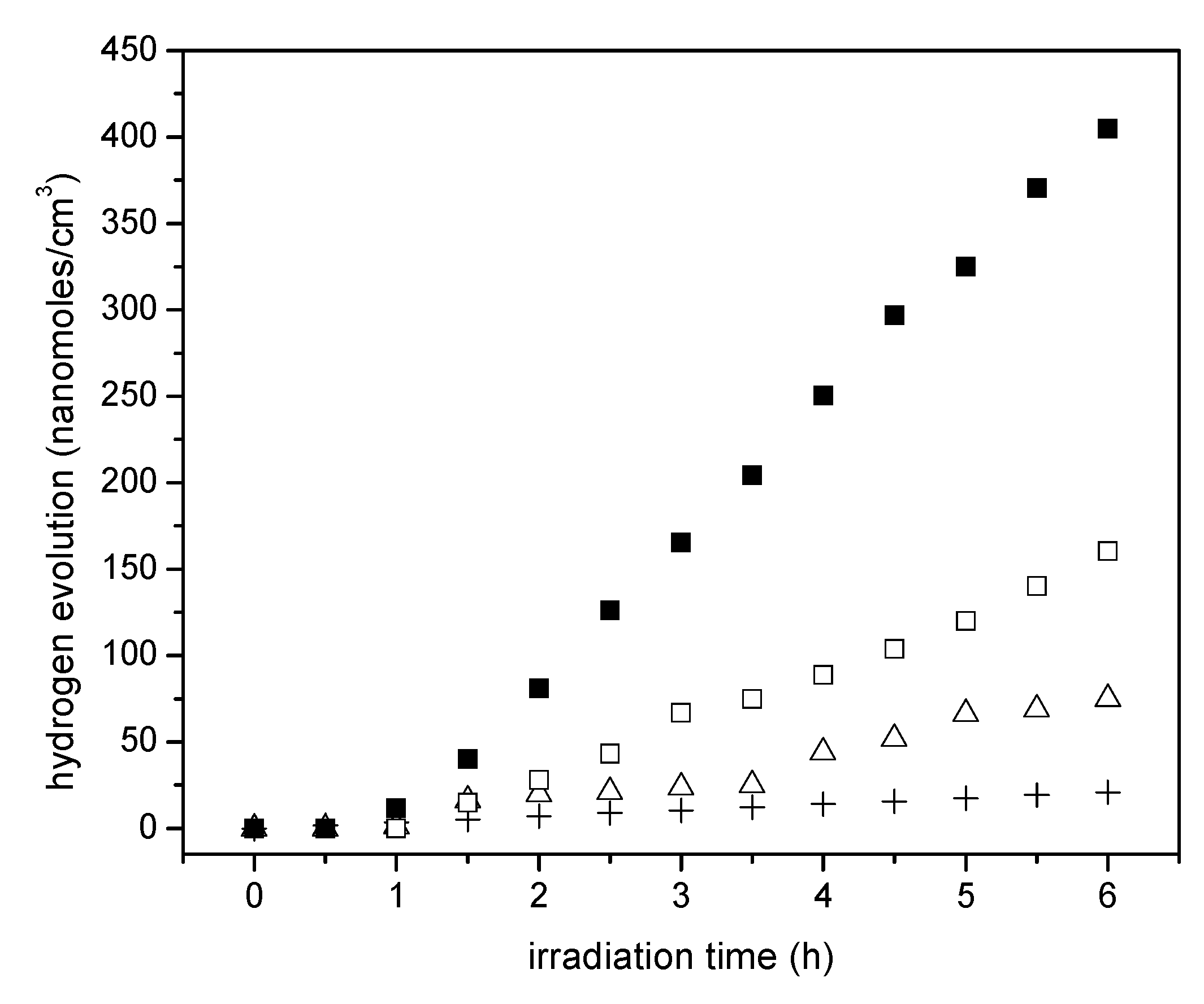
Figure 9 reports the cumulative hydrogen produced using the wet impregnated photocatalyst with platinum loadings in the 0.42 to 1.70 wt % range. Photocatalyst loading in all experiments was 0.15 g/L.
It can be noticed in
Figure 9 that the cumulative hydrogen production increases both with irradiation time and platinum loading. Thus, it appears that augmenting the Pt loadings from 0.82 wt % to 1.70 wt % leads to higher Pt surface density sites and provides increased electron storage capacity. This augmented electron storage capacity helps in reducing electron-hole recombination, contributing to H
+ conversion into H
•.
Furthermore, one can observe from these experiments that there is a consistent linear trend with irradiation time and the cumulative hydrogen being produced. This shows zero-order hydrogen formation kinetics with no detectable activity decay. Thus, it can be concluded that all the prepared photocatalysts are very stable, with performance unaffected by the extent of irradiation.
Figure 10 reports the cumulative hydrogen production for TiO
2 prepared using sol-gel method A. One can observe an increase of hydrogen production when using TiO
2 prepared via the sol-gel A method and compared to that obtained with Degussa P25. Furthermore, one can notice a further enhancement of the hydrogen production with the Pt–TiO
2 prepared via sol-gel A with 1.70 wt % of Pt loadings. This cumulative hydrogen production is again of zero order in all cases, with no observable photocatalyst deactivation.
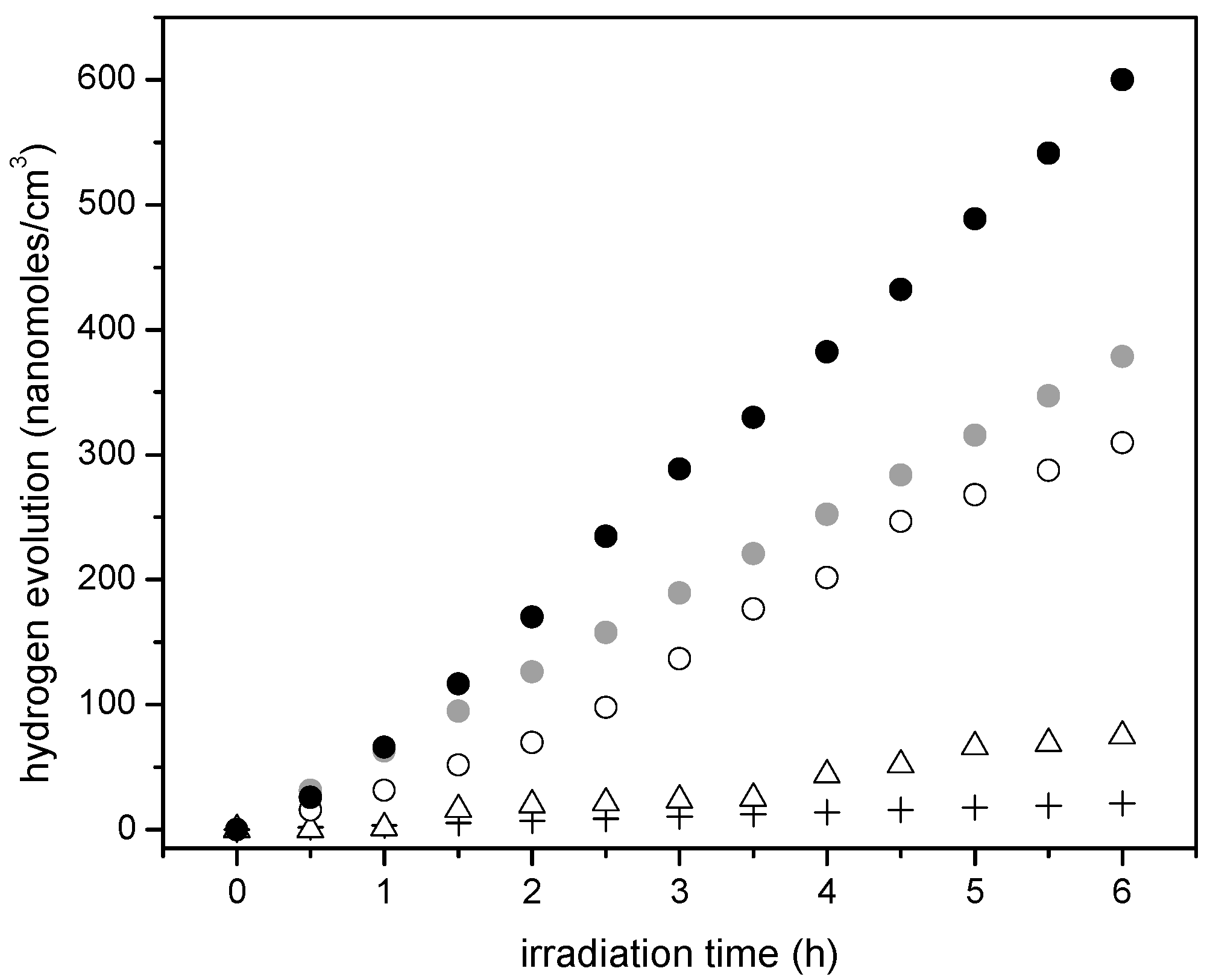
Figure 11 shows the cumulative hydrogen production for the Pt–TiO
2 synthesized photocatalysts using the sol-gel Method B. One can recognize that the observed linear trend is consistent with the photocatalyst prepared using the sol-gel Method A and with respect to Degussa P25.
Additionally, one can notice that Pt addition leads to a further improvement of the hydrogen production. Hence, consistent increase of hydrogen production with Pt loadings is observed. This cumulative hydrogen production is again of zero order with no observable photocatalyst deactivation.
If one compares the cumulative hydrogen production of the 1.70 wt % Pt–TiO
2 of
Figure 9 with respect to that of
Figure 10, one can observe that sol-gel A displayed a mildly increased hydrogen production versus the one prepared via incipient impregnation: from 350 nanomoles/cm
3 to 400 nanomoles/cm
3.
Furthermore, if one considers the hydrogen production when using the 1.00 wt % Pt–TiO
2 sol-gel B (refer to
Figure 11), one can see that this photocatalyst yields essentially the same amount of hydrogen as the one with a higher Pt loading (1.70 wt % Pt–TiO
2 sol-gel Method A, refer to
Figure 10). As well, after 5 h of irradiation, a further increase of Pt up to 2.50 wt % in sol-gel B gives a valuable 600 nanomoles/cm
3 of cumulative hydrogen.
2.6. Hydrogen Formation with Ethanol Scavenger
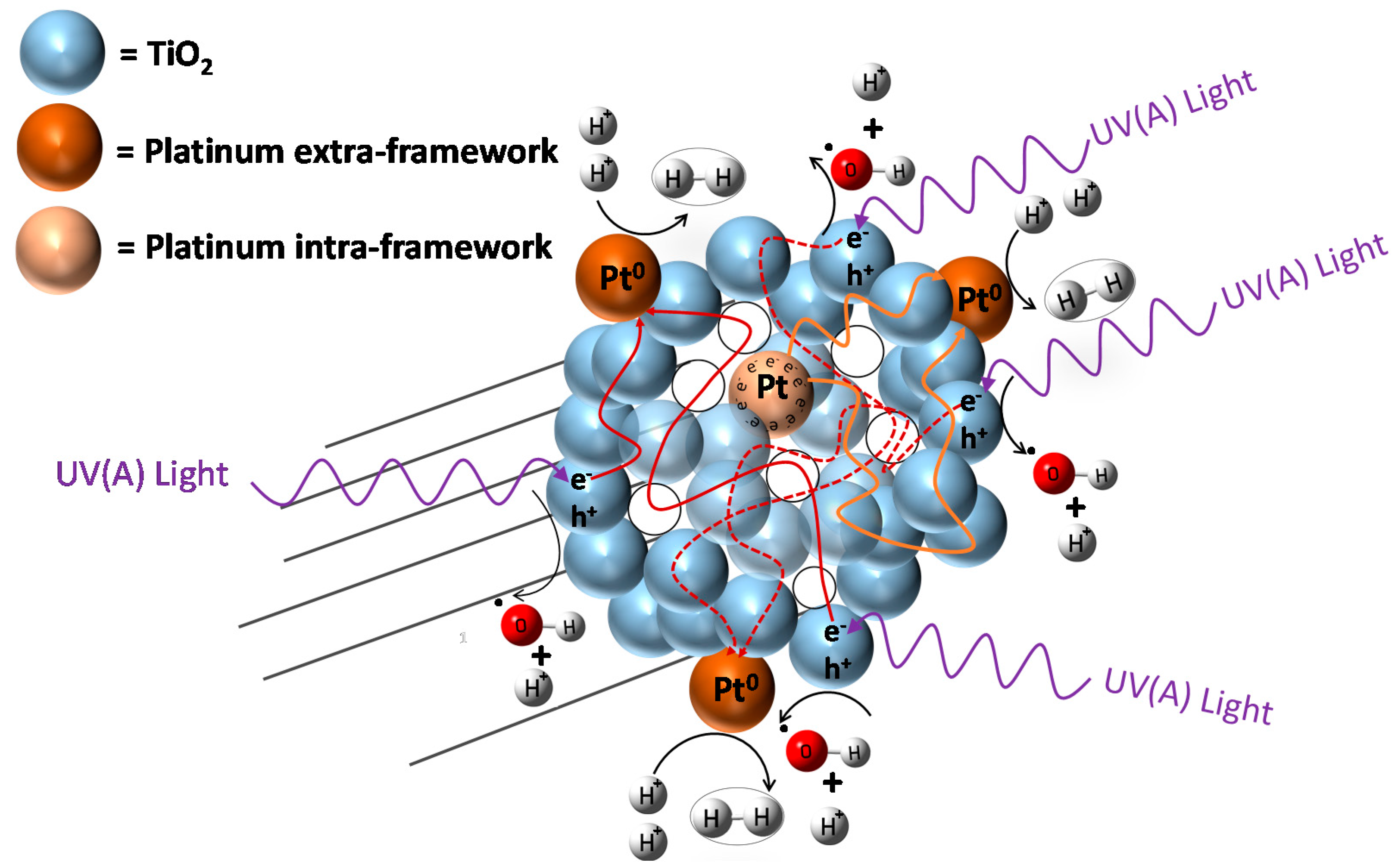
Hydrogen formation with the consumption of an ethanol scavenger, using a Pt–TiO2 photocatalyst, can be described as the contribution of two photocatalyst sites: (a) a TiO2 site promoting oxidation reactions; and (b) a TiO2 site enhanced by Pt promoting hydrogenation (reduction) reactions. Hence, both oxidation and reduction networks are of series-parallel type. This series-parallel network character can be assigned to: (a) the variability of irradiated photons and thus the h+ changing density on TiO2; and (b) the changing extent of stored electron density in the Pt sites.
Therefore, as described in
Figure 12, the resulting series-parallel network is of the redox type [
10]. Included in this process is the formation of hydrogen peroxide. Additional details of various oxidation and reduction steps are provided in
Table 4 and
Table 5, respectively.
Figure 13 reports the progressive and consistent methane formation for various Pt-doped photocatalysts prepared using incipient impregnation, wet impregnation and sol-gel. The Degussa P25 photocatalyst without Pt addition is reported as a reference.
One can see in
Figure 13 that methane formation augments yet tends to stabilize with irradiation time. This is a consistent trend for all the photocatalysts of the present study. Thus, these findings highlight the photocatalytic reduction character of the proposed reaction network. Furthermore, it is observed that 1.70 wt % wet impregnation, and 1.70 wt % Pt sol-gel A photocatalysts yield comparable methane concentrations. These methane concentrations are however, surpassed when 2.50 wt % Pt on TiO
2 (sol-gel B) is considered. This case shows an enhanced hydrogenating activity at higher Pt loadings, with this being consistent with the results reported in
Figure 10.
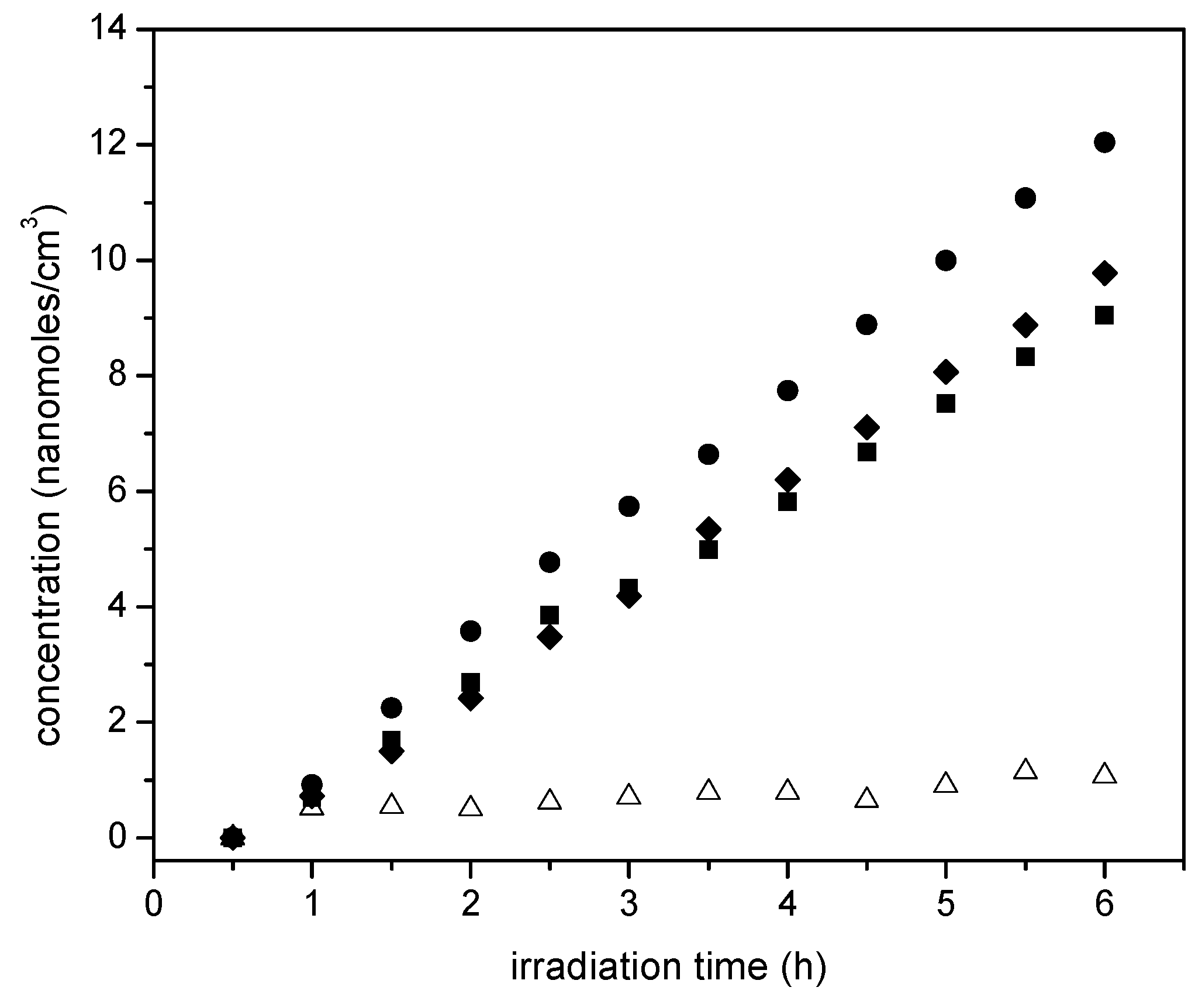
Figure 14 reports ethane cumulative formation with irradiation time for the various photocatalysts of the present study.
Figure 14 shows that ethane increases linearly with irradiation time. Thus, it appears that ethane is a main reduction final product of the proposed series-parallel network. Again, here the highest ethane concentration levels were obtained using the sol-gel B with the 2.50 wt % Pt loadings. In other words, it can be concluded that the photocatalytic conversion of the ethanol scavenger is significantly affected by several reduction steps leading to the formation of both methane and ethane.
On the other hand,
Figure 14 reports the acetaldehyde obtained, with acetaldehyde formation increasing progressively with irradiation time. This described trend was observed consistently for all the studied photocatalysts with added Pt.
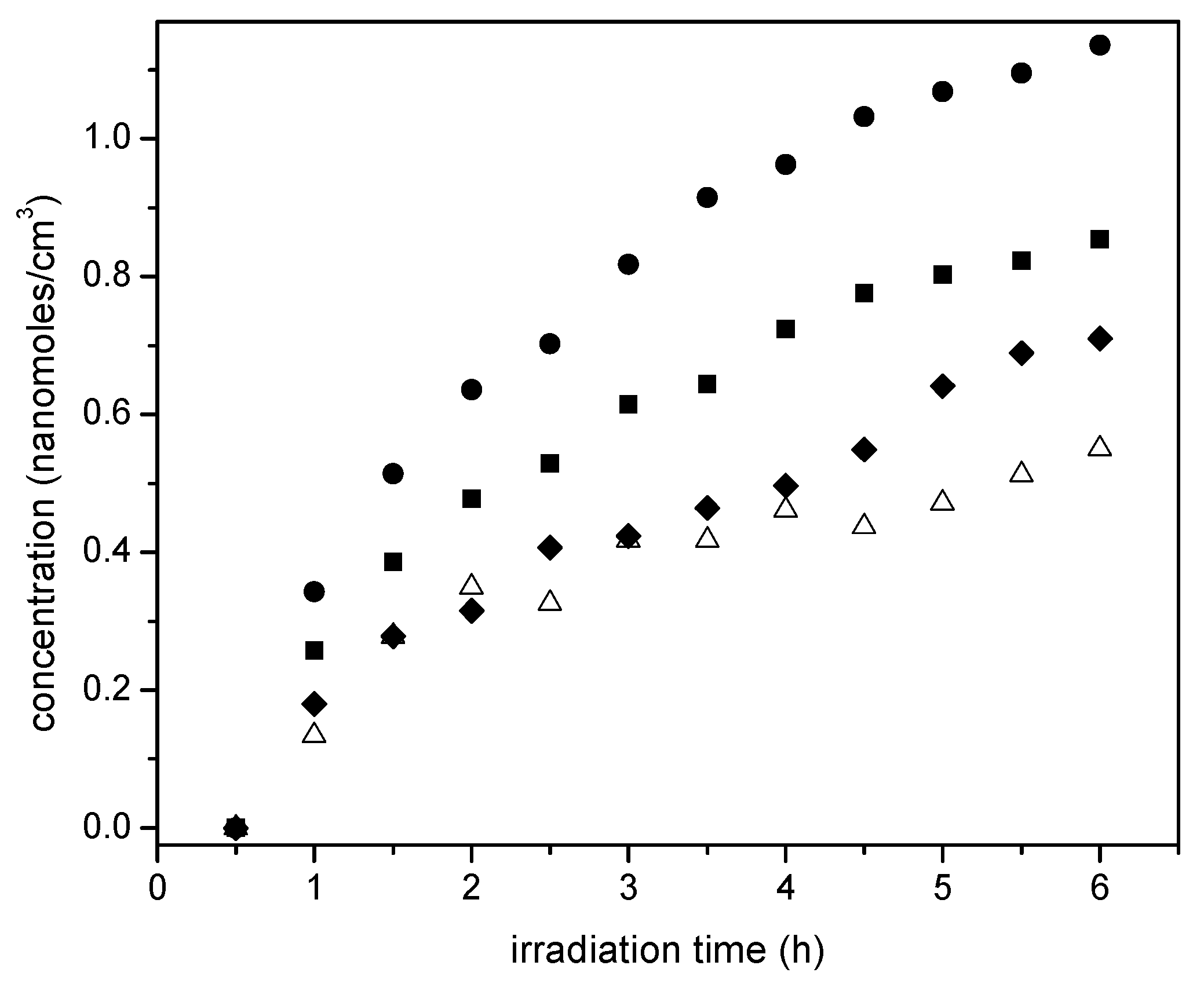
Thus, as shown in
Figure 15, photocatalysis with loaded Pt leads to the oxidation of the ethanol scavenger. One should notice in this respect, that acetaldehyde is a characteristic representative species of primary oxidation products of the network oxidation branch, as described in
Table 4. One can notice in
Figure 15 that the 1.70 wt % Pt-sol-gel A yields the highest acetaldehyde concentrations, consistent with higher overall photocatalytic activity.
In order to validate the proposed redox mechanism, various detectable chemical species (methane, ethane, acetaldehyde, ethanol, hydrogen peroxide, hydrogen) were identified and quantified both in the gas phase as well in the liquid phase. This was done at various stages (every 30 min of the 6-h irradiation period). This approach was consistently used in all runs utilizing the various photocatalysts of the present study.
Furthermore,
Figure 16 reports carbon dioxide concentrations augmenting steadily with irradiation. This carbon dioxide presence in the product gases points to a complete oxidation of some ethanol contained carbons during the photocatalytic reaction.
One can notice in
Figure 16 the formation of CO
2 even at the early stages of irradiation. This observation, combined with the concurrent formation of acetaldehyde at short irradiation times, points towards a series-parallel network for the photoconversion of water and air pollutants as reported by our research group in earlier studies [
5,
9].
Furthermore, and to establish the total amount of CO
2 at every stage of the irradiation process, vapour–liquid equilibrium calculations for CO
2 shall be considered, as described in
Appendix E.
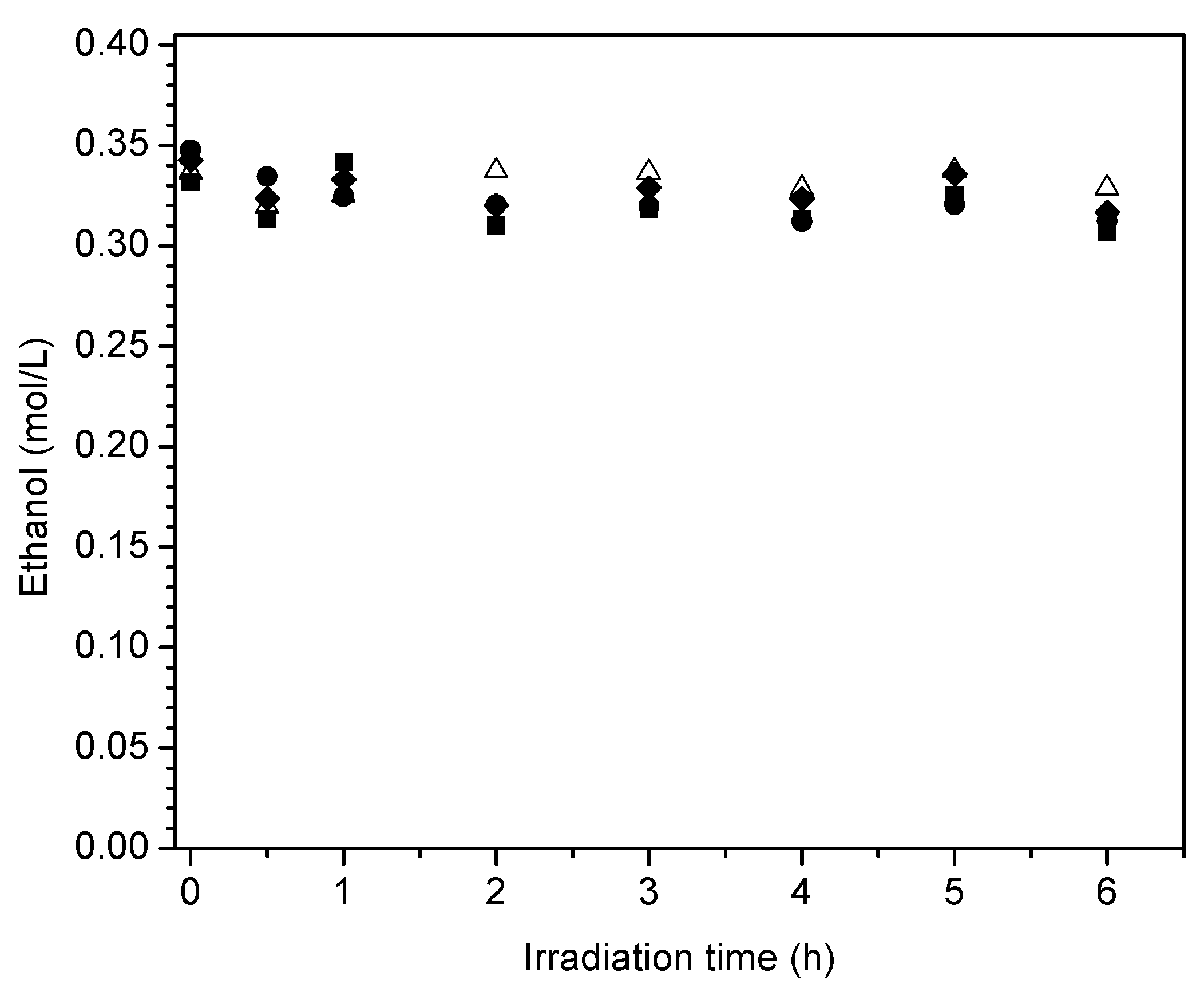
Figure 17 reports the ethanol scavenger concentration changes in the liquid phase with radiation time. These changes of ethanol with irradiation time are reported for the various Pt-doped photocatalysts of the present study.
One can observe in
Figure 17 a progressive and mild decline of the ethanol scavenger concentration with irradiation time. This tendency for net ethanol to decrease is in line with an oxidation–reduction network where ethanol may be consumed and formed simultaneously.























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}