Racemization of Serine Residues Catalyzed by Dihydrogen Phosphate Ion: A Computational Study


Abstract
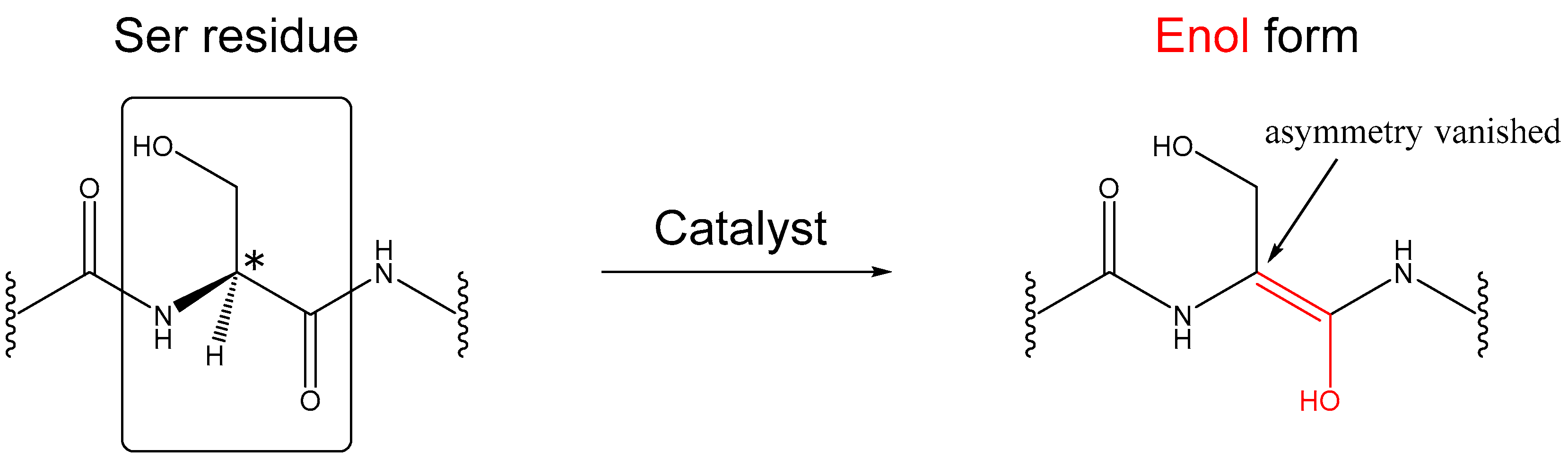
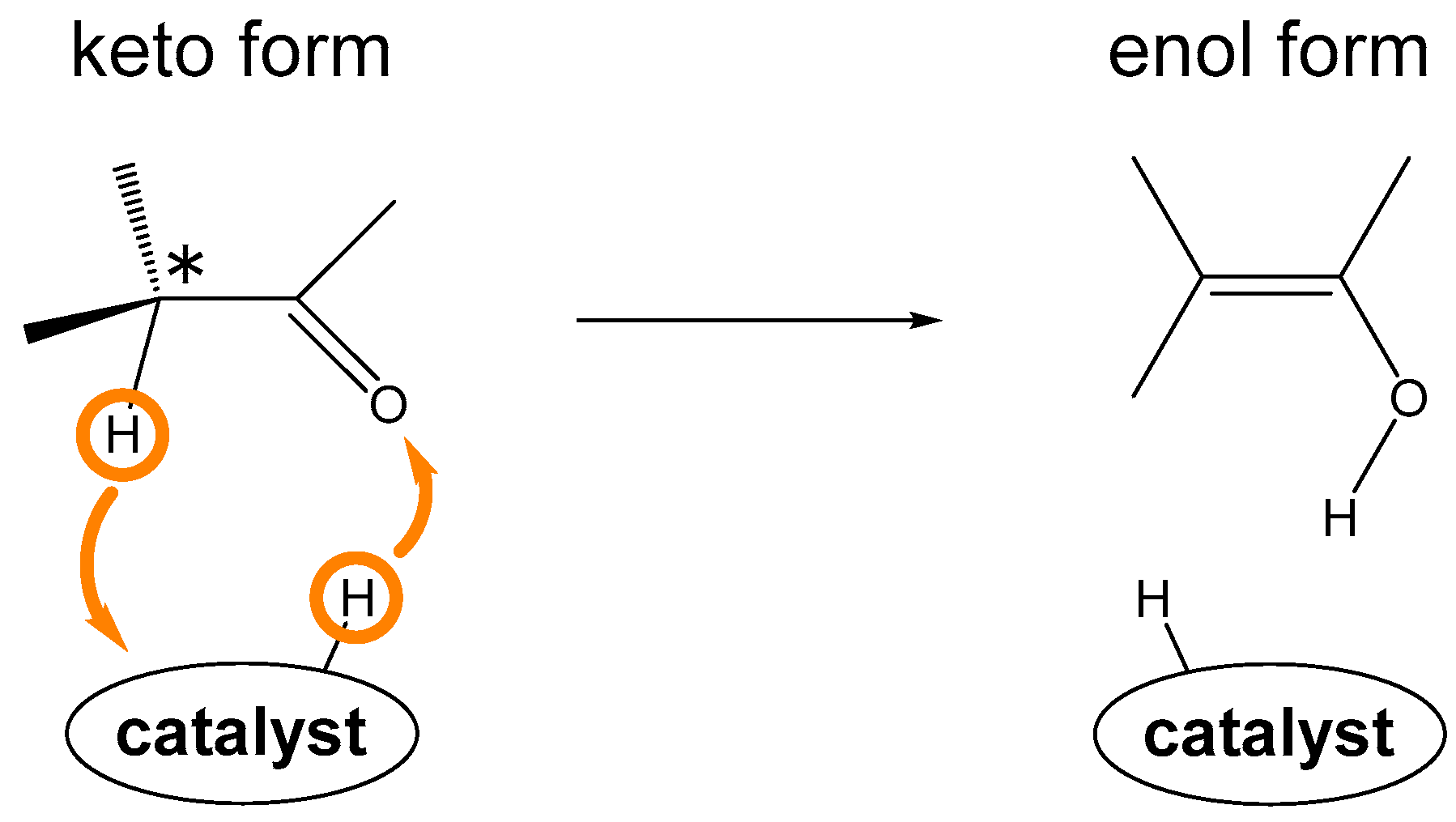
:1. Introduction
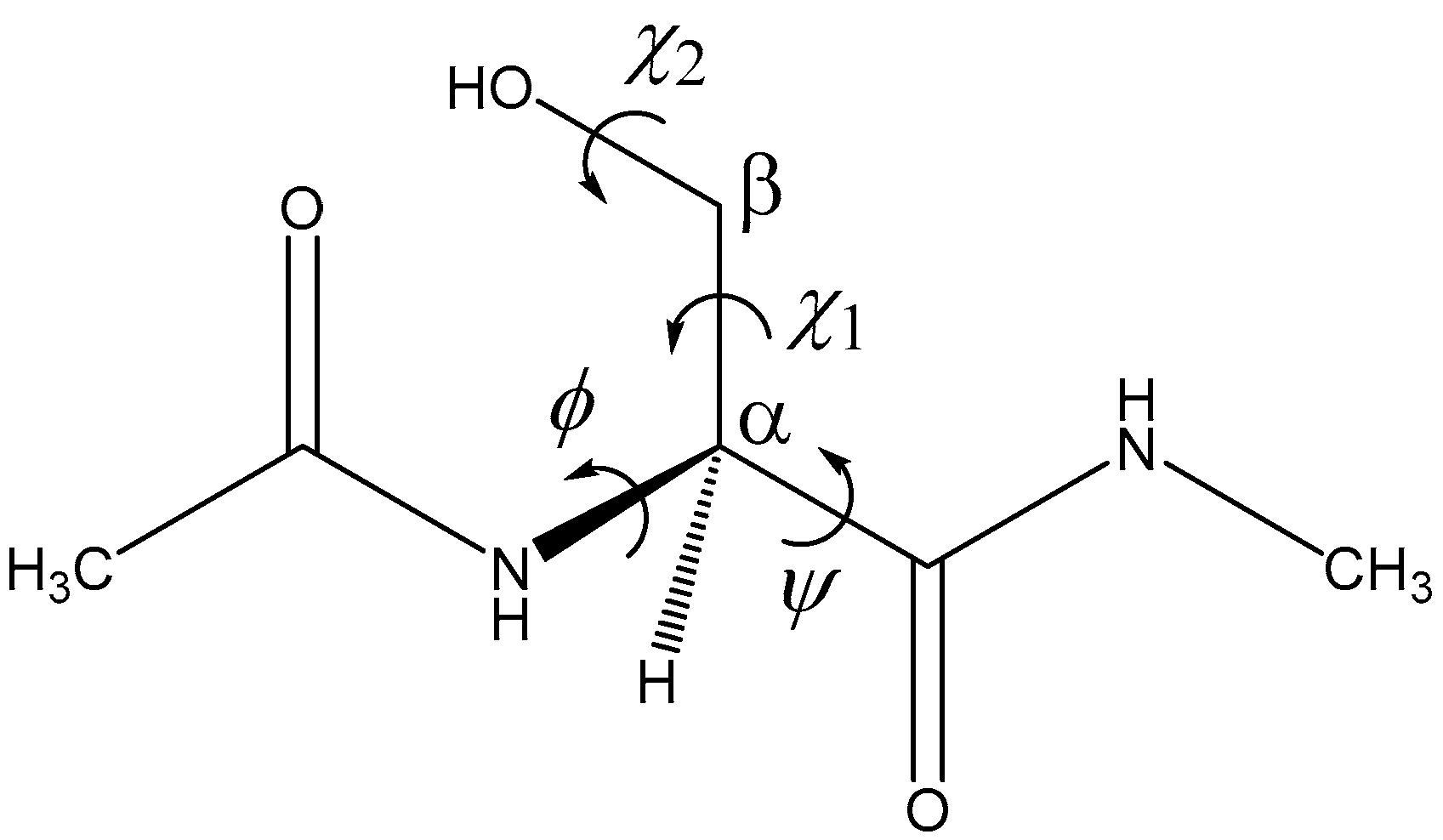
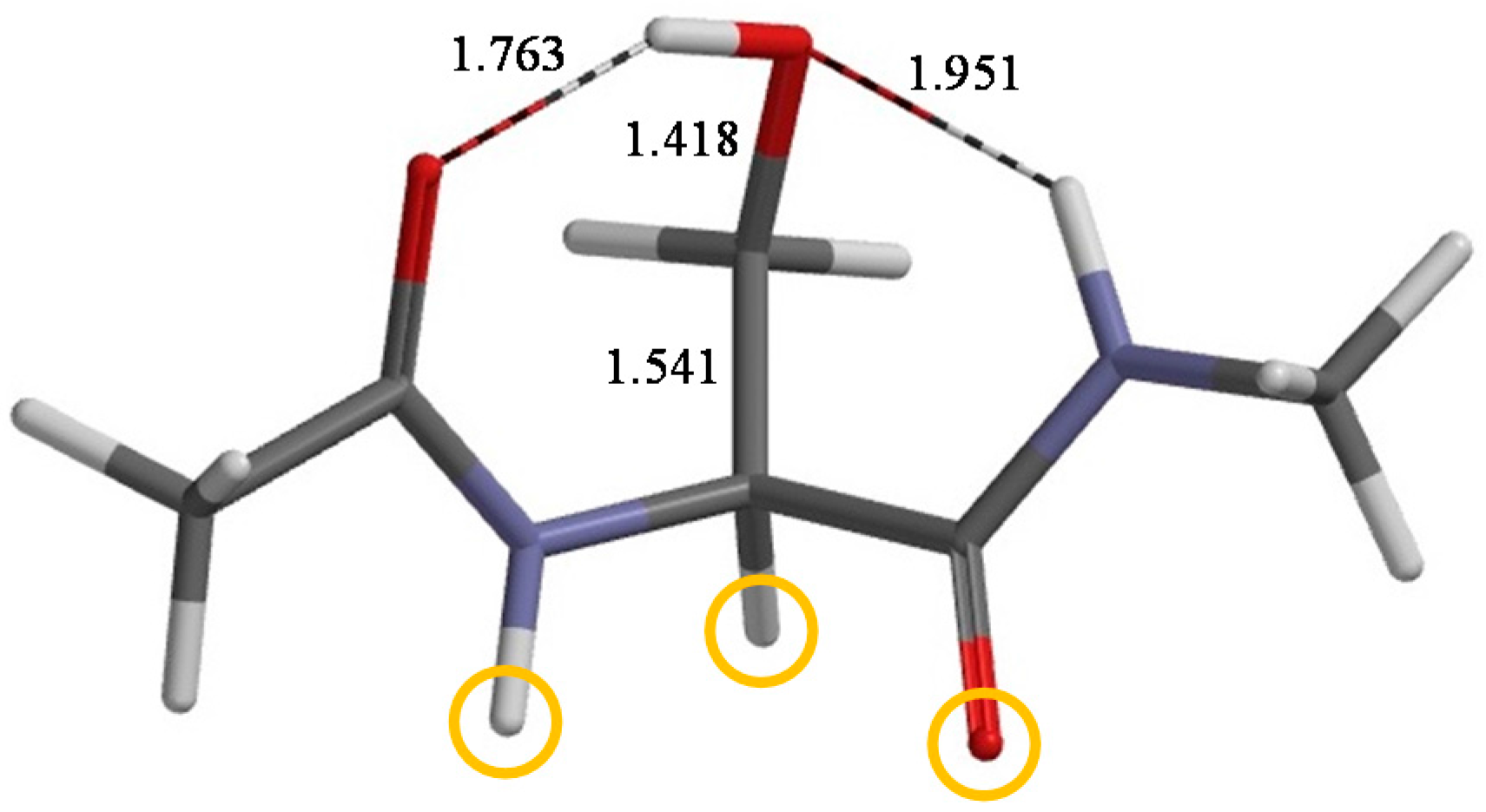
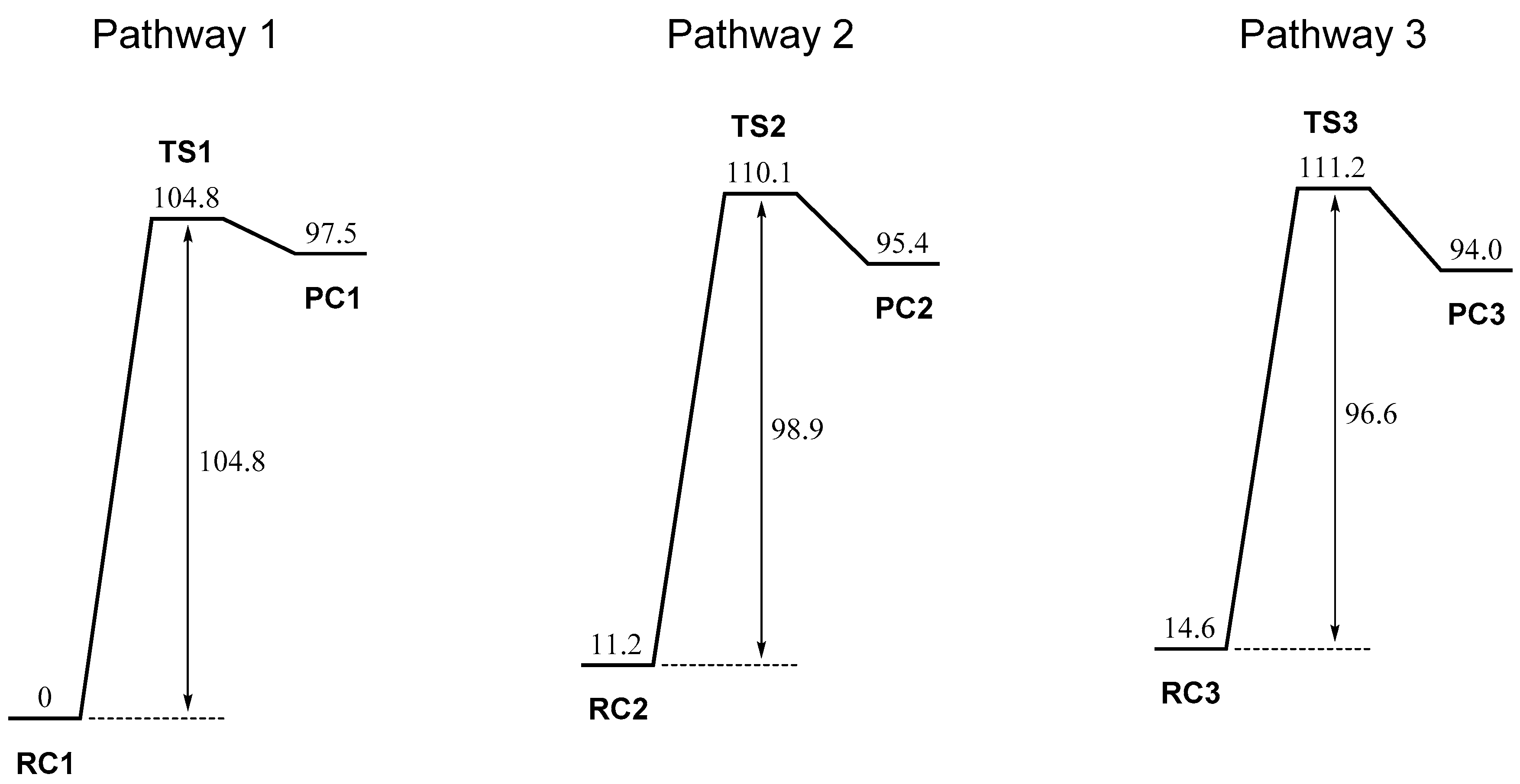
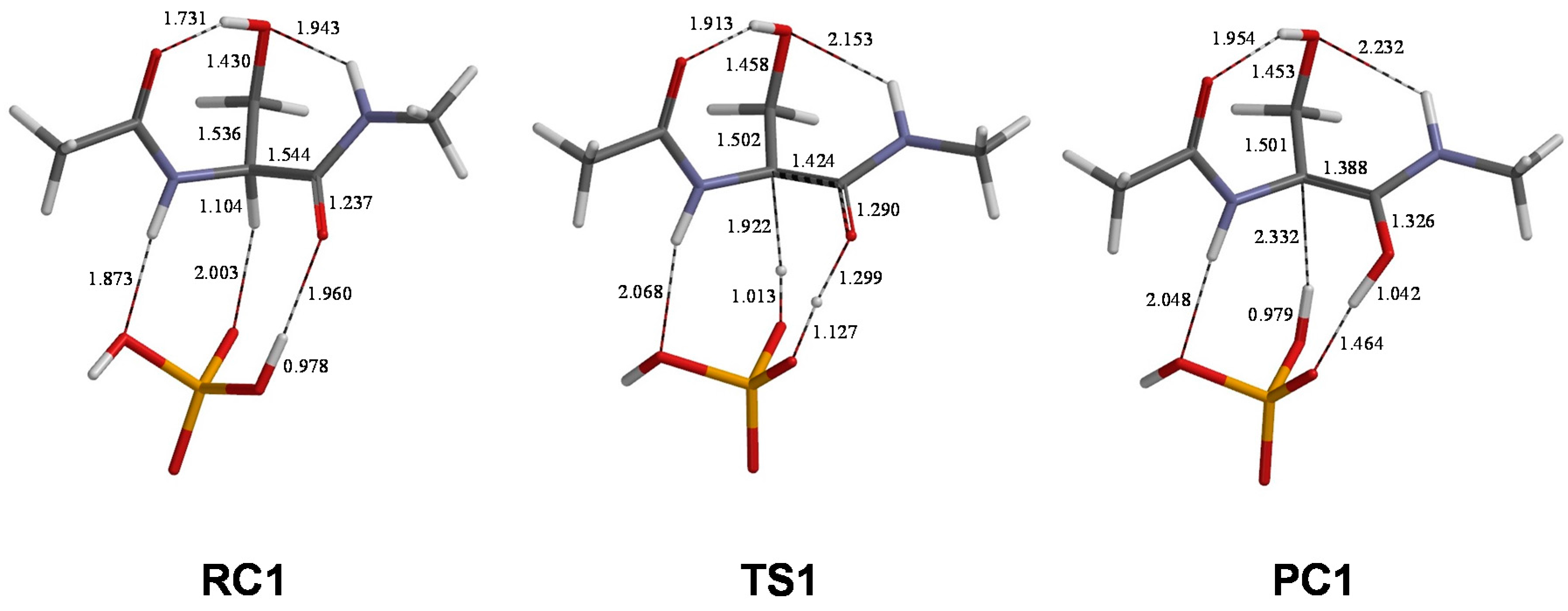
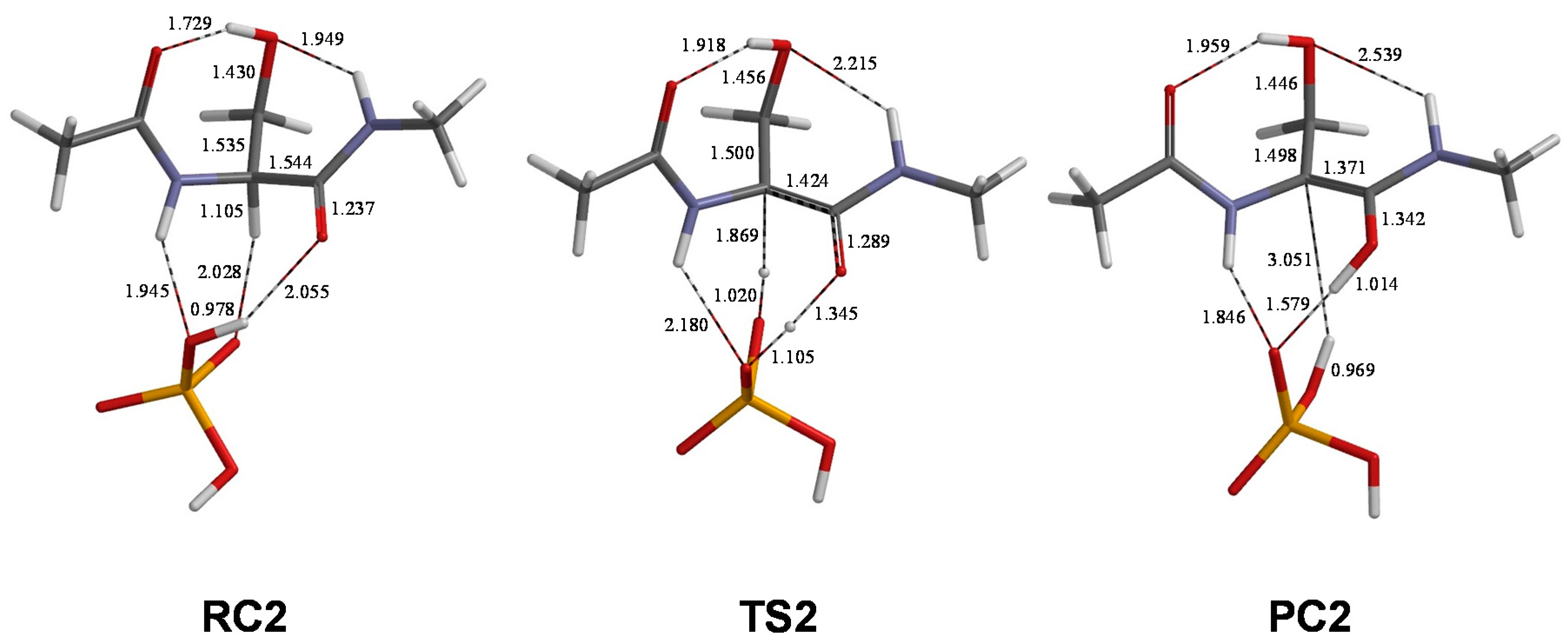
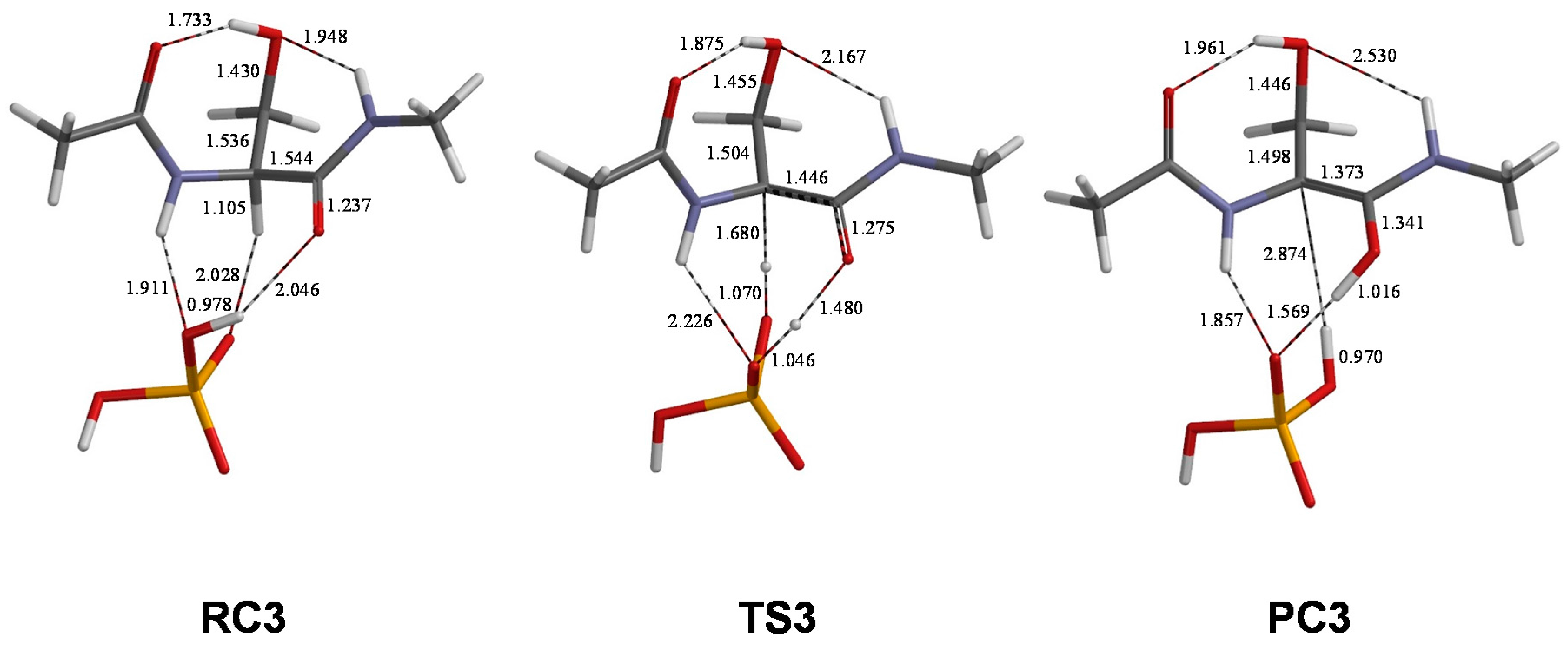
2. Results and Discussion
3. Computational Details
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cloos, P.A.C.; Christgau, S. Non-enzymatic covalent modifications of proteins: Mechanisms, physiological consequences and clinical applications. Matrix Biol. 2002, 21, 39–52. [Google Scholar] [CrossRef]
- Hipkiss, A.R. Non-oxidative modification of DNA and proteins. In Aging at the Molecular Level; von Zglinicki, T., Ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2003; pp. 145–177. ISBN 1-4020-1738-3. [Google Scholar]
- Hipkiss, A.R. Accumulation of altered proteins and ageing: Causes and effects. Exp. Gerontol. 2006, 41, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Truscott, R.J.W. Are ancient proteins responsible for the age-related decline in health and fitness? Rejuvenation Res. 2010, 13, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Truscott, R.J.W.; Friedrich, M.G. The etiology of human age-related cataract. Proteins don’t last forever. Biochim. Biophys. Acta 2016, 1860, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Truscott, R.J.W.; Schey, K.L.; Friedrich, M.G. Old proteins in man: A field in its infancy. Trends Biochem. Sci. 2016, 41, 654–664. [Google Scholar] [CrossRef] [PubMed]
- Ritz-Timme, S.; Collins, M.J. Racemization of aspartic acid in human proteins. Ageing Res. Rev. 2002, 1, 43–59. [Google Scholar] [CrossRef]
- Ritz-Timme, S.; Laumeier, I.; Collins, M.J. Aspartic acid racemization: Evidence for marked longevity of elastin in human skin. Br. J. Dermatol. 2003, 149, 951–959. [Google Scholar] [CrossRef] [PubMed]
- Fujii, N. d-Amino acid in elderly tissues. Biol. Pharm. Bull. 2005, 28, 1585–1589. [Google Scholar] [CrossRef] [PubMed]
- Fujii, N.; Kaji, Y.; Fujii, N. d-Amino acids in aged proteins: Analysis and biological relevance. J. Chromatogr. B 2011, 879, 3141–3147. [Google Scholar] [CrossRef] [PubMed]
- Fujii, N.; Takata, T.; Fujii, N.; Aki, K.; Sakaue, H. d-Amino acid residues in proteins related to aging and age-related diseases and a new analysis of the isomers in proteins. In d-Amino Acids: Physiology, Metabolism, and Application; Yoshimura, T., Nishikawa, T., Homma, H., Eds.; Springer: Tokyo, Japan, 2016; pp. 241–254. ISBN 978-4-431-56075-3. [Google Scholar]
- Geiger, T.; Clarke, S. Deamidation, isomerization, and racemization at asparaginyl and aspartyl residues in peptides. Succinimide-linked reactions that contribute to protein degradation. J. Biol. Chem. 1987, 262, 785–794. [Google Scholar] [PubMed]
- Stephenson, R.C.; Clarke, S. Succinimide formation from aspartyl and asparaginyl peptides as a model for the spontaneous degradation of proteins. J. Biol. Chem. 1989, 264, 6164–6170. [Google Scholar] [PubMed]
- Radkiewicz, J.L.; Zipse, H.; Clarke, S.; Houk, K.N. Accelerated racemization of aspartic acid and asparagine residues via succinimide intermediates: An ab initio theoretical exploration of mechanism. J. Am. Chem. Soc. 1996, 118, 9148–9155. [Google Scholar] [CrossRef]
- Aki, K.; Fujii, N.; Fujii, N. Kinetics of isomerization and inversion of aspartate 58 of αA-crystallin peptide mimics under physiological conditions. PLoS ONE 2013, 8, e58515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapira, R.; Chou, C.-H.J. Differential racemization of aspartate and serine in human myelin basic protein. Biochem. Biophys. Res. Commun. 1987, 146, 1342–1349. [Google Scholar] [CrossRef]
- Shapira, R.; Austin, G.E.; Mirra, S.S. Neuritic plaque amyloid in Alzheimer’s disease is highly racemized. J. Neurochem. 1988, 50, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Roher, A.E.; Lowenson, J.D.; Clarke, S.; Wolkow, C.; Wang, R.; Cotter, R.J.; Reardon, I.M.; Zürcher-Neely, H.A.; Heinrikson, R.L.; Ball, M.J.; et al. Structural alterations in the peptide backbone of β-amyloid core protein may account for its deposition and stability in Alzheimer’s disease. J. Biol. Chem. 1993, 268, 3072–3083. [Google Scholar] [PubMed]
- Lowenson, J.D.; Clarke, S.; Roher, A.E. Chemical modifications of deposited amyloid-β peptides. Methods Enzymol. 1999, 309, 89–105. [Google Scholar] [PubMed]
- Kaneko, I.; Yamada, N.; Sakuraba, Y.; Kamenosono, M.; Tutumi, S. Suppression of mitochondrial succinate dehydrogenase, a primary target of β-amyloid, and its derivative racemized at Ser residue. J. Neurochem. 1995, 65, 2585–2593. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, I.; Yamada, N.; Usui, Y.; Oda, T. Possible involvement of β-amyloids racemized at Ser residue in Alzheimer’s disease. In Alzheimer’s Disease: Biology, Diagnosis and Therapeutics; Iqbal, K., Winblad, B., Nishimura, T., Takeda, M., Wisniewski, H.M., Eds.; Wiley: Chichester, UK, 1997; pp. 519–528. ISBN 0-471-96964-8. [Google Scholar]
- Kaneko, I.; Morimoto, K.; Kubo, T. Drastic neuronal loss in vivo by β-amyloid racemized at Ser26 residue: Conversion of non-toxic [d-Ser26]β-amyloid 1–40 to toxic and proteinase-resistant fragments. Neuroscience 2001, 104, 1003–1011. [Google Scholar] [CrossRef]
- Kubo, T.; Nishimura, S.; Kumagae, Y.; Kaneko, I. In vivo conversion of racemized β-amyloid ([d-Ser26]Aβ1–40) to truncated and toxic fragments ([d-Ser26]Aβ25–35/40) and fragment presence in the brains of Alzheimer’s patients. J. Neurosci. Res. 2002, 70, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Kubo, T.; Kumagae, Y.; Miller, C.A.; Kaneko, I. β-Amyloid racemized at the Ser26 residue in the brains of patients with Alzheimer disease: Implications in the pathogenesis of Alzheimer disease. J. Neuropathol. Exp. Neurol. 2003, 62, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Hooi, M.Y.S.; Truscott, R.J.W. Racemisation and human cataract. d-Ser, d-Asp/Asn and d-Thr are higher in the lifelong proteins of cataract lenses than in age-matched normal lenses. AGE 2011, 33, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Hooi, M.Y.S.; Raftery, M.J.; Truscott, R.J.W. Age-dependent racemization of serine residues in a human chaperone protein. Protein Sci. 2013, 22, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Julian, R.R. Identification of amino acid epimerization and isomerization in crystallin proteins by tandem LC-MS. Anal. Chem. 2014, 86, 9733–9741. [Google Scholar] [CrossRef] [PubMed]
- Lyon, Y.A.; Sabbah, G.M.; Julian, R.R. Identification of sequence similarities among isomerization hotspots in crystallin proteins. J. Proteosome Res. 2017, 16, 1797–1805. [Google Scholar] [CrossRef] [PubMed]
- Stabler, T.V.; Byers, S.S.; Zura, R.D.; Kraus, V.B. Amino acid racemization reveals differential protein turnover in osteoarthritic articular and meniscal cartilages. Arthritis Res. Ther. 2009, 11, R34. [Google Scholar] [CrossRef] [PubMed]
- Cloos, P.A.C.; Jensen, A.L. Age-related de-phosphorylation of proteins in dentin: A biological tool for assessment of protein age. Biogerontology 2000, 1, 341–356. [Google Scholar] [CrossRef] [PubMed]
- Demarchi, B.; Collins, M.; Bergström, E.; Dowle, A.; Penkman, K.; Thomas-Oates, J.; Wilson, J. New experimental evidence for in-chain amino acid racemization of serine in a model peptide. Anal. Chem. 2013, 85, 5835–5842. [Google Scholar] [CrossRef] [PubMed]
- Lyons, B.; Jamie, J.F.; Truscott, R.J.W. Separate mechanisms for age-related truncation and racemisation of peptide-bound serine. Amino Acids 2014, 46, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, O.; Kobayashi, K.; Oda, A. Modeling the enolization of succinimide derivatives, a key step of racemization of aspartic acid residues: Importance of a two-H2O mechanism. Chem. Biodiv. 2010, 7, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, O.; Kobayashi, K.; Oda, A. Computational insight into the mechanism of serine residue racemization. Chem. Biodiv. 2010, 7, 1625–1629. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, O. Two-water-assisted racemization of the succinimide intermediate formed in proteins. A computational model study. Health 2013, 5, 2018–2021. [Google Scholar] [CrossRef]
- Takahashi, O.; Kirikoshi, R.; Manabe, N. Racemization of the succinimide intermediate formed in proteins and peptides: A computational study of the mechanism catalyzed by dihydrogen phosphate ion. Int. J. Mol. Sci. 2016, 17, 1698. [Google Scholar] [CrossRef] [PubMed]
- Tomizawa, H.; Yamada, H.; Wada, K.; Imoto, T. Stabilization of lysozyme against irreversible inactivation by suppression of chemical reactions. J. Biochem. 1995, 117, 635–640. [Google Scholar] [CrossRef] [PubMed]
- Šponer, J.E.; Šponer, J.; Fuentes-Cabrera, M. Prebiotic routes to nucleosides: A quantum chemical insight into the energetics of the multistep reaction pathways. Chem. Eur. J. 2011, 17, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Szabla, R.; Šponer, J.E.; Šponer, J.; Góra, R.W. Theoretical studies of the mechanism of 2-aminooxazole formation under prebiotically plausible conditions. Phys. Chem. Chem. Phys. 2013, 15, 7812–7818. [Google Scholar] [CrossRef] [PubMed]
- Bohne, C.; MacDonald, I.D.; Dunford, H.B. Measurement of rates and equilibria for keto–enol tautomerism of aldehydes using horseradish peroxidase compound I. J. Am. Chem. Soc. 1986, 108, 7867–7868. [Google Scholar] [CrossRef] [PubMed]
- Bora, P.P.; Bez, G. Henry reaction in aqueous media at neutral pH. Eur. J. Org. Chem. 2013, 2013, 2922–2929. [Google Scholar] [CrossRef]
- Miyamoto, T.; Takahashi, N.; Sekine, M.; Ogawa, T.; Hidaka, M.; Homma, H.; Masaki, H. Transition of serine residues to the d-form during the conversion of ovalbumin into heat stable S-ovalbumin. J. Pharm. Biomed. Anal. 2015, 116, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Fujii, N.; Momose, Y.; Harada, K. Kinetic study of racemization of aspartyl residues in model peptides of αA-crystallin. Int. J. Pept. Protein Res. 1996, 48, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Borchardt, R.T. Chemical pathways of peptide degradation. II. Kinetics of deamidation of an asparaginyl residue in a model hexapeptide. Pharm. Res. 1990, 7, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Borchardt, R.T. Chemical pathways of peptide degradation. III. Effect of primary sequence on the pathways of deamidation of asparaginyl residues in hexapeptides. Pharm. Res. 1990, 7, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Connolly, B.D.; Tran, B.; Moore, J.M.R.; Sharma, V.K.; Kosky, A. Specific catalysis of asparaginyl deamidation by carboxylic acids: Kinetic, thermodynamic, and quantitative structure–property relationship analyses. Mol. Pharm. 2014, 11, 1345–1358. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Olson, R.M.; Kelly, C.P.; Cramer, C.J.; Truhlar, D.G. Self-consistent reaction field model for aqueous and nonaqueous solutions based on accurate polarized partial charges. J. Chem. Theory Comput. 2007, 3, 2011–2033. [Google Scholar] [CrossRef] [PubMed]
- Cramer, C.J.; Truhlar, D.G. A universal approach to solvation modeling. Acc. Chem. Res. 2008, 41, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Klamt, A.; Mennucci, B.; Tomasi, J.; Barone, V.; Curutchet, C.; Orozco, M.; Luque, F.J. On the performance of continuum solvation methods. A comment on “universal approaches to solvation modeling”. Acc. Chem. Res. 2009, 42, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Wavefunction, Inc. Spartan ’14; Version 1.1.4; Wavefunction, Inc.: Irvine, CA, USA, 2014. [Google Scholar]
- Hayes, D.M.; Kollman, P.A.; Rothenberg, S. A conformational analysis of H3PO4, H2PO4−, HPO42− and related model compounds. J. Am. Chem. Soc. 1977, 99, 2150–2154. [Google Scholar] [CrossRef]
- Ma, B.; Xie, Y.; Shen, M.; Schleyer, P.V.R.; Schaefer, H.F., III. Isomerization of PO3−·(H2O)n clusters to H2PO4−·(H2O)n−1: Transition states and barrier heights. J. Am. Chem. Soc. 1993, 115, 11169–11179. [Google Scholar] [CrossRef]
- Wang, X.-B.; Vorpagel, E.R.; Yang, X.; Wang, L.-S. Experimental and theoretical investigations of the stability, energetics, and structures of H2PO4−, H2P2O72−, and H3P3O102− in the gas phase. J. Phys. Chem. A 2001, 105, 10468–10474. [Google Scholar] [CrossRef]
- Brandán, S.A.; Díaz, S.B.; González, J.J.L.; Disalvo, E.A.; Altabef, A.B. Experimental and theoretical study of the hydration of phosphate groups in esters of biological interest. Spectrochim. Acta Part A 2007, 66, 884–897. [Google Scholar] [CrossRef] [PubMed]
- Chamberlin, A.C.; Cramer, C.J.; Truhlar, D.G. Performance of SM8 on a test to predict small-molecule solvation free energies. J. Phys. Chem. B 2008, 112, 8651–8655. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | Geometry | φ | ψ | χ1 | χ2 |
|---|---|---|---|---|---|
| 1 | RC1 | 80 | −125 | 79 | −51 |
| TS1 | 90 | −151 | 89 | −47 | |
| PC1 | 101 | −157 | 94 | −50 | |
| 2 | RC2 | 86 | −126 | 79 | −55 |
| TS2 | 98 | −152 | 89 | −52 | |
| PC2 | 122 | −168 | 94 | −55 | |
| 3 | RC3 | 85 | −127 | 79 | −55 |
| TS3 | 92 | −148 | 86 | −51 | |
| PC3 | 121 | −167 | 94 | −56 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takahashi, O.; Kirikoshi, R.; Manabe, N. Racemization of Serine Residues Catalyzed by Dihydrogen Phosphate Ion: A Computational Study. Catalysts 2017, 7, 363. https://doi.org/10.3390/catal7120363
Takahashi O, Kirikoshi R, Manabe N. Racemization of Serine Residues Catalyzed by Dihydrogen Phosphate Ion: A Computational Study. Catalysts. 2017; 7(12):363. https://doi.org/10.3390/catal7120363
Chicago/Turabian StyleTakahashi, Ohgi, Ryota Kirikoshi, and Noriyoshi Manabe. 2017. "Racemization of Serine Residues Catalyzed by Dihydrogen Phosphate Ion: A Computational Study" Catalysts 7, no. 12: 363. https://doi.org/10.3390/catal7120363
APA StyleTakahashi, O., Kirikoshi, R., & Manabe, N. (2017). Racemization of Serine Residues Catalyzed by Dihydrogen Phosphate Ion: A Computational Study. Catalysts, 7(12), 363. https://doi.org/10.3390/catal7120363