
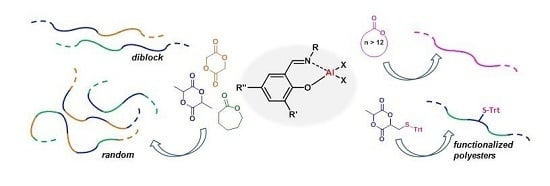
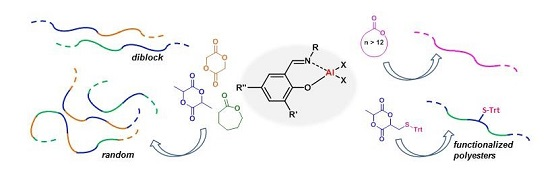
Aluminum Alkyl Complexes Bearing Salicylaldiminato Ligands: Versatile Initiators in the Ring-Opening Polymerization of Cyclic Esters


Abstract
:
1. Introduction
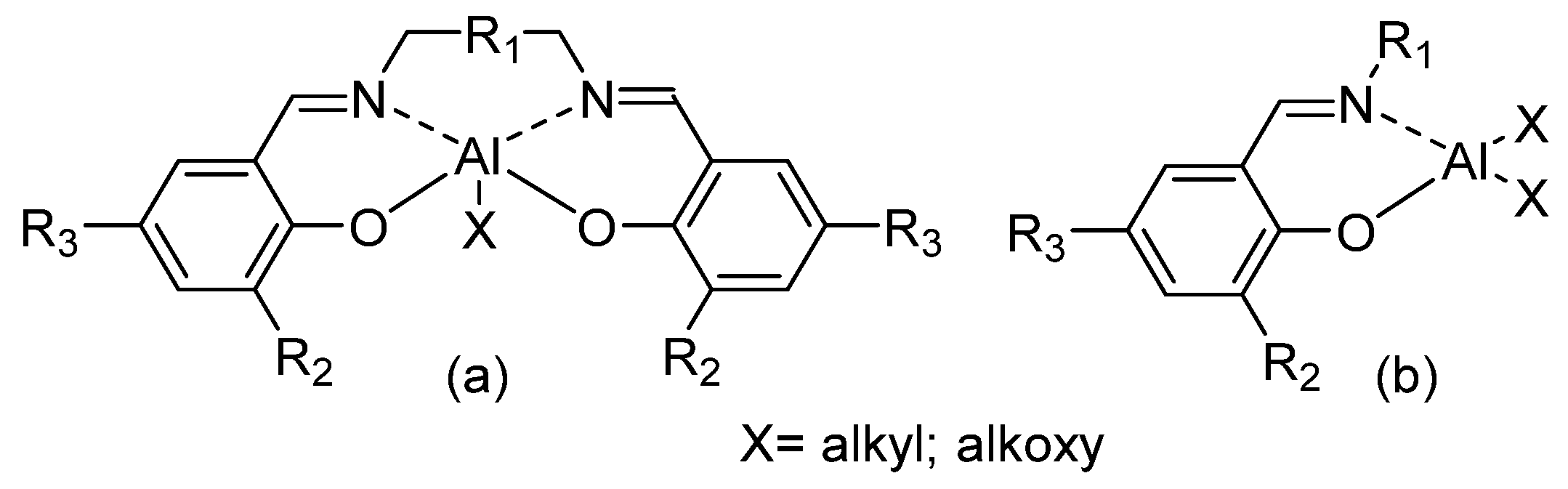
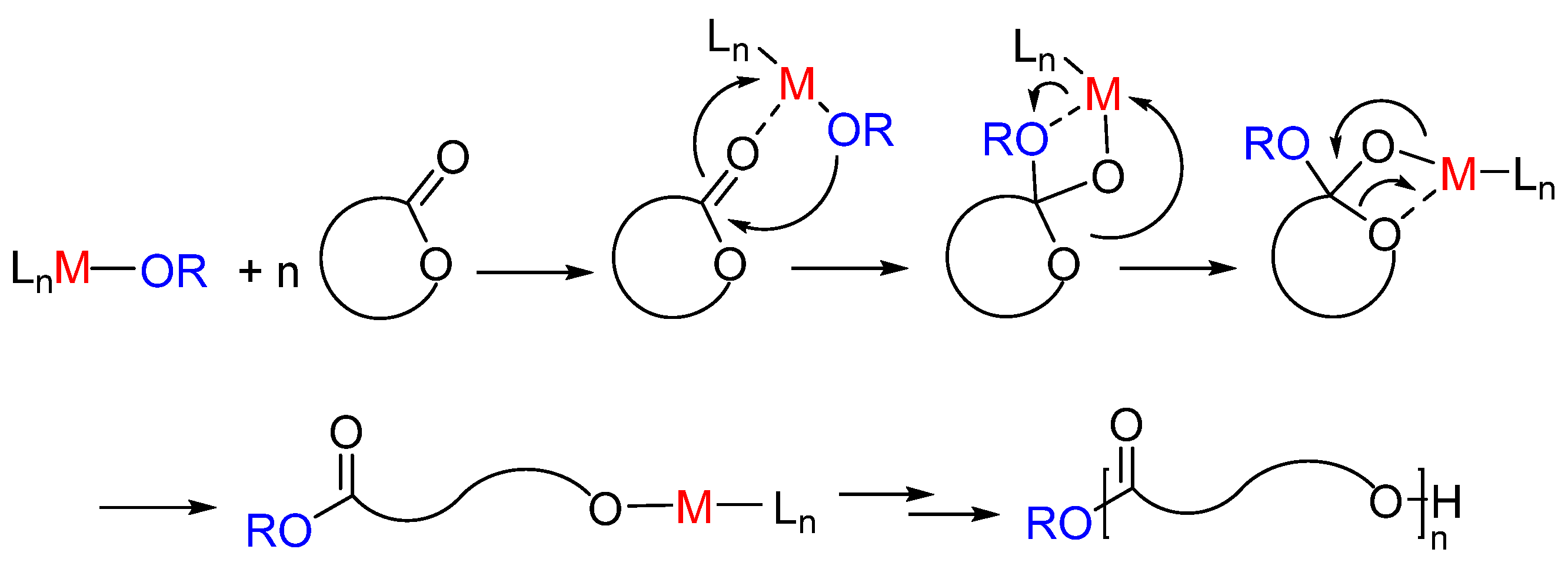
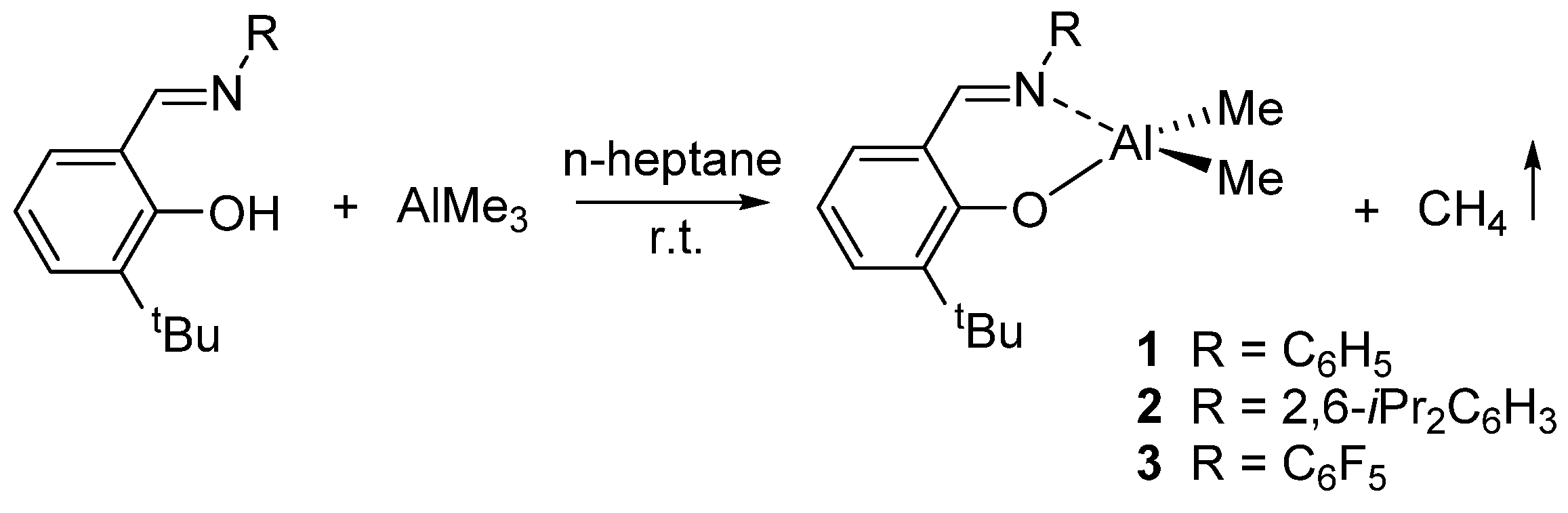
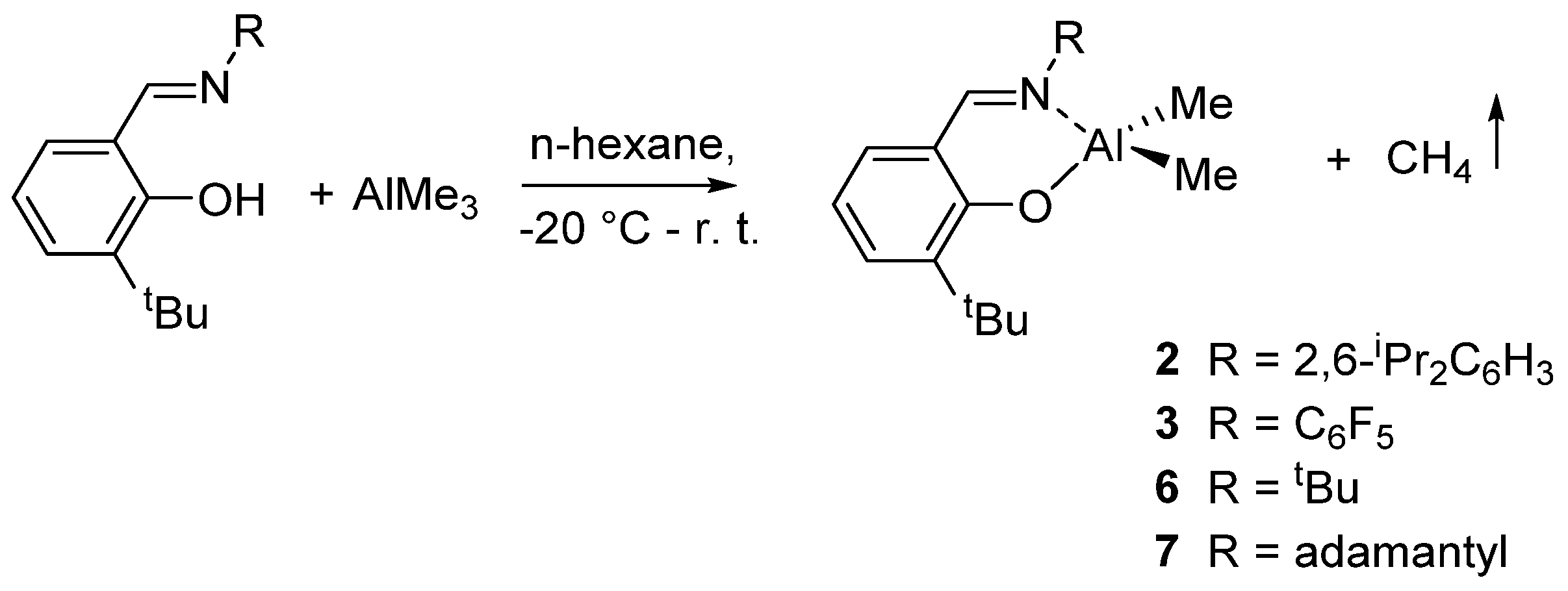
2. Dimethyl(salicilaldiminato)aluminum Complexes
3. Salicylaldiinato-Aluminum Complexes in the Ring-Opening Polymerization of ε-Caprolactone and Other Small Ring Size Lactones
4. Salicylaldiminato-Aluminum Complexes in the Ring-Opening Polymerization of Lactide and in the Copolymerization of ε-Caprolactone with Lactide
4.1. Polymerization of Lactide
4.2. Copolymerization of ε-Caprolactone and Lactide
5. Salicylaldiminato-Aluminum Complexes in the Ring-Opening Polymerization of Glycolide
6. Co-Polymerizations of Glycolide with Other Cyclic Esters
6.1. Copolymerization of Lactide and Glycolide
6.2. Glycolide, ε-Caprolactone, and rac-Lactide co- and ter-Polymerization
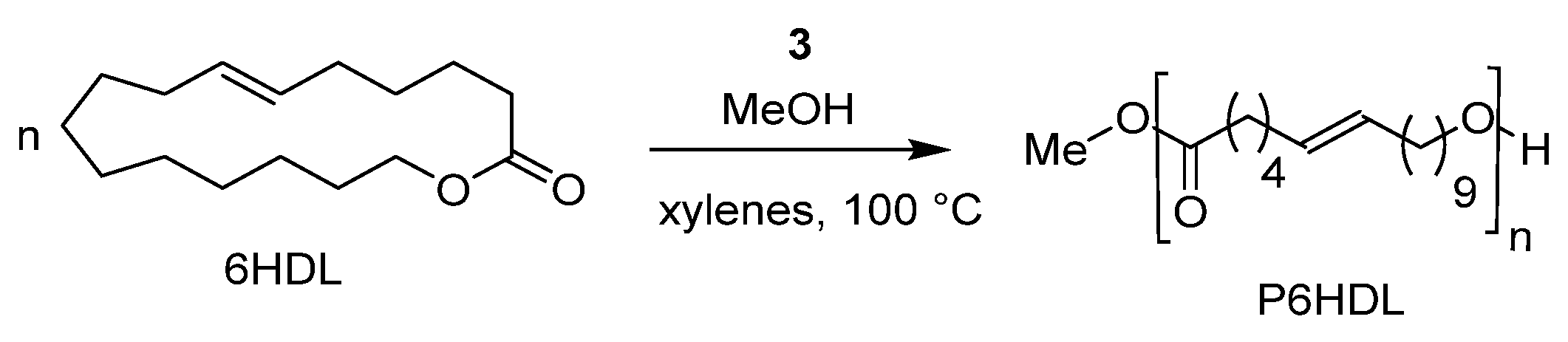
7. Ring-Opening Polymerization of Macrolactones
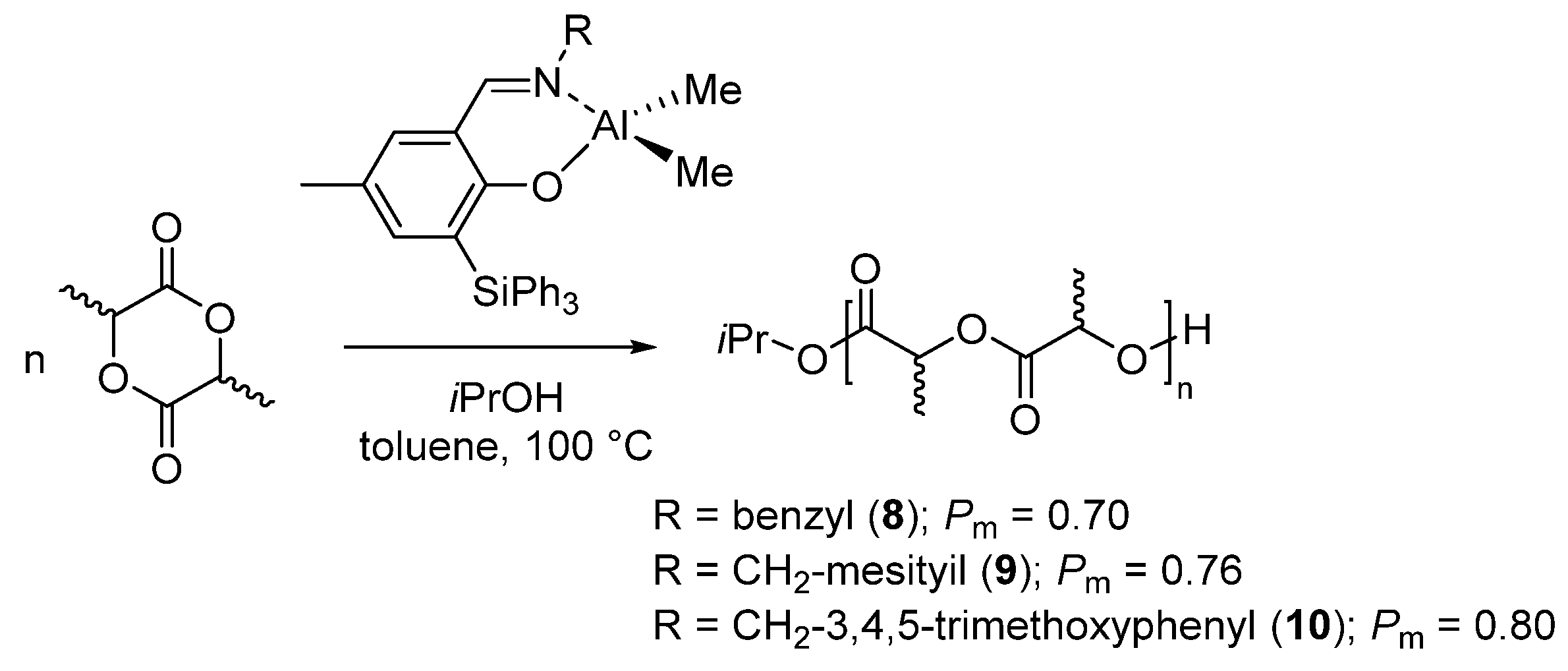
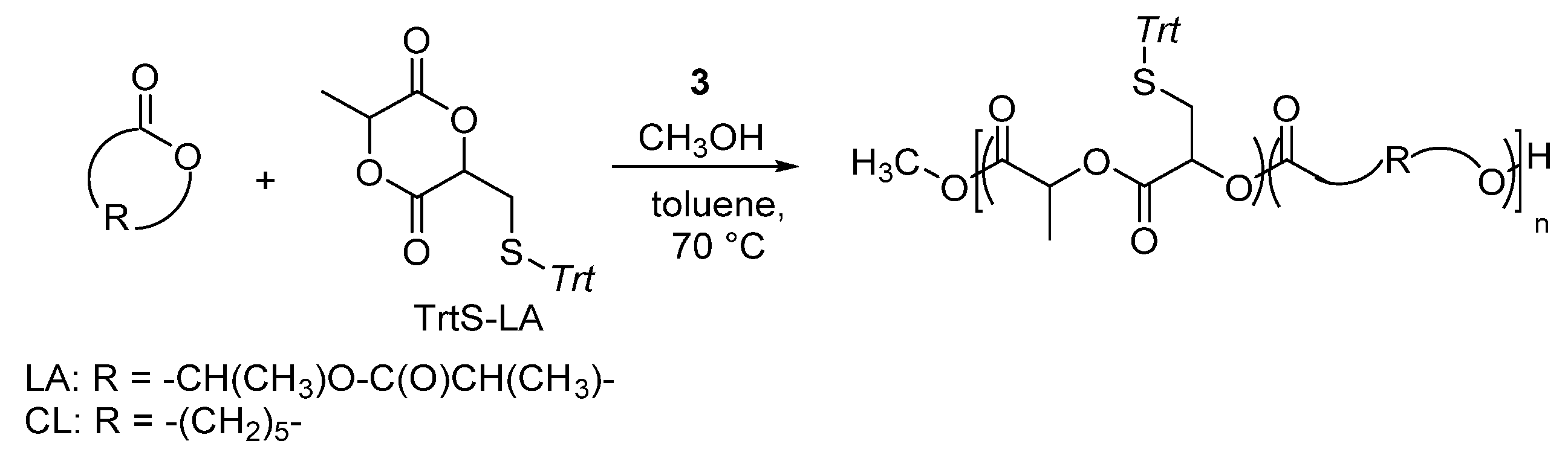
8. Ring-Opening Polymerization of Functionalized Monomers
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Scott, G.; Gilead, D. Degradable polymers. Principles and Applications; Chapman & Hall: London, UK, 1995. [Google Scholar]
- Jarrett, P.; Benedict, C.; Bell, J.P.; Cameron, J.A.; Huang, S.J. Mechanism of the biodegradation of polycaprolactone. In Polymers as Biomaterials; Shalaby, S.W., Hoffman, A.S., Ratner, B.D., Horbett, T.A., Eds.; Plenum Press: New York, NY, USA, 1984. [Google Scholar]
- Raquez, J.-M.; Mincheva, R.; Coulembier, O.; Dubois, P. Ring-opening polymerization of cyclic esters: Industrial synthesis, properties, applications, and perspectives. In Polymer Science: A Comprehensive Reference; Elsevier: Amsterdam, The Netherlands, 2012; pp. 761–777. [Google Scholar]
- Ikada, Y.; Tsuji, H. Biodegradable polyesters for medical and ecological applications. Macromol. Rapid Commun. 2000, 21, 117–132. [Google Scholar] [CrossRef]
- Albertsson, A.-C.; Varma, I.K. Recent developments in ring opening polymerization of lactones for biomedical applications. Biomacromolecules 2003, 4, 1466–1486. [Google Scholar] [CrossRef] [PubMed]
- Dechy-Cabaret, O.; Martin-Vaca, B.; Bourissou, D. Controlled ring-opening polymerization of lactide and glycolide. Chem. Rev. 2004, 104, 6147–6176. [Google Scholar] [CrossRef] [PubMed]
- Gibson, C.; Marshall, E.L. Metal complexes as catalysts for polymerization reactions. In Comprehensive Coordination Chemistry II; Ward, M.D., Ed.; Elsevier: Amsterdam, The Netherlands, 2004; Volume 9, pp. 1–74. [Google Scholar]
- Wu, J.; Yu, T.-L.; Chen, C.-T.; Lin, C.-C. Recent developments in main group metal complexes catalyzed/initiated polymerization of lactides and related cyclic esters. Coord. Chem. Rev. 2006, 250, 602–626. [Google Scholar] [CrossRef]
- Thomas, C.M. Stereocontrolled ring-opening polymerization of cyclic esters: Synthesis of new polyester microstructures. Chem. Soc. Rev. 2010, 39, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Dijkstra, P.J.; Du, H.; Feijen, J. Single site catalysts for stereoselective ring-opening polymerization of lactides. Polym. Chem. 2011, 2, 520–527. [Google Scholar] [CrossRef]
- MacDonald, J.P.; Shaver, M.P. Aluminum salen and salan polymerization catalysts: from monomer scope to macrostructure control. In Green Polymer Chemistry: Biobased Materials and Biocatalysis; American Chemical Society: Washington, DC, USA, 2015; Volume 1192, pp. 147–167. [Google Scholar]
- Jianming, R.; Anguo, X.; Hongwei, W.; Hailin, Y. Review—Recent development of ring-opening polymerization of cyclic esters using aluminum complexes. Des. Monomers Polym. 2014, 1, 345–355. [Google Scholar] [CrossRef]
- Wei, Y.; Wang, S.; Zhou, S. Aluminum alkyl complexes: Synthesis, structure, and application in ROP of cyclic esters. Dalton Trans. 2016, 45, 4471–4485. [Google Scholar] [CrossRef] [PubMed]
- Atwood, D.A.; Harvey, M.J. Group 13 compounds incorporating salen ligands. Chem. Rev. 2001, 101, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Cameron, P.A.; Gibson, V.C.; Irvine, D.J. Nickel-catalyzed generation of Schiff base aluminum enolate initiators for controlled methacrylate polymerization. Angew. Chem. Int. Ed. 2000, 39, 2141–2144. [Google Scholar] [CrossRef]
- Cameron, P.A.; Gibson, V.C.; Irvine, D.J. A Living Polymerisation Process. World Pat. Appl. WO 00/00525, 6 January 2000. [Google Scholar]
- Cameron, P.A.; Jhurry, D.; Gibson, V.C.; White, A.J.P.; Williams, D.J.; Williams, S. Controlled polymerization of lactides at ambient temperature using [5-Cl-salen]AlOMe. Macromol. Rapid Commun. 1999, 20, 616–618. [Google Scholar] [CrossRef]
- Irvine, D.J.; Gibson, V.C.; Cameron, P.A. Aluminium Compounds for Producing Vinylic Polymers. World Pat. Appl. WO 00/00496, 6 January 2000. [Google Scholar]
- Le Borgne, A.; Vincens, V.; Jouglard, M.; Spassky, N. Ring-opening oligomerization reactions using aluminium complexes of Schiff’s bases as initiators. Macromol. Symp. 1993, 73, 37–46. [Google Scholar] [CrossRef]
- Ovitt, T.M.; Coates, G.W. Stereoselective ring-opening polymerization of rac-lactide with a single-site, racemic aluminum alkoxide catalyst: Synthesis of stereoblock poly(lactic acid). J. Polym. Sci. Part A 2000, 38, 4686–4692. [Google Scholar] [CrossRef]
- Spassky, N.; Wisniewski, M.; Pluta, C.; Le Borgne, A. Highly stereoelective polymerization of rac-(d,l)-lactide with a chiral Schiff’s base/aluminium alkoxide initiator. Macromol. Chem. Phys. 1996, 197, 2627–2637. [Google Scholar] [CrossRef]
- Nomura, N.; Ishii, R.; Yamamoto, Y.; Kondo, T. Stereoselective ring-opening polymerization of a racemic lactide by using achiral salen- and homosalen-aluminum complexes. Chem. Eur. J. 2007, 13, 4433–4451. [Google Scholar] [CrossRef] [PubMed]
- Aeilts, S.L.; Coles, M.P.; Swenson, D.C.; Jordan, R.F.; Young, V.G. Aluminum alkyl complexes containing guanidinate ligands. Organometallics 1998, 17, 3265–3270. [Google Scholar] [CrossRef]
- Coles, M.P.; Swenson, D.C.; Jordan, R.F.; Young, V.G. Aluminum complexes incorporating bulky nitrogen and sulfur donor ligands. Organometallics 1998, 17, 4042–4048. [Google Scholar] [CrossRef]
- Ong, C.M.; Stephan, D.W. 1,2-Cyclopentadienyl diimine—Group 13 complexes. Inorg. Chem. 1999, 38, 5189–5191. [Google Scholar] [CrossRef] [PubMed]
- Gibson, V.C.; Redshaw, C.; White, A.J.P.; Williams, D.J. Synthesis and structural characterization of aluminium imino-amide and pyridyl-amide complexes: Bulky monoanionic N,N chelate ligands via methyl group transfer. J. Organomet. Chem. 1998, 550, 453–456. [Google Scholar] [CrossRef]
- Wissing, E.; Jastrzebski, J.T.B.H.; Boersma, J.; van Koten, G. An unexpected 1,2-alkyl shift within a chelate bonded organoaluminium-enamine. J. Organomet. Chem. 1993, 459, 11–16. [Google Scholar] [CrossRef]
- Radzewich, C.E.; Guzei, I.A.; Jordan, R.F. Three-coordinate cationic aluminum alkyl complexes incorporating β-diketiminate ligands. J. Am. Chem. Soc. 1999, 121, 8673–8674. [Google Scholar] [CrossRef]
- Cameron, P.A.; Gibson, V.C.; Redshaw, C.; Segal, J.A.; Solan, G.A.; White, A.J.P.; Williams, D.J. Synthesis and characterisation of neutral dialkylaluminium complexes stabilised by salicylaldiminato ligands, and their conversion to monoalkylaluminium cations. J. Chem. Soc. Dalton Trans. 2001. [Google Scholar] [CrossRef]
- Hogerheide, M.P.; Wesseling, M.; Jastrzebski, J.T.B.H.; Boersma, J.; Kooijman, H.; Spek, A.L.; van Koten, G. Versatility in phenolate bonding in organoaluminum complexes containing mono- and bis-ortho-chelating phenolate ligands. X-ray structures of Al{OC6H2(CH2NMe2)2-2,6-Me-4}3, Al(Me)2{OC6H2(CH2NMe2)2-2,6-Me-4}·N-AlMe3, and Al(Me)2{OC6H2(CH2NMe2)2-2,6-Me-4}·N-AlMe3·O-AlMe3. Organometallics 1995, 14, 4483–4492. [Google Scholar]
- Tian, J.; Hustad, P.D.; Coates, G.W. A new catalyst for highly syndiospecific living olefin polymerization: Homopolymers and block copolymers from ethylene and propylene. J. Am. Chem. Soc. 2001, 123, 5134–5135. [Google Scholar] [CrossRef] [PubMed]
- Saito, J.; Mitani, M.; Mohri, J.-i.; Ishii, S.-i.; Yoshida, Y.; Matsugi, T.; Kojoh, S.-i.; Kashiwa, N.; Fujita, T. Highly syndiospecific living polymerization of propylene using a titanium complex having two phenoxy-imine chelate ligands. Chem. Lett. 2001, 30, 576–577. [Google Scholar] [CrossRef]
- Pappalardo, D.; Tedesco, C.; Pellecchia, C. New neutral and cationic dialkylaluminium complexes bearing imino-amide or imino-phenoxide ligands: Synthesis, characterization and reactivity with olefins. Eur. J. Inorg. Chem. 2002, 2002, 621–628. [Google Scholar] [CrossRef]
- Annunziata, L.; Pappalardo, D.; Tedesco, C.; Pellecchia, C. Octahedral bis(phenoxy-imine)tin(IV) alkyl complexes: Synthesis, characterization, and reactivity toward ionizing species and ethylene. Organometallics 2005, 24, 1947–1952. [Google Scholar] [CrossRef]
- Baugh, L.S.; Sissano, J.A. Polymerization of methyl methacrylate and other polar monomers with alkylaluminum initiators bearing bidentate and tridentate N- and O-donor ligands. J. Polym. Sci. Part A 2002, 40, 1633–1651. [Google Scholar] [CrossRef]
- Pappalardo, D.; Annunziata, L.; Pellecchia, C. Living ring-opening homo- and copolymerization of ε-caprolactone and l- and d,l-lactides by dimethyl(salicylaldiminato)aluminum compounds. Macromolecules 2009, 42, 6056–6062. [Google Scholar] [CrossRef]
- Meduri, A.; Fuoco, T.; Lamberti, M.; Pellecchia, C.; Pappalardo, D. Versatile copolymerization of glycolide and rac-lactide by dimethyl(salicylaldiminato)aluminum compounds. Macromolecules 2014, 47, 534–543. [Google Scholar] [CrossRef]
- Fuoco, T.; Meduri, A.; Lamberti, M.; Venditto, V.; Pellecchia, C.; Pappalardo, D. Ring-opening polymerization of ω-6-hexadecenlactone by a salicylaldiminato aluminum complex: A route to semicrystalline and functional poly(ester)s. Polym. Chem. 2015, 6, 1727–1740. [Google Scholar] [CrossRef]
- Fuoco, T.; Finne-Wistrand, A.; Pappalardo, D. A route to aliphatic poly(ester)s with thiol pendant groups: From monomer design to editable porous scaffolds. Biomacromolecules 2016, 17, 1383–1394. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Iwasa, N.; Nomura, K. Synthesis of Al complexes containing phenoxy-imine ligands and their use as the catalyst precursors for efficient living ring-opening polymerisation of ε-caprolactone. Dalton Trans. 2008. [Google Scholar] [CrossRef] [PubMed]
- Iwasa, N.; Katao, S.; Liu, J.; Fujiki, M.; Furukawa, Y.; Nomura, K. Notable effect of fluoro substituents in the imino group in ring-opening polymerization of ε-caprolactone by Al complexes containing phenoxyimine ligands. Organometallics 2009, 28, 2179–2187. [Google Scholar] [CrossRef]
- Iwasa, N.; Fujiki, M.; Nomura, K. Ring-opening polymerization of various cyclic esters by Al complex catalysts containing a series of phenoxy-imine ligands: Effect of the imino substituents for the catalytic activity. J. Mol. Catal. A 2008, 292, 67–75. [Google Scholar] [CrossRef]
- Iwasa, N.; Liu, J.; Nomura, K. Notable effect of imino substituent for the efficient ring-opening polymerization of ε-caprolactone initiated by Al complexes containing phenoxy-imine ligand of type, Me2Al(L) [L: O-2-tBu-6-(RNCH)C6H3; R: 2,6-iPr2C6H3, tBu, adamantyl, C6F5]. Catal. Commun. 2008, 9, 1148–1152. [Google Scholar] [CrossRef]
- Fuoco, T.; Meduri, A.; Lamberti, M.; Pellecchia, C.; Pappalardo, D. Copolymerization and terpolymerization of glycolide with lactones by dimethyl(salicylaldiminato)aluminum compounds. J. Appl. Polym. Sci. 2015, 132, 42567. [Google Scholar] [CrossRef]
- Nomura, N.; Aoyama, T.; Ishii, R.; Kondo, T. Salicylaldimine−aluminum complexes for the facile and efficient ring-opening polymerization of ε-caprolactone. Macromolecules 2005, 38, 5363–5366. [Google Scholar] [CrossRef]
- Normand, M.; Dorcet, V.; Kirillov, E.; Carpentier, J.-F. {Phenoxy-imine}aluminum versus -indium complexes for the immortal ROP of lactide: Different stereocontrol, different mechanisms. Organometallics 2013, 32, 1694–1709. [Google Scholar] [CrossRef]
- Normand, M.; Roisnel, T.; Carpentier, J.-F.; Kirillov, E. Dinuclear vs. mononuclear complexes: Accelerated, metal-dependent ring-opening polymerization of lactide. Chem. Commun. 2013, 49, 11692–11694. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.-C.; Lu, W.-Y.; Chang, H.-Y.; Lai, Y.-C.; Chiang, M.Y.; Chen, H.-Y.; Chen, H.-Y. Comparative study of aluminum complexes bearing N,O- and N,S-Schiff base in ring-opening polymerization of ε-caprolactone and l-lactide. Inorg. Chem. 2015, 54, 11292–11298. [Google Scholar] [CrossRef] [PubMed]
- Partridge, M.; Davidson, M.; Eade, G. Polymerisation Reaction and Catalyst Therefor. World Pat. Appl. WO 2004052980, 24 June 2004. [Google Scholar]
- Nomura, N.; Ishii, R.; Akakura, M.; Aoi, K. Stereoselective ring-opening polymerization of racemic lactide using aluminum-achiral ligand complexes: Exploration of a chain-end control mechanism. J. Am. Chem. Soc. 2002, 124, 5938–5939. [Google Scholar] [CrossRef] [PubMed]
- Montes de Oca, H.; Ward, I.M. Structure and mechanical properties of PGA crystals and fibres. Polymer 2006, 47, 7070–7077. [Google Scholar] [CrossRef]
- Chujo, K.; Kobayashi, H.; Suzuki, J.; Tokuhara, S.; Tanabe, M. Ring-opening polymerization of glycolide. Makromol. Chem. 1967, 100, 262–266. [Google Scholar] [CrossRef]
- Pan, Z.; Ding, J. Poly(lactide-co-glycolide) porous scaffolds for tissue engineering and regenerative medicine. Inter. Focus 2012, 2, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Stayshich, R.M.; Meyer, T.Y. New insights into poly(lactic-co-glycolic acid) microstructure: Using repeating sequence copolymers to decipher complex NMR and thermal behavior. J. Am. Chem. Soc. 2010, 132, 10920–10934. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Stayshich, R.M.; Meyer, T.Y. Exploiting sequence to control the hydrolysis behavior of biodegradable PLGA copolymers. J. Am. Chem. Soc. 2011, 133, 6910–6913. [Google Scholar] [CrossRef] [PubMed]
- Dobrzynski, P.; Kasperczyk, J.; Janeczek, H.; Bero, M. Synthesis of biodegradable copolymers with the use of low toxic zirconium compounds. 1. Copolymerization of glycolide with l-lactide initiated by Zr(Acac)4. Macromolecules 2001, 34, 5090–5098. [Google Scholar] [CrossRef]
- Hodge, P.; Colquhoun, H.M. Recent work on entropically-driven ring-opening polymerizations: Some potential applications. Polym. Adv. Technol. 2005, 16, 84–94. [Google Scholar] [CrossRef]
- Strandman, S.; Gautrot, J.E.; Zhu, X.X. Recent advances in entropy-driven ring-opening polymerizations. Polym. Chem. 2011, 2, 791–799. [Google Scholar] [CrossRef]
- Hodge, P. Entropically driven ring-opening polymerization of strainless organic macrocycles. Chem. Rev. 2014, 114, 2278–2312. [Google Scholar] [CrossRef] [PubMed]
- Dove, A.P. Organic catalysis for ring-opening polymerization. ACS Macro Lett. 2012, 1, 1409–1412. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Karroonnirun, O. Stereoselective ring-opening polymerization of rac-lactides catalyzed by chiral and achiral aluminum half-salen complexes. Organometallics 2010, 29, 5627–5634. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Karroonnirun, O.; Wilson, S.J. Ring-opening polymerization of cyclic esters and trimethylene carbonate catalyzed by aluminum half-salen complexes. Inorg. Chem. 2011, 50, 6775–6787. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Zhao, K.-Q.; Prior, T.J.; Hughes, D.L.; Arbaoui, A.; Elsegood, M.R.J.; Redshaw, C. Structural studies of Schiff-base [2 + 2] macrocycles derived from 2,2′-oxydianiline and the ROP capability of their organoaluminium complexes. Dalton Trans. 2016, 45, 11990–12005. [Google Scholar] [CrossRef] [PubMed]
- Redshaw, C.; Wang, X.; Zhao, K.-Q.; Al-Khafaji, Y.; Mo, S.; Prior, T.J.; Elsegood, M.R.J. Organoaluminium complexes derived from Anilines or Schiff bases for ring opening polymerization of epsilon-caprolactone, delta-valerolactone and rac-lactide. Eur. J. Inorg. Chem. 2017. [Google Scholar] [CrossRef]
- Pradeepa, C.P.; Dasb, S.K. Coordination and supramolecular aspects of the metal complexes of chiral N-salicyl-β-amino alcohol Schiff base ligands: Towards understanding the roles of weak interactions in their catalytic reactions. Coordin. Chem. Rev. 2013, 257, 1699–1715. [Google Scholar] [CrossRef]
- Gorrasi, G.; Meduri, A.; Rizzarelli, P.; Carroccio, S.; Curcuruto, G.; Pellecchia, C.; Pappalardo, D. Preparation of poly(glycolide-co-lactide)s through a green process: Analysis of structural, thermal, and barrier properties. React. Funct. Polym. 2016, 109, 70–78. [Google Scholar] [CrossRef]
- Lu, W.-Y.; Hsiao, M.-W.; Hsu, S.C.N.; Peng, W.-T.; Chang, Y.-J.; Tsou, Y.-C.; Wu, T.-Y.; Lai, Y.-C.; Chen, Y.; Chen, H.-Y. Synthesis, characterization and catalytic activity of lithium and sodium iminophenoxide complexes towards ring-opening polymerization of l-lactide. Dalton Trans. 2012, 41, 3659–3667. [Google Scholar] [CrossRef] [PubMed]










| Complex Number a | R1 | R2 | R3 | X | Reference |
|---|---|---|---|---|---|
| 1 | Ph | tBu | H | Me | [33,36,40,41,42] |
| 2 | 2,6-iPr2C6H3 | tBu | H | Me | [33,36,40,41,43] |
| 3 | C6F5 | tBu | H | Me | [33,36,38,39,40,41,42,43,44] |
| 4 | 2,4,6-tBu3C6H2 | Me | H | Et | [45] |
| 5 | 2,4,6-tBu3C6H2 | Me | Me | Et | [45] |
| 6 | tBu | tBu | H | Me | [40,42,43] |
| 7 | Adamantyl | tBu | H | Me | [40,42,43] |
| 8 | Benzyl | SiPh3 | Me | Me | [46] |
| 9 | CH2-mesityl | SiPh3 | Me | Me | [46] |
| 10 | CH2-3,4,5-trymethoxyphenil | SiPh3 | Me | Me | [46] |
| 11 | C6F5 | H | H | Me | [37] |
| 12 | C6F5 | Cumyl | Cumyl | Me | [37] |
| 13 | Ph | H | H | Et | [45] |
| 14 | 2,6-Me2C6H3 | H | H | Et | [45] |
| 15 | 2,6-iPr2C6H3 | H | H | Et | [45] |
| 16 | 2,6-Ph2C6H3 | H | H | Et | [45] |
| 17 | 2,6-tBu2C6H3 | H | H | Et | [45] |
| 18 | 2,6-F2C6H3 | H | H | Et | [45] |
| 19 | 4-ClC6H4 | H | H | Et | [45] |
| 20 | 2,6-Cl2C6H3 | H | H | Et | [45] |
| 21 | 2,4,6-tBu3C6H2 | H | Me | Et | [45] |
| 22 | 2,4,6-tBu3C6H2 | iPr | H | Et | [45] |
| 23 | 2,4,6-tBu3C6H2 | Ph | H | Et | [45] |
| 24 | 2,4,6-tBu3C6H2 | tBu | tBu | Et | [45] |
| 25 | 2,4,6-tBu3C6H2 | Br | Br | Et | [45] |
| 26 | 2,4,6-tBu3C6H2 | Me | Me | Et | [45] |
| 27 | Ph | tBu | tBu | Et | [45] |
| 28 | 2,6-iPr2C6H3 | Me | H | Me | [40] |
| 29 | tBu | Me | H | Me | [40] |
| 30 | Cyclohexyl | tBu | H | Me | [40,42] |
| 31 | 2,6-Me2C6H3 | tBu | H | Me | [40,41,42] |
| 32 | 2,4,6-Me3C6H2 | tBu | H | Me | [42] |
| 33 | 2,4,6-tBu3C6H2 | tBu | H | Me | [42] |
| 34 | 2,6-F2C6H3 | tBu | H | Me | [41] |
| 35 | 2,4-F2C6H3 | tBu | H | Me | [41] |
| 36 | 3,4-F2C6H3 | tBu | H | Me | [41] |
| 37 | C6H5 | tBu | H | Et | [41] |
| 38 | C6H5 | tBu | H | Me,Cl | [41] |
| 39 | 8-Quinolyl | SiPh3 | Me | Me | [46] |
| 40 | 2,6-iPr2C6H3 | SiPh3 | Me | Me | [46] |
| 41 | 3,5-(CF3)2C6H3 | SiPh3 | Me | Me | [46] |
| 42 | 2,6-[(3,5)-(CF3)2C6H3]2C6H3 | SiPh3 | Me | Me | [46] |
| 43 | 2-MorpholineC6H4 | SiPh3 | Me | Me | [46] |
| 44 | CH2(2-pyridyl) | SiPh3 | Me | Me | [46] |
| 45 | Diphenylmethyl | SiPh3 | Me | Me | [46] |
| 46 | Trityl | SiPh3 | Me | Me | [46] |
| 47 | (R)-3,3-(Me3)2-2-butyl | SiPh3 | Me | Me | [46] |
| 48 | N-benzyl-4-piperidinyl | SiPh3 | Me | Me | [46] |
| 49 | N-benzyl-4-piperidinyl | Ph | H | Me | [47] |
| 50 | Benzyl | H | H | Me | [48] |
| 51 | tBu | H | H | Me | [48] |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
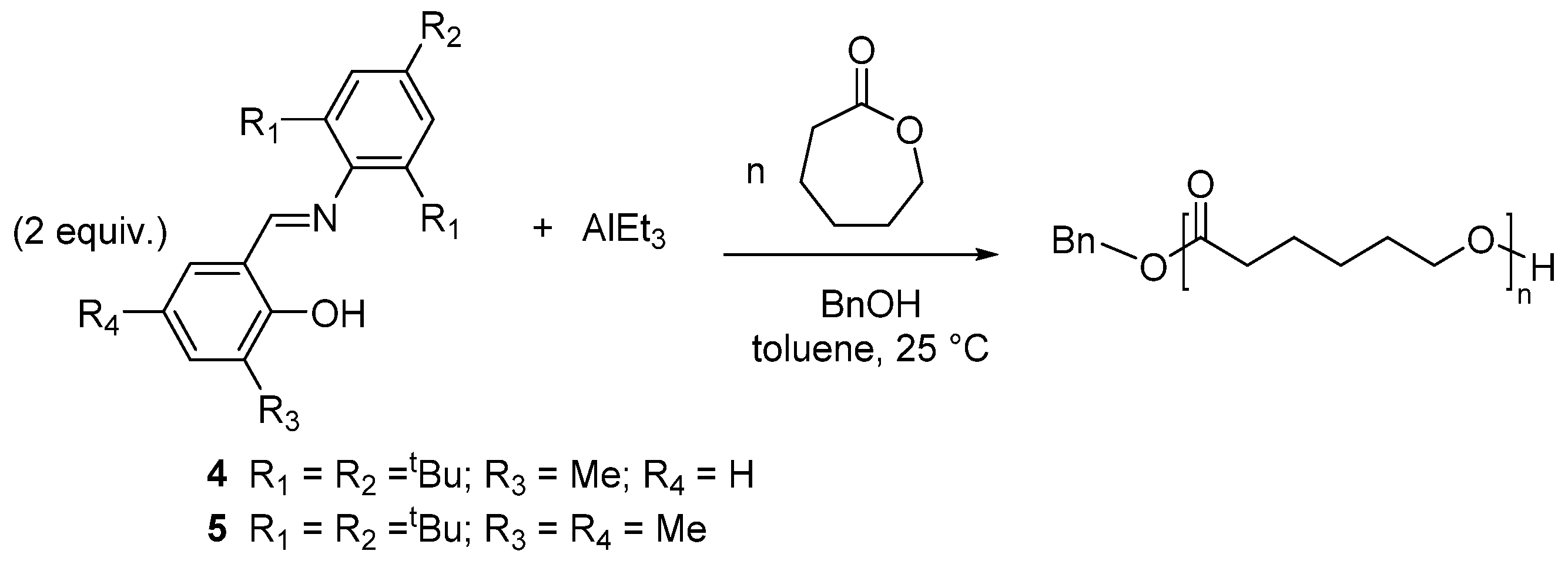{kind=link}
{kind=link}
{kind=link}
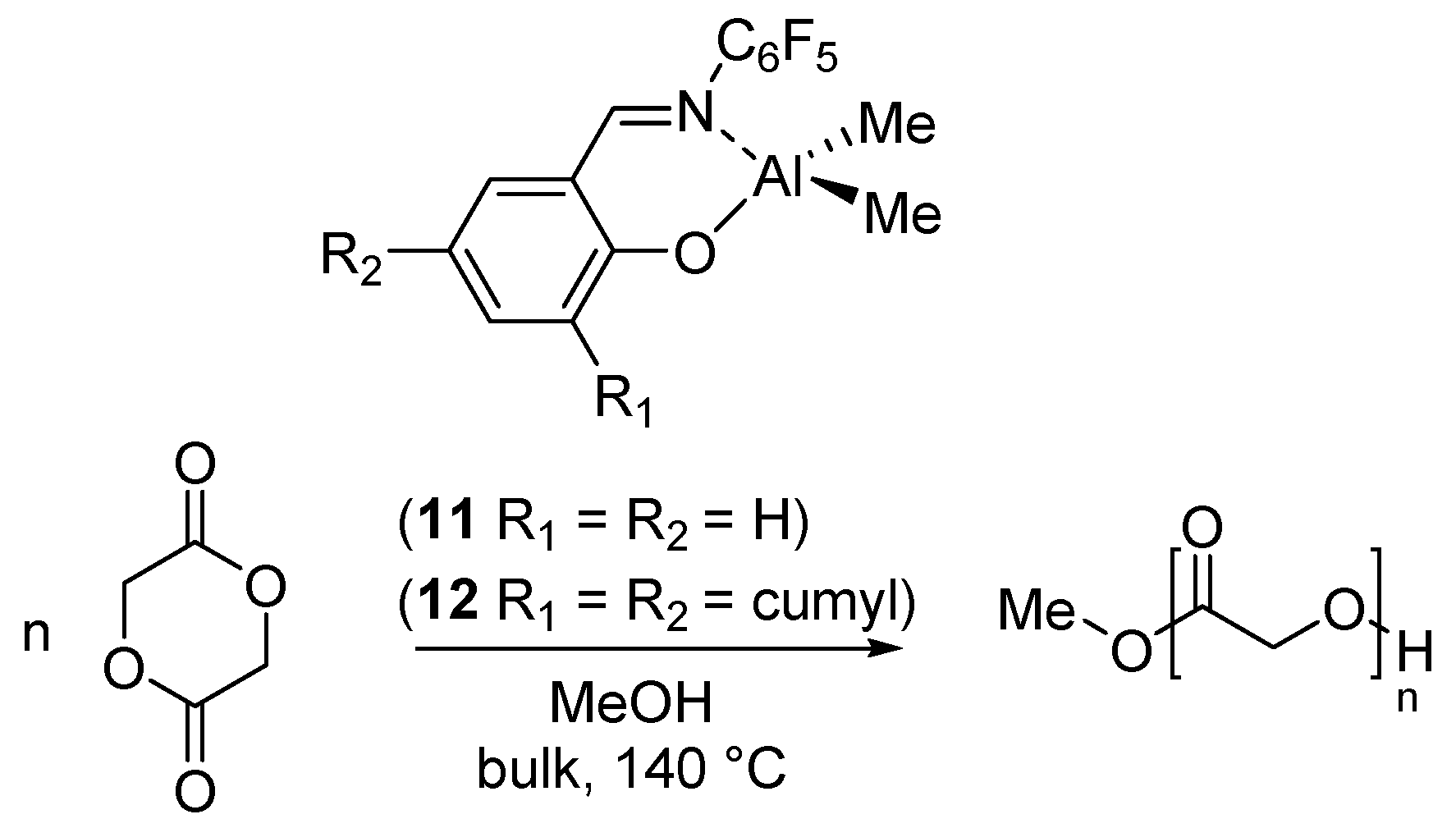{kind=link}
{kind=link}
{kind=link}
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fuoco, T.; Pappalardo, D. Aluminum Alkyl Complexes Bearing Salicylaldiminato Ligands: Versatile Initiators in the Ring-Opening Polymerization of Cyclic Esters. Catalysts 2017, 7, 64. https://doi.org/10.3390/catal7020064
Fuoco T, Pappalardo D. Aluminum Alkyl Complexes Bearing Salicylaldiminato Ligands: Versatile Initiators in the Ring-Opening Polymerization of Cyclic Esters. Catalysts. 2017; 7(2):64. https://doi.org/10.3390/catal7020064
Chicago/Turabian StyleFuoco, Tiziana, and Daniela Pappalardo. 2017. "Aluminum Alkyl Complexes Bearing Salicylaldiminato Ligands: Versatile Initiators in the Ring-Opening Polymerization of Cyclic Esters" Catalysts 7, no. 2: 64. https://doi.org/10.3390/catal7020064
APA StyleFuoco, T., & Pappalardo, D. (2017). Aluminum Alkyl Complexes Bearing Salicylaldiminato Ligands: Versatile Initiators in the Ring-Opening Polymerization of Cyclic Esters. Catalysts, 7(2), 64. https://doi.org/10.3390/catal7020064