
Catalytic Dehydration of Glycerol to Acrolein over a Catalyst of Pd/LaY Zeolite and Comparison with the Chemical Equilibrium

 ,
, 
Abstract
:
1. Introduction
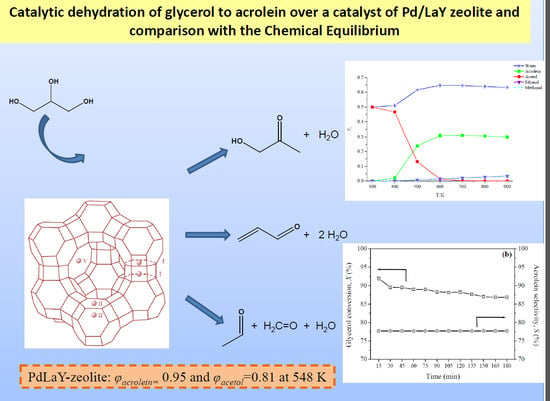
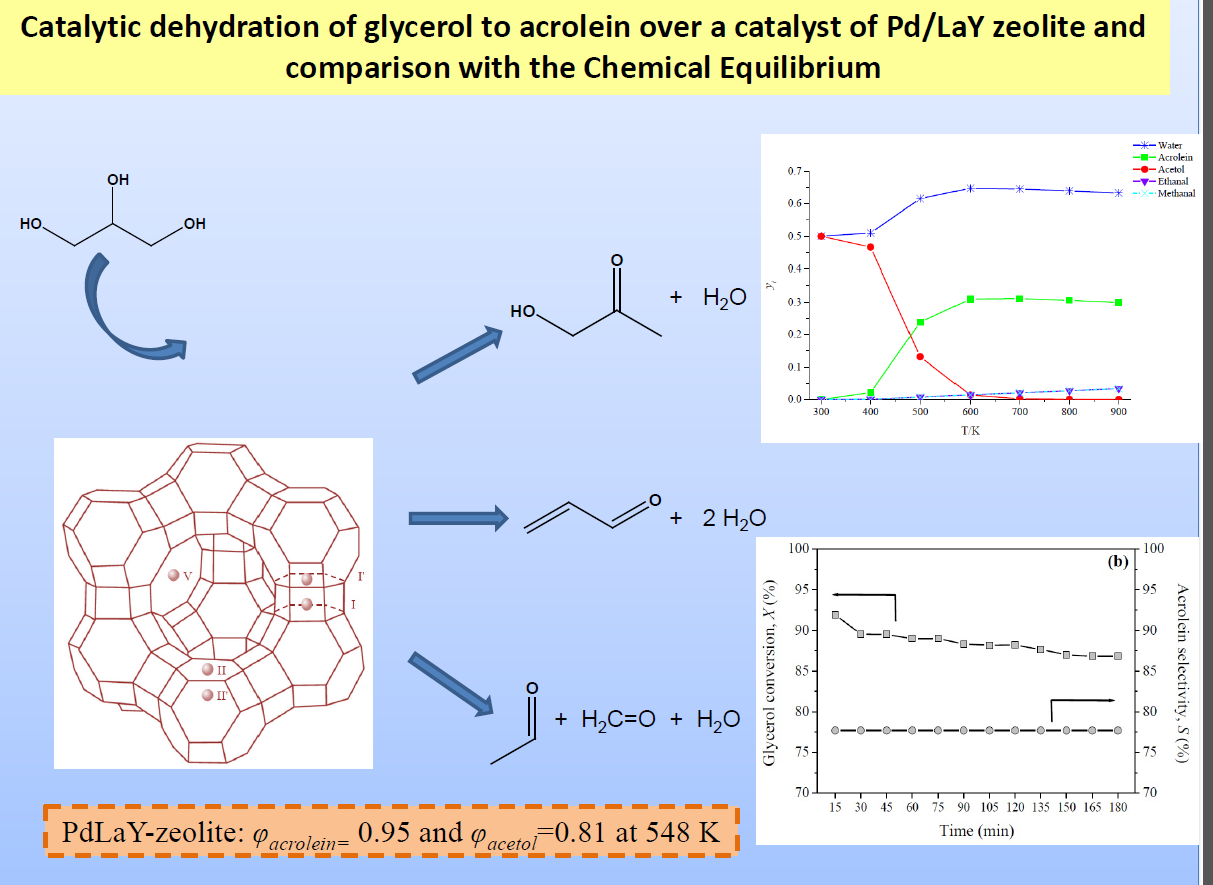
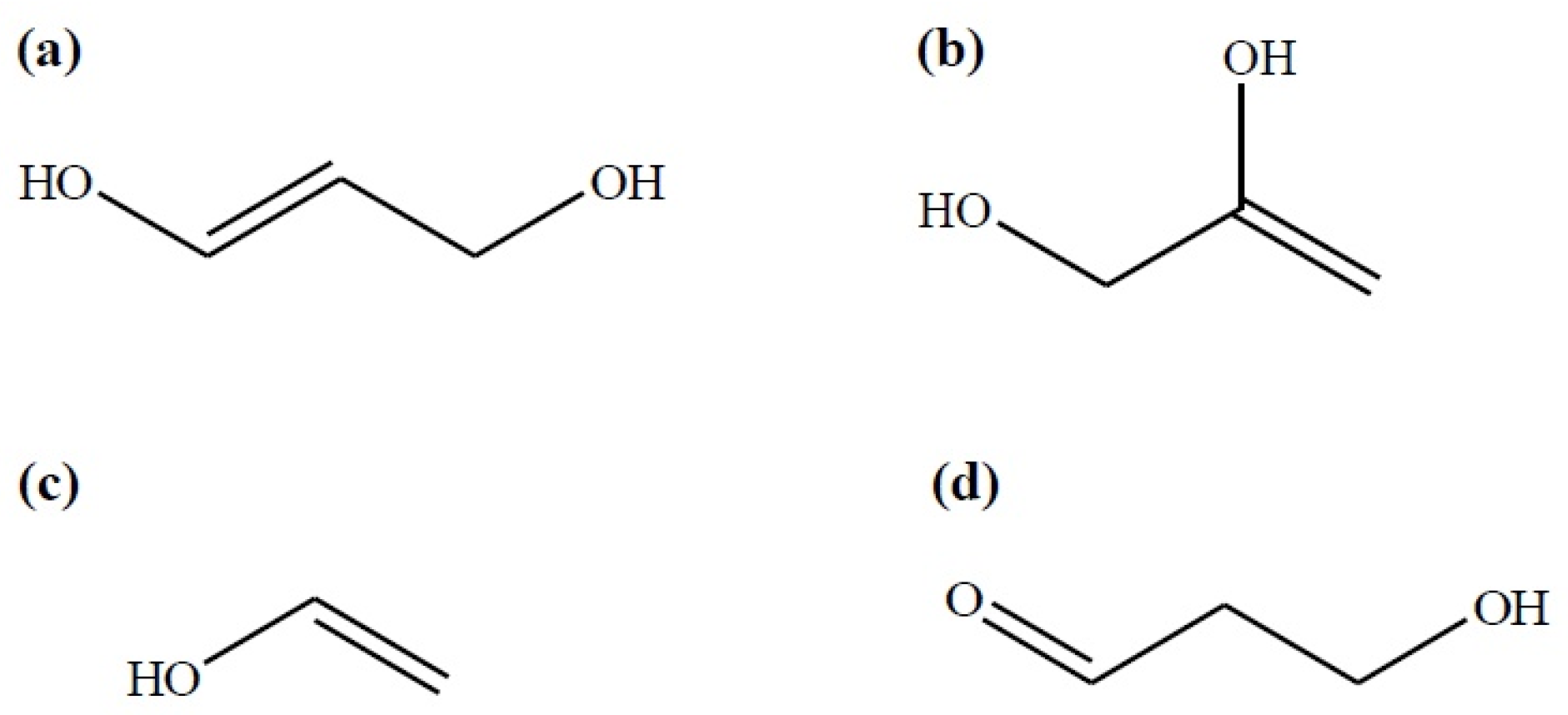
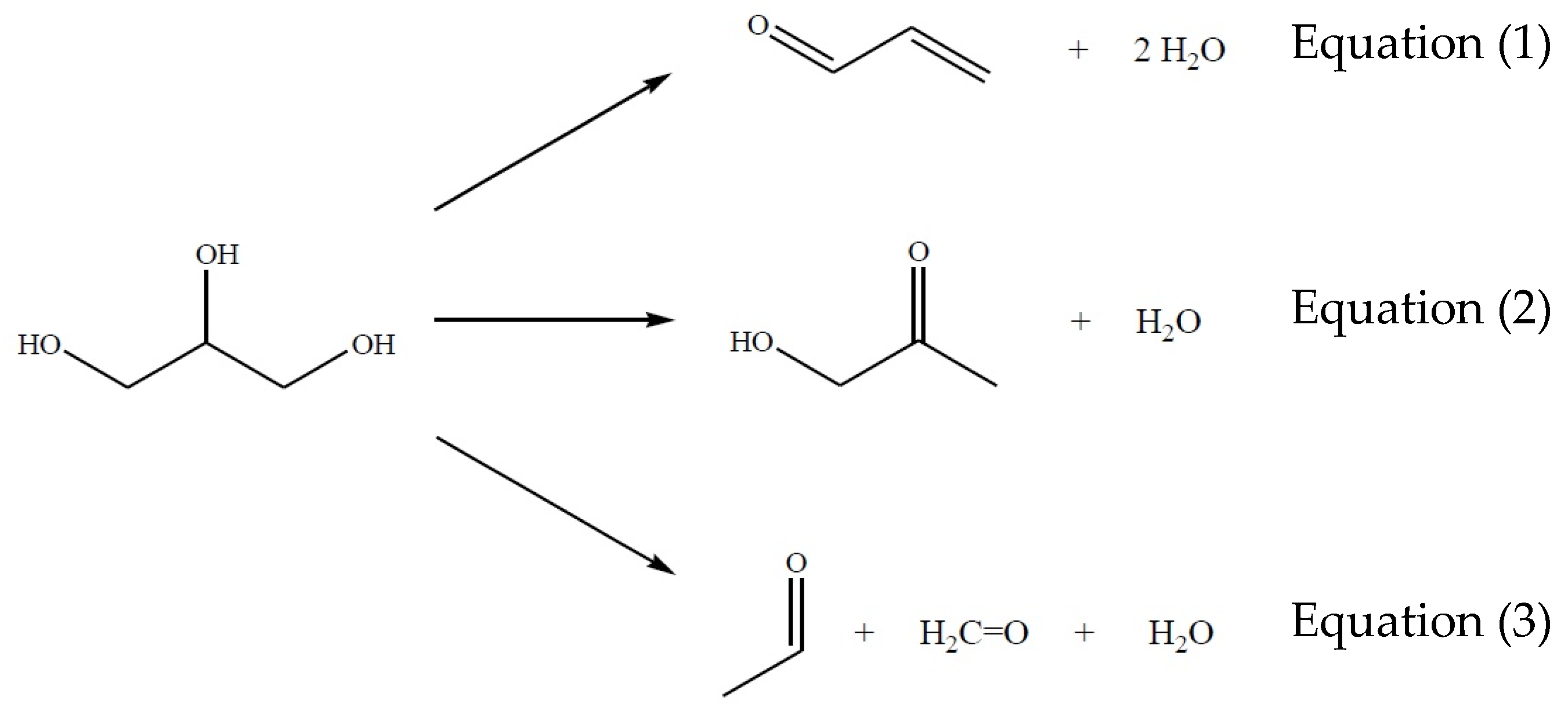
2. The Main Reactions
3. Results and Discussion
3.1. Catalyst Characterization
3.1.1. Concentration by Atomic Absorption
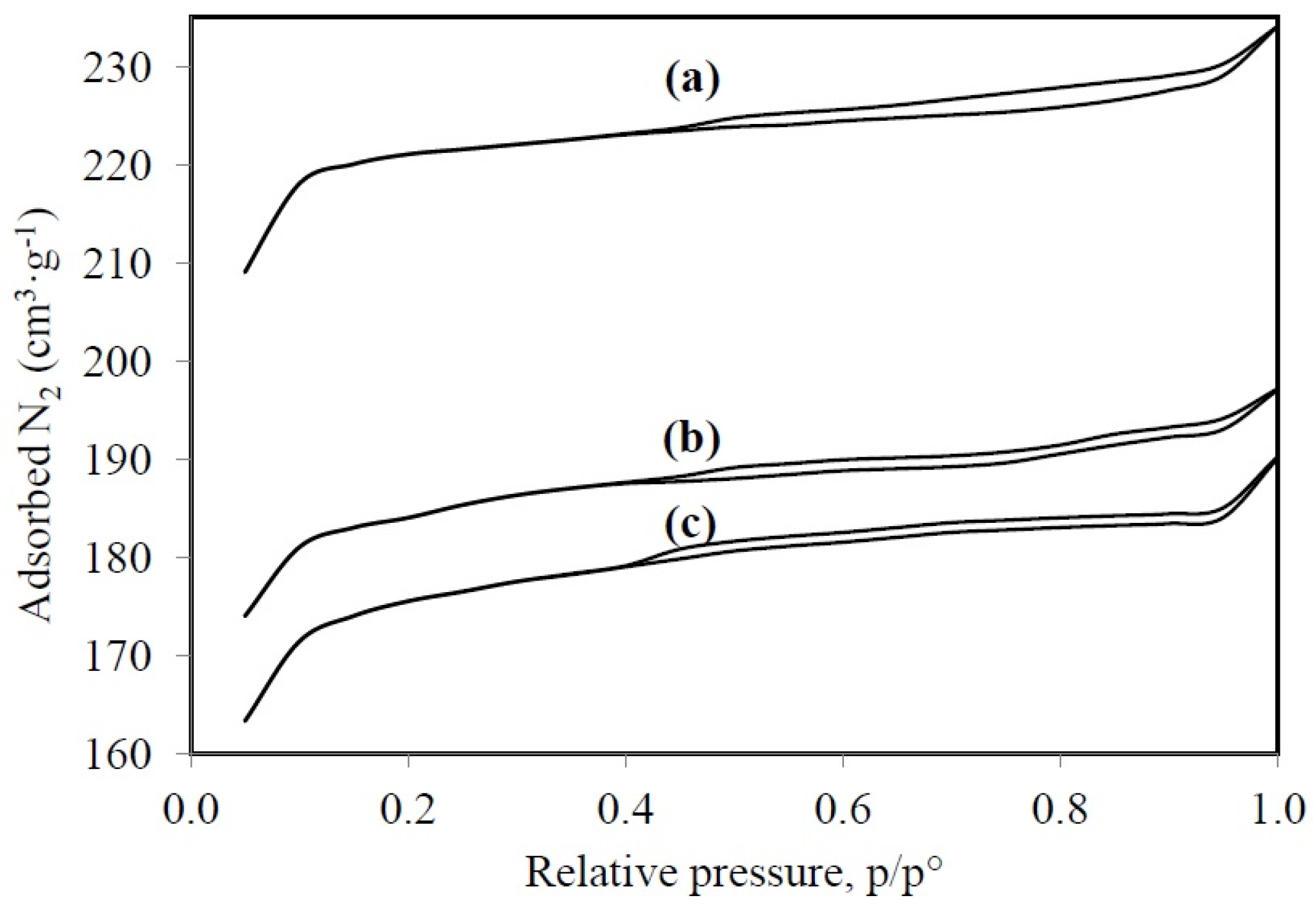
3.1.2. N2 Physisorption
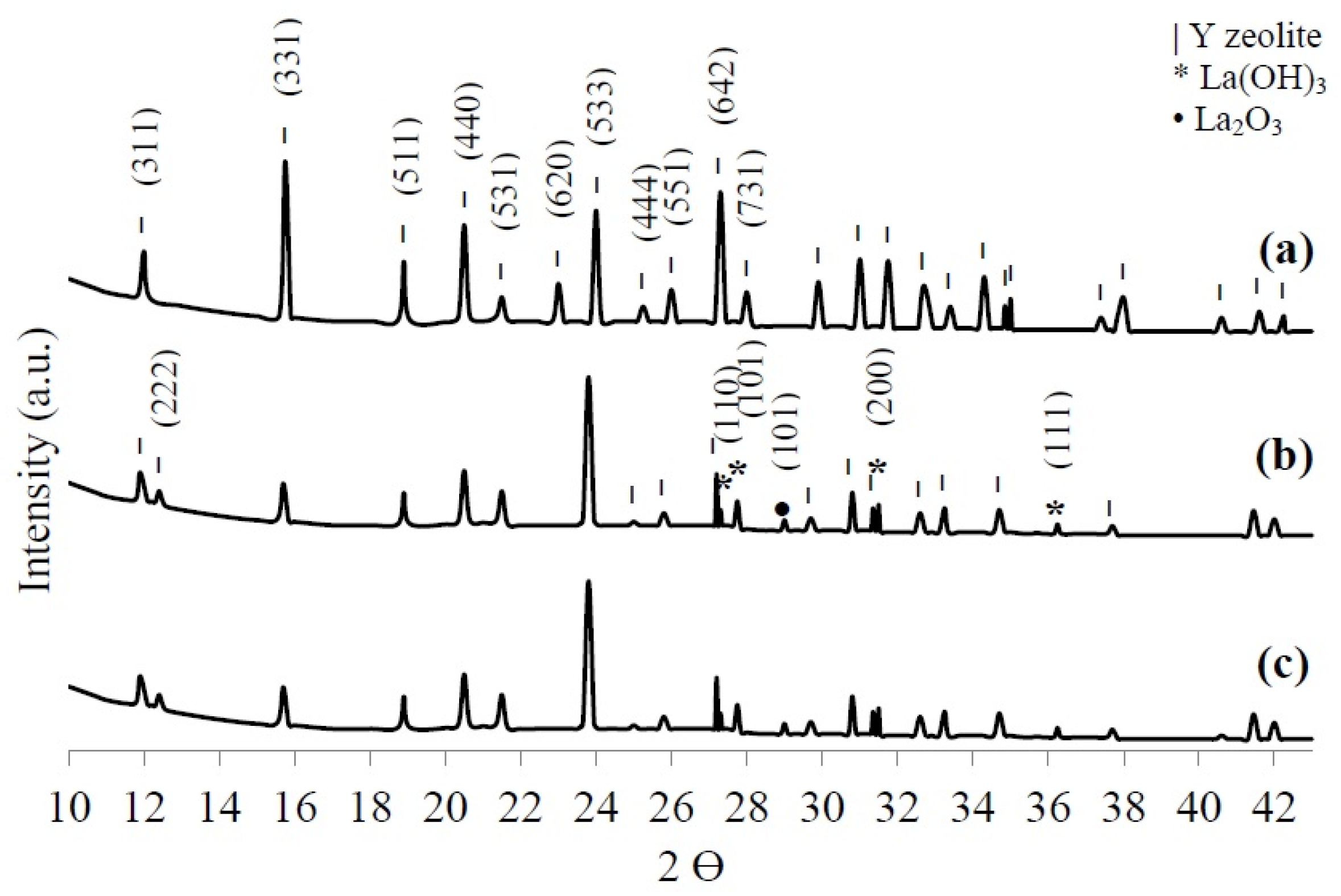
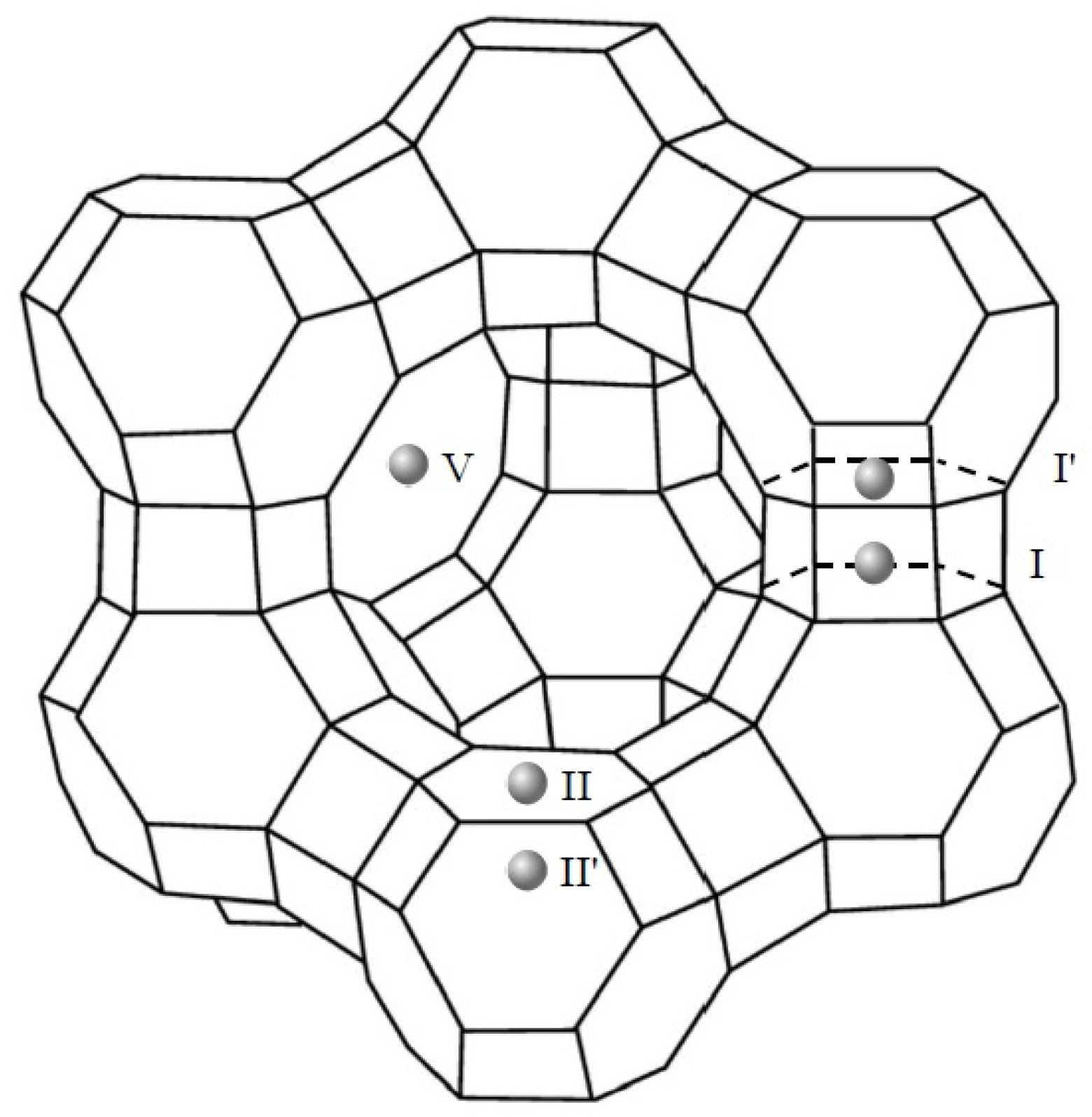
3.1.3. X-ray Diffraction
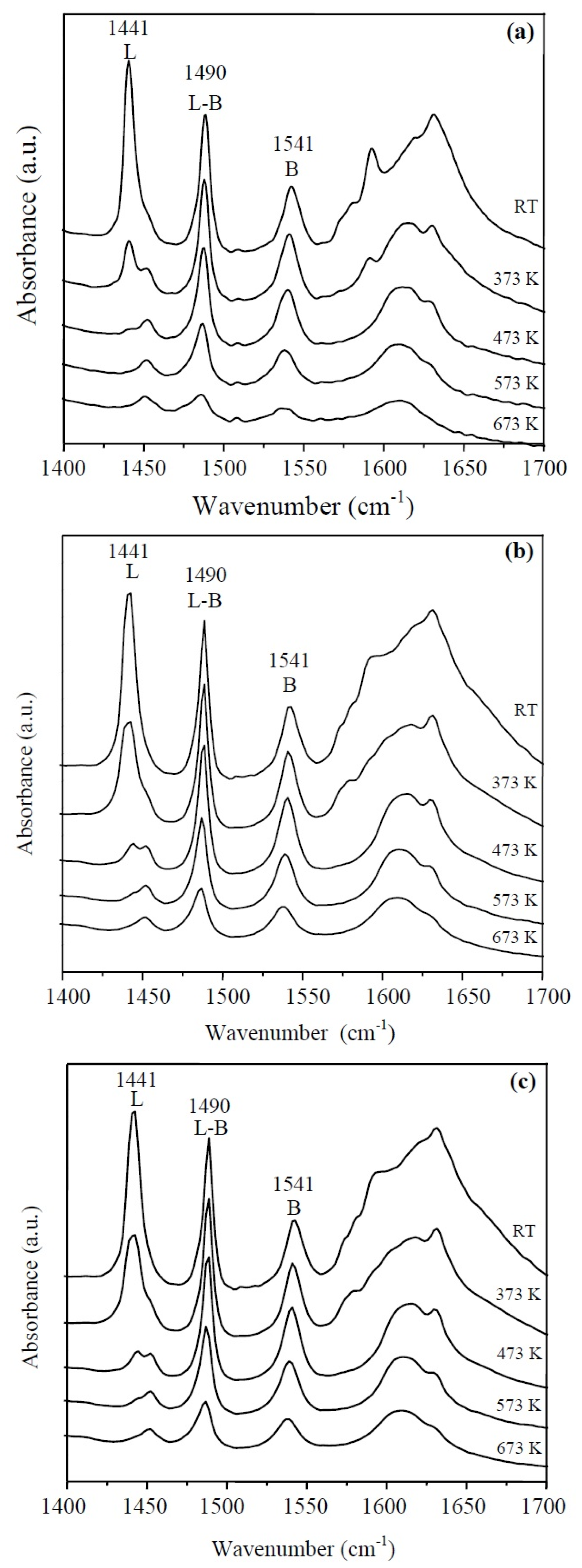
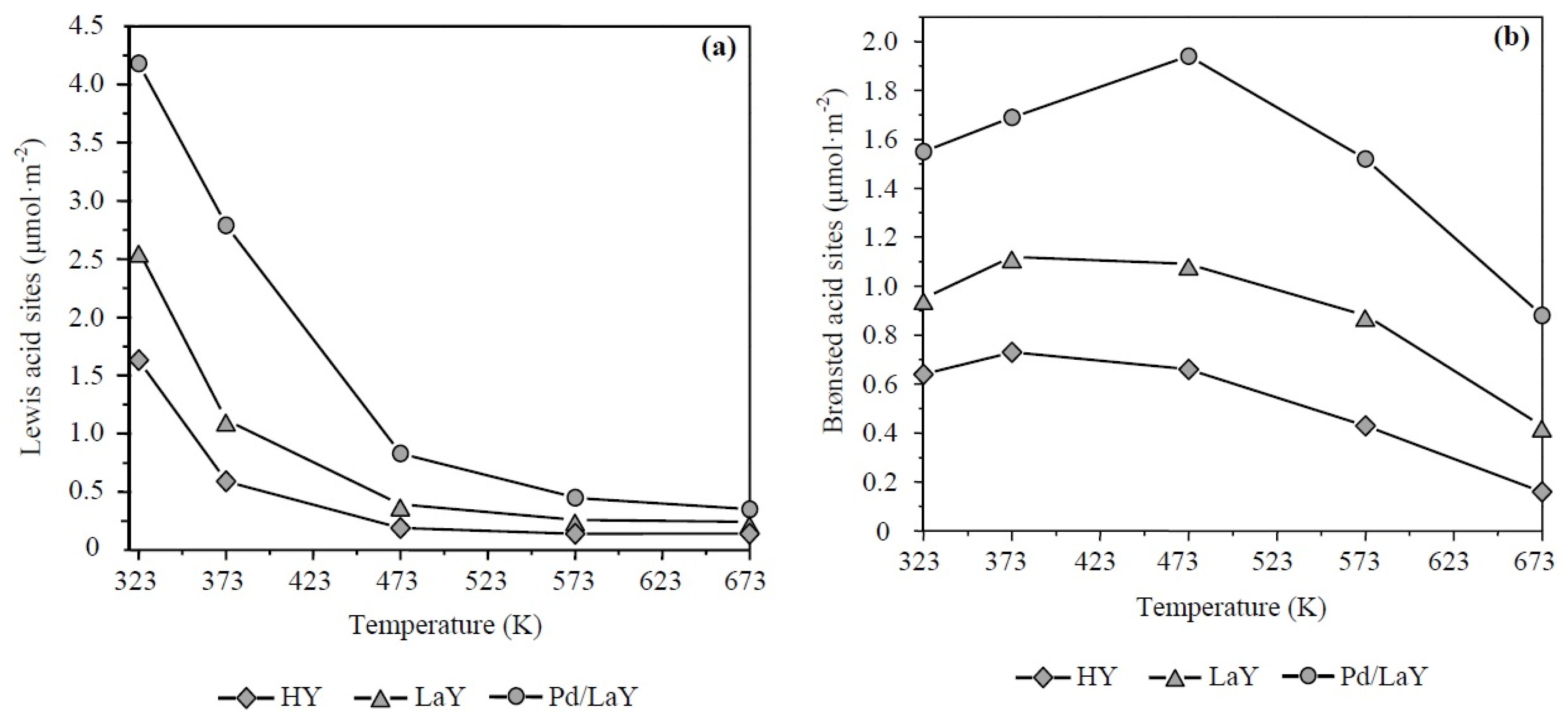
3.1.4. IR Spectroscopy with Adsorption of Pyridine (IR-Py)
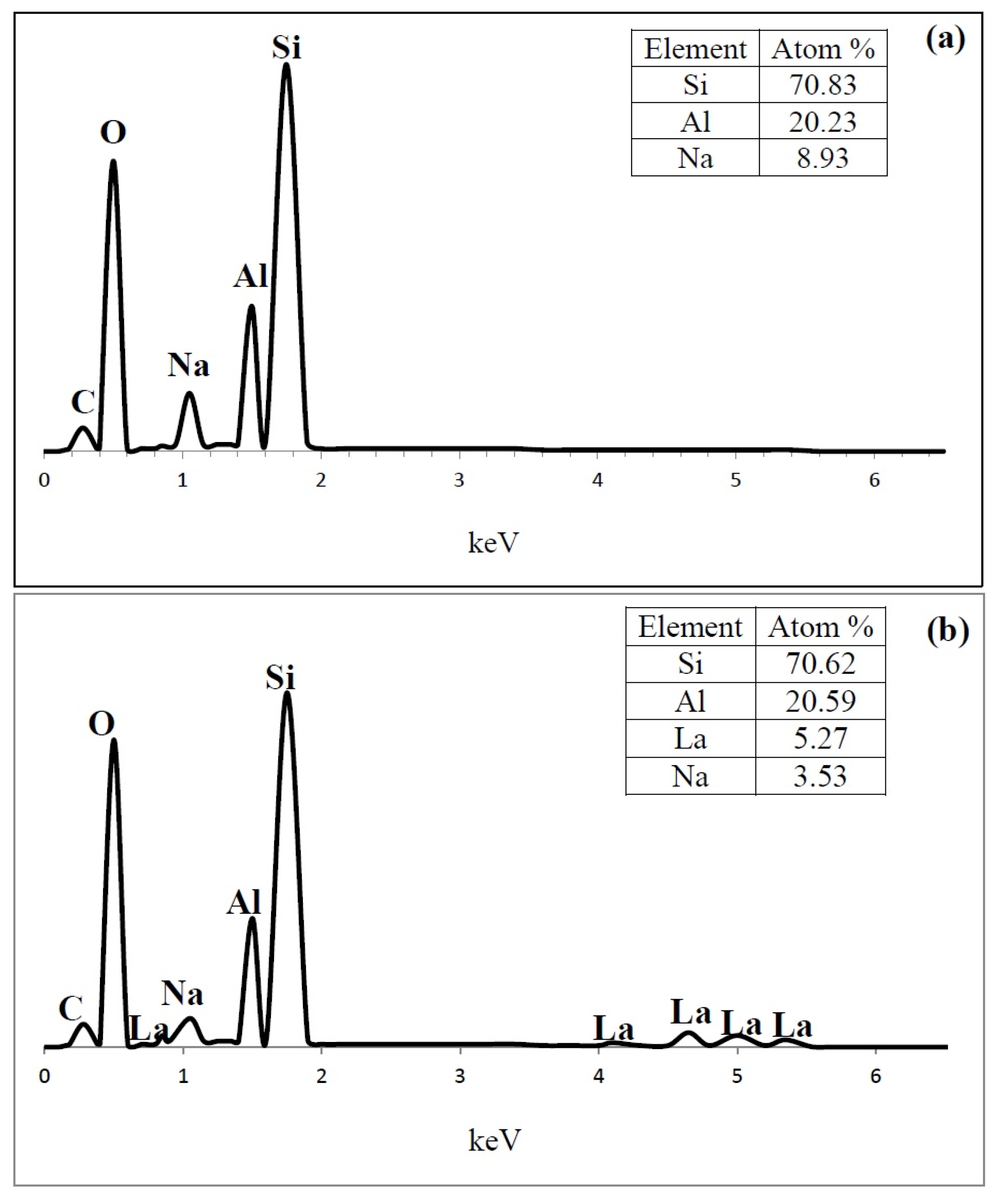
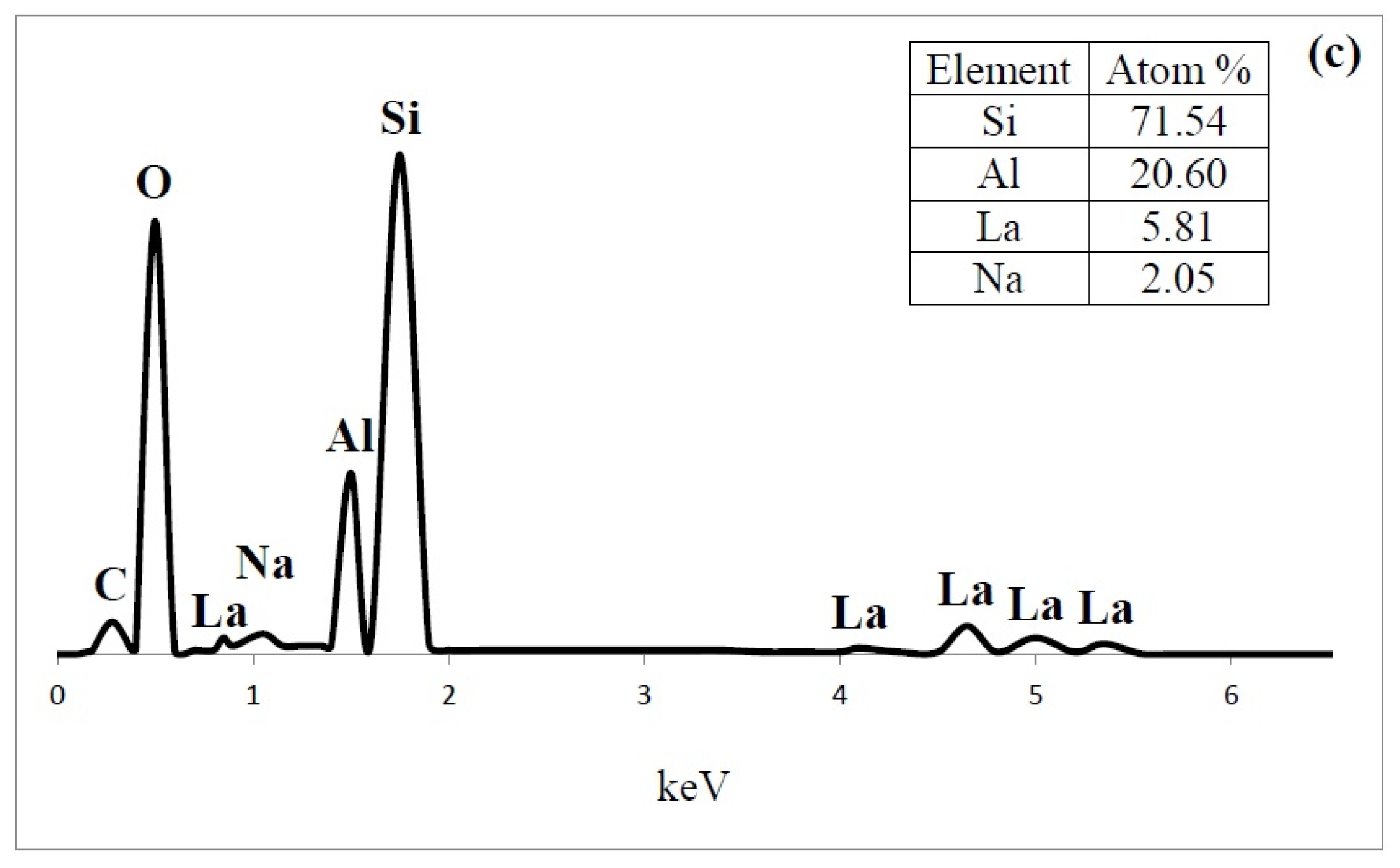
3.1.5. Scanning Electron Microscopy (SEM) and Energy-Dispersive X-ray Spectroscopy (EDS)
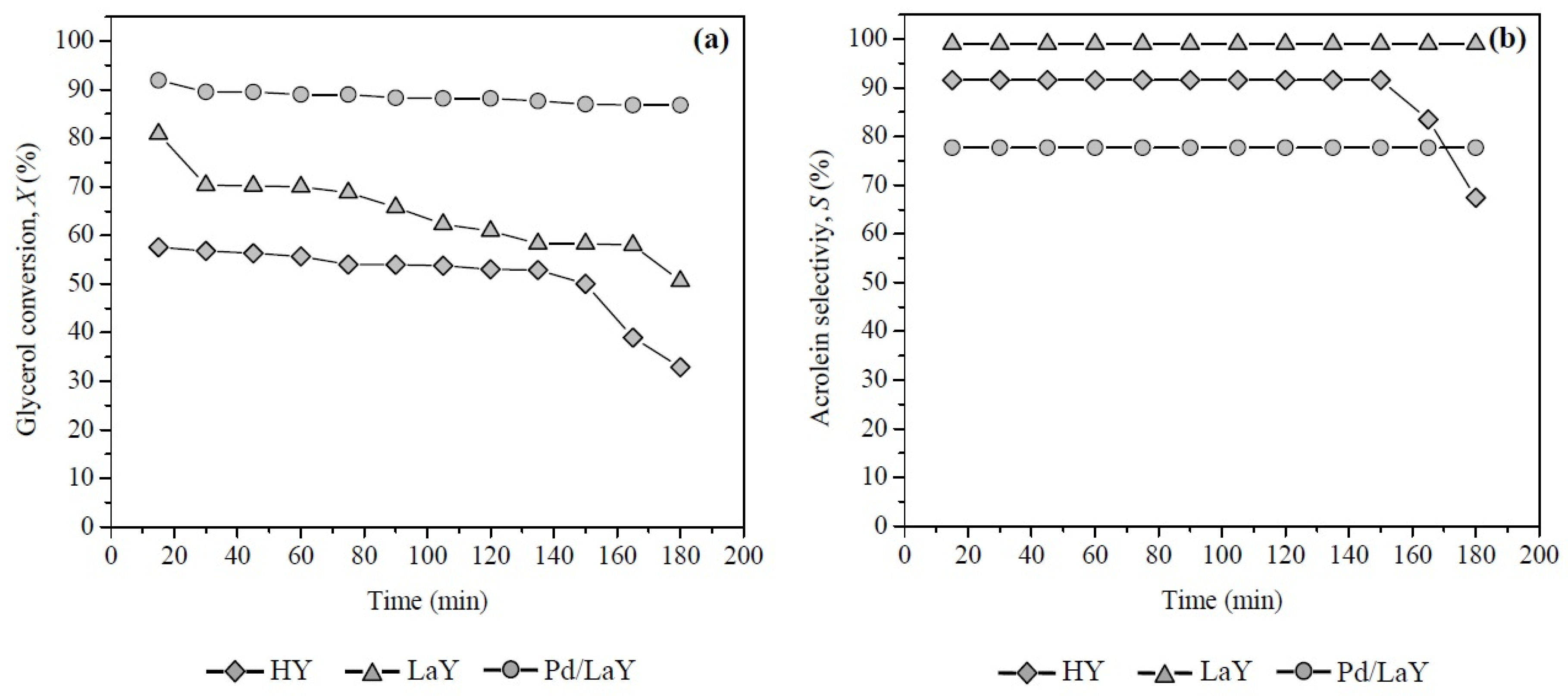
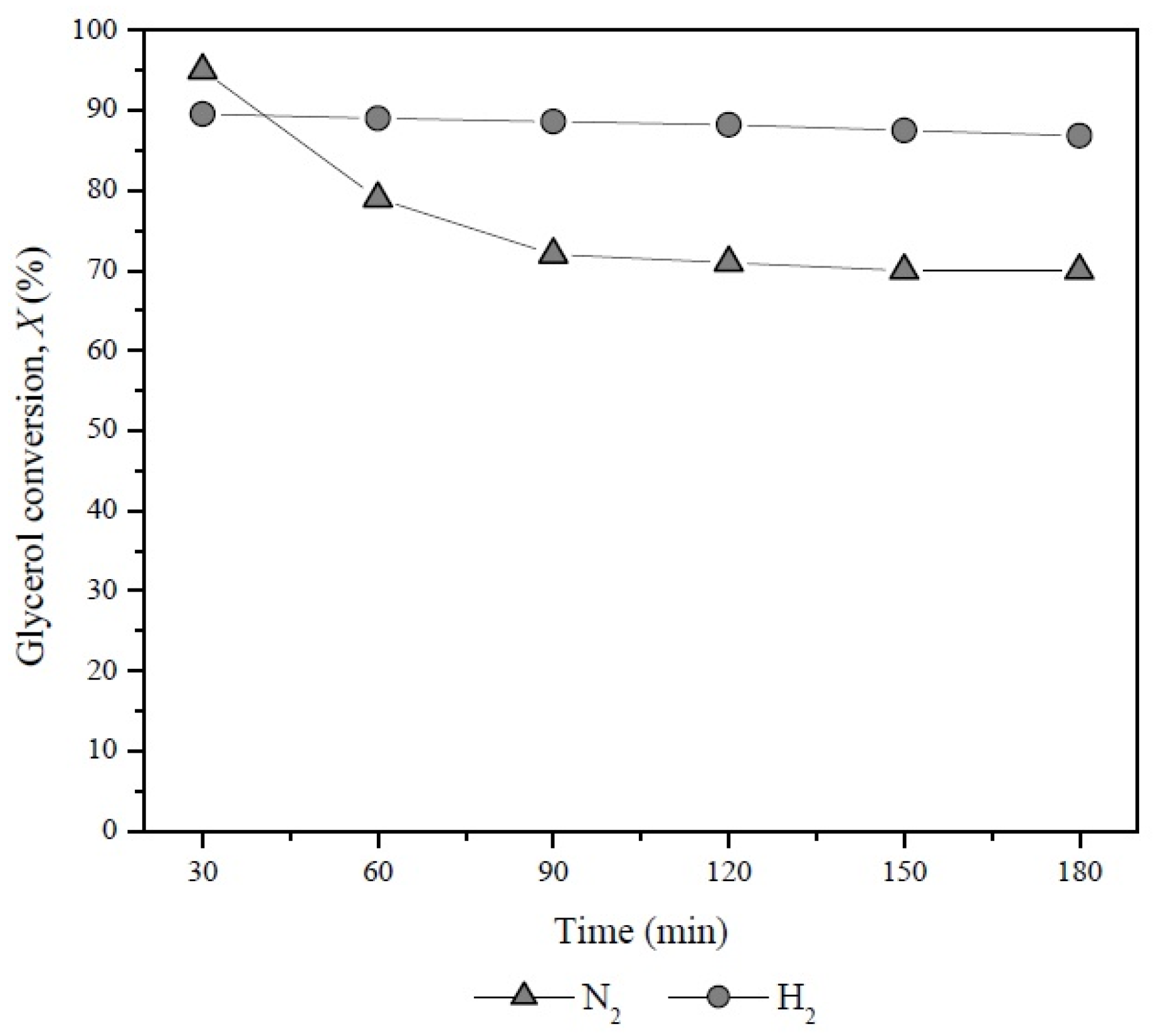
3.2. Catalytic Activity
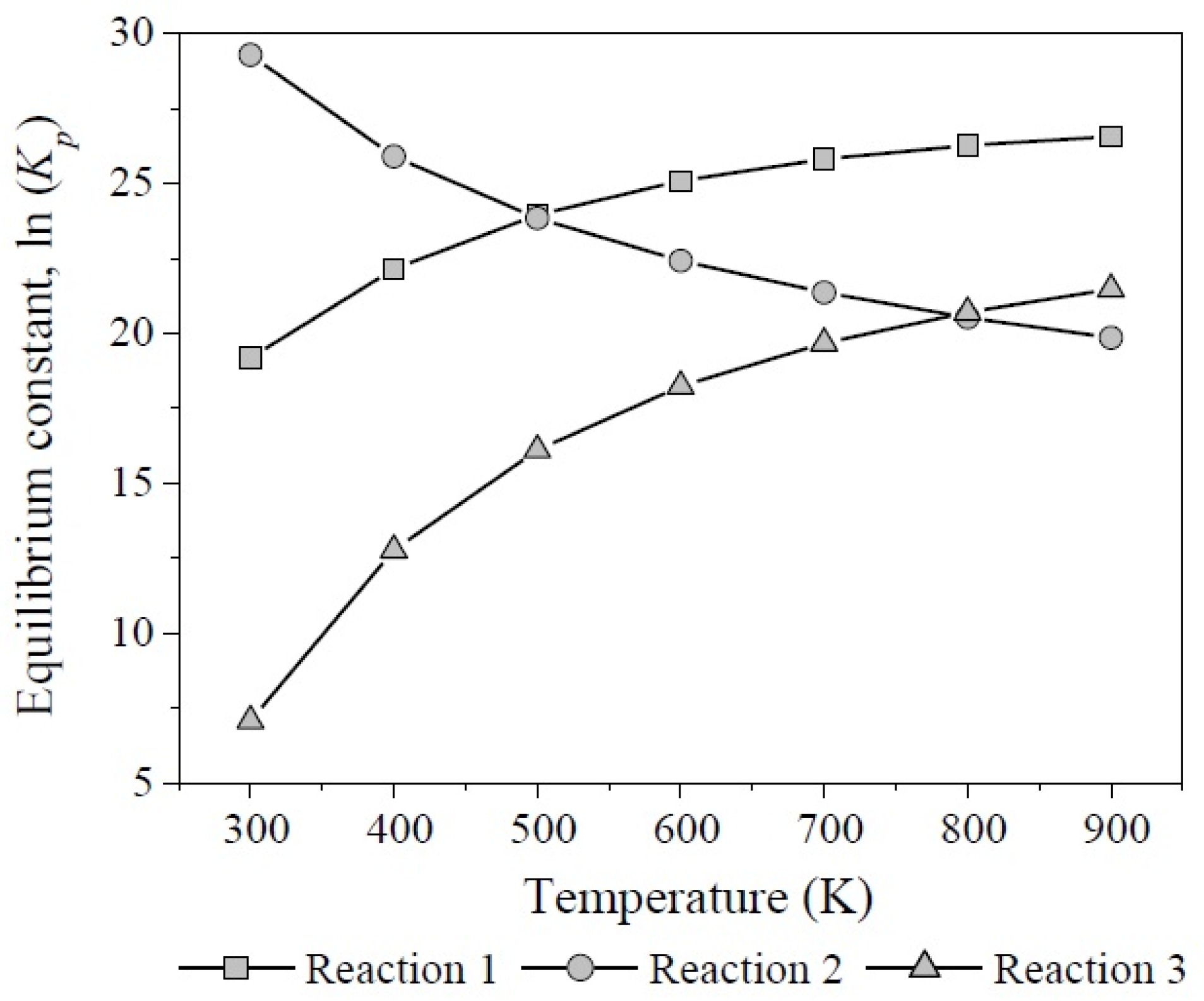
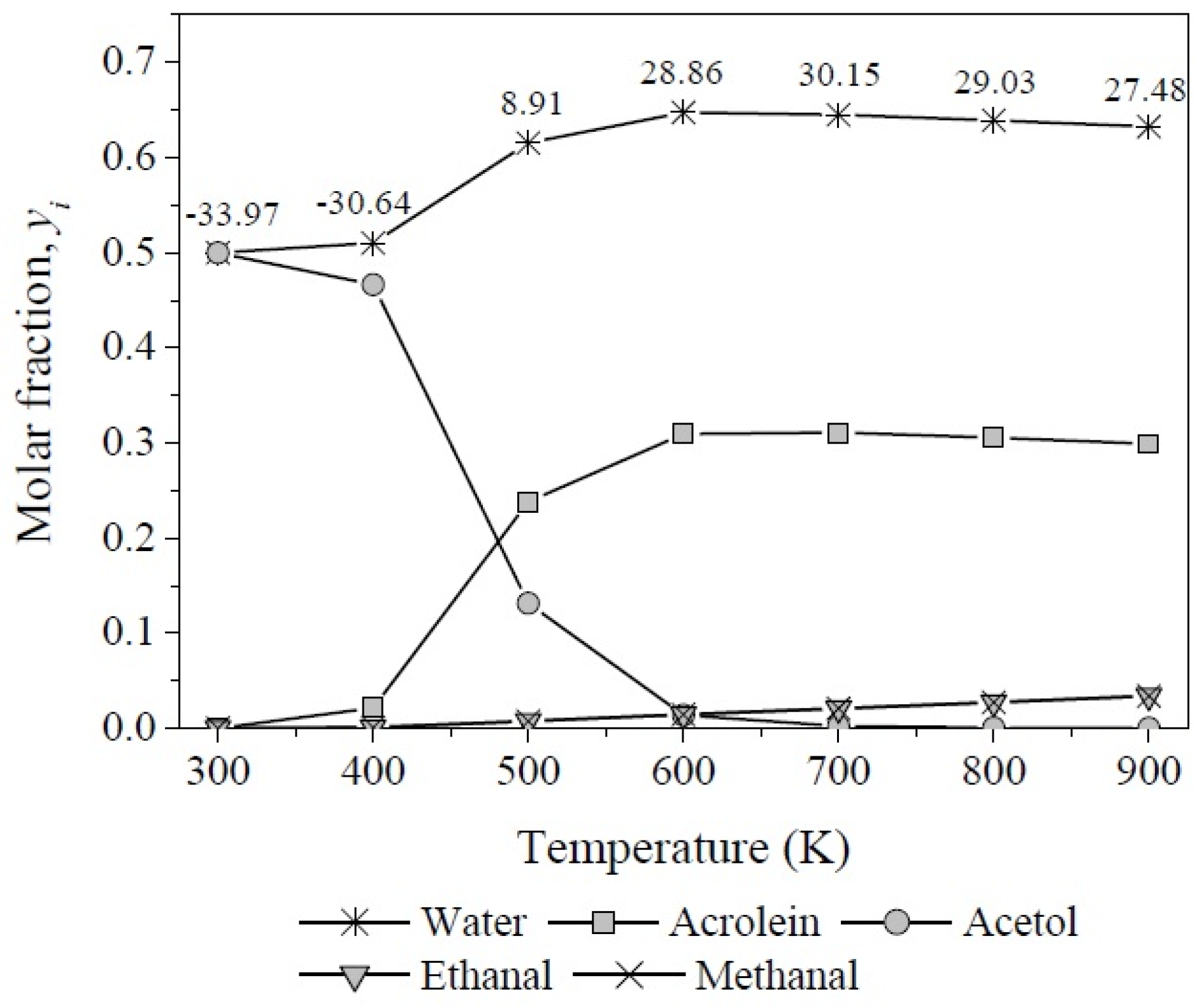
3.3. The Chemical Equilibrium
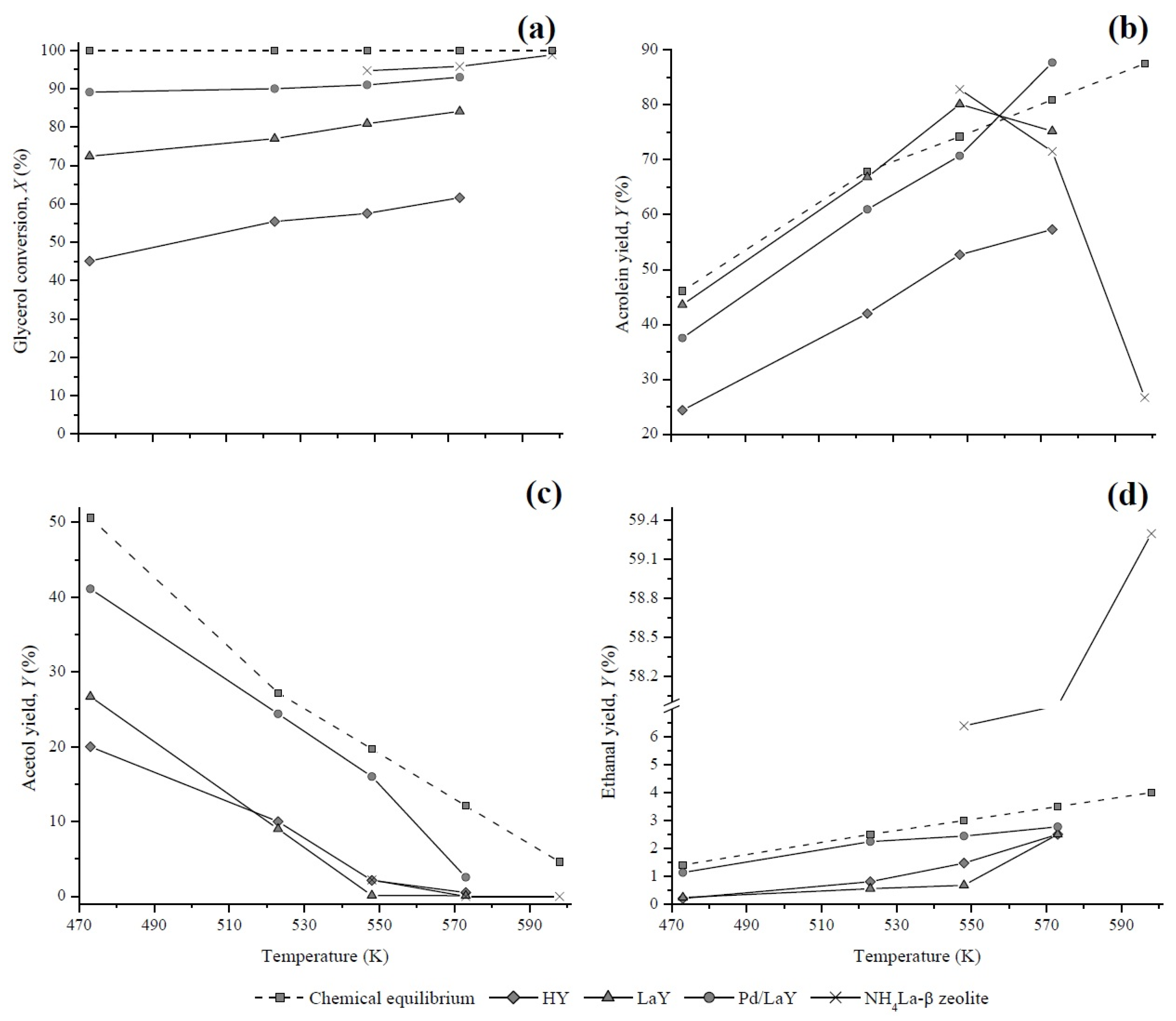
3.4. Comparison of Experimental and Calculated Yields
4. Materials and Methods
4.1. Catalyst Preparation
4.2. Catalyst Characterization
4.3. Catalytic Activity
4.4. The Chemical Equilibrium
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
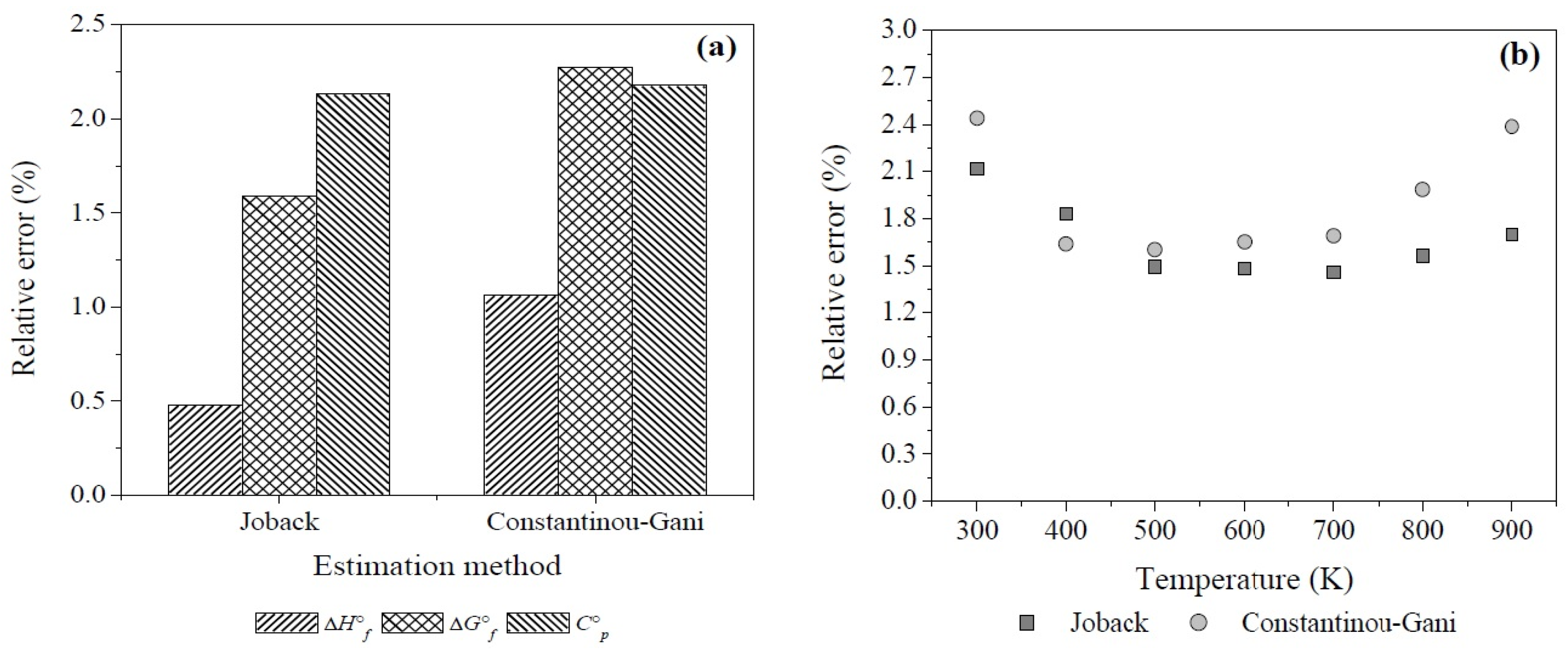
Appendix A. Estimation of Thermodynamic Properties of Pure Organic Compounds Using the Group Contribution Method

| Constantinou-Gani Method [81] | Joback Method [82] |
|---|---|
| With |
| Group | hf,1i | gf,1i | CpA,1i | CpB,1i | CpC,1i |
|---|---|---|---|---|---|
| CH3 | −45.947 | −8.030 | 35.1152 | 39.5923 | −9.9232 |
| CH2 | −20.763 | 8.231 | 22.6346 | 45.0933 | −15.7033 |
| CH | −3.766 | 19.848 | 8.9272 | 59.9786 | −29.5143 |
| CH2=CH | 53.712 | 84.926 | 49.2506 | 59.384 | −21.7908 |
| CH2=C | 64.145 | 88.402 | 37.6299 | 62.1285 | −26.0637 |
| CH=CH | 69.939 | 92.900 | 35.2248 | 62.1924 | −24.8152 |
| OH | −181.422 | −158.589 | 27.2107 | 2.7609 | 1.3060 |
| CH3CO | −182.329 | −131.366 | 59.3032 | 67.8149 | −20.9948 |
| CHO | −129.158 | −107.858 | 40.7501 | 19.6990 | −5.4360 |
| COOH | −396.242 | −349.439 | 46.5577 | 48.2322 | −20.4868 |
| Group | hf,i | gf,i | CpA,i | CpB,i | CpC,i | CpD,i |
|---|---|---|---|---|---|---|
| CH3 | −76.45 | −43.96 | 19.5 | −8.08 × 10−3 | 1.53 × 10−4 | −9.67 × 10−8 |
| CH2 | −20.64 | 8.42 | −0.909 | 9.50 × 10−2 | −5.44 × 10−5 | 1.19 × 10−8 |
| CH | 29.89 | 58.36 | −23.0 | 2.04 × 10−1 | −2.65 × 10−4 | 1.20 × 10−7 |
| H2C= | −9.63 | 3.77 | 23.6 | −3.81 × 10−2 | 1.72 × 10−4 | −1.03 × 10−7 |
| HC= | 37.97 | 48.53 | −8.0 | 1.05 × 10−1 | −9.63 × 10−5 | 3.56 × 10−8 |
| C= | 83.99 | 92.36 | −2.81 | 2.08 × 10−1 | −3.06 × 10−4 | 1.46 × 10−7 |
| C=O | −133.22 | −120.5 | 6.45 | 6.70 × 10−2 | −3.57 × 10−5 | 2.86 × 10−9 |
| CH=O | −162.03 | −143.48 | 30.9 | −3.36 x 10−2 | 1.60 x 10−4 | −9.88 x 10−8 |
| COOH | −426.72 | −387.87 | 24.1 | 4.27 x 10−2 | 8.04 x 10−5 | −6.87 x 10−8 |
| OH | −208.04 | −189.2 | 25.7 | −6.91 x 10−2 | 1.77 x 10−4 | −9.88 x 10−8 |
| Compound | ΔH°f (kJ·mol−1) | ΔG°f (kJ·mol−1) | C°p (J·mol−1·K−1) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Experimental | AEi,Joback | AEi,C-G | Experimental | AEi,Joback | AEi,C-G | Experimental | AEi,Joback | AEi,C-G | |
| Ethanol | −234 | −2.84 | −3.30 | −167.8 | −3.01 | −5.37 | 65.21 | −0.59 | −0.004 |
| Ethanal | −170.7 | 0.51 | 6.39 | −133.1 | −0.46 | 2.38 | 55.32 | 0.68 | 0.78 |
| Ethanoic acid | −432.9 | −1.98 | 1.55 | −374.6 | −3.35 | 2.30 | 63.44 | 2.23 | −1.52 |
| 1,2-Ethanediol | −390.3 | 1.23 | −2.87 | −302.6 | −5.08 | −9.14 | 77.99 | −0.39 | 1.95 |
| 1-Propanol | −256.3 | −1.18 | −1.76 | −159.9 | −2.54 | −5.08 | 85.56 | 1.96 | 2.29 |
| Propanal | −188.7 | −2.13 | 3.63 | −124.6 | −0.54 | 2.11 | 80.73 | −1.84 | −1.98 |
| 2-Propanone | −218.5 | 0.67 | 1.06 | −151.3 | −3.24 | −2.92 | 75.02 | −0.05 | −0.35 |
| Acrylic acid | −330.7 | 0.61 | 0.28 | −271 | −10.69 | −6.19 | 81.8 | −3.77 | −5.74 |
| n-Butanol | −277 | −1.12 | −1.82 | −150.3 | −3.72 | −6.45 | 108.03 | 2.38 | 2.46 |
| 2-Butanone | −238.6 | 0.13 | 1.37 | −147 | 0.88 | 1.08 | 101.68 | −3.82 | −0.57 |
| AE (kJ·mol−1) | 1.24 | 2.40 | AE (kJ·mol−1) | 3.35 | 4.30 | AE (J·mol−1·K−1) | 1.77 | 1.77 | |
| σ (kJ·mol−1) | 1.48 | 2.91 | σ (kJ·mol−1) | 4.39 | 4.93 | σ (J·mol−1·K−1) | 2.18 | 2.35 | |

| Compound | ΔH°f|298.15 K (kJ·mol−1) | ΔG°f|298.15 K (kJ·mol−1) | Cp (J·mol−1·K−1) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 298 K | 300 K | 400 K | 500 K | 600 K | 700 K | 800 K | 900 K | |||
| Glycerol | −577.90 | −447.10 | 113.26 | 113.76 | 139.04 | 160.92 | 179.74 | 195.79 | 209.42 | 220.93 |
| Water (a) | −241.83 | −228.59 | 33.41 | 33.45 | 34.37 | 35.38 | 36.47 | 37.60 | 38.73 | 39.94 |
| Acrolein | −65.40 | −37.30 | 71.28 | 71.59 | 87.72 | 101.33 | 112.42 | 121.45 | 128.90 | 135.0 |
| Acetol | −370.06 | −291.36 | 87.95 | 88.34 | 108.18 | 125.60 | 140.76 | 153.81 | 164.90 | 174.17 |
| Methanal (a) | −108.60 | −102.60 | 35.39 | 35.44 | 39.24 | 43.74 | 48.18 | 52.28 | 55.94 | 59.16 |
| Ethanal (a) | −170.70 | −133.10 | 55.32 | 55.51 | 66.28 | 76.68 | 85.94 | 94.04 | 101.07 | 107.19 |
| 1,3-DHP (b) | −292.49 | −219.04 | 86.08 | 86.51 | 107.69 | 125.40 | 140.16 | 152.54 | 163.09 | 172.34 |
| 2,3-DHP (b) | −294.07 | −219.97 | 115.38 | 115.78 | 136.02 | 153.26 | 167.90 | 180.31 | 190.86 | 199.95 |
| Vinyl Alcohol | −111.41 | −83.02 | 54.09 | 54.34 | 66.91 | 77.67 | 86.86 | 94.71 | 101.47 | 107.38 |
| 3-HP | −343.06 | −261.96 | 91.87 | 92.26 | 111.78 | 128.82 | 143.58 | 156.23 | 166.98 | 176.03 |
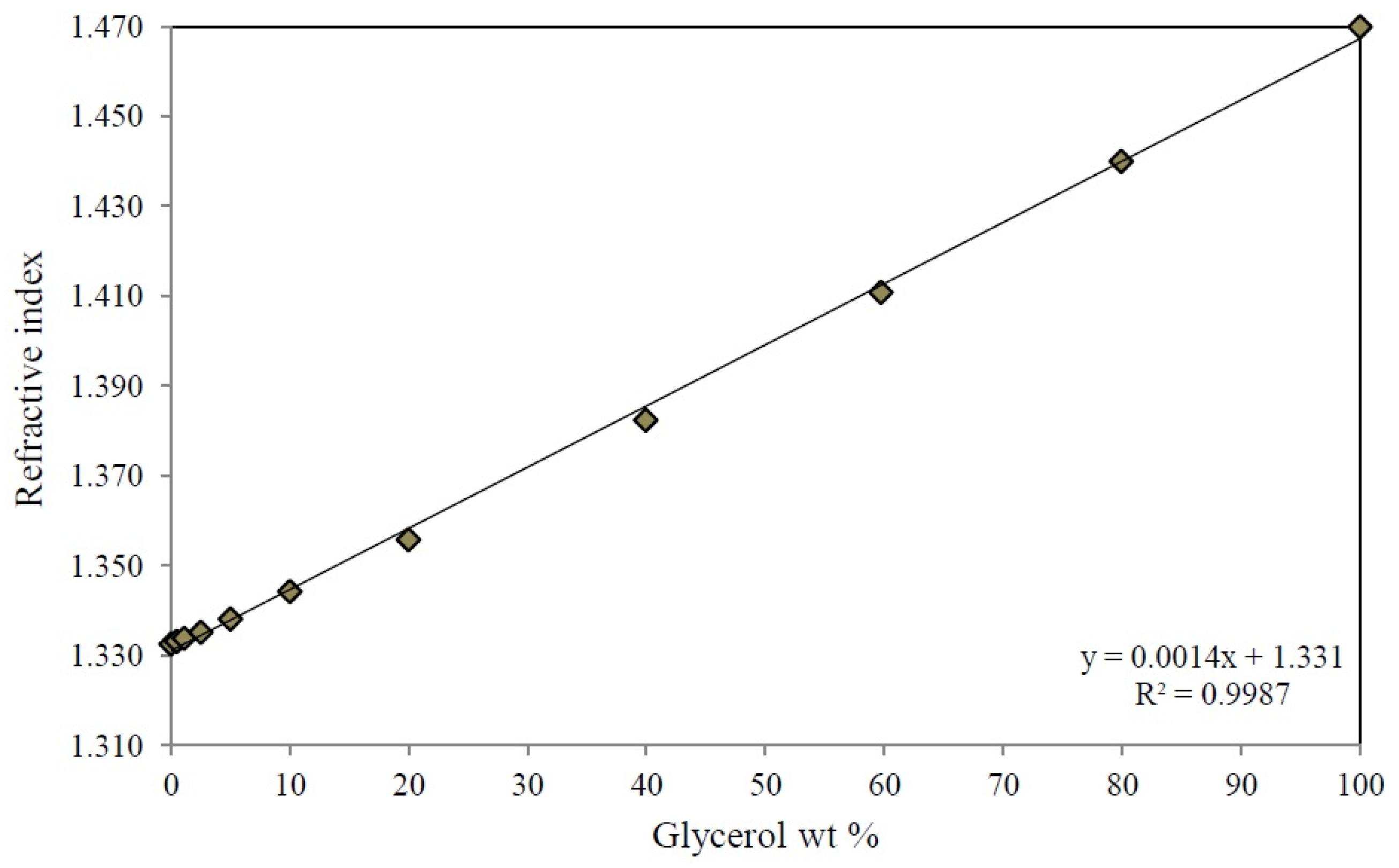
Appendix B. Calibration Curve of Refractive Index of Glycerol-Water Mixtures at 298 K

References
- Knothe, G.; van Gerpen, J.; Krahl, J. The Biodiesel Handbook; AOCS Press: Champaign, IL, USA, 2005; pp. 9–11. [Google Scholar]
- Di Serio, M.; Cozzolino, M.; Giordano, M.; Tesser, R.; Patrono, P.; Santacesaria, E. From homogeneous to heterogeneous catalysts in biodiesel production. Ind. Eng. Chem. Res. 2007, 46, 6379–6384. [Google Scholar] [CrossRef]
- Helwani, Z.; Othman, M.R.; Aziz, N.; Fernando, W.J.N.; Kim, J. Technologies for production of biodiesel focusing on green catalytic techniques: A review. Fuel Process. Technol. 2009, 90, 1502–1514. [Google Scholar] [CrossRef]
- Talebian-Kiakalaieh, A.; Saidina Amin, N.A.; Mazaheri, H. A review on novel processes of biodiesel production from waste cooking oil. Appl. Energy 2013, 104, 683–710. [Google Scholar] [CrossRef]
- Atadashi, I.M.; Aroua, M.K.; Abdul Aziz, A.R.; Sulaiman, N.M.N. Membrane biodiesel production and refining technology: A critical review. Renew. Sustain. Energy Rev. 2011, 15, 5051–5062. [Google Scholar] [CrossRef]
- Berrios, M.; Skelton, R.L. Comparison of purification methods for biodiesel. Chem. Eng. J. 2008, 144, 459–465. [Google Scholar] [CrossRef]
- Cao, P.; Dubé, M.A.; Tremblay, A.Y. Methanol recycling in the production of biodiesel in a membrane reactor. Fuel 2008, 87, 825–833. [Google Scholar] [CrossRef]
- Helwani, Z.; Othman, M.R.; Aziz, N.; Kim, J.; Fernando, W.J.N. Solid heterogeneous catalysts for transesterification of triglycerides with methanol: A review. Appl. Catal. A Gen. 2009, 363, 1–10. [Google Scholar] [CrossRef]
- Kulkarni, M.G.; Dalai, A.K. Waste Cooking Oil—An Economical Source for Biodiesel: A Review. Ind. Eng. Chem. Res. 2006, 45, 2901–2913. [Google Scholar] [CrossRef]
- Sanli, H.; Canakci, M. Effects of Different Alcohol and Catalyst Usage on Biodiesel Production from Different Vegetable Oils. Energy Fuel 2008, 22, 2713–2719. [Google Scholar] [CrossRef]
- Sérgi Gomes, M.C.; Curvelo Pereira, N.; Davantel de Barros, S.T. Separation of biodiesel and glycerol using ceramic membranes. J. Membrane. Sci. 2010, 352, 271–276. [Google Scholar] [CrossRef]
- Quispe, C.A.G.; Coronado, C.J.R.; Carvalho, J.A., Jr. Glycerol: Production, consumption, prices, characterization and new trends in combustion. Renew. Sustain. Energy Rev. 2013, 27, 475–493. [Google Scholar] [CrossRef]
- OCDE/FAO. OCDE-FAO Perspectivas Agrícolas 2015; OECD Publishing: Paris, France, 2015. [Google Scholar]
- Markočič, E.; Kramberger, B.; van Bennekom, J.G.; Heeres, H.J.; Vos, J.; Knez, Ž. Glycerol reforming in supercritical water: A short review. Renew. Sustain. Energy Rev. 2013, 23, 40–48. [Google Scholar] [CrossRef]
- Martin, A.; Richter, M. Oligomerization of glycerol—A critical review. Eur. J. Lipid Sci. Technol. 2011, 113, 100–117. [Google Scholar] [CrossRef]
- Nanda, M.R.; Zhang, Y.; Yuan, Z.; Qin, W.; Ghaziaskar, H.S.; Xu, C. Catalytic conversion of glycerol for sustainable production of solketal as a fuel additive: A review. Renew. Sustain. Energy Rev. 2016, 56, 1022–1031. [Google Scholar] [CrossRef]
- Okoye, P.U.; Hameed, B.H. Review on recent progress in catalytic carboxylation and acetylation of glycerol as a byproduct of biodiesel production. Renew. Sustain. Energy Rev. 2016, 53, 558–574. [Google Scholar] [CrossRef]
- Tagliapietra, S.; Orio, L.; Palmisano, G.; Penoni, A.; Cravotto, G. Catalysis in glycerol: A survey of recent advances. Chem. Pap. 2015, 69, 1519–1531. [Google Scholar] [CrossRef]
- Teng, W.K.; Ngoh, G.C.; Yusoff, R.; Aroua, M.K. A review on the performance of glycerol carbonate production via catalytic transesterification: Effects of influencing parameters. Energy Convers. Manag. 2014, 88, 484–497. [Google Scholar] [CrossRef]
- Viana, M.B.; Freitas, A.V.; Leitao, R.C.; Pinto, G.A.S.; Santaella, S.T. Anaerobic digestion of crude glycerol: A review. Environ. Technol. Rev. 2012, 1, 81–92. [Google Scholar] [CrossRef]
- Weissermel, K.; Arpe, H.-J. Chapter 11 Propene conversion products. In Industrial Organic Chemistry, 3rd ed.; VCH Publisher, Inc.: Weinheim, Germany, 1997. [Google Scholar]
- Liu, L.; Ye, X.P.; Bozell, J.J. A Comparative Review of Petroleum-Based and Bio-Based Acrolein Production. ChemSusChem 2012, 5, 1162–1180. [Google Scholar] [CrossRef] [PubMed]
- Etzkorn, W.G. Kirk-Othmer Encyclopedia of Chemical Technology; John Wiley & Sons, Inc.: New York, NY, USA, 2009; pp. 1–29. [Google Scholar]
- Mohamad, M.H.; Awang, R.; Yunus, W.M.Z.W. A Review of Acetol: Application and Production. Am. J. Appl. Sci. 2011, 8, 1135–1139. [Google Scholar] [CrossRef]
- Kongpatpanich, K.; Nanok, T.; Boekfa, B.; Probste, M.; Limtrakul, J. Structures and reaction mechanisms of glycerol dehydration over H-ZSM-5 zeolite: A density functional theory study. Phys. Chem. Chem. Phys. 2011, 13, 6462–6470. [Google Scholar] [CrossRef] [PubMed]
- Laino, T.; Tuma, C.; Curioni, A.; Jochnowitz, E.; Stolz, S. A Revisited Picture of the Mechanism of Glycerol Dehydration. J. Phys. Chem. A 2011, 115, 3592–3595. [Google Scholar] [CrossRef] [PubMed]
- Nimlos, M.R.; Blanksby, S.J.; Qian, X.; Himmel, M.E.; Johnson, D.K. Mechanisms of Glycerol Dehydration. J. Phys. Chem. A 2006, 110, 6145–6156. [Google Scholar] [CrossRef] [PubMed]
- Chai, S.H.; Wang, H.-P.; Liang, Y.; Xu, B.-Q. Sustainable production of acrolein: Gas-phase dehydration of glycerol over Nb2O5 catalyst. J. Catal. 2007, 250, 342–349. [Google Scholar] [CrossRef]
- Lauriol-Garbay, P.; Millet, J.M.; Loridant, S.; Bellière-Baca, V.; Rey, P. New efficient and long-life catalyst for gas-phase glycerol dehydration to acrolein. J. Catal. 2011, 280, 68–76. [Google Scholar] [CrossRef]
- Mane, R.B.; Rode, C.V. Continuous Dehydration and Hydrogenolysis of Glycerol over Non-Chromium Copper Catalyst: Laboratory-Scale Process Studies. Org. Process Res. Dev. 2012, 16, 1043–1052. [Google Scholar] [CrossRef]
- Massa, M.; Andersson, A.; Finocchio, E.; Busca, G. Gas-phase dehydration of glycerol to acrolein over Al2O3-, SiO2-, and TiO2-supported Nb- and W-oxide catalysts. J. Catal. 2013, 307, 170–184. [Google Scholar] [CrossRef]
- Suprun, W.; Papp, H. Gas-phase conversion of glycerol over mixed metal oxide catalysts. Catal. Ind. 2011, 3, 70–75. [Google Scholar] [CrossRef]
- Talebian-Kiakalaieh, A.; Amin, N.A.S.; Hezaveh, H. Glycerol for renewable acrolein production by catalytic dehydration. Renew. Sustain. Energy Rev. 2014, 40, 28–59. [Google Scholar] [CrossRef]
- Tao, L.-Z.; Chai, S.-H.; Zuo, Y.; Zheng, W.-T.; Liang, Y.; Xu, B.-Q. Sustainable production of acrolein: Acidic binary metal oxide catalysts for gas-phase dehydration of glycerol. Catal. Today 2010, 158, 310–316. [Google Scholar] [CrossRef]
- Kim, Y.T.; Jung, K.-D.; Park, E.D. Gas-phase dehydration of glycerol over ZSM-5 catalysts. Microporous Mesoporous Mater. 2010, 131, 28–36. [Google Scholar] [CrossRef]
- Kim, Y.T.; Jung, K.-D.; Park, E.D. A comparative study for gas-phase dehydration of glycerol over H zeolites. Appl. Catal. A Gen. 2011, 393, 275–287. [Google Scholar] [CrossRef]
- Yue, C.-J.; Gan, M.-M.; Gu, L.-P.; Zhuang, Y.-F. In situ synthesized nano-copper over ZSM-5 for the catalytic dehydration of glycerol under mild conditions. J. Taiwan Inst. Chem. Eng. 2014, 45, 1443–1448. [Google Scholar] [CrossRef]
- Dalla Costa, B.O.; Peralta, M.A.; Querini, C.A. Gas phase dehydration of glycerol over, lanthanum modified beta-zeolite. Appl. Catal. A Gen. 2014, 472, 53–63. [Google Scholar] [CrossRef]
- Atia, H.; Armbruster, U.; Martin, A. Dehydration of glycerol in gas phase using heteropolyacid catalysts as active compounds. J. Catal. 2008, 258, 71–82. [Google Scholar] [CrossRef]
- Atia, H.; Armbruster, U.; Martin, A. Influence of alkaline metal on performance of supported silicotungstic acid catalysts in glycerol dehydration towards acrolein. Appl. Catal. A Gen. 2011, 393, 331–339. [Google Scholar] [CrossRef]
- Haider, M.H.; Dummer, N.F.; Zhang, D.; Miedziak, P.; Davies, T.E.; Taylor, S.H.; Willock, D.J.; Knight, D.W.; Chadwick, D.; Hutchings, G.J. Rubidium- and caesium-doped silicotungstic acid catalysts supported on alumina for the catalytic dehydration of glycerol to acrolein. J. Catal. 2012, 286, 206–213. [Google Scholar] [CrossRef]
- Kraleva, E.; Palcheva, R.; Dimitrov, L.; Armbruster, U.; Brückner, A.; Spojakina, A. Solid acid catalysts for dehydration of glycerol to acrolein in gas phase. J. Mater. Sci. 2011, 46, 7160–7168. [Google Scholar] [CrossRef]
- Liu, R.; Wang, T.; Liu, C.; Jin, Y. Highly selective and stable CsPW/Nb2O5 catalysts for dehydration of glycerol to acrolein. Chin. J. Catal. 2013, 34, 2174–2182. [Google Scholar] [CrossRef]
- Shen, L.; Feng, Y.; Yin, H.; Wang, A.; Yu, L.; Jiang, T.; Shen, Y.; Wu, Z. Gas phase dehydration of glycerol catalyzed by rutile TiO2-supported heteropolyacids. J. Ind. Eng. Chem. 2011, 17, 484–492. [Google Scholar] [CrossRef]
- Talebian-Kiakalaieh, A.; Amin, N.A.S. Kinetic Modeling, Thermodynamic, and Mass Transfer Studies of Gas-Phase Glycerol Dehydration to Acrolein over Supported Silicotungstic Acid Catalyst. Ind. Eng. Chem. Res. 2015, 54, 8113–8121. [Google Scholar] [CrossRef]
- Tsukuda, E.; Sato, S.; Takahashi, R.; Sodesawa, T. Production of acrolein from glycerol over silica-supported heteropoly acids. Catal. Commun. 2007, 8, 1349–1353. [Google Scholar] [CrossRef]
- Viswanadham, B.; Pavankumar, V.; Chary, K.V.R. Vapor Phase Dehydration of Glycerol to Acrolein Over Phosphotungstic Acid Catalyst Supported on Niobia. Catal. Lett. 2014, 144, 744–755. [Google Scholar] [CrossRef]
- Gates, B.C.; Katzer, J.R.; Schuit, G.C.A. Chemistry of Catalytic Processes; McGraw-Hill, Inc.: New York, NY, USA, 1979. [Google Scholar]
- Weitkamp, J. Zeolites and catalysis. Solid State Ionics 2000, 131, 175–188. [Google Scholar] [CrossRef]
- Lutz, W. Zeolite Y: Synthesis, modification, and properties—A case revisited. Adv. Mater. Sci. Eng. 2014, 2014, 724248. [Google Scholar] [CrossRef]
- Smith, J.M.; van Ness, H.C.; Abbott, M.M. Chapter 13 Chemical-reaction equilibria. In Introduction to Chemical Engineering Thermodynamics, 7nd ed.; McGraw-Hill Higher Education: New York, NY, USA, 2005. [Google Scholar]
- Levenspiel, O. Chapter 9. Temperature and pressure effects. In Chemical Reaction Engineering, 3rd ed.; John Wiley & Sons, Inc.: New York, NY, USA, 1999. [Google Scholar]
- Martinuzzi, I.; Azizi, Y.; Devaux, J.-F.; Tretjak, S.; Zahraa, O.; Leclerc, J.-P. Reaction mechanism for glycerol dehydration in the gas phase over a solid acid catalyst determined with on-line gas chromatography. Chem. Eng. Sci. 2014, 116, 118–127. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmet, P.H.; Teller, E. Adsorption of Gases in Multimolecular Layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Lippens, B.C.; Linsen, B.G.; de Boer, J.H. Studies on pore systems in catalysts I. The adsorption of nitrogen; Apparatus and calculation. J. Catal. 1964, 3, 32–37. [Google Scholar] [CrossRef]
- Sing, K.S.W.; Everett, D.H.; Haul, R.A.W.; Moscou, L.; Pierotti, R.A.; Rouquerol, J.; Siemieniewska, T. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity. Pure Appl. Chem. 1985, 57, 603–619. [Google Scholar] [CrossRef]
- De Angelis, A.; Flego, C.; Pollesel, P.; Tagliabue, M. Pd/La H-Y a novel and promising catalyst for hydrocracking of heavy Pygas. Stud. Surf. Sci. Catal. 2005, 1701–1708. [Google Scholar]
- Kaduk, J.A.; Faber, J. Crystal structure of zeolite Y as function of ion exchange. Rigaku J. 1995, 12, 14–34. [Google Scholar]
- Du, X.; Zhang, H.; Li, X.; Tan, Z.; Liu, H.; Gao, X. Cation location and migration in lanthanum exchanged NaY zeolite. Chin. J. Catal. 2013, 34, 1599–1607. [Google Scholar] [CrossRef]
- Azaroff, L.V.; Buerger, M.J. The Powder Method in X-ray Crystallography; McGraw-Hill: New York, NY, USA, 1958. [Google Scholar]
- Füglein, E.; Walter, D. Thermal analysis of lanthanum hydroxide. J. Therm. Anal. Calorim. 2012, 110, 199–202. [Google Scholar] [CrossRef]
- Parry, E.P. An infrared study of pyridine adsorbed on acidic solids. Characterization of surface acidity. J. Catal. 1963, 2, 371–379. [Google Scholar] [CrossRef]
- Oliveira, A.S.; Vasconcelos, S.J.S.; Sousa, J.R.; Sousa, F.F.; Filho, M.; Oliveira, A.C. Catalytic conversion of glycerol to acrolein over modified molecular sieves: Activity and deactivation studies. Chem. Eng. J. 2011, 168, 765–774. [Google Scholar] [CrossRef]
- Tynjälä, P.; Pakkanen, T.T. Acidic properties of ZSM-5 zeolite modified with Ba2+, Al3+ and La3+ ion-exchange. J. Mol. Catal. A Chem. 1996, 110, 153–161. [Google Scholar] [CrossRef]
- Kim, J.-G.; Kompany, T.; Ryoo, R.; Ito, T.; Fraissard, J. 129Xe n.m.r. of Y3+-, La3+-, and Ce3+-exchanged X zeolites. Zeolites 1994, 14, 427–432. [Google Scholar] [CrossRef]
- Li, D.; Li, F.; Ren, J.; Sun, Y. Rare earth-modified bifunctional Ni/HY catalysts. Appl. Catal. A Gen. 2003, 241, 15–24. [Google Scholar] [CrossRef]
- Gu, Y.; Cui, N.; Yu, Q.; Li, C.; Cui, Q. Study on the influence of channel structure properties in the dehydration of glycerol to acrolein over H-zeolite catalysts. Appl. Catal. A Gen. 2012, 429–430, 9–16. [Google Scholar] [CrossRef]
- Alhanash, A.; Kozhevnikona, E.F.; Kozhevnikov, I.V. Gas-phase dehydration of glycerol to acrolein catalysed by caesium heteropoly salt. Appl. Catal. A Gen. 2010, 378, 11–18. [Google Scholar] [CrossRef]
- Ma, T.; Yun, Z.; Xu, W.; Chen, L.; Li, L.; Ding, J.; Shao, R. Pd-H3PW12O40/Zr-MCM-41: An efficient catalyst for the sustainable dehydration of glycerol to acrolein. Chem. Eng. J. 2016, 294, 343–352. [Google Scholar] [CrossRef]
- Sabater Prieto, S. Optimization of the Dehydration of Glycerol to Acrolein and a Scale up in a Pilot Plant. Ph.D. Dissertation, University of Technology, RWTH Aachen, Aachen, Germany, 2007. [Google Scholar]
- Meunier, F.C.; Scalbert, J.; Thibault-Starzyk, F. Unraveling the mechanism of chemical reactions through thermodynamic analyses: A short review. Appl. Catal. A Gen. 2015, 504, 220–227. [Google Scholar] [CrossRef]
- Ahmed Ali, S. Chapter 4 Thermodynamics of hydroprocessing reactions. In Hydroprocessing of Heavy Oils and Residua; Ancheyta, J., Speigh, J., Eds.; CRC Press: New York, NY, USA, 2007. [Google Scholar]
- Vasiliu, M.; Guynn, K.; Dixon, D.A. Prediction of the Thermodynamic Properties of Key Products and Intermediates from Biomass. J. Phys. Chem. C 2011, 115, 15686–15702. [Google Scholar] [CrossRef]
- Verevkin, S.P.; Zaitsau, D.H.; Emel’yanenko, V.N.; Zhabina, A.A. Thermodynamic properties of glycerol: Experimental and theoretical study. Fluid Phase Equilib. 2015, 397, 87–94. [Google Scholar] [CrossRef]
- Linstrom, P.J.; Mallard, W.G. (Eds.) NIST Chemistry WebBook, NIST Standard Reference Database Number 69, National Institute of Standards and Technology, Gaithersburg MD, 20899, 2015. Available online: http://webbook.nist.gov/ (accessed on 15 July 2016).
- Stumm, W.; Morgan, J.J. Chapter 2 Chemical thermodynamics and kinetics. In Aquatic Chemistry, 3rd ed.; John Wiley and Sons Inc.: New York, NY, USA, 1996. [Google Scholar]
- Contreras, J.L.; Tapia, C.; Fuentes, G.A.; Nuño, L.; Quintana, B.; Salmones, J.; Zeifert, B.; Córdova, I. Equilibrium composition of ethanol steam reforming reaction to produce H2 applied to Ni, Co and Pt/hydrotalcite–WOx catalysts. Int. J. Hydrog. Energy 2014, 39, 16608–16618. [Google Scholar] [CrossRef]
- Bondioli, P.; Della Bella, L. An Alternative Spectrophotometric Method for the Determination of free Glycerol in Biodiesel. Eur. J. Lipid Sci. Technol. 2005, 107, 153–157. [Google Scholar] [CrossRef]
- Walas, S.M. Chapter 10 Chemical equilibria. In Phase Equilibria in Chemical Engineering; Butterworth-Heinemann: Oxford, UK, 1985. [Google Scholar]
- De Nevers, N. Chapter 12 Chemical equilibrium. In Physical and Chemical Equilibrium for Chemical Engineers; John Wiley and Sons, Inc.: New York, NY, USA, 2002. [Google Scholar]
- Constantinou, L.; Gani, R. New group contribution method for estimating properties of pure compounds. AIChE J. 1994, 40, 1697–1710. [Google Scholar] [CrossRef]
- Joback, K.G. Unified Approach to Physical Property Estimation Using Multivariate Statistical Techniques. Master’s Thesis, Massachusetts Institute of Technology (MIT), Cambridge, MA, USA, 1984. [Google Scholar]
- Poling, B.E.; Prausnitz, J.M.; O’connell, J.P. Chapter 3 Thermodynamic properties of ideal gases. In The Properties of Gases and Liquids, 5th ed.; McGraw-Hill Professional: New York, NY, USA, 2000. [Google Scholar]
- Lewars, E.; Liebman, J.F. What are the enthalpy of formation and the stabilization energy of acrolein? Struct. Chem. 2013, 24, 741–744. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










| Sample | Zeolite Y | La(OH)3 | La2O3 | |||
|---|---|---|---|---|---|---|
| % | L(533) (nm) | % | L(101) (nm) | % | L(101) (nm) | |
| HY | 100 | 19.05 | - | - | - | - |
| LaY | 88.30 | 32.54 | 10.25 | 47.89 | 1.45 | 103.10 |
| Pd/LaY | 88.18 | 32.47 | 10.18 | 48.00 | 1.64 | 103.16 |
| T(K) | HY | LaY | Pd/LaY | ||||||
|---|---|---|---|---|---|---|---|---|---|
| XGlyc | SAcro | SAcet | XGlyc | SAcro | SAcet | XGlyc | SAcro | SAcet | |
| 473 | 45.1 | 54.1 | 44.3 | 72.4 | 60.2 | 36.9 | 89.1 | 42.1 | 46.1 |
| 523 | 55.4 | 75.8 | 18.0 | 77 | 86.7 | 11.7 | 90.0 | 67.7 | 27.1 |
| 548 | 57.5 | 91.6 | 3.72 | 80.9 | 99.0 | 0.15 | 91.0 | 77.7 | 17.6 |
| 573 | 61.6 | 93.0 | 0.81 | 84.1 | 89.41 | 0.09 | 93.0 | 94.2 | 2.75 |
| T (K) | Reaction 1 | Reaction 2 | Reaction 3 | |||
|---|---|---|---|---|---|---|
| ΔHr (kJ·mol−1) | ΔGr (kJ·mol−1) | ΔHr (kJ·mol−1) | ΔGr (kJ·mol−1) | ΔHr (kJ·mol−1) | ΔGr (kJ·mol−1) | |
| 298.15 | 28.84 | −47.38 | −33.99 | −72.85 | 56.77 | −17.19 |
| 300 | 28.89 | −47.85 | −33.98 | −73.09 | 56.79 | −17.65 |
| 400 | 30.61 | −73.69 | −33.63 | −86.18 | 56.86 | −42.47 |
| 500 | 31.09 | −99.61 | −33.98 | −99.16 | 55.73 | −66.97 |
| 600 | 30.54 | −125.21 | −34.75 | −111.90 | 54.01 | −90.99 |
| 700 | 29.18 | −150.28 | −35.75 | −124.37 | 52.00 | −114.55 |
| 800 | 27.30 | −174.79 | −36.90 | −136.59 | 49.90 | −137.74 |
| 900 | 25.20 | −198.86 | −38.09 | −148.62 | 47.96 | −160.74 |
| Compound | ΔH°f (kJ·mol−1) | ΔG°f (kJ·mol−1) | ||||
|---|---|---|---|---|---|---|
| (a) | (b) | (c) | (a) | (b) | (c) | |
| Glycerol | −583 | −577.9 | −669.3 b | −449 | −447 | −478.3 b |
| Acrolein | −81 | −65.4 | −98.9 a | −56.2 | −37.3 | −104.3 a |
| Water | −242 | −241.8 | −285.8 c | −243 | −228.6 | −306.7 c |
| T (K) | ΔHr (kJ·mol−1) | ΔGr (kJ·mol−1) | K | |||
|---|---|---|---|---|---|---|
| (a) | (b) | (a) | (b) | (a) | (b) | |
| 298.15 | 18.1 | 28.84 | −92.6 | −47.38 | 1.64 × 1016 | 2.00 × 108 |
| 553.15 | 20.4 | 30.79 | −163 | −113.39 | 2.66 × 1015 | 5.41 × 1010 |
| 563.15 | 20.5 | 30.74 | −166 | −115.95 | 2.59 × 1015 | 5.94 × 1010 |
| 573.15 | 20.6 | 30.68 | −169 | −118.37 | 2.53 × 1015 | 6.49 × 1010 |
| 593.15 | 20.7 | 30.57 | −175 | −123.58 | 2.42 × 1015 | 7.57 × 1010 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pala Rosas, I.; Contreras, J.L.; Salmones, J.; Tapia, C.; Zeifert, B.; Navarrete, J.; Vázquez, T.; García, D.C. Catalytic Dehydration of Glycerol to Acrolein over a Catalyst of Pd/LaY Zeolite and Comparison with the Chemical Equilibrium. Catalysts 2017, 7, 73. https://doi.org/10.3390/catal7030073
Pala Rosas I, Contreras JL, Salmones J, Tapia C, Zeifert B, Navarrete J, Vázquez T, García DC. Catalytic Dehydration of Glycerol to Acrolein over a Catalyst of Pd/LaY Zeolite and Comparison with the Chemical Equilibrium. Catalysts. 2017; 7(3):73. https://doi.org/10.3390/catal7030073
Chicago/Turabian StylePala Rosas, Israel, José Luis Contreras, José Salmones, Carlos Tapia, Beatriz Zeifert, Juan Navarrete, Tamara Vázquez, and Diana Carolina García. 2017. "Catalytic Dehydration of Glycerol to Acrolein over a Catalyst of Pd/LaY Zeolite and Comparison with the Chemical Equilibrium" Catalysts 7, no. 3: 73. https://doi.org/10.3390/catal7030073
APA StylePala Rosas, I., Contreras, J. L., Salmones, J., Tapia, C., Zeifert, B., Navarrete, J., Vázquez, T., & García, D. C. (2017). Catalytic Dehydration of Glycerol to Acrolein over a Catalyst of Pd/LaY Zeolite and Comparison with the Chemical Equilibrium. Catalysts, 7(3), 73. https://doi.org/10.3390/catal7030073