Catalytic Performance of MgO-Supported Co Catalyst for the Liquid Phase Oxidation of Cyclohexane with Molecular Oxygen

Abstract
:1. Introduction
2. Results and Discussion
2.1. Effects of the Carrier and Co Loading
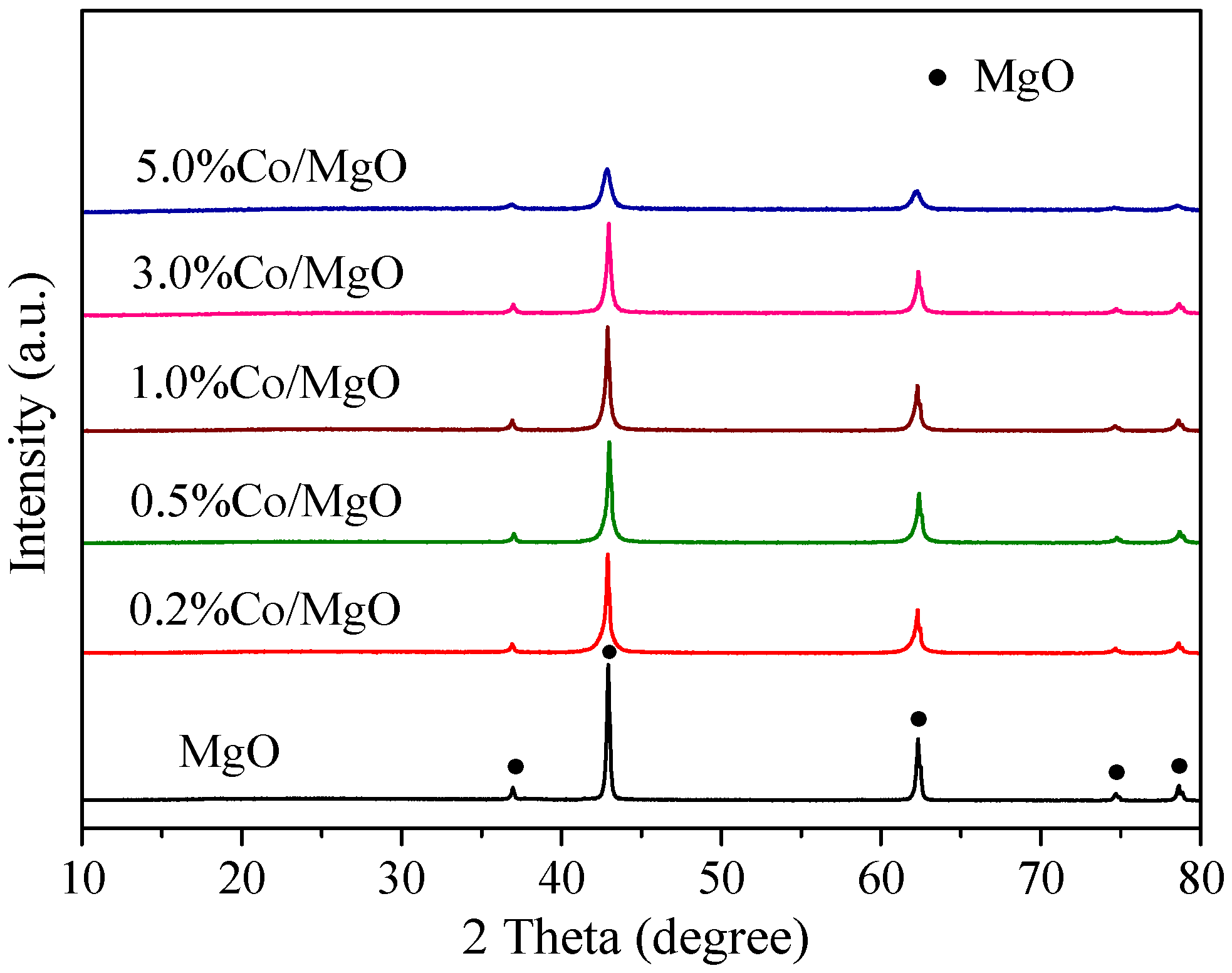
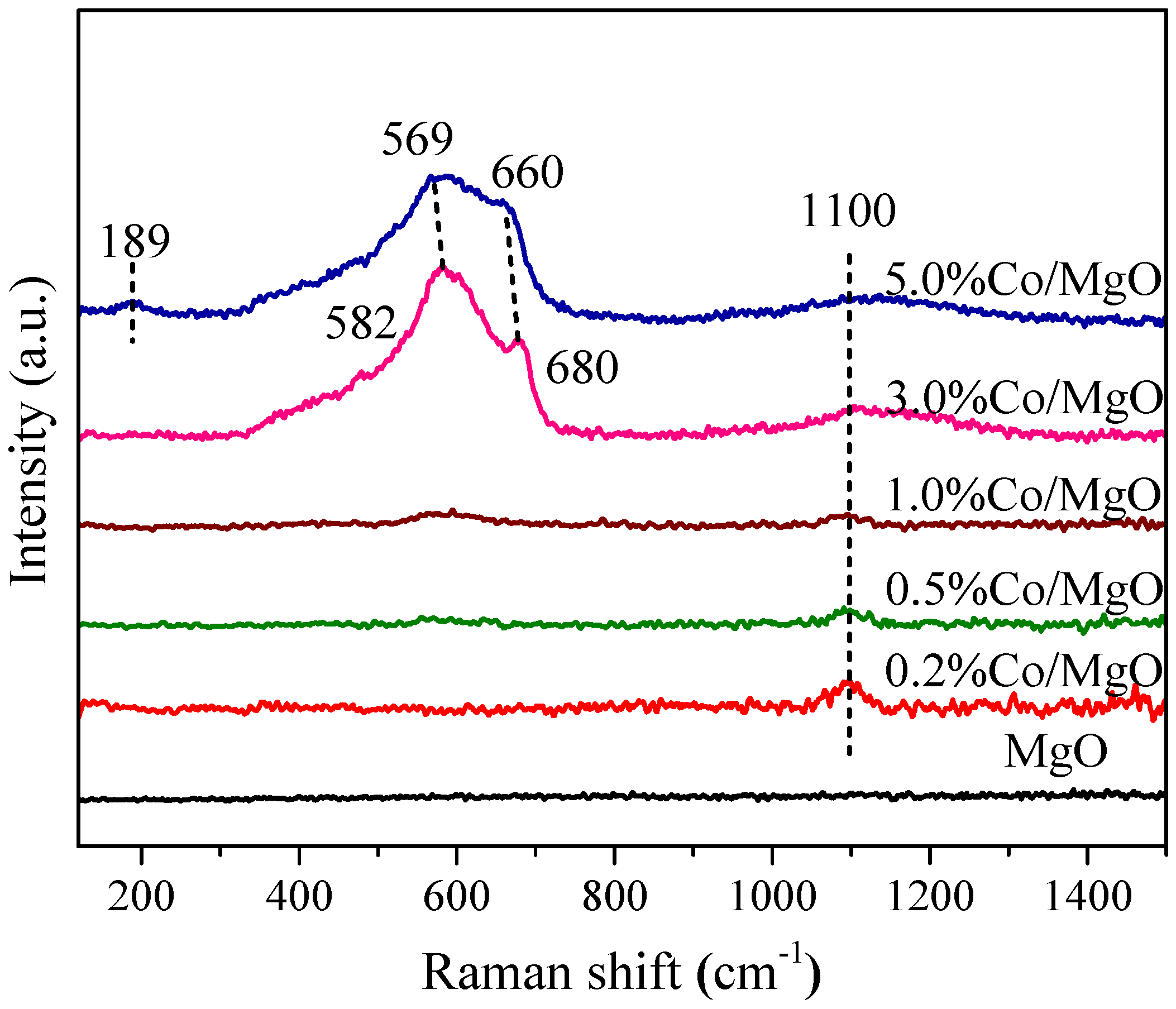
2.2. Structure and Surface Properties
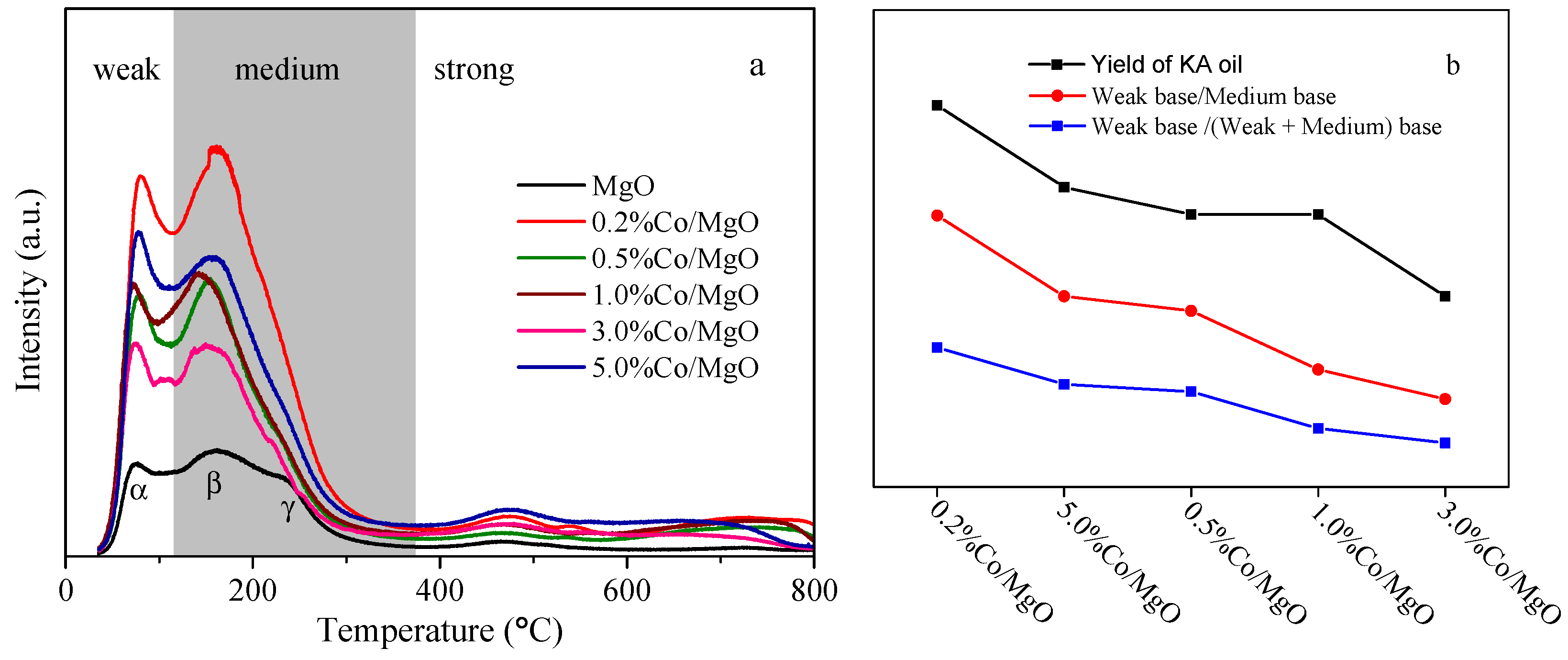
2.3. Surface Basic Property (Temperature Programmed Desorption of CO2 (CO2-TPD))
2.4. Redox Property (H2 Temperature-Programmed Reduction (H2-TPR) and Temperature Programmed Desorption of O2 (O2-TPD))
2.5. SEM (scanning electron microscope) and TEM (transmission electron microscope) Images
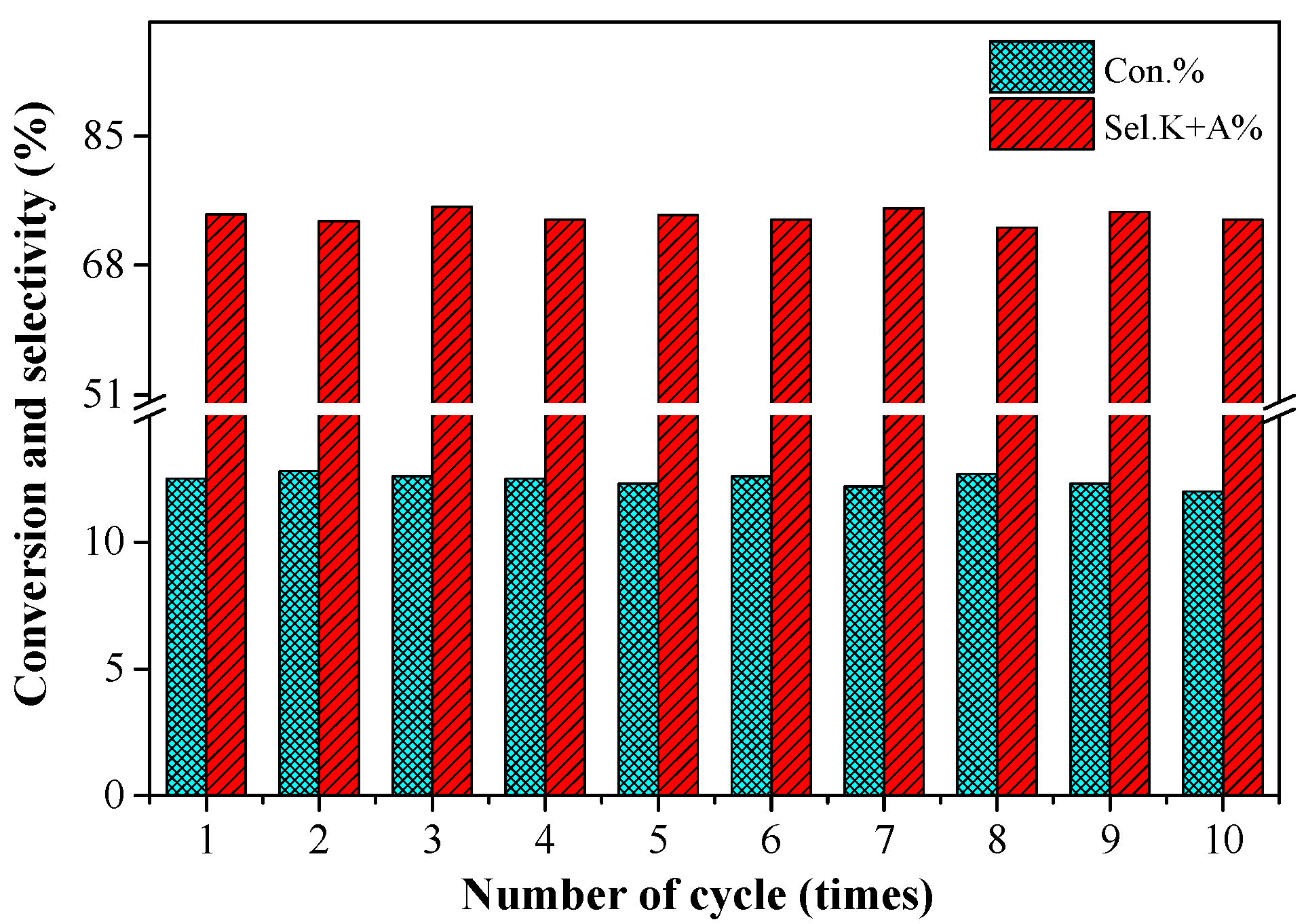
2.6. The Stability of the 0.2%Co/MgO Catalyst
3. Materials and Methods
3.1. Synthesis of Catalysts
3.2. Characterization of Catalysts
3.3. Catalytic Activity Testing
3.4. Repeated Usage of the Catalyst
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Clerici, M.G.; Kholdeeva, O.A. Liquid Phase Oxidation via Heterogeneous Catalysis: Organic Synthesis and Industrial Applications; John Wiley & Sons: Hoboken, NJ, USA, 2013; ISBN 1118356756. [Google Scholar]
- Schuchardt, U.; Carvalho, W.A.; Spinacé, E.V. Why is it interesting to study cyclohexane oxidation? Synlett 1993, 1993, 713–718. [Google Scholar] [CrossRef]
- Sakthivel, A.; Selvam, P. Mesoporous (Cr)MCM-41: A mild and efficient heterogeneous catalyst for solvent oxidation of cyclohexane. J. Catal. 2002, 211, 134–143. [Google Scholar] [CrossRef]
- Gómez-Hortigüela, L.; Corà, F.; Catlow, C.R.A. Aerobic oxidation of hydrocarbons catalyzed by Mn-doped nanoporous aluminophosphates(I): Preactivation of the Mn sites. ACS Catal. 2010, 1, 18–28. [Google Scholar] [CrossRef]
- Schuchardt, U.; Cardoso, D.; Sercheli, R.; Pereira, R.; de Cruz, R.S.; Guerreiro, M.C.; Mandelli, D.; Spinace, E.V.; Fires, E.L. Cyclohexane oxidation continues to be a challenge. Appl. Catal. A Gen. 2001, 211, 1–17. [Google Scholar] [CrossRef]
- Kirillov, A.M.; Haukka, M.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Preparation and crystal structures of benzoylhydrazido- and-diazenidorhenium complexes with N,O-ligands and their catalytic activity towards peroxidative oxidation of cycloalkanes. Eur. J. Inorg. Chem. 2005, 11, 2071–2080. [Google Scholar] [CrossRef]
- Gupta, S.; Kirillova, M.V.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L.; Kirillov, A.M. Alkali metal directed assembly of heterometallic Vv/M (M = Na, K, Cs) coordination polymers: Structures, topological analysis, and oxidation catalytic properties. Inorg. Chem. 2013, 52, 8601–8611. [Google Scholar] [CrossRef] [PubMed]
- Dias, S.S.P.; Kirillova, M.V.; André, V.; Kłak, J.; Kirillov, A.M. New tetracopper(II) cubane cores driven by a diamino alcohol: Self-assembly synthesis, structural and topological features, and magnetic and catalytic oxidation properties. Inorg. Chem. 2015, 54, 5204–5212. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, T.A.; Santos, C.I.M.; André, V.; Kłak, J.; Kirillova, M.V.; Kirillov, A.M. Copper(II) coordination polymers self-assembled from aminoalcohols and pyromellitic acid: Highly active precatalysts for the mild water-promoted oxidation of alkanes. Inorg. Chem. 2016, 55, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Kirillov, A.M.; Kirillova, M.V.; Pombeiro, A.J.L. Chapter one—Homogeneous multicopper catalysts for oxidation and hydrocarboxylation of alkanes. In Advances in Inorganic Chemistry; Rudi van, E., Colin, D.H., Eds.; Academic Press: Cambridge, MA, USA, 2013; Volume 65, pp. 1–31. ISBN 0898-8838. [Google Scholar]
- Long, J.L.; Liu, H.L.; Wu, S.J.; Liao, S.J.; Li, Y.W. Selective oxidation of saturated hydrocarbons using Au-Pd alloy nanoparticles supported on metal-organic frameworks. ACS Catal. 2013, 3, 647–654. [Google Scholar] [CrossRef]
- Wang, L.; Zhao, S.; Liu, C.; Li, C.; Li, X.; Li, H.; Wang, Y.; Ma, C.; Li, Z.; Zeng, J. Aerobic oxidation of cyclohexane on catalysts based on twinned and single-crystal Au75Pd25 bimetallic nanocrystals. Nano Lett. 2015, 15, 2875–2880. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.M. Gold loaded titanium dioxide-carbon nanotube composites as active photocatalysts for cyclohexane oxidation at ambient conditions. RSC Adv. 2015, 5, 46405–46414. [Google Scholar] [CrossRef]
- Carabineiro, S.A.C.; Martins, L.M.D.R.S.; Avalos-Borja, M.; Buijnsters, J.G.; Pombeiro, A.J.L.; Figueiredo, J.L. Gold nanoparticles supported on carbon materials for cyclohexane oxidation with hydrogen peroxide. Appl. Catal. A Gen. 2013, 467, 279–290. [Google Scholar] [CrossRef]
- De Almeida, M.P.; Martins, L.M.D.R.S.; Carabineiro, S.A.C.; Lauterbach, T.; Rominger, F.; Hashmi, A.S.K.; Pombeiro, A.J.L.; Figueiredo, J.L. Homogeneous and heterogenised new gold C-scorpionate complexes as catalysts for cyclohexane oxidation. Catal. Sci. Technol. 2013, 3, 3056–3069. [Google Scholar] [CrossRef]
- Martins, L.M.D.R.S.; Carabineiro, S.A.C.; Wang, J.; Rocha, B.G.M.; Maldonado-Hódar, F.J.; Latourrette de Oliveira Pombeiro, A.J. Supported gold nanoparticles as reusable catalysts for oxidation reactions of industrial significance. ChemCatChem 2017, 9, 1211–1221. [Google Scholar] [CrossRef]
- Jiang, Y.X.; Su, T.M.; Qin, Z.Z.; Huang, G. A zinc sulfide-supported iron tetrakis (4-carboxyl phenyl) porphyrin catalyst for aerobic oxidation of cyclohexane. RSC Adv. 2015, 5, 24788–24794. [Google Scholar] [CrossRef]
- Feng, Z.; Xie, Y.J.; Hao, F.; Liu, P.L.; Luo, H.A. Catalytic oxidation of cyclohexane to KA oil by zinc oxide supported manganese 5,10,15,20-tetrakis(4-nitrophenyl)porphyrin. J. Mol. Catal. A Chem. 2015, 410, 221–225. [Google Scholar] [CrossRef]
- Mishra, G.S.; Alegria, E.C.B.; Martins, L.M.D.R.S.; Fraústo da Silva, J.J.R.; Pombeiro, A.J.L. Cyclohexane oxidation with dioxygen catalyzed by supported pyrazole rhenium complexes. J. Mol. Catal. A Chem. 2008, 285, 92–100. [Google Scholar] [CrossRef]
- Martins, L.M.D.R.S.; de Almeida, M.P.; Carabineiro, S.A.C.; Figueiredo, J.L.; Pombeiro, A.J.L. Heterogenisation of a C-scorpionate feii complex on carbon materials for cyclohexane oxidation with hydrogen peroxide. ChemCatChem 2013, 5, 3847–3856. [Google Scholar] [CrossRef]
- Devika, S.; Palanichamy, M.; Murugesan, V. Vapour phase oxidation of cyclohexane over CeAlPO-5 molecular sieves. J. Mol. Catal. A Chem. 2011, 351, 136–142. [Google Scholar] [CrossRef]
- Anand, R.; Hamdy, M.S.; Hanefeld, U.; Maschmeyer, T. Liquid-Phase oxidation of cyclohexane over Co-TUD-1. Catal. Lett. 2004, 95, 113–117. [Google Scholar] [CrossRef]
- Li, J.; Shi, Y.; Xu, L.; Lu, G.Z. Selective oxidation of cyclohexane over transition-metal-incorporated HMS in a solvent-free system. Ind. Eng. Chem. Res. 2010, 49, 5392–5399. [Google Scholar] [CrossRef]
- Zhang, P.F.; Lu, H.F.; Zhou, Y.; Zhang, L.; Wu, Z.L.; Yang, S.Z.; Shi, H.L.; Zhu, Q.L.; Chen, Y.F.; Dai, S. Mesoporous MnCeOx solid solutions for low temperature and selective oxidation of hydrocarbons. Nat. Commun. 2015, 6, 8446. [Google Scholar] [CrossRef] [PubMed]
- Zabihi, M.; Khorasheh, F.; Shayegan, J. Supported copper and cobalt oxides on activated carbon for simultaneous oxidation of toluene and cyclohexane in air. RSC Adv. 2015, 5, 5107–5122. [Google Scholar] [CrossRef]
- Aboelfetoh, E.F.; Pietschnig, R. Preparation, characterization and catalytic activity of MgO/SiO2 supported vanadium oxide based catalysts. Catal. Lett. 2014, 144, 97–103. [Google Scholar] [CrossRef]
- Liu, D.C.; Zhang, B.Q.; Liu, X.F.; Li, J. Cyclohexane oxidation over AFI molecular sieves: Effects of Cr, Co incorporation and crystal size. Catal. Sci. Technol. 2015, 5, 3394–3402. [Google Scholar] [CrossRef]
- Masters, A.F.; Beattie, J.K.; Roa, A.L. Synthesis of a CrCoAPO-5(AFI) molecular sSieve and its activity in cyclohexane oxidation in the liquid phase. Catal. Lett. 2001, 75, 159–162. [Google Scholar] [CrossRef]
- Zhao, R.; Ji, D.; Lv, G.M.; Qian, G.; Yan, L.; Wang, X.L.; Suo, J.S. A highly efficient oxidation of cyclohexane over Au/ZSM-5 molecular sieve catalyst with oxygen as oxidant. Chem. Commun. 2004, 7, 904–905. [Google Scholar] [CrossRef] [PubMed]
- Modén, B.; Oliviero, L.; Dakka, J.; Santiesteban, J.G.; Iglesia, E. Structural and functional characterization of redox mn and co sites in alpo materials and their role in alkane oxidation catalysis. J. Phys. Chem. B 2004, 108, 5552–5563. [Google Scholar] [CrossRef]
- Qian, G.; Ji, D.; Lu, G.M.; Zhao, R.; Qi, Y.X.; Suo, J.S. Bismuth-containing MCM-41: Synthesis, characterization, and catalytic behavior in liquid-phase oxidation of cyclohexane. J. Catal. 2005, 232, 378–385. [Google Scholar] [CrossRef]
- Perkas, N.; Koltypin, Y.; Palchik, O.; Gedanken, A.; Chandrasekaran, S. Oxidation of cyclohexane with nanostructured amorphous catalysts under mild conditions. Appl. Catal. A Gen. 2001, 209, 125–130. [Google Scholar] [CrossRef]
- Perkas, N.; Wang, Y.; Koltypin, Y.; Gedanken, A.; Chandrasekaran, S. Mesoporous iron-titania catalyst for cyclohexane oxidation. Chem. Commun. 2001, 11, 988–989. [Google Scholar] [CrossRef]
- Zhou, L.P.; Xu, J.; Miao, H.; Wang, F.; Li, X.Q. Catalytic oxidation of cyclohexane to cyclohexanol and cyclohexanone over Co3O4 nanocrystals with molecular oxygen. Appl. Catal. A Gen. 2005, 292, 223–228. [Google Scholar] [CrossRef]
- Conte, M.; Liu, X.; Murphy, D.M.; Whiston, K.; Hutchings, G.J. Cyclohexane oxidation using Au/MgO: An investigation of the reaction mechanism. Phys. Chem. Chem. Phys. 2012, 14, 16279–16285. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Z.; Zhan, W.C.; Guo, Y.L.; Guo, Y.; Wang, Y.S.; Wang, L.; Lu, G.Z. An effective Mn-Co mixed oxide catalyst for the solvent-free selective oxidation of cyclohexane with molecular oxygen. Appl. Catal. A Gen. 2016, 523, 97–106. [Google Scholar] [CrossRef]
- Aboelfetoh, E.F.; Pietschnig, R. Preparation and catalytic performance of Al2O3, TiO2 and SiO2 supported vanadium based-catalysts for C–H activation. Catal. Lett. 2009, 127, 83–94. [Google Scholar] [CrossRef]
- Masteri-Farahani, M. Investigation of catalytic activities of new heterogeneous molybdenum catalysts in epoxidation of olefins. J. Mol. Catal. A Chem. 2010, 316, 45–51. [Google Scholar] [CrossRef]
- Kim, P.S.; Kim, M.K.; Cho, B.K.; Nam, I.S. Autocatalytic synergism observed during lean-NOx reduction with a bifunctional reductant over Ag/Al2O3 catalyst. J. Catal. 2012, 292, 44–52. [Google Scholar] [CrossRef]
- Ramanathan, A.; Hamdy, M.S.; Parton, R.; Maschmeyer, T.; Jansen, J.C.; Hanefeld, U. Co-TUD-1 catalysed aerobic oxidation of cyclohexane. Appl. Catal. A Gen. 2009, 355, 78–82. [Google Scholar] [CrossRef]
- Tolman, C.A.; Druliner, J.D.; Nappa, M.J.; Herron, C. Activation and Functionalisation of Alkanes; Hill, C.L., Ed.; Wiley: New York, NY, USA, 1989. [Google Scholar]
- Cazzanelli, E.; Kuzmin, A.; Mariotto, G.; Mironova-Ulmane, N. Study of vibrational and magnetic excitations in Nic Mg1− cO solid solutions by Raman spectroscopy. J. Phys. Condens. Matter 2003, 15, 2045. [Google Scholar] [CrossRef]
- Mironova-Ulmane, N.; Kuzmin, A.; Steins, I.; Grabis, J.; Sildos, I.; Pärs, M. Raman scattering in nanosized nickel oxide NiO. J. Phys. Conf. Ser. 2007, 93, 012039. [Google Scholar] [CrossRef]
- Wu, L.B.; Jiao, D.M.; Wang, J.A.; Chen, L.F.; Cao, F.H. The role of MgO in the formation of surface active phases of CoMo/Al2O3-MgO catalysts for hydrodesulfurization of dibenzothiophene. Catal. Commun. 2009, 11, 302–305. [Google Scholar] [CrossRef]
- Wang, G.X.; Shen, X.P.; Horvat, J.; Wang, B.; Liu, H.; Wexler, D.; Yao, J. Hydrothermal synthesis and optical, magnetic, and supercapacitance properties of nanoporous cobalt oxide nanorods. J. Phys. Chem. C 2009, 113, 4357–4361. [Google Scholar] [CrossRef]
- Jiang, J.; Li, L.C. Synthesis of sphere-like Co3O4 nanocrystals via a simple polyol route. Mater. Lett. 2007, 61, 4894–4896. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, L.C.; Chen, M.; Cao, Y.; He, H.Y.; Fan, K.N. Dry citrate-precursor synthesized nanocrystalline cobalt oxide as highly active catalyst for total oxidation of propane. J. Catal. 2009, 263, 104. [Google Scholar] [CrossRef]
- Li, J.; Lu, G.Z.; Wu, G.S.; Mao, D.S.; Guo, Y.L.; Wang, Y.Q.; Guo, Y. The role of iron oxide in the highly effective Fe-modified Co3O4 catalyst for low-temperature CO oxidation. RSC Adv. 2013, 3, 12409–12416. [Google Scholar] [CrossRef]
- Liu, F.D.; He, H.; Ding, Y.; Zhang, C.B. Effect of manganese substitution on the structure and activity of iron titanate catalyst for the selective catalytic reduction of NO with NH3. Appl. Catal. B Environ. 2009, 93, 194–204. [Google Scholar] [CrossRef]
- Wang, X.Y.; Kang, Q.; Li, D. Catalytic combustion of chlorobenzene over MnOx-CeO2 mixed oxide catalysts. Appl. Catal. B Environ. 2009, 86, 166–175. [Google Scholar] [CrossRef]
- Zhang, W.; Tay, H.L.; Lim, S.S.; Wang, Y.S.; Zhong, Z.Y.; Xu, R. Supported cobalt oxide on MgO: Highly efficient catalysts for degradation of organic dyes in dilute solutions. Appl. Catal. B Environ. 2010, 95, 93–99. [Google Scholar] [CrossRef]
- Riva, R.; Miessner, H.; Vitali, R.; Del Piero, G. Metal-support interaction in Co/SiO2 and Co/TiO2. Appl. Catal. A Gen. 2000, 196, 111–123. [Google Scholar] [CrossRef]
- Todorova, S.; Kolev, H.; Holgado, J.P.; Kadinov, G.; Bonev, C.; Pereñíguez, R.; Caballero, A. Complete n-hexane oxidation over supported Mn–Co catalysts. Appl. Catal. B Environ. 2010, 94, 46–54. [Google Scholar] [CrossRef]
- Sun, C.H.; Berg, J.C. A review of the different techniques for solid surface acid-base characterization. Colloid Interface Sci. 2003, 105, 151–175. [Google Scholar] [CrossRef]
- Skofronick, J.G.; Toennies, J.P.; Traeger, F.; Weiss, H. Helium atom scattering studies of the structure and vibrations of H2 physisorbed on MgO(001) single crystals. Phys. Rev. B 2003, 67, 035413. [Google Scholar] [CrossRef]
- Mei, J.; Zhao, S.J.; Xu, H.M.; Qu, Z.; Yan, N.Q. The performance and mechanism for the catalytic oxidation of dibromomethane (CH2Br2) over Co3O4/TiO2 catalysts. RSC Adv. 2016, 6, 31181–31190. [Google Scholar] [CrossRef]
- Li, C.; Domen, K.; Maruya, K.; Onishi, T. Dioxygen adsorption on well-outgassed and partially reduced cerium oxide studied by FT-IR. J. Am. Chem. Soc. 1989, 111, 7683–7687. [Google Scholar] [CrossRef]
- Huang, H.; Gu, Y.F.; Zhao, J.; Wang, X.Y. Catalytic combustion of chlorobenzene over VOx/CeO2 catalysts. J. Catal. 2015, 326, 54–68. [Google Scholar] [CrossRef]
- Liang, H.; Hong, Y.X.; Zhu, C.Q.; Li, S.H.; Chen, Y.; Liu, Z.L.; Ye, D.Q. Influence of partial Mn-substitution on surface oxygen species of LaCoO3 catalysts. Catal. Today 2013, 201, 98–102. [Google Scholar] [CrossRef]
- Xie, X.W.; Li, Y.; Liu, Z.Q.; Haruta, M.; Shen, W.J. Low-temperature oxidation of CO catalysed by Co3O4 nanorods. Nature 2009, 458, 746–749. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.P.; Yu, J.S.; Xu, H.M.; Chen, W.X.; Hu, W.; Chen, G.L. Interfacial effects of the Cu2O nano-dots decorated Co3O4 nanorods array and its photocatalytic activity for cleaving organic molecules. Appl. Surf. Sci. 2016, 382, 249–259. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Wallau, M.; Arends, I.W.C.E.; Schuchardt, U. Heterogeneous catalysts for liquid-phase oxidations: Philosophers’ stones or Trojan horses? Acc. Chem. Res. 1998, 31, 485–493. [Google Scholar] [CrossRef]
- Wang, Q.; Peng, Y.; Fu, J.; Kyzas, G.Z.; Billah, S.M.R.; An, S.Q. Synthesis, characterization, and catalytic evaluation of Co3O4/γ-Al2O3 as methane combustion catalysts: Significance of Co species and the redox cycle. Appl. Catal. B Environ. 2015, 168–169, 42–50. [Google Scholar] [CrossRef]
- Wen, Y.; Potter, O.E.; Sridhar, T. Uncatalysed oxidation of cyclohexane in a continuous reactor. Chem. Eng. Sci. 1997, 52, 4593–4605. [Google Scholar] [CrossRef]
- Shul’pin, G.B. Metal-catalyzed hydrocarbon oxygenations in solutions: The dramatic role of additives: A review. J. Mol. Catal. A Chem. 2002, 189, 39–66. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Conversion (%) | Selectivity (%) | Yield of KA (%) | |||
|---|---|---|---|---|---|---|
| -nol (A) b | -none (K) b | KA oil | Acids | |||
| Co/MgO | 12.5 | 38.1 | 36.6 | 74.7 | 5.2 | 8.8 |
| Co/BaCO3 | 10.1 | 40.5 | 37.9 | 78.4 | 5.0 | 7.9 |
| ZrO2 | 5.3 | 39.3 | 36.8 | 76.1 | 5.5 | 4,0 |
| Co/ZrO2 | 10.8 | 35.7 | 33.3 | 69.0 | 7.1 | 7.5 |
| Co/SAPO-34 | 10.2 | 37.2 | 35.6 | 72.8 | 9.2 | 7.4 |
| Al2O3 | 4.8 | 32.3 | 35.0 | 67.3 | 4.8 | 3.2 |
| Co/Al2O3 | 9.5 | 34.9 | 35.4 | 70.3 | 8.3 | 6.7 |
| TiO2 | 1.5 | 39.1 | 43.5 | 82.6 | 5.9 | 1.2 |
| Co/TiO2 | 8.1 | 37.8 | 39.2 | 77.0 | 7.9 | 6.2 |
| SiO2 | 1.1 | 41.6 | 45.4 | 87.0 | 3,1 | 1.0 |
| Co/SiO2 | 6.8 | 39.5 | 41.2 | 80.7 | 9.5 | 5.5 |
| Catalyst | Temp./Time (°C h−1) | Pressure (MPa) | Conversion (%) | Selectivity (%) | YKA (%) | Mass Balance c (%) | ||
|---|---|---|---|---|---|---|---|---|
| -nol (A) | -none (K) | Acids b | ||||||
| Blank | 140/5 | O2/0.5 | 1.2 | 43.0 | 49.2 | 4.1 | 1.1 | 95 |
| MgO | 140/4 | O2/0.5 | 1.3 | 40.2 | 47.9 | 3.4 | 1.1 | 94 |
| MgO | 140/4 | N2/0.5 | - | - | - | - | - | - |
| 0.1%Co/MgO | 140/4 | O2/0.5 | 8.2 | 40.3 | 38.1 | 4.2 | 6.4 | 95 |
| 0.2%Co/MgO | 140/4 | air/0.5 | 3.2 | 47.5 | 42.6 | 2.7 | 2.9 | 96 |
| 0.2%Co/MgO | 140/4 | O2/0.5 | 12.5 | 38.1 | 36.6 | 5.2 | 9.3 | - |
| 0.2%Co + MgO d | 140/4 | O2/0.5 | 6.1 | 35.7 | 28.8 | 5.0 | 4.6 | - |
| 0.2%Co/MgO e | 140/4 | O2/0.5 | 15.6 | 34.7 | 32.5 | 6.3 | 10.5 | - |
| 0.2%Co/MgO f | 140/4 | O2/0.5 | - | - | - | - | - | - |
| 0.5%Co/MgO | 140/4 | O2/0.5 | 12.0 | 36.4 | 34.6 | 9.6 | 8.5 | 95 |
| 1.0%Co/MgO | 140/4 | O2/0.5 | 12.6 | 34.4 | 33.0 | 6.3 | 8.5 | 95 |
| 3.0%Co/MgO | 140/4 | O2/0.5 | 11.7 | 34.2 | 32.8 | 4.5 | 7.9 | 94 |
| 5.0%Co/MgO | 140/4 | O2/0.5 | 13.3 | 33.2 | 31.9 | 5.3 | 8.7 | 95 |
| Co3O4 | 140/4 | O2/0.5 | 7.7 | 44.2 | 22.4 | 10.2 | 5.1 | 95 |
| Au/MgO g of [35] | 140/17 | O2/0.3 | 5.0 | 50 | 29 | 19 | 4.0 | - |
| Co-TUD-1 h [40] | 120/24 | 8%O2/N2/1.5 | 3.8 | 56 | 31 | - | 3.3 | - |
| Sample | SBET (m2 g−1) | Co Content a (wt %) | Crystallite Size b (nm) | d200 Spacing (nm) | a0 c (nm) |
|---|---|---|---|---|---|
| MgO | 15.0 | - | 36.6 | 2.106 | 4.21 |
| 0.2%Co/MgO | 58.7 | 0.2 | 21.1 | 2.105 | 4.21 |
| 0.5%Co/MgO | 49.3 | 0.5 | 22.8 | 2.103 | 4.21 |
| 1.0%Co/MgO | 43.4 | 1.0 | 24.5 | 2.108 | 4.22 |
| 3.0%Co/MgO | 41.2 | 3.0 | 23.4 | 2.104 | 4.21 |
| 5.0%Co/MgO | 51.0 | 5.0 | 12.4 | 2.109 | 4.22 |
| Sample | Surface Atom Concentration (%) | Mg/O (Atom) | Co3+/Co Atomic Ratio | Co3+/Co2+ Atomic Ratio | Oads/(Oads + Olat) | |
|---|---|---|---|---|---|---|
| Mg | O | |||||
| 0.2%Co/MgO | 29.6 | 49.5 | 0.60 | - | - | 0.63 |
| 0.5%Co/MgO | 28.8 | 51.8 | 0.56 | - | - | 0.47 |
| 1.0%Co/MgO | 34.6 | 65.4 | 0.53 | - | - | 0.63 |
| 3.0%Co/MgO | 33.4 | 66.6 | 0.50 | 0.6 | 2.0 | 0.65 |
| 5.0%Co/MgO | 22.8 | 51.9 | 0.44 | 0.7 | 2.0 | 0.60 |
| Sample | Weak Base (a.u. m−2) | Medium Base (a.u. m−2) | Weak Base/Medium Base | Weak Base/(Weak + Medium) Base |
|---|---|---|---|---|
| MgO | 4.7 | 23.2 | 0.20 | 0.17 |
| 0.2%Co/MgO | 4.7 | 9.0 | 0.52 | 0.34 |
| 0.5%Co/MgO | 4.5 | 11.4 | 0.39 | 0.28 |
| 1.0%Co/MgO | 4.3 | 14.1 | 0.31 | 0.23 |
| 3.0%Co/MgO | 3.2 | 11.9 | 0.27 | 0.21 |
| 5.0%Co/MgO | 5.2 | 12.7 | 0.41 | 0.29 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, M.; Fu, Y.; Zhan, W.; Guo, Y.; Guo, Y.; Wang, Y.; Lu, G. Catalytic Performance of MgO-Supported Co Catalyst for the Liquid Phase Oxidation of Cyclohexane with Molecular Oxygen. Catalysts 2017, 7, 155. https://doi.org/10.3390/catal7050155
Wu M, Fu Y, Zhan W, Guo Y, Guo Y, Wang Y, Lu G. Catalytic Performance of MgO-Supported Co Catalyst for the Liquid Phase Oxidation of Cyclohexane with Molecular Oxygen. Catalysts. 2017; 7(5):155. https://doi.org/10.3390/catal7050155
Chicago/Turabian StyleWu, Mingzhou, Yu Fu, Wangcheng Zhan, Yanglong Guo, Yun Guo, Yunsong Wang, and Guanzhong Lu. 2017. "Catalytic Performance of MgO-Supported Co Catalyst for the Liquid Phase Oxidation of Cyclohexane with Molecular Oxygen" Catalysts 7, no. 5: 155. https://doi.org/10.3390/catal7050155
APA StyleWu, M., Fu, Y., Zhan, W., Guo, Y., Guo, Y., Wang, Y., & Lu, G. (2017). Catalytic Performance of MgO-Supported Co Catalyst for the Liquid Phase Oxidation of Cyclohexane with Molecular Oxygen. Catalysts, 7(5), 155. https://doi.org/10.3390/catal7050155