
A Theoretical Insight into Enhanced Catalytic Activity of Au by Multiple Twin Nanoparticles


Abstract
:1. Introduction
2. Computational Details
3. Results
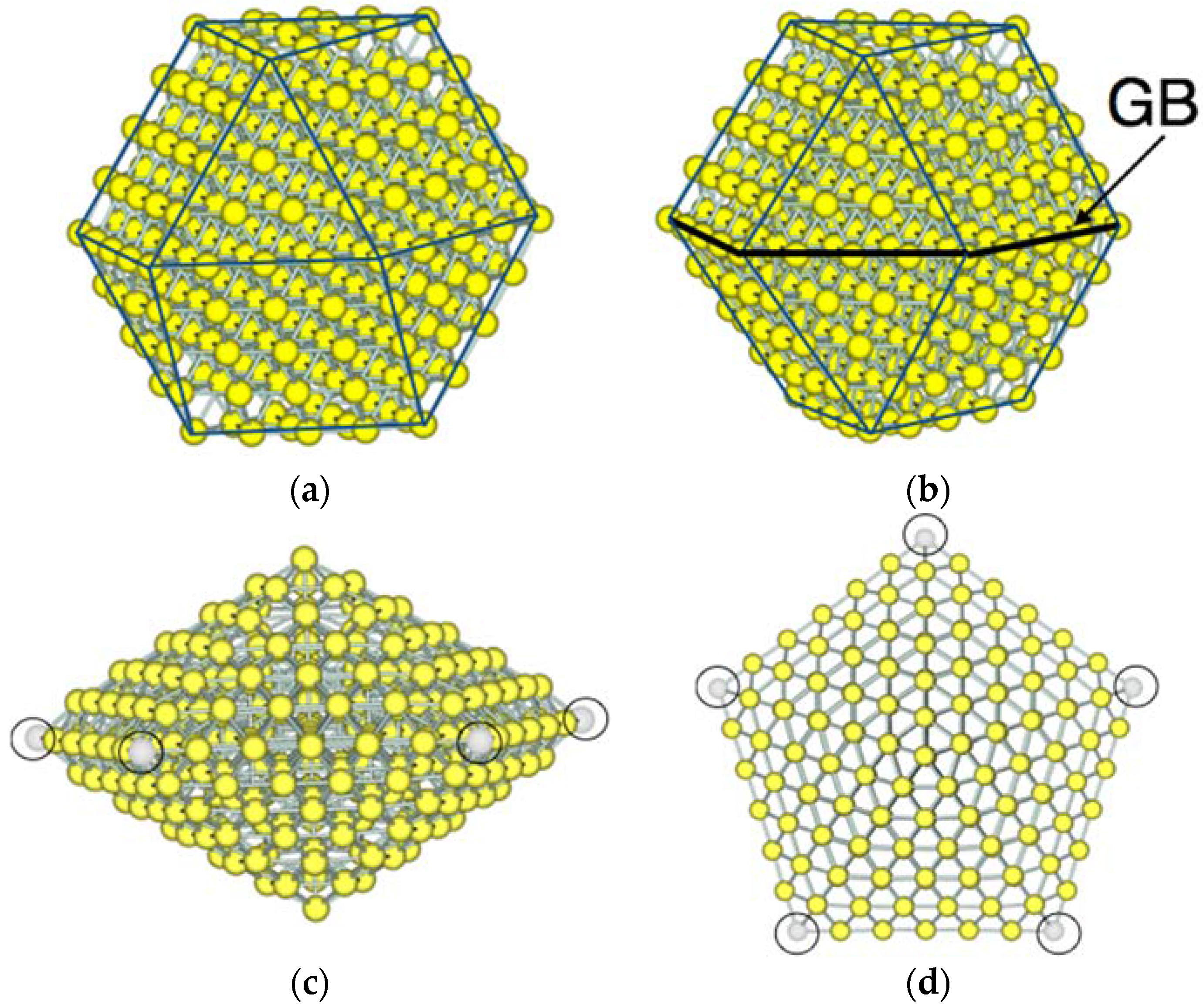
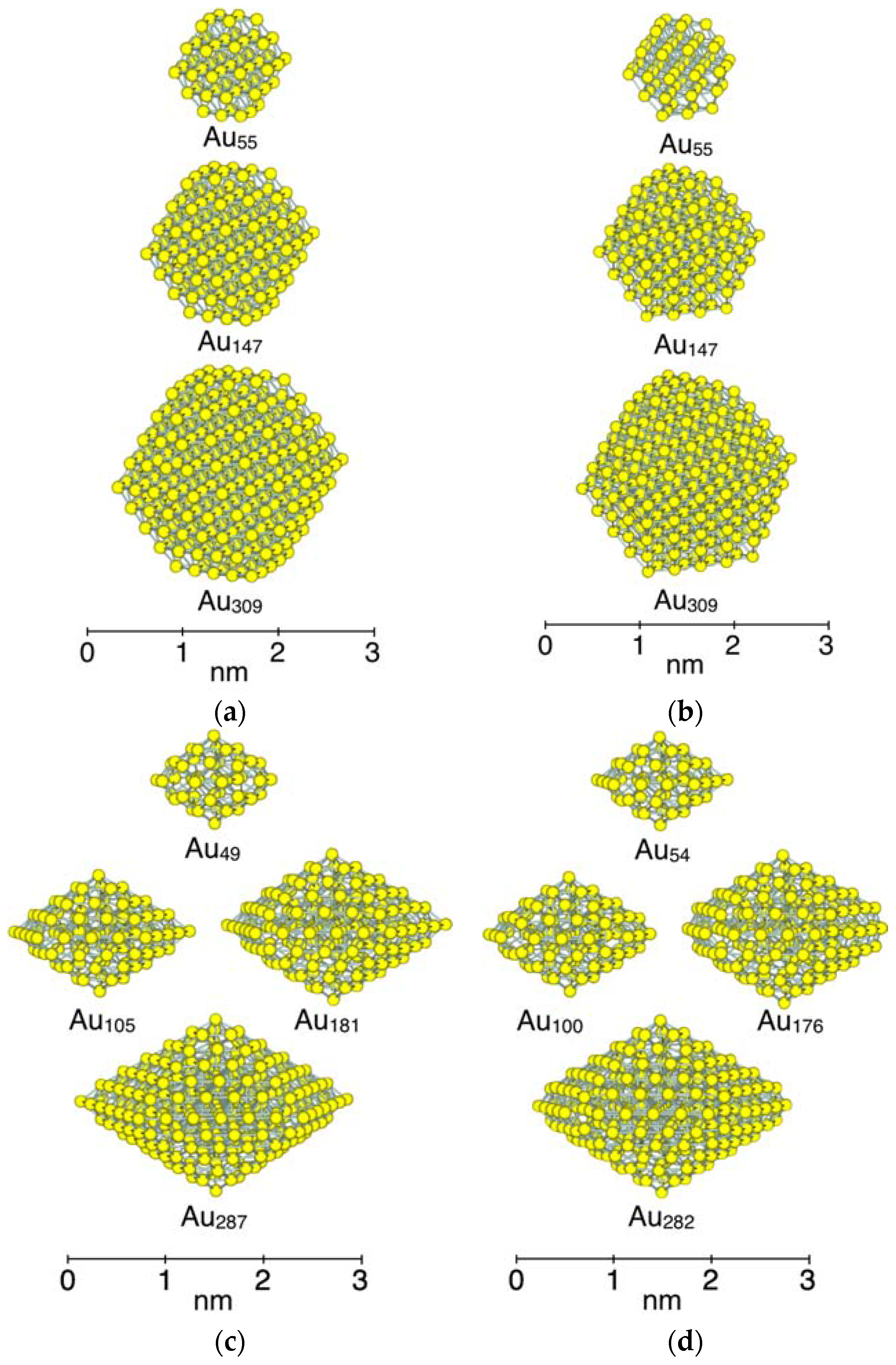
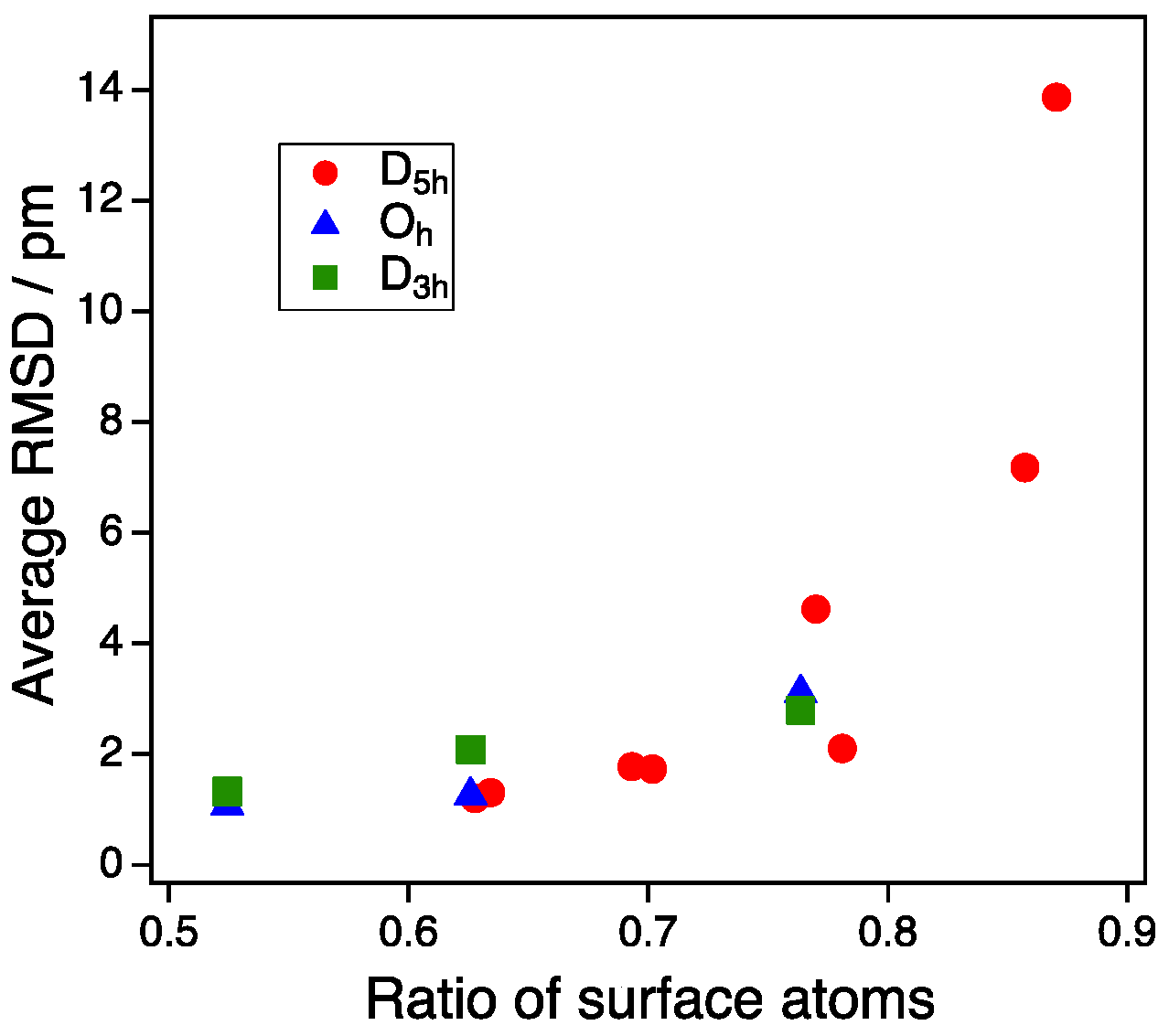
3.1. Model Nanoparticles
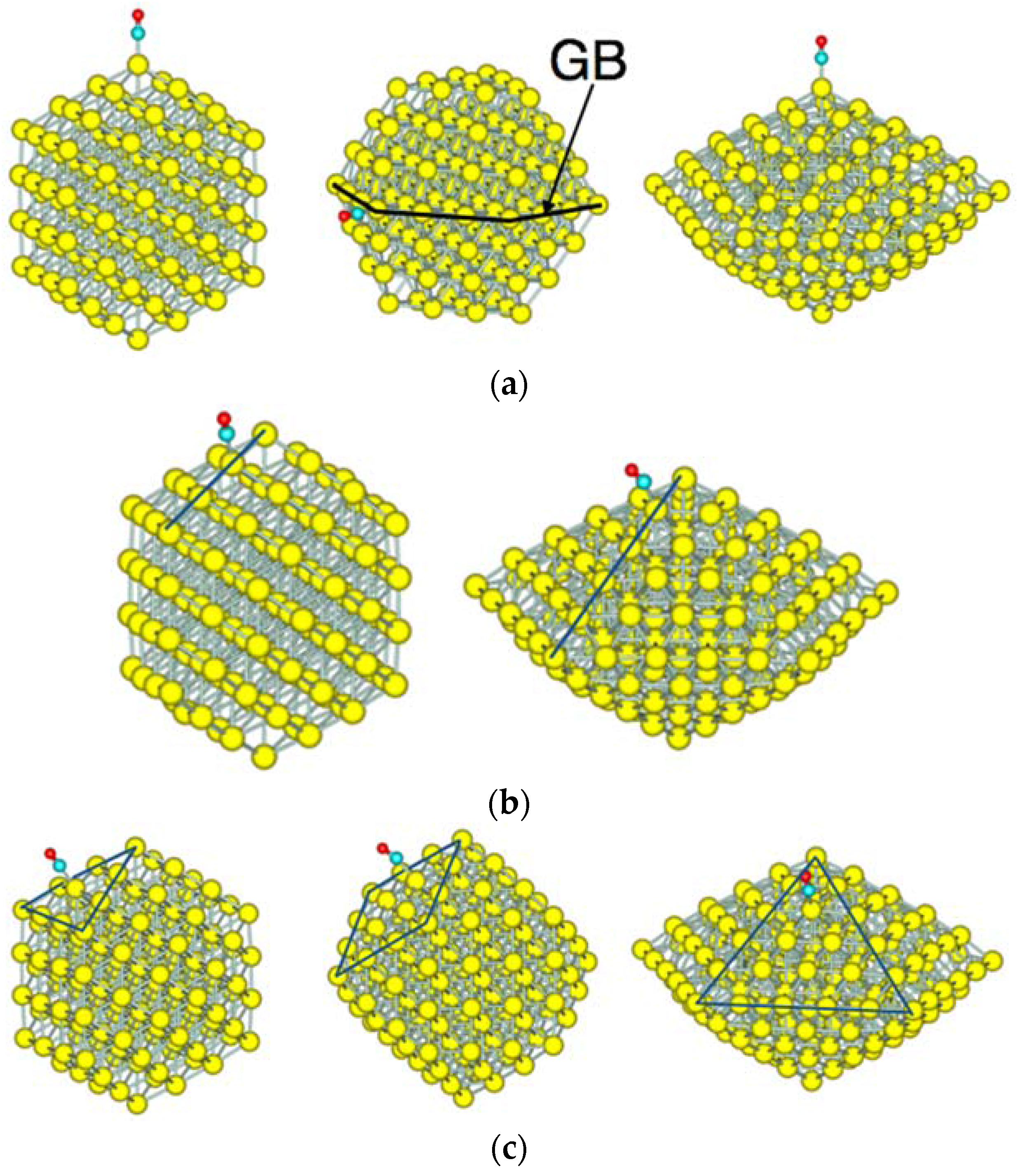
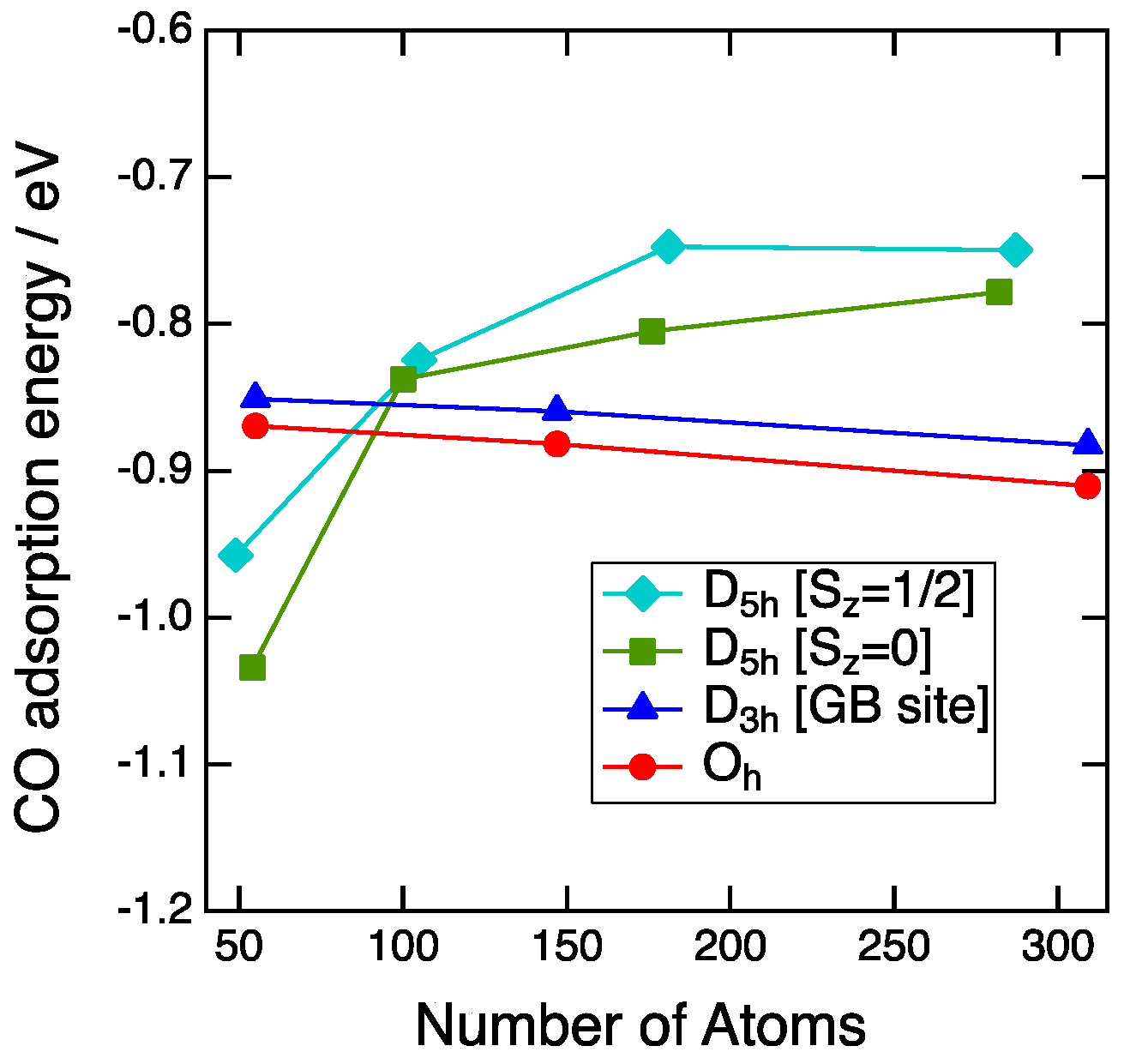
3.2. Size Dependency of CO Adsoprtion
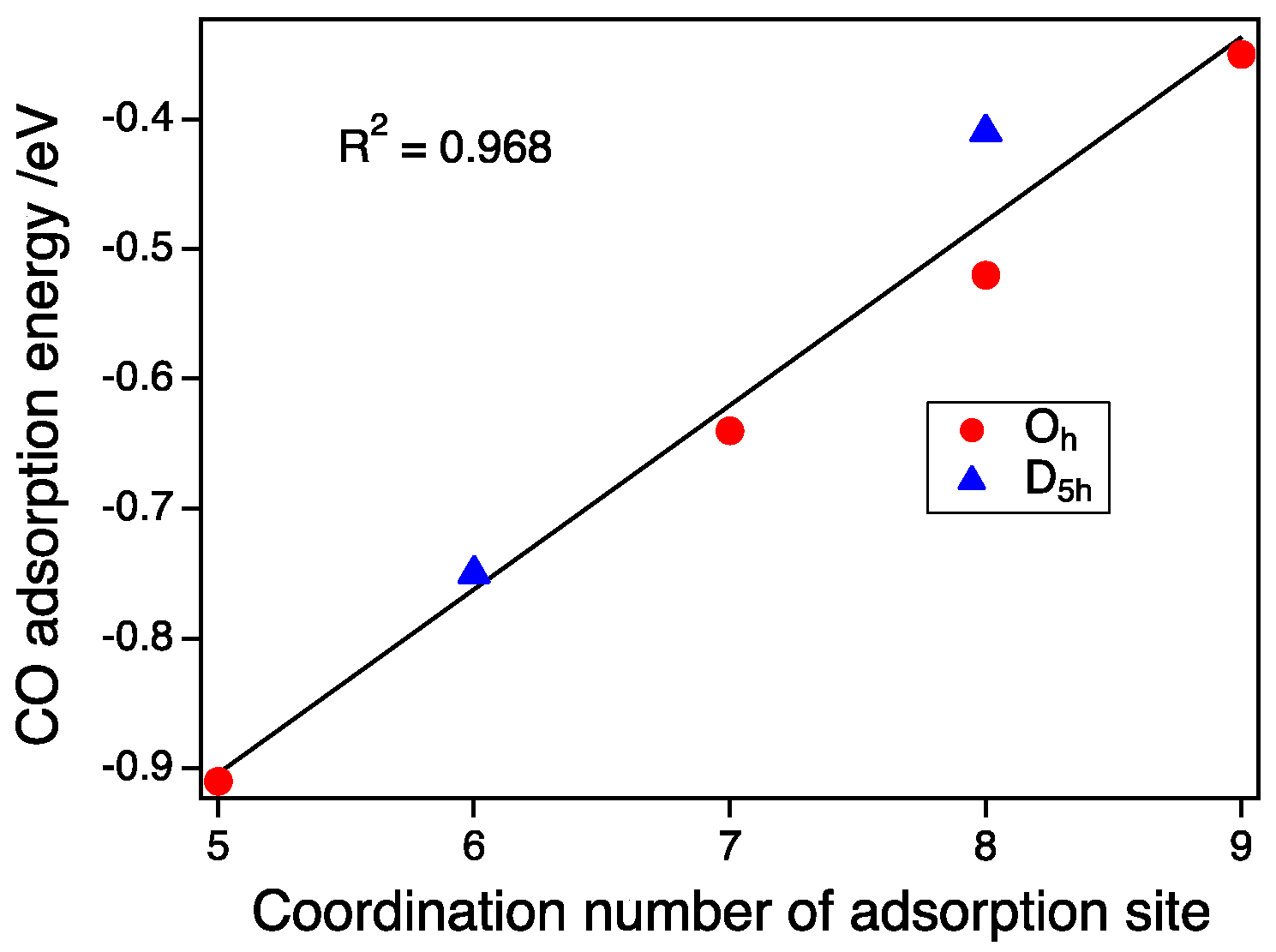
3.3. CO Adsorption vs. the Coordination Number
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Haruta, M. Chance and necessity: My encounter with gold catalysts. Angew. Chem. Int. Ed. Engl. 2014, 53, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Koga, H.; Okumura, M.; Haruta, M. Advances in Gold Catalysis and Understanding the Catalytic Mechanism. Chem. Rec. 2016, 16, 2278–2293. [Google Scholar] [CrossRef] [PubMed]
- Min, B.K.; Friend, C.M. Heterogeneous gold-based catalysis for green chemistry: Low-temperature CO oxidation and propene oxidation. Chem. Rev. 2007, 107, 2709–2724. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Tokunaga, T.; Zhang, L.; Li, D.W.; Chen, L.Y.; Arai, S.; Yamamoto, Y.; Hirata, A.; Tanaka, N.; Ding, Y.; et al. Atomic Observation of Catalysis-Induced Nanopore Coarsening of Nanoporous Gold. Nano Lett. 2014, 14, 1172–1177. [Google Scholar] [CrossRef] [PubMed]
- Brodersen, S.H.; Grønbjerg, U.; Hvolbæk, B.; Schiøtz, J. Understanding the catalytic activity of gold nanoparticles through multi-scale simulations. J. Catal. 2011, 284, 34–41. [Google Scholar] [CrossRef]
- Lemire, C.; Meyer, R.; Shaikhutdinov, S.; Freund, H.J. Do quantum size effects control CO adsorption on gold nanoparticles? Angew. Chem. Int. Ed. Engl. 2004, 43, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.G.; Austin, N.; Gounaris, C.E.; Mpourmpakis, G. Catalyst Design Based on Morphology- and Environment-Dependent Adsorption on Metal Nanoparticles. ACS Catal. 2015, 5, 6296–6301. [Google Scholar] [CrossRef]
- Choudhary, T.V.; Goodman, D.W. Catalytically active gold: The role of cluster morphology. Appl. Catal. A Gen. 2005, 291, 32–36. [Google Scholar] [CrossRef]
- Cunningham, D.A.H.; Vogel, W.; Sanchez, R.M.T.; Tanaka, K.; Haruta, M. Structural analysis of Au/TiO2 catalysts by Debye function analysis. J. Catal. 1999, 183, 24–31. [Google Scholar] [CrossRef]
- Feng, X.F.; Jiang, K.L.; Fan, S.S.; Kanan, M.W. Grain-Boundary-Dependent CO2 Electroreduction Activity. J. Am. Chem. Soc. 2015, 137, 4606–4609. [Google Scholar] [CrossRef] [PubMed]
- Kartusch, C.; Krumeich, F.; Safonova, O.; Hartfelder, U.; Makosch, M.; Sa, J.; van Bokhoven, J.A. Redispersion of Gold Multiple Twinned Particles during Liquid-Phase Hydrogenation. ACS Catal. 2012, 2, 1394–1403. [Google Scholar] [CrossRef]
- Lin, Y.; Wu, Z.; Wen, J.; Ding, K.; Yang, X.; Poeppelmeier, K.R.; Marks, L.D. Adhesion and Atomic Structures of Gold on Ceria Nanostructures: The Role of Surface Structure and Oxidation State of Ceria Supports. Nano Lett. 2015, 15, 5375–5381. [Google Scholar] [CrossRef] [PubMed]
- Mohr, C.; Hofmeister, H.; Claus, P. The influence of real structure of gold catalysts in the partial hydrogenation of acrolein. J. Catal. 2003, 213, 86–94. [Google Scholar] [CrossRef]
- Ohyama, J.; Koketsu, T.; Yamamoto, Y.; Arai, S.; Satsuma, A. Preparation of TiO2-supported twinned gold nanoparticles by CO treatment and their CO oxidation activity. Chem. Commun. 2015, 51, 15823–15826. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.D.; Guttel, R.; Leoni, M.; Schuth, F.; Weidenthaler, C. Influence of the Microstructure of Gold-Zirconia Yolk-Shell Catalysts on the CO Oxidation Activity. J. Phys. Chem. C 2010, 114, 19386–19394. [Google Scholar] [CrossRef]
- Quintanilla, A.; Butselaar-Orthlieb, V.C.L.; Kwakernaak, C.; Sloof, W.G.; Kreutzer, M.T.; Kapteijn, F. Weakly bound capping agents on gold nanoparticles in catalysis: Surface poison? J. Catal. 2010, 271, 104–114. [Google Scholar] [CrossRef]
- Marks, L. Experimental studies of small particle structures. Rep. Prog. Phys. 1994, 57, 603. [Google Scholar] [CrossRef]
- Falsig, H.; Hvolbæk, B.; Kristensen, I.S.; Jiang, T.; Bligaard, T.; Christensen, C.H.; Nørskov, J.K. Trends in the catalytic CO oxidation activity of nanoparticles. Angew. Chem. 2008, 120, 4913–4917. [Google Scholar] [CrossRef]
- Lopez, N.; Janssens, T.V.W.; Clausen, B.S.; Xu, Y.; Mavrikakis, M.; Bligaard, T.; Nørskov, J.K. On the origin of the catalytic activity of gold nanoparticles for low-temperature CO oxidation. J. Catal. 2004, 223, 232–235. [Google Scholar] [CrossRef]
- Meier, D.C.; Goodman, D.W. The influence of metal cluster size on adsorption energies: CO adsorbed on Au clusters supported on TiO(2). J. Am. Chem. Soc. 2004, 126, 1892–1899. [Google Scholar] [CrossRef] [PubMed]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple [Phys. Rev. Lett. 77, 3865 (1996)]. Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Methfessel, M.; Paxton, A.T. High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B 1989, 40, 3616–3621. [Google Scholar] [CrossRef]
- Gruber, M.; Heimel, G.; Romaner, L.; Brédas, J.-L.; Zojer, E. First-principles study of the geometric and electronic structure of Au13 clusters: Importance of the prism motif. Phys. Rev. B 2008, 77, 165411. [Google Scholar] [CrossRef]
- Pereiro, M.; Baldomir, D.; Arias, J.E. Unexpected magnetism of small silver clusters. Phys. Rev. A 2007, 75, 063204. [Google Scholar] [CrossRef]
- Sawabe, K.; Hiro, T.; Shimizu, K.-i.; Satsuma, A. Density functional theory calculation on the promotion effect of H2 in the selective catalytic reduction of NOx over Ag–MFI zeolite. Catal. Today 2010, 153, 90–94. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Mpourmpakis, G.; Andriotis, A.N.; Vlachos, D.G. Identification of descriptors for the CO interaction with metal nanoparticles. Nano Lett. 2010, 10, 1041–1045. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Samano, E.; Koel, B.E. CO adsorption and reaction on clean and oxygen-covered Au(211) surfaces. J. Phys. Chem. B 2006, 110, 17512–17517. [Google Scholar] [CrossRef] [PubMed]
- Hvolbæk, B.; Janssens, T.V.W.; Clausen, B.S.; Falsig, H.; Christensen, C.H.; Nørskov, J.K. Catalytic activity of Au nanoparticles. Nano Today 2007, 2, 14–18. [Google Scholar] [CrossRef]
- Shimizu, K.-i.; Sawabe, K.; Satsuma, A. Self-Regenerative Silver Nanocluster Catalyst for CO Oxidation. ChemCatChem 2011, 3, 1290–1293. [Google Scholar] [CrossRef]
- Zhang, W.; Cheng, D.; Zhu, J. Theoretical study of CO catalytic oxidation on free and defective graphene-supported Au–Pd bimetallic clusters. RSC Adv. 2014, 4, 42554–42561. [Google Scholar] [CrossRef]
- Uchiyama, T.; Yoshida, H.; Kamiuchi, N. Correlation of catalytic activity with the morphology change of supported Au nanoparticles in gas. Surf. Sci. 2017, 659, 16–19. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sz | PBE/Plane Wave | M06L/Def2-TZVPP(SDD) * |
|---|---|---|
| 1/2 | - | - |
| 3/2 | 0.26 | 0.25 |
| 5/2 | 1.00 | 0.99 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sawabe, K.; Koketsu, T.; Ohyama, J.; Satsuma, A. A Theoretical Insight into Enhanced Catalytic Activity of Au by Multiple Twin Nanoparticles. Catalysts 2017, 7, 191. https://doi.org/10.3390/catal7060191
Sawabe K, Koketsu T, Ohyama J, Satsuma A. A Theoretical Insight into Enhanced Catalytic Activity of Au by Multiple Twin Nanoparticles. Catalysts. 2017; 7(6):191. https://doi.org/10.3390/catal7060191
Chicago/Turabian StyleSawabe, Kyoichi, Taiki Koketsu, Junya Ohyama, and Atsushi Satsuma. 2017. "A Theoretical Insight into Enhanced Catalytic Activity of Au by Multiple Twin Nanoparticles" Catalysts 7, no. 6: 191. https://doi.org/10.3390/catal7060191
APA StyleSawabe, K., Koketsu, T., Ohyama, J., & Satsuma, A. (2017). A Theoretical Insight into Enhanced Catalytic Activity of Au by Multiple Twin Nanoparticles. Catalysts, 7(6), 191. https://doi.org/10.3390/catal7060191