Selective Acetylation of Small Biomolecules and Their Derivatives Catalyzed by Er(OTf)3

 ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 

Abstract
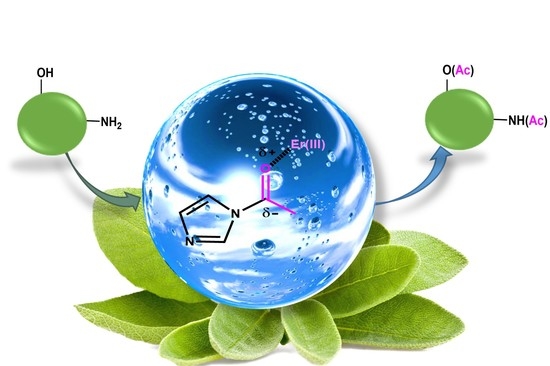
:
1. Introduction
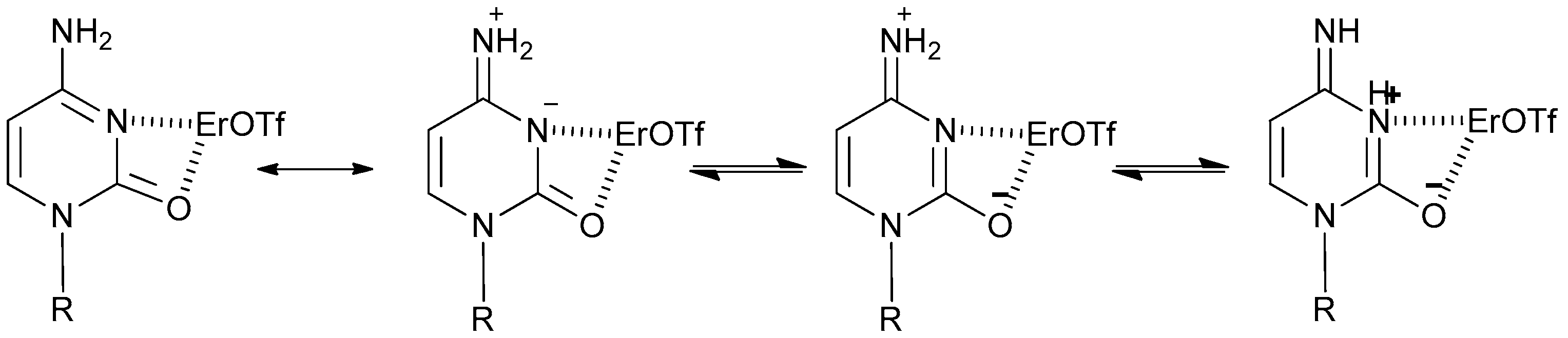
2. Results
3. Materials and Methods
3.1. General Methods
3.2. General Experimental Procedure for Microwave-Assisted Acetylation
- Methyl 6-O-acetyl α-d-glucopyranoside (1a): 1H- and 13C-NMR (CDCl3, 300 MHz and 75.5 MHz, respectively) [63].
- Methyl 6-O-acetyl α-d-mannopyranoside (2a): 1H- and 13C-NMR (CDCl3, 300 MHz and 75.5 MHz, respectively) [64].
- Methyl 6-O-acetyl α-d-galattopyranoside (3a): 1H- and 13C-NMR (CDCl3, 300 MHz and 75.5 MHz, respectively) [65].
- Methyl 6-O-acetyl β-d-glucopyranoside (4a): 1H- and 13C-NMR (CDCl3, 300 MHz and 75.5 MHz, respectively) [65].
- Methyl 6-O-acetyl β-d-mannopyranoside (5a): 1H- and 13C-NMR (CDCl3, 300 MHz and 75.5 MHz, respectively) [64].
- Methyl 6-O-acetyl β-d-galattopyranoside (6a): 1H- and 13C-NMR (CDCl3, 300 MHz and 75.5 MHz, respectively) [66].
- Phenyl 6-O-acetyl α-d-glucopyranoside (7a): 1H- and 13C-NMR (CDCl3, 300 MHz and 75.5 MHz, respectively) [67].
- Phenyl 6-O-acetyl β-d-glucopyranoside (8a): 1H- and 13C-NMR (CDCl3, 300 MHz and 75.5 MHz, respectively) [67].
- N-acetyl-5′-O-acetyl adenosine (9a): 1H-NMR (CDCl3, 300 MHz): δ 2.02 (s, 3H, CH3COO), 2.18 (s, 3H, CH3CON), 3.85 (d, 1H, H5′, J = 13.15), 3.98 (d, 1H, H5′, Jgem = 13.15 Hz), 4.37 (s, 1H, H3′), 5.69 (s, 1H, H2′), 6.01–6.03 (m, 4H, H4′, H1′ 2OH), 7.84 (s, 1H, H8), 8.34 (s, 1H, H2). 13C-NMR (CDCl3, 75.5 MHz). HRMS (ESI) for [(C14H17N5O7) + Na]+: calcd 390.1026, found 390.1019 [M + Na]+.
- 5′-O-acetyl thymidine (10a): 1H-NMR (DMSO, 300 MHz): δ 1.93 (s, 3H, CH3), 2.09 (s, 1H, H2′), 2.13 (s, 3H, CH3COO), 2.18 (s, 1H, H2′), 2.46–2.44 (br, 1H, OH), 4.19 (t, 1H, H3′, J = 3.40 Hz), 4.27 (t, 1H, H4′, J = 3.29 Hz), 4.30 (dd, H5′, J = 11.84 Hz, J = 3.29 Hz), 4.36–4.40 (m, 1H, H5′), 3.98 (d, 1H, H5′, Jgem = 13.15 Hz), 6.30 (t, 1H, H1′, J = 6.47 Hz), 7.32 (s, 1H, H6), 9.78 (s, 1H, NH). 13C-NMR (DMSO, 75.5 MHz): 19.8, 21.5, 41.0, 63.9, 69.3, 82.7, 86.6, 109.8, 137.2, 150.6, 163.8, 170.3. HRMS (ESI) for [(C12H16N2O6) + Na]+: calcd 307.0906, found 307.0892 [M + Na]+.
- 5′-O-acetyl-2′-deoxyguanosine (11a): 1H-NMR (DMSO, 300 MHz): δ 2.07 (s, 3H, CH3COO), 2.36 (m, 1H, H2′), 2.77 (m, 1H, H2′), 3.57–3.58 (m br, 2H, H3′ OH), 3.99–4.02 (m, 2H, H5′), 5.28 (m, 1H, H4′), 6.10 (m, 1H, H1′), 6.72 (s, 2H, NH2), 7.94 (s, 1H, H8). 13C-NMR (DMSO, 75.5 MHz): 21.0, 40.7, 61.8, 75.3, 82.9, 85.2, 116.9, 135.4, 150.1, 154.2, 157.1, 170.3. HRMS (ESI) for [(C12H15N5O2) + Na]+: calcd 332.0971, found 332.0965 [M + Na]+.
- 2′-deoxy-3′,5′-di-O-acetylguanosine (11b): 1H-NMR (DMSO, 300 MHz): δ 2.03 (s, 3H, CH3COO), 2.08 (s, 3H, CH3COO), 2.41 (1H, ddd, 2J = 14.26 Hz, 3J1 = 5.47 Hz, 3J2 = 2.1 Hz, H-2b′) (m, 1H, H2′), 2.92 (1H, ddd, 2J = 14.27 Hz, 3J1 = 8.7 Hz, 3J2 = 6.4 Hz, H-2a′), 4.15-4.30 (m, 3H, H5′, H3′), 5.28 (d, 1H, H4′, J = 6.83), 6.08–6.15 (m, 1H, H1′), 6.52 (2H, s, NH2), 7.92 (1H, s, H8), 10.68 (1H, s, NH, 13C-NMR (DMSO, 75.5 MHz): 20.4, 20.7, 35.4, 63.6, 74.4, 81.4, 82.9, 116.7, 151.0, 153.7, 156.9, 169.9, 170.1.45. HRMS (ESI) for [(C14H17N5O6) + Na]+: calcd 374.1077, found 374.1071 [M + Na]+.
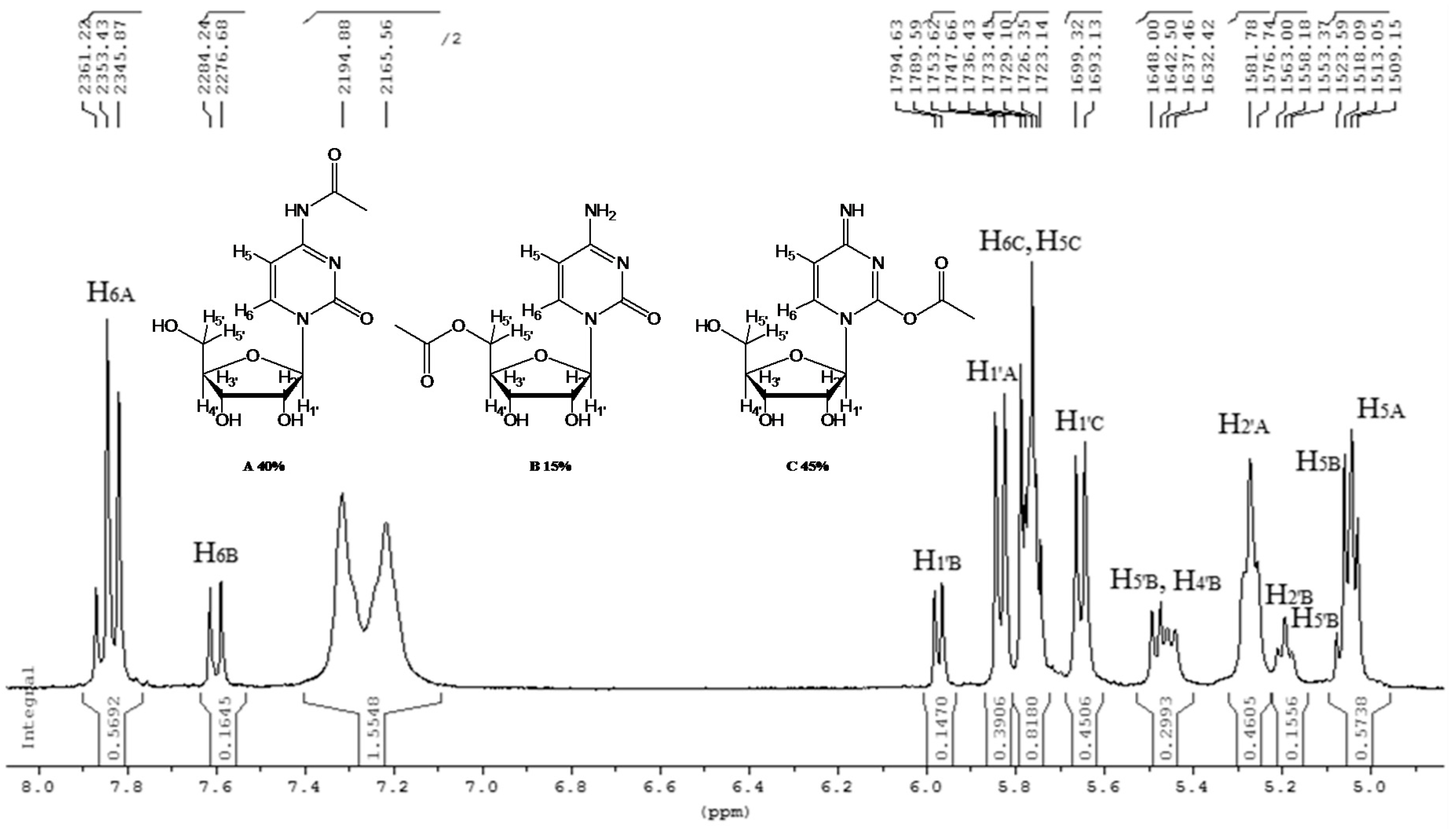
- N-acetyl-cytidine (12a), 5′-O-acetyl-cytidine (12b), 6-O-acetyl-cytidine (12c): 1H-NMR (DMSO, 300 MHz): δ 2.05 (s, 3H, CH3A), 2.08 (s, 6H, CH3B + CH3C), 3.51–3.71 (m, 6H, H5′A, H5′C, H3′C, H2′C), 3.82–4.01 (m, 3H, H5′A, H5′C, H4′A), 4.11–4.29 (m, 4H, H3′A, H4′A, H4′C, H3′B), 5.03–5.07 (m, 3H, H5A, H5B, H5′B), 5.19 (t, 1H, H2′B, J = 4.81 Hz), 5.27 (t, 1H, H2′A, J = 5.04 Hz), 5.45 (d, H4′B, J = 5.04 Hz), 5.48 (d, 1H, H5′B), 5.65 (d, 1H, H1′C, J = 6.19 Hz), 5.76 (m, 2H, H6C, H5C), 5.83 (d, 1H, H1′A, J = 5.96), 5.97 (d, 1H, H1′B), 7.60 (d, 1H, H6B, J = 7.56 Hz), 7.83 (d, 1H, H6A, J = 7.56 Hz). 13C-NMR (DMSO, 75.5 MHz): 60.5, 60.7, 68.1, 69.6, 71.8, 72.4, 73.2, 75.3, 79.1, 80.4, 81.9, 84.7, 86.7, 88.7, 90.0, 94.2, 94.3, 94.4, 141.3, 155.2, 155.4, 155.4, 165.5, 165.5, 169.6, 169.7, 178.3. HRMS (ESI) for [(C11H18N3O6) + Na]+: calcd 311.1093, found 311.1093 [M + Na]+, 311.1095 [M + Na]+, 311.1086 [M + Na]+.
- 4-[2-(Acetyloxy)ethyl]phenyl acetate (13a): 1H-NMR (DMSO, 300 MHz): δ 1.98 (s, 3H, CH3), 2.25 (s, 3H, CH3), 2.88 (t, 2H, CH2, J = 7.50 Hz), 4.20 (t, 2H, CH2, J = 7.11 Hz), 7.05 (d, 2H, Har, J = 8.49 Hz), 7.28 (d, 2H, Har, J = 8.46 Hz), 13C-NMR (DMSO, 75.5 MHz): Spectroscopic data compared to those reported in the literature [68].
- 3,4-dihydroxyphenethyl acetate (14a): 1H-NMR (DMSO, 300 MHz): δ 1.97 (s, 3H, CH3), 2.68 (t, 2H, CH2, J = 6.87 Hz), 4.10 (t, 2H, CH2, J = 7.11 Hz), 6.45–6.48 (m, 1H, Har), 6.48–6.62 (m, 2H, Har). 13C-NMR (DMSO, 75.5 MHz): 20.4, 34.3, 65.2, 115.0, 116.9, 121.1, 128.9, 144.2, 145.6, 170.6. HRMS (ESI) for [(C10H12O4) + H]+: calcd 197.0814, found 197.0818, 219.0635 [M + Na]+.
- 4-hydroxy-3-methoxyphenethyl acetate (15a): 1H-NMR (CDCl3, 300 MHz): δ 1.99 (s, 3H, CH3), 2.25 (s, 3H, CH3), 2.85 (t, 2H, CH2, J = 7.50 Hz), 3.73 (s, 3H, CH3), 4.33 (t, 2H, CH2, J = 7.11 Hz), 7.03 (d, 2H, Har, J = 8.43 Hz), 7.25 (d, 2H, Har, J = 8.43 Hz). 13C-NMR (CDCl3, 75.5 MHz): 20.4, 34.8, 56.9, 64.8, 113.7, 151.8, 121.5, 133.1, 142.9, 151.1, 170.0. HRMS (ESI) for [(C11H14O4) + H]+: calcd 211.0970, found 211.0978, 233.0781 [M + Na]+.
- Acetyl-3,5-dihydroxybenzyl alcohol (16a): 1H-NMR (DMSO, 300 MHz): 2.05 (s, 3H, CH3), 4.88 (s, 2H, CH2), 6.14–6.18 (m, 3H, Har). 13C-NMR (DMSO, 75.5 MHz): 20.6, 35.1, 64.8, 121.5, 128.3, 134.1, 143.2, 169.1, 170.0. 13C-NMR (DMSO, 75.5 MHz): 20.6, 65.3, 101.9, 105.6, 105.6, 137.9, 158.3, 170.0. HRMS (ESI) for [(C9H11O4) + H]+: calcd 183.0657, found 183.0655, 205.0480 [M + Na]+.
- Glycerol 1,3-diacetate (17a): 1H- and 13C-NMR (CDCl3, 300 MHz, and 75.5 MHz). Spectroscopic data compared to those reported in the literature [69].
- n-Butyl acetate (18a): 1H- and 13C-NMR (CDCl3, 300 MHz, and 75.5 MHz) [70].
- 1,4-Butanediol, diacetate (19a): 1H- and 13C-NMR (CDCl3, 300 MHz, and 75.5 MHz). Spectroscopic data compared to those of the pure product. GC-MS (EI): 114.0 (11), 73.0 (20), 71.0 (30), 54.0 (45), 43.0 (100) [71].
- n-Octyl acetate (20a): 1H- and 13C-NMR (CDCl3, 300 MHz, and 75.5 MHz). Spectroscopic data compared to those of the pure product. GC-MS (EI): 112.0 (10), 84.0 (30), 70.0 (36), 56.0 (30), 43.0 (100) [71].
- Fmoc-Ser(Ac)-OH (21a): 1H- and 13C-NMR (CDCl3, 300 MHz, and 75.5 MHz). Spectroscopic data compared to those of the pure product.
- Fmoc-Lys(Ac)-OH (22a): 1H- and 13C-NMR (CDCl3, 300 MHz, and 75.5 MHz). Spectroscopic data compared to those of the pure product.
4. Conclusions
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- Lu, K.; Hsieh, S.; Patkar, L.N.; Chen, C.; Lin, C. Simple and efficient Per-O-acetylation of Carbohydrates by lithium perchlorate catalyst. Tetrahedron 2004, 60, 8967–8973. [Google Scholar] [CrossRef]
- Kasiganesan, H.; Wright, G.L.; Chiacchio, M.A.; Gumina, G. Novel l-adenosine analogs as cardioprotective agents. Bioorg. Med. Chem. 2009, 17, 5347–5352. [Google Scholar] [CrossRef] [PubMed]
- Ferry, A.; Guinchard, X.; Retailleau, P.; David, C. Synthesis, characterization, and coupling reactions of six-membered cyclic p-chiral ammonium phosphonite–boranes; reactive H-phosphinateequivalents for the stereoselective synthesis of glycomimetics. J. Am. Chem. Soc. 2012, 134, 12289–12301. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.X.; Rahm, M.; Wu, B.; Dong, H. H-bonding activation in highly regioselective acetylation of diols. J. Org. Chem. 2013, 78, 11618–11622. [Google Scholar] [CrossRef] [PubMed]
- Ballini, R.; Bosica, G.; Carloni, L.; Maggi, R.; Sartori, G. Zeolite HSZ-360 as a new reusable catalyst for the direct acetylation of alcohols and phenols under solventless conditions. Tetrahedron Lett. 1998, 39, 6049–6052. [Google Scholar] [CrossRef]
- Bartoli, G.; Bosco, M.; Dalpozzo, R.; Marcantoni, E.; Massaccesi, M.; Rinaldi, S.; Sambri, L. Mg(ClO4) as a powerful catalyst for the acylation of alcohols under solvent-free conditions. Synlett 2003, 2003, 39–42. [Google Scholar] [CrossRef]
- Bartoli, G.; Bosco, M.; Dalpozzo, R.; Marcantoni, E.; Massaccesi, M.; Sambri, L. Zn(ClO4) 6H2O as a powerful catalyst for a practical acylation of alcohols with acid anhydrides. Eur. J. Org. Chem. 2003, 23, 4611–4617. [Google Scholar] [CrossRef]
- Chandra, K.L.; Saravanan, P.; Singh, R.K.; Singh, V.K. Lewis acid catalyzed acylation reactions: Scope and limitations. Tetrahedron 2002, 58, 1369–1374. [Google Scholar] [CrossRef]
- Dalpozzo, R.; Nino, A.D.; Maiuolo, L.; Procopio, A.; Nardi, M.; Bartoli, G.; Romeo, R. Highly efficient and versatile acetylation of alcohols catalyzed by cerium (III) triflate. Tetrahedron Lett. 2003, 44, 5621–5624. [Google Scholar] [CrossRef]
- Danieli, B.; Luisetti, M.; Sampognato, G.; Carrea, G.; Riva, S. Regioselective acylation of polyhydroxylated natural compounds catalyzed by Candida Antarctica lipase B (Novozym 435) in organic solvents. J. Mol. Catal. B Enzym. 1997, 3, 193–201. [Google Scholar] [CrossRef]
- Sugahara, K.; Satake, N.; Kamata, K.; Nakajima, T.; Mizuno, N. A basic germanodecatungstate with a −7 charge: Efficient chemoselective acylation of primary alcohols. Angew. Chem. Int. Ed. 2014, 53, 13248–13252. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, W.; William, J.M.; Onomura, O. Selective monobenzoylation of 1,2- and 1,3-diols catalyzed by Me2SnCl2in water (organic solvent free) under mild conditions. J. Org. Chem. 2012, 77, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Lemieux, R.U.; Driguez, H. Chemical synthesis of 2-acetamido-2-deoxy-4-O-(alpha-l-fucopyranosyl)-3-O-(beta-d-galactopyranosyl)-d-glucose. Lewis a blood-group antigenic determinant. J. Am. Chem. Soc. 1975, 97, 4063–4069. [Google Scholar] [CrossRef]
- Heller, S.T.; Sarpong, R. Chemoselectiveesterification and amidation of carboxylic acids with imidazole carbamates and ureas. Org. Lett. 2010, 12, 4572–4575. [Google Scholar] [CrossRef] [PubMed]
- Heller, S.T.; Schultz, E.E.; Sarpong, R. Chemoselective N-acylation of indoles and oxazolidinones with carbonylazoles. Angew. Chem. Int. Ed. 2012, 51, 8304–8308. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Hou, C.; Ren, J.; Xin, X.; Xu, H.; Pei, Y.; Dong, H.; Pei, Z. Regioselectivebenzoylation of diols and carbohydrates by catalytic amounts of organobase. Molecules 2016, 21, 641. [Google Scholar] [CrossRef] [PubMed]
- Clarke, D.; Ali, M.A.; Clifford, A.A. Reactions in unusual media. Curr. Top. Med. Chem. 2004, 4, 729–771. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Dou, R.; Song, G.; Jiang, J. Dramatically accelerated synthesis of aminoketones via aqueous mannich reaction under combined microwave and ultrasound irradiation. Synlett 2005, 14, 2245–2247. [Google Scholar] [CrossRef]
- Shi, L.; Wang, M.; Zhang, F.M.; Fan, C.A. Microwave-promoted three-component coupling of aldehyde, alkyne, and amine via C-H activation catalyzed by copper in water. Org. Lett. 2004, 6, 1001–1003. [Google Scholar] [CrossRef] [PubMed]
- Cherng, Y.J. Efficient nucleophilic substitution reaction of aryl halides with amino acids under focused microwave irradiation. Tetrahedron 2000, 56, 8287–8289. [Google Scholar] [CrossRef]
- Kaval, N.; Dehaen, W.; Matyus, P.; Van der Eycken, E. Convenient and rapid microwave-assisted synthesis of pyrido-fused ring systems applying the tert-amino effect. Green Chem. 2004, 6, 125–127. [Google Scholar] [CrossRef]
- Pironti, V.; Colonna, S. Microwave-promoted synthesis of β-hydroxy sulfides and β-hydroxysulfoxides in water. Green Chem. 2005, 7, 43–45. [Google Scholar] [CrossRef]
- Lindsay, K.B.; Pyne, S.G. Studies on the synthesis of croomine: Synthesis of the tricyclic B,C,D-ring core structure. Synlett 2004, 5, 779–782. [Google Scholar]
- Chen, I.H.; Young, J.N.; Yu, S.J. Recyclable organotungsten Lewis acid and microwave assisted Diels-Alder reactions in water and in ionic liquids. Tetrahedron 2004, 60, 11903–11909. [Google Scholar] [CrossRef]
- Kranjc, K.; Kocevar, M.; Iosif, F.; Coman, S.M.; Parvulescu, V.I.; Genin, E.; Genet, J.P.; Michelet, V. Efficient and green access to functionalized and highly constrained heteropolycyclic derivatives via a microwave accelerated Diels-Alder cycloaddition and hetereogeneous hydrogenation sequence. Synlett 2006, 2006, 1075–1079. [Google Scholar]
- Molteni, V.; Hamilton, M.M.; Mao, L.; Crane, C.M.; Termin, A.P.; Wilson, D.M. Aqueous one-pot synthesis of pyrazoles, pyrimidines and isoxazoles promoted by microwave irradiation. Synthesis 2002, 12, 1669–1674. [Google Scholar] [CrossRef]
- Bryson, T.A.; Stewart, J.J.; Gibson, J.M.; Thomas, P.S.; Berch, J.K. Green heterocycle synthesis, isochromenones and artemidin. Green Chem. 2003, 5, 174–176. [Google Scholar] [CrossRef]
- Kaiser, N.F.K.; Hallberg, A.; Larhed, M. In situ generation of carbon monoxide from solid molybdenum hexacarbonyl. A convenient and fast route to palladium-catalyzed carbonylation reactions. J. Comb. Chem. 2002, 4, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Georgsson, J.; Hallberg, A.; Larhed, M. Rapid palladium-catalyzed synthesis of esters from aryl halides utilizing Mo(CO)6as a solid carbon monoxide source. J. Comb. Chem. 2003, 5, 350–352. [Google Scholar] [CrossRef] [PubMed]
- Wannberg, J.; Larhed, M. Increasing rates and scope of reactions: Sluggish amines in microwave-heated aminocarbonylation reactions under air. J. Comb. Chem. 2003, 68, 5750–5753. [Google Scholar]
- Leadbeater, N.E.; Marco, M. Rapid and amenable suzuki coupling reaction in water using microwave and conventional heating. J. Org. Chem. 2003, 68, 888–892. [Google Scholar] [CrossRef] [PubMed]
- Capek, P.; Pohl, R.; Hocek, M. Cross-coupling reactions of unprotected halopurine bases, nucleosides, nucleotidesand nucleoside triphosphates with 4-boronophenylalanineinwater. Synthesis of (purin-8-yl)- and (purin-6-yl)phenylalanines. Org. Biomol. Chem. 2006, 4, 2278–2284. [Google Scholar] [CrossRef] [PubMed]
- Arvela, R.K.; Leadbeater, N.E. Microwave-promoted heck coupling using ultralow metal catalyst concentrations. J. Org. Chem. 2005, 70, 1786–1790. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wei, P.; Pei, Y.; Xu, H.; Xina, X.; Pei, Z. Regioselective acetylation of carbohydrates and diols catalyzed by tetramethyl-ammonium hydroxide in water. Green Chem. 2014, 16, 4510–4514. [Google Scholar] [CrossRef]
- Kobayashi, S.; Nagayama, S.; Busujima, T. Lewis acid catalysts stable in water. Correlation between catalytic activity in water and hydrolysis constants and exchange rate constants for substitution of inner-sphere water ligands. J. Am. Chem. Soc. 1998, 120, 8287–8288. [Google Scholar] [CrossRef]
- Di Gioia, M.L.; Barattucci, A.; Bonaccorsi, P.; Leggio, A.; Minuti, L.; Romio, E.; Temperini, A.; Siciliano, C. Deprotection/reprotection of the amino group in α-amino acids and peptides. A one-pot procedure in [Bmim][BF4] ionic liquid. RSC Adv. 2014, 4, 2678–2686. [Google Scholar] [CrossRef]
- Di Gioia, M.L.; Gagliardi, A.; Leggio, A.; Leotta, V.; Romio, E.; Liguori, A. N-Urethane protection of amines and amino acids in an ionic liquid. RSC Adv. 2015, 5, 63407–63420. [Google Scholar] [CrossRef]
- Di Gioia, M.L.; Costanzo, P.; De Nino, A.; Maiuolo, L.; Nardi, M.; Olivito, F.; Procopio, A. Simple and efficient Fmoc removal in ionic liquid. RSC Adv. 2017, 7, 36482–36491. [Google Scholar] [CrossRef]
- Procopio, A.; Dalpozzo, R.; De Nino, A.; Maiuolo, L.; Nardi, M.; Russo, B. Synthesis of acetonides from epoxides catalyzed by erbium(III) triflate. Adv. Synth. Catal. 2005, 347, 1447–1450. [Google Scholar] [CrossRef]
- Dalpozzo, R.; De Nino, A.; Nardi, M.; Russo, B.; Procopio, A. 1,2-Diacetates by epoxide ring opening promoted by erbium(III) triflate. Arkivoc 2006, VI, 67–73. [Google Scholar]
- Dalpozzo, R.; Nardi, M.; Oliverio, M.; Paonessa, R.; Procopio, A. Erbium(III) triflate is a highly efficient catalyst for the synthesis of β-alkoxy alcohols, 1,2-diols and β-hydroxysulfides by ring opening of epoxides. Synthesis 2009, 2009, 3433–3438. [Google Scholar]
- Oliverio, M.; Nardi, M.; Cariati, L.; Vitale, E.; Bonacci, S.; Procopio, A. “On Water” MW-Assisted Synthesis of Hydroxytyrosol Fatty Esters. ACS Sustain. Chem. Eng. 2016, 4, 4661–4665. [Google Scholar] [CrossRef]
- Procopio, A.; Das, G.; Nardi, M.; Oliverio, M.; Pasqua, L. A mesoporous ErIII-MCM-41 catalyst for the cyanosilylation of aldehydes and ketones under solvent-free conditions. ChemSusChem 2008, 1, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Nardi, M.; Cozza, A.; De Nino, A.; Oliverio, M.; Procopio, A. One-pot synthesis of dibenzo [b,e] [1,4] diazepin-1-ones. Synthesis 2012, 44, 800–804. [Google Scholar] [CrossRef]
- Nardi, M.; Cozza, A.; Maiuolo, L.; Oliverio, M.; Procopio, A. 1,5-Benzoheteroazepines through eco-friendly general condensation reactions. Tetrahedron Lett. 2011, 52, 4827–4834. [Google Scholar] [CrossRef]
- Procopio, A.; Costanzo, P.; Curini, M.; Nardi, M.; Oliverio, M.; Sindona, G. Erbium(III) chloride in ethyl lactate as a smart ecofriendly system for efficient and rapid stereoselective synthesis of trans-4,5-diaminocyclopent-2-enones. ACS Sustain. Chem. Eng. 2013, 1, 541–544. [Google Scholar] [CrossRef]
- Nardi, M.; Oliverio, M.; Costanzo, P.; Sindona, G.; Procopio, A. Eco-friendly stereoselective reduction of α,β-unsaturated carbonyl compounds by Er(OTf)3/NaBH4in 2-MeTHF. Tetrahedron 2015, 71, 1132–1135. [Google Scholar] [CrossRef]
- Oliverio, M.; Nardi, M.; Cariati, L.; Vitale, E.; Bonacci, S.; Procopio, A. Facile ecofriendly synthesis of monastrol and its structural isomers via biginelli reaction. ACS Sustain. Chem. Eng. 2016, 2, 1228–1233. [Google Scholar] [CrossRef]
- De Nino, A.; Maiuolo, L.; Merino, P.; Nardi, M.; Procopio, A.; Roca-López, D.; Russo, B.; Algieri, V. New efficient organocatalyst supported on simple ionic liquid as recoverable system for asymmetric diels-alder reaction in presence of water. ChemCatChem 2015, 7, 830–835. [Google Scholar] [CrossRef]
- Nardi, M.; Herrera Cano, N.; Costanzo, P.; Oliverio, M.; Sindona, G.; Procopio, A. Aqueous MW eco-friendly protocol for amino group protection. RSC Adv. 2015, 5, 18751–18760. [Google Scholar] [CrossRef]
- Procopio, A.; Cravotto, G.; Oliverio, M.; Costanzo, P.; Nardi, M.; Paonessa, R. An eco-sustainable erbium(III)-catalyzed method for formation/cleavage of O-tert-butoxy carbonates. Green Chem. 2011, 13, 436–443. [Google Scholar] [CrossRef]
- Musikas, C. Solvent extraction for the chemical separation of the 5f elements. Inorg. Chem. Acta 1987, 140, 197–206. [Google Scholar] [CrossRef]
- Musikas, C.; Hubert, H. Extraction by N,N′-tetraalkylmalonamides II. Sol. Extr. Ion Exch. 1987, 5, 877–893. [Google Scholar] [CrossRef]
- Narita, H.; Yaita, T.; Tachimori, S. Solvent extraction of trivalent lanthanoid ions with N,N’-Dimethyl-N,N′-Diphenyl-3-Oxapentanediamide. Radiochim. Acta 1998, 81, 223–226. [Google Scholar] [CrossRef]
- Narita, H.; Yaita, T.; Tachimori, S. Study on the extraction of trivalent lanthanide ions with N,N′-dimethyl-N,N′-diphenyl-malonamide and diglycolamide. Radioanal. J. Nucl. Chem. 1999, 239, 381–384. [Google Scholar] [CrossRef]
- Cepeda, J.; Balda, R.; Beobide, G.; Castillo, O.; Fernández, J.; Luque, A.; Pérez-Yáñez, S.; Román, P.; Sánchez, D.V. Lanthanide(III)/Pyrimidine-4,6-dicarboxylate/Oxalate extended frameworks: A detailed study based on the lanthanide contraction and temperature effects. Inorg. Chem. 2011, 50, 8437–8451. [Google Scholar] [CrossRef] [PubMed]
- Kocienski, P.J. Protecting Groups; Georg ThiemeVerlag: Stuttgart, Germany, 2005. [Google Scholar]
- Di Gioia, M.L.; Leggio, A.; Liguori, A.; Perri, F.; Siciliano, C.; Viscomi, M.C. A preparation of N-Fmoc-N-methyl-alpha-amino acids and N-nosyl-N-methyl-alpha-amino acids. Amino Acids 2010, 38, 133–143. [Google Scholar] [CrossRef] [PubMed]
- De Marco, R.; Di Gioia, M.L.; Leggio, A.; Liguori, A.; Perri, F.; Siciliano, C.; Viscomi, M.C. A new non-natural arginine-like amino acid derivative with a sulfamoyl group in the side-chain. Amino Acids 2010, 38, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Di Gioia, M.L.; Leggio, A.; Malagrinò, F.; Romio, E.; Siciliano, C.; Liguori, A. N-Methylated α-Amino acids and peptides: Synthesis and biological activity. Mini Rev. Med. Chem. 2016, 16, 683–690. [Google Scholar] [CrossRef] [PubMed]
- De Marco, R.; Di Gioia, M.L.; Liguori, A.; Perri, F.; Siciliano, C.; Spinella, M. N-alkylation of N-aryl-α-amino acid methyl esters by trialkyloxonium tetrafluoroborates. Tetrahedron 2011, 67, 9708–9714. [Google Scholar] [CrossRef]
- Siciliano, C.; Barattucci, A.; Bonaccorsi, P.; Di Gioia, M.L.; Leggio, A.; Minuti, L.; Romio, E.; Temperini, A. Synthesis of d-erythro-sphinganine through serine-derived α-amino epoxides. J. Org. Chem. 2014, 79, 5320–5326. [Google Scholar] [CrossRef] [PubMed]
- Bianco, A.; Brufani, M.; Melchioni, C.; Romagnoli, P. Protection of primary alcoholic function with rare-earths salts. Tetrahedron Lett. 1997, 38, 651–652. [Google Scholar] [CrossRef]
- Jansson, P.E.; Kenne, L.; Schweda, E. Nuclear magnetic resonance and conformational studies on monoacetylated methyl d-gluco- and d-galacto-pyranosides. J. Chem. Soc. Perkin Trans. 1987, 1, 377–383. [Google Scholar] [CrossRef]
- Osada, M.; Kikuta, K.; Yoshida, K.; Oqata, M.; Usui, T. Non-catalytic synthesis of Chromogen I and III from N-acetyl-d-glucosamine in high-temperature water. Green Chem. 2013, 15, 2960–2966. [Google Scholar] [CrossRef]
- McClure, M.S.; Berry, M.B.; Caine, D.; Crawford, C.; Crump, B.C.; Glover, B.N.; Kedia, S.B.; Millar, A.; Mitchell, M.B.; Nichols, C.J.; et al. Highly selective primary alkoxycarboxylation and esterification of unprotected pyranose derivatives mediated by scandium(III) triflate catalysis. Eur. J. Org. Chem. 2012, 19, 3561–3565. [Google Scholar] [CrossRef]
- Ciuffreda, P.; Casati, S.; Manzocchi, A. Complete (1)H and (13)C NMR spectral assignment of alpha- and beta-adenosine, 2’-deoxyadenosine and their acetate derivatives. Magn. Reson. Chem. 2007, 45, 781–784. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, B.; Phukan, P. A new ferrocene-based bulky pyridine as an efficient reusable homogeneous catalyst. RSC Adv. 2013, 3, 15327–15336. [Google Scholar] [CrossRef]
- Lange, K.; Koenig, A.; Roegler, C.; Seeling, A.; Lehmann, J. NO donors—Part 18: Bioactive metabolites of GTN and PETN—Synthesis and vasorelaxant properties. Bioorg. Med. Chem. Lett. 2009, 19, 3141–3144. [Google Scholar] [CrossRef] [PubMed]
- Qiu, R.; Zhang, G.; Ren, X.; Xu, X.; Yang, R.; Luo, S.; Yin, S. Air-stable titanocenebis(perfluorooctanesulfonate) as a new catalyst for acylation of alcohols, phenols, thiols, and amines under solvent-free condition. J. Organomet. Chem. 2010, 695, 1182–1188. [Google Scholar] [CrossRef]
- Ruijie, Z.; Hongting, S.; Yongcang, Z.; Yan, F.; Zhi, C.; Junfeng, W.; Man, C.; Manzhou, Z.; Qingxiang, G. Heterobimetallicdinuclear lanthanide alkoxide complexes as acid-basedifunctional catalysts for transesterification. J. Org. Chem. 2014, 79, 9246–9252. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}

| Entry | 1-Acetylimidazole (equiv) | Er(OTf)3 (mol %) | Time (min) | Yield (%) b |
|---|---|---|---|---|
| 1 | 1.2 | 10 | 10 | trace |
| 2 | 1.2 | 10 | 20 | 5 |
| 3 | 1.2 | 10 | 30 | 26 |
| 4 c | 1.2 | 10 | 30 | 14 |
| 5 | 2.0 | 10 | 10 | 15 |
| 6 | 2.0 | 10 | 30 | 28 |
| 7 | 3.0 | 10 | 15 | 52 |
| 8 | 3.0 | 10 | 30 | 67 |
| 9 | 3.0 | 10 | 60 | 40 |
| 10 | 3.0 | 0 | 40 | - |
| 11 | 3.0 | 20 | 30 | 68 |
| 12 | 3.0 | 5 | 30 | 26 |
| 13 | 3.0 | 20 | 60 | 66 |
| Entry | Substrate | Product | Yield (%) b |
|---|---|---|---|
| 1 |  1 |  1a | 67 |
| 2 |  2 |  2a | 63 |
| 3 |  3 |  3a | 64 |
| 4 |  4 |  4a | 65 |
| 5 |  5 |  5a | 62 |
| 6 |  6 |  6a | 65 |
| 7 |  7 |  7a | 60 |
| 8 |  8 |  8a | 63 |
| 9 |  9 |  9a | 67 |
| 10 |  10 |  10a | 64 |
| 11 |  11 |  11a | 50 |
| 12 |  12 |  (12a,b,c) | 60 a: 40 b: 15 c: 45 |
| 13 |  13 |  13a | 59 |
| 14 |  14 |  14a | 80 |
| 15 |  15 |  15a | 80 |
| 16 |  16 |  16a | 75 |
| 17 d |  17 |  17a | 95 |
| 18 |  18 |  18a | 65 |
| 19 |  19 |  19a | 80 |
| 20 c |  20 |  20a | 95 |
| 21 |  21 |  21a | 92 |
| 22 |  22 |  22a | 93 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nardi, M.; Gioia, M.L.D.; Costanzo, P.; Nino, A.D.; Maiuolo, L.; Oliverio, M.; Olivito, F.; Procopio, A. Selective Acetylation of Small Biomolecules and Their Derivatives Catalyzed by Er(OTf)3. Catalysts 2017, 7, 269. https://doi.org/10.3390/catal7090269
Nardi M, Gioia MLD, Costanzo P, Nino AD, Maiuolo L, Oliverio M, Olivito F, Procopio A. Selective Acetylation of Small Biomolecules and Their Derivatives Catalyzed by Er(OTf)3. Catalysts. 2017; 7(9):269. https://doi.org/10.3390/catal7090269
Chicago/Turabian StyleNardi, Monica, Maria Luisa Di Gioia, Paola Costanzo, Antonio De Nino, Loredana Maiuolo, Manuela Oliverio, Fabrizio Olivito, and Antonio Procopio. 2017. "Selective Acetylation of Small Biomolecules and Their Derivatives Catalyzed by Er(OTf)3" Catalysts 7, no. 9: 269. https://doi.org/10.3390/catal7090269
APA StyleNardi, M., Gioia, M. L. D., Costanzo, P., Nino, A. D., Maiuolo, L., Oliverio, M., Olivito, F., & Procopio, A. (2017). Selective Acetylation of Small Biomolecules and Their Derivatives Catalyzed by Er(OTf)3. Catalysts, 7(9), 269. https://doi.org/10.3390/catal7090269