Hydrogen-Etched TiO2−x as Efficient Support of Gold Catalysts for Water–Gas Shift Reaction


Abstract
:
1. Introduction
2. Results
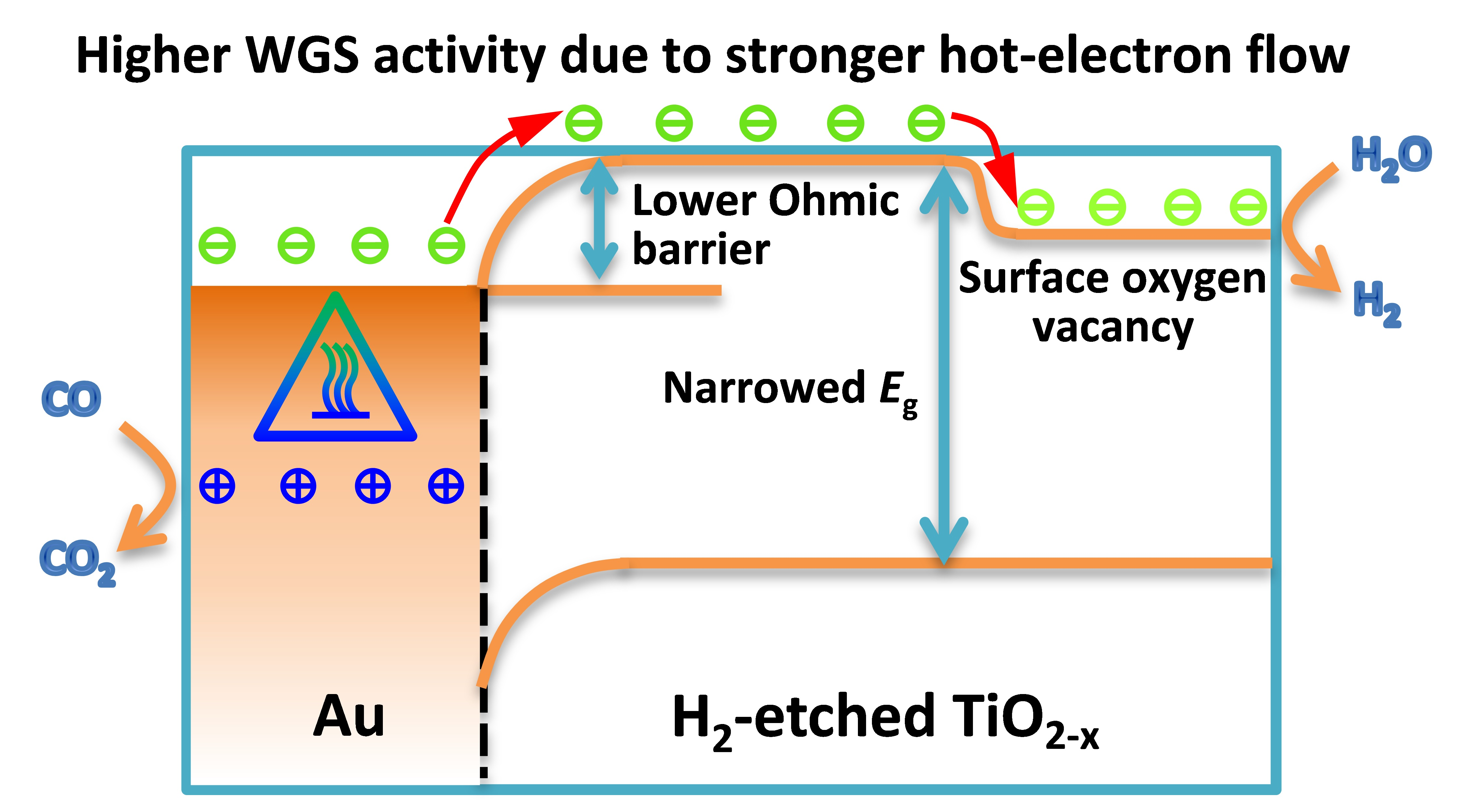
2.1. Catalytic Activities and Stabilities
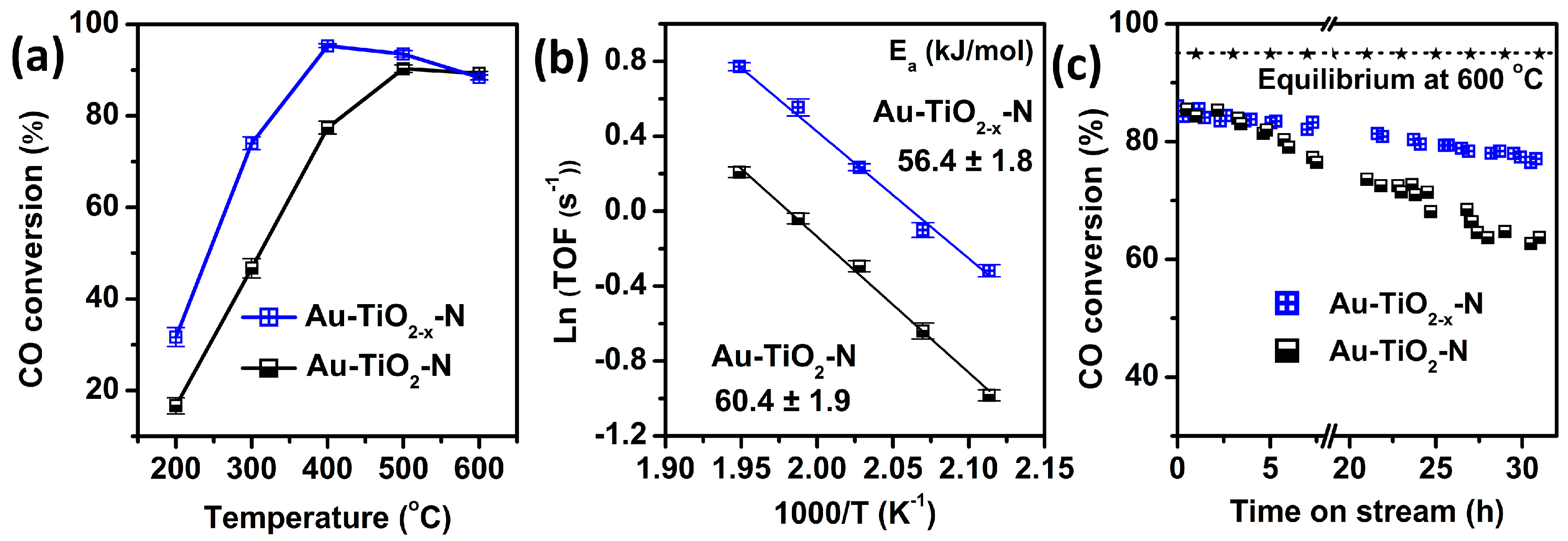
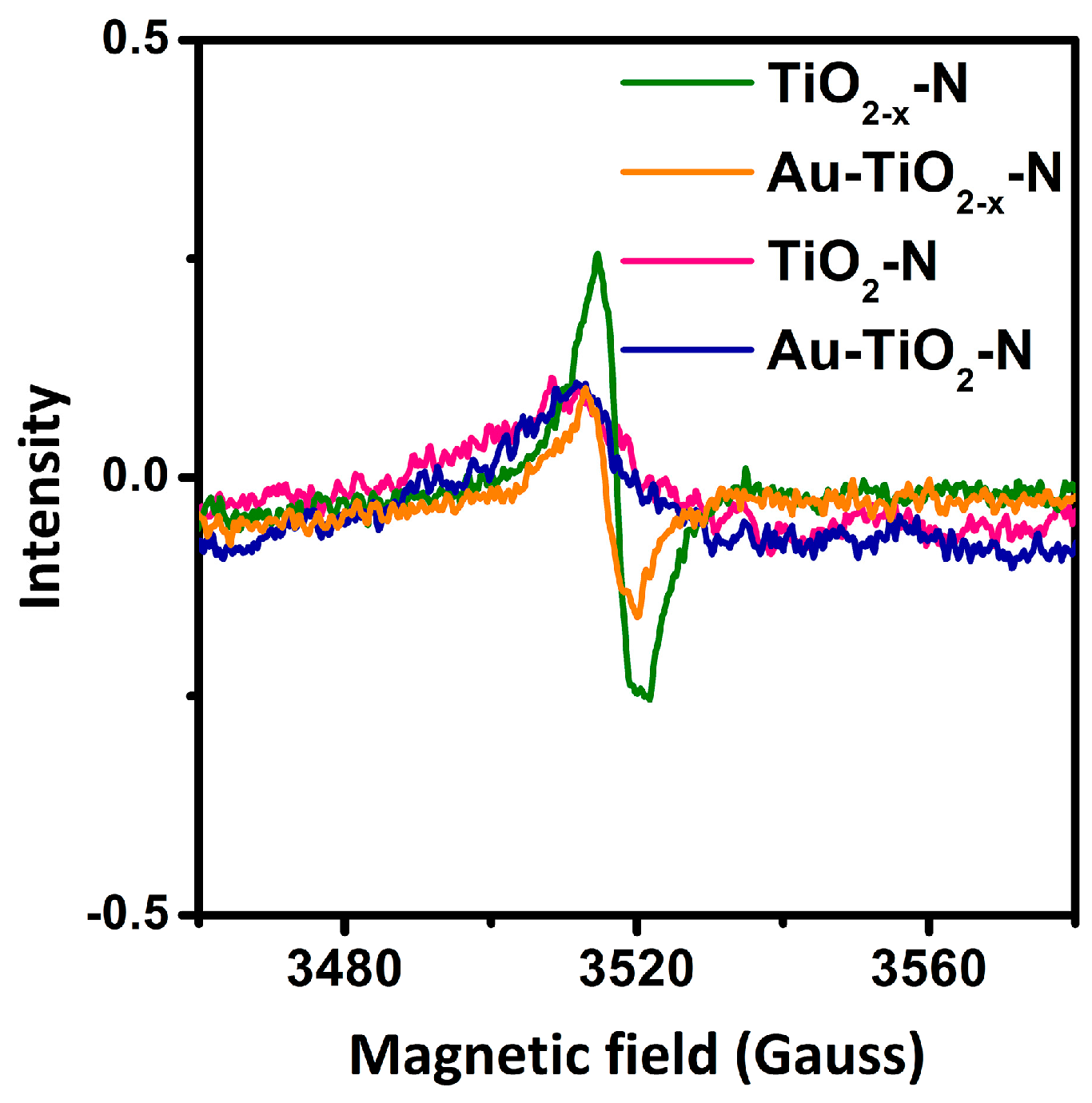
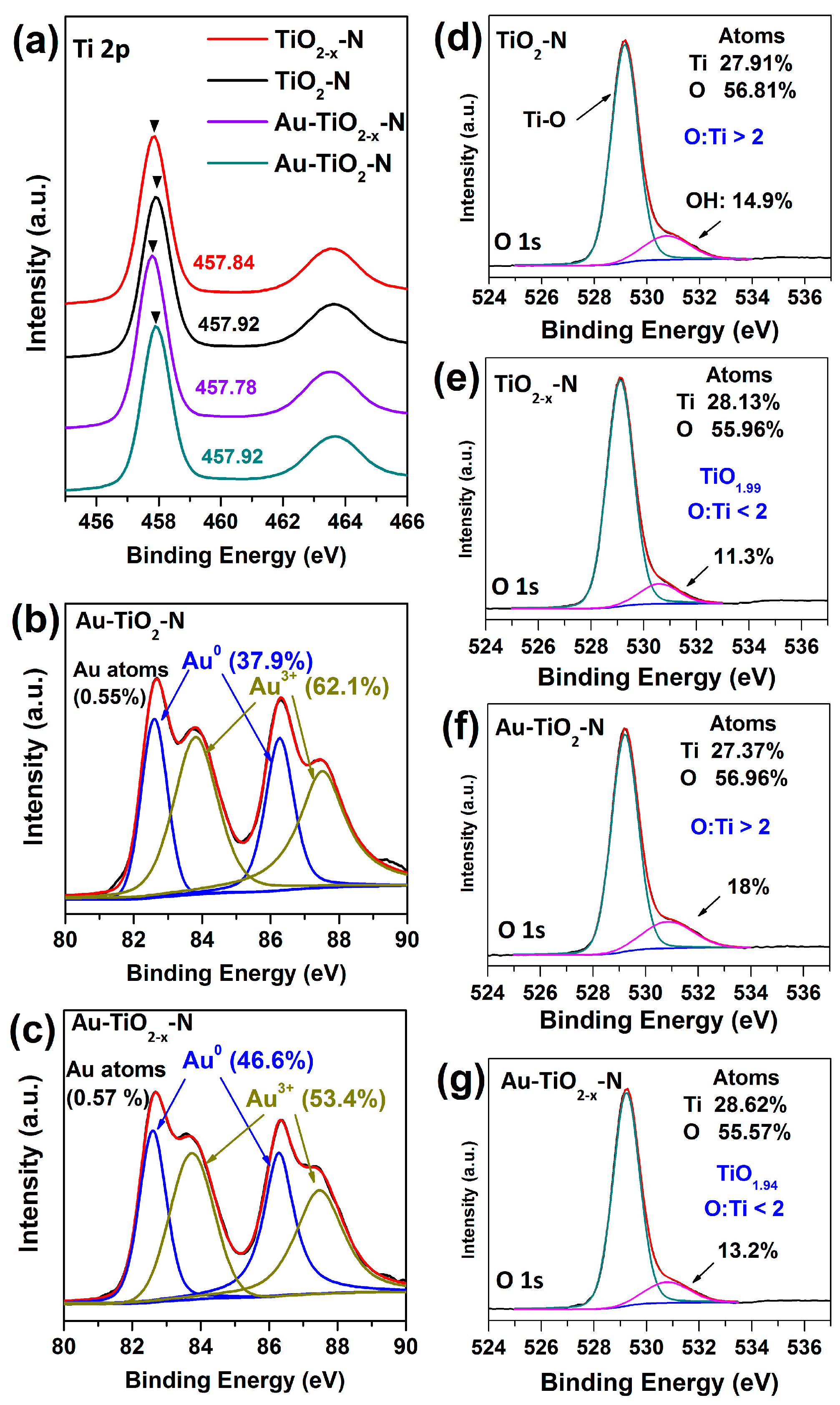
2.2. Structural Properties
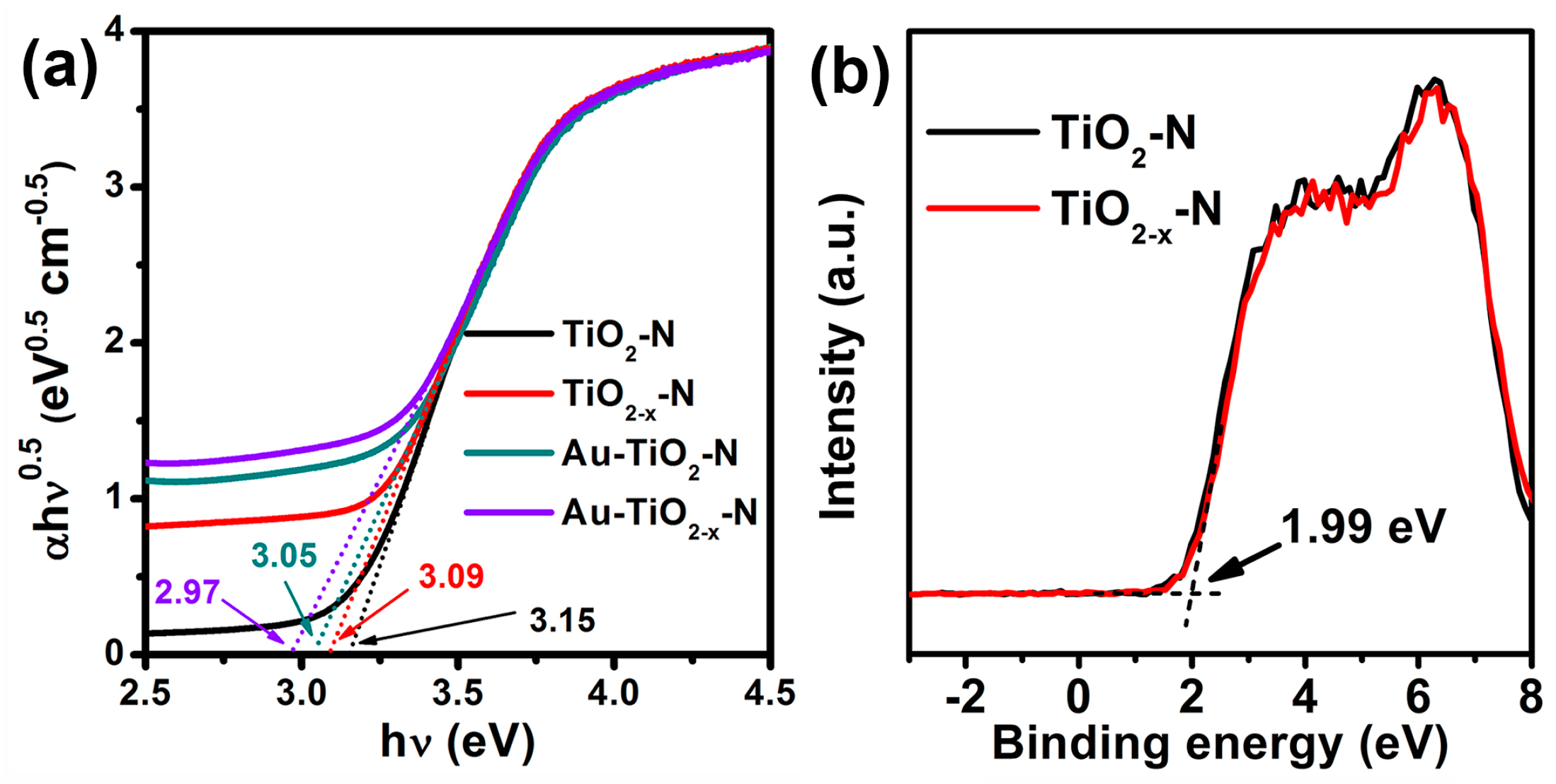
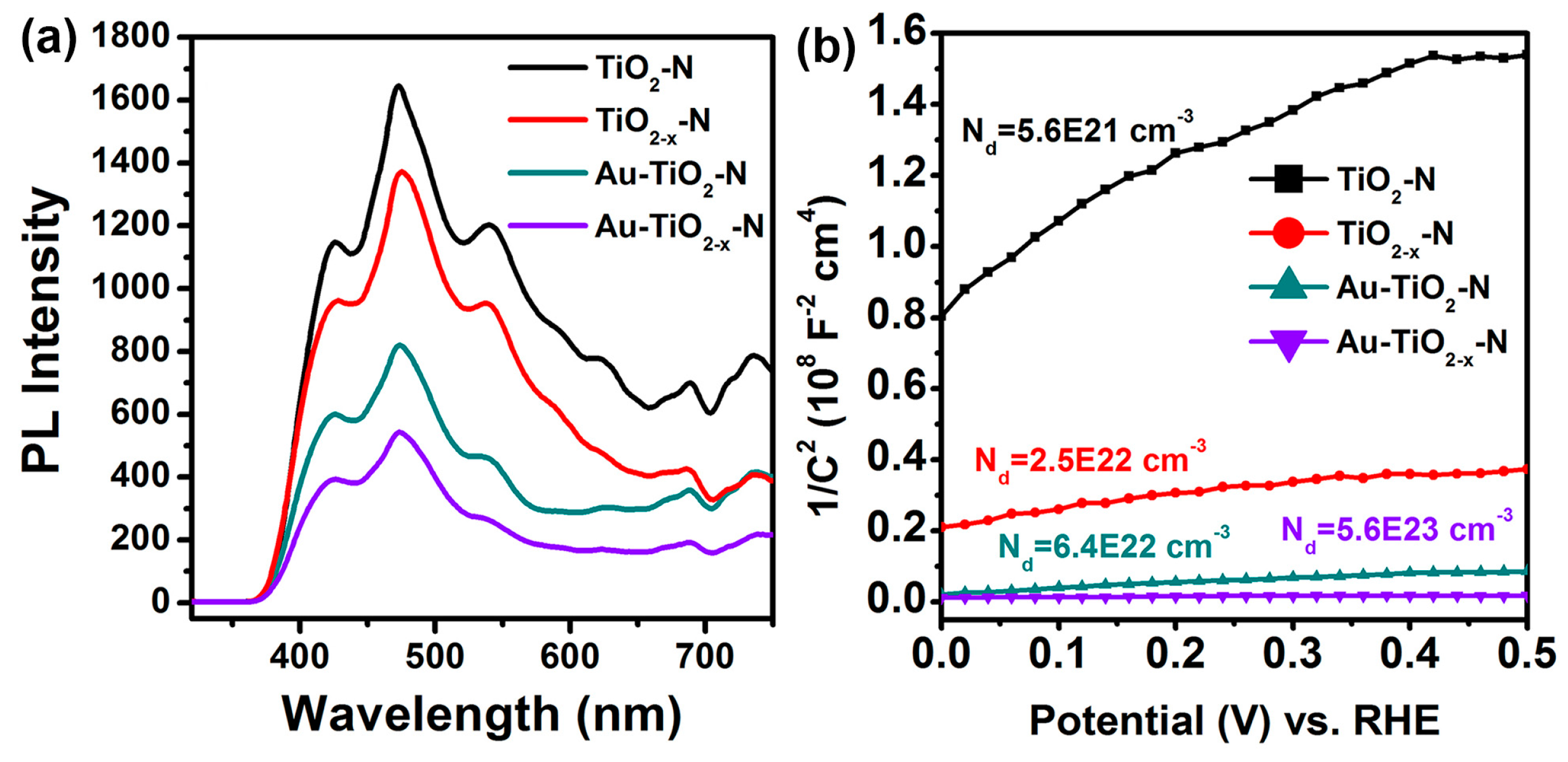
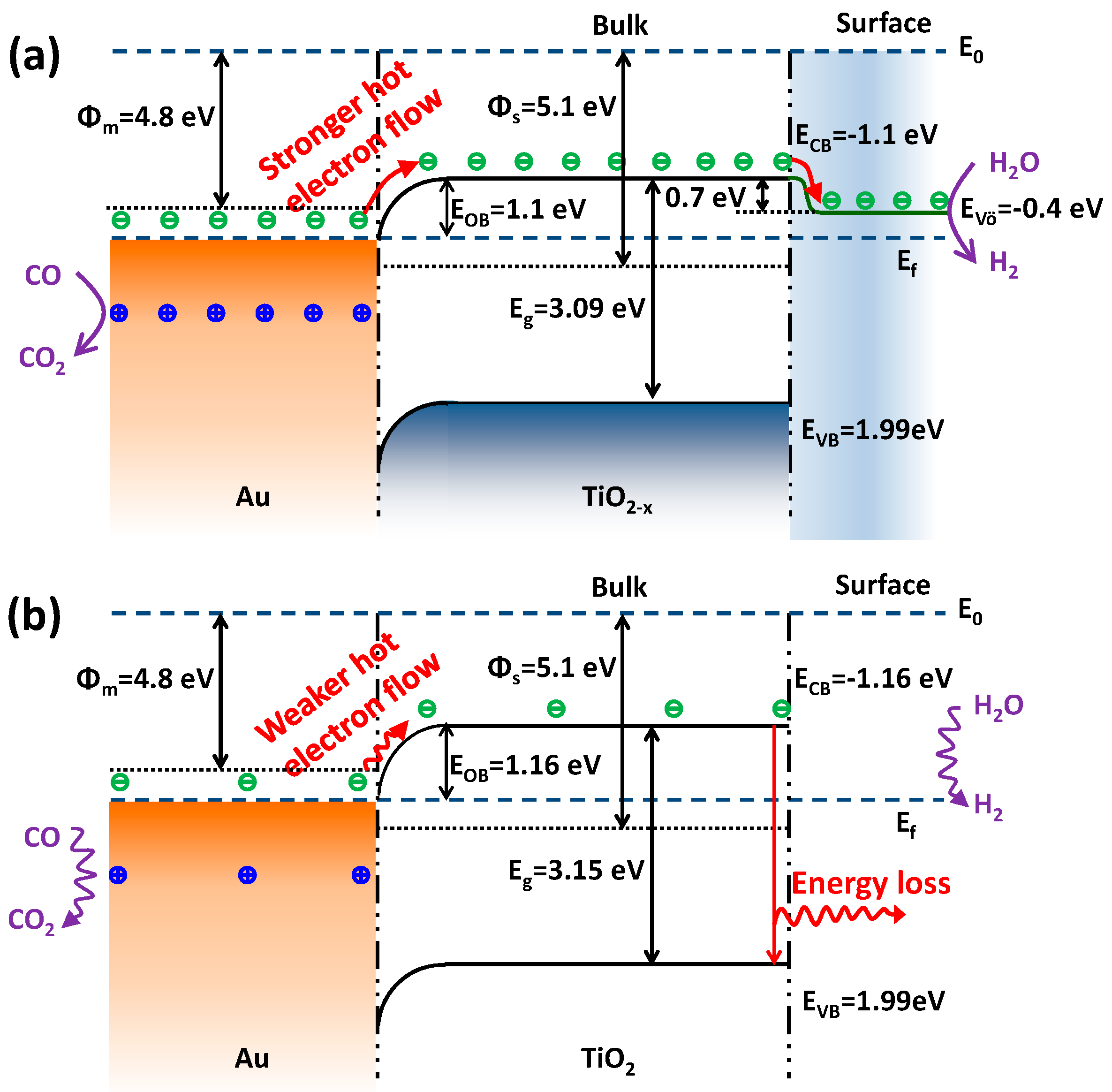
2.3. Optoelectronic Properties
3. Discussion
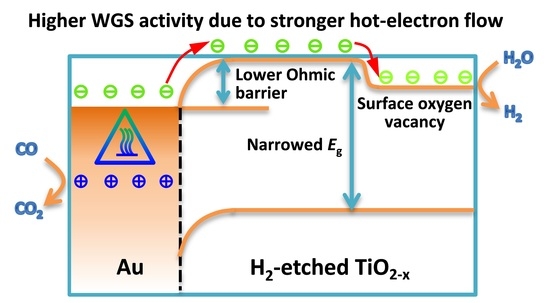
Proposed Electron Flow Process
4. Materials and Methods
4.1. Preparation of White TiO2-N and Blue-Black TiO2−x-N Supports
4.2. Preparation of Au-TiO2-N and Au-TiO2−x-N Catalysts
4.3. Evaluation of Catalytic Activity and Stability
4.4. Characterization
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Flytzani-Stephanopoulos, M. Gold atoms stabilized on various supports catalyze the water-gas shift reaction. Acc. Chem. Res. 2013, 47, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Bunluesin, T.; Gorte, R.J.; Graham, G.W. Studies of the water-gas-shift reaction on ceria-supported Pt, Pd, and Rh: Implications for oxygen-storage properties. Appl. Catal. B Environ. 1998, 15, 107–114. [Google Scholar] [CrossRef]
- Hakeem, A.A.; Vásquez, R.S.; Rajendran, J.; Li, M.; Berger, R.J.; Delgado, J.J.; Kapteijn, F.; Makkee, M. The role of rhodium in the mechanism of the water–gas shift over zirconia supported iron oxide. J. Catal. 2014, 313, 34–45. [Google Scholar] [CrossRef]
- Li, Y.; Fu, Q.; Flytzani-Stephanopoulos, M. Low-temperature water-gas shift reaction over Cu- and Ni-loaded cerium oxide catalysts. Appl. Catal. B Environ. 2000, 27, 179–191. [Google Scholar] [CrossRef]
- Ren, Z.; Peng, F.; Li, J.; Liang, X.; Chen, B. Morphology-Dependent Properties of Cu/CeO2 Catalysts for the Water-Gas Shift Reaction. Catalysts 2017, 7, 48. [Google Scholar] [CrossRef]
- Sakurai, H.; Ueda, A.; Kobayashi, T.; Haruta, M. Low-temperature water-gas shift reaction over gold deposited on TiO2. Chem. Commun. 1997, 0, 271–272. [Google Scholar] [CrossRef]
- Ma, Z.; Yin, H.F.; Dai, S. Performance of Au/MxOy/TiO2 Catalysts in water-gas shift reaction. Catal. Lett. 2010, 136, 83–91. [Google Scholar] [CrossRef]
- Shi, J.; Mahr, C.; Mangir Murshed, M.; Zielasek, V.; Rosenauer, A.; Gesing, T.M.; Bäumer, M.; Wittstock, A. A versatile sol–gel coating for mixed oxides on nanoporous gold and their application in the water gas shift reaction. Catal. Sci. Technol. 2016, 6, 5311–5319. [Google Scholar] [CrossRef]
- Faust, M.; Dinkel, M.; Bruns, M.; Bräse, S.; Seipenbusch, M. Support Effect on the Water Gas Shift Activity of Chemical Vapor Deposition-Tailored-Pt/TiO2 Catalysts. Ind. Eng. Chem. Res. 2017, 56, 3194–3203. [Google Scholar] [CrossRef]
- Yang, M.; Allard, L.F.; Flytzani-Stephanopoulos, M. Atomically Dispersed Au−(OH)x Species Bound on Titania Catalyze the Low-Temperature Water-Gas Shift Reaction. J. Am. Chem. Soc. 2013, 135, 3768–3771. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kispersky, V.F.; Nicholas Delgass, W.; Ribeiro, F.H. Determination of the Au active site and surface active species via operando transmission FTIR and isotopic transient experiments on 2.3 wt. % Au/TiO2 for the WGS reaction. J. Catal. 2012, 289, 171–178. [Google Scholar] [CrossRef]
- Shekhar, M.; Wang, J.; Lee, W.S.; Williams, W.D.; Kim, S.M.; Stach, E.A.; Miller, J.T.; Delgass, W.N.; Ribeiro, F.H. Size and support effects for the water-gas shift catalysis over gold nanoparticles supported on model Al2O3 and TiO2. J. Am. Chem. Soc. 2012, 134, 4700–4708. [Google Scholar] [CrossRef] [PubMed]
- Panagiotopoulou, P.; Kondarides, D.I. Effects of alkali promotion of TiO2 on the chemisorptive properties and water-gas shift activity of supported noble metal catalysts. J. Catal. 2009, 267, 57–66. [Google Scholar] [CrossRef]
- Pérez, P.; Soria, M.A.; Carabineiro, S.A.C.; Maldonado-Hódar, F.J.; Mendes, A.; Madeira, L.M. Application of Au/TiO2 catalysts in the low-temperature water–gas shift reaction. Int. J. Hydrogen Energy 2016, 41, 4670–4681. [Google Scholar] [CrossRef]
- Hinojosa-Reyes, M.; Rodríguez-González, V.; Zanella, R. Gold nanoparticles supported on TiO2–Ni as catalysts for hydrogen purification via water–gas shift reaction. RSC Adv. 2014, 4, 4308–4316. [Google Scholar] [CrossRef]
- Zhu, X.; Shen, M.; Lobban, L.L.; Mallinson, R.G. Structural effects of Na promotion for high water gas shift activity on Pt-Na/TiO2. J. Catal. 2011, 278, 123–132. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Ramírez, P.J.; Asara, G.G.; Viñes, F.; Evans, J.; Liu, P.; Ricart, J.M.; Illas, F. Charge Polarization at a Au–TiC Interface and the Generation of Highly Active and Selective Catalysts for the Low-Temperature Water-Gas Shift Reaction. Angew. Chem. Int. Ed. 2014, 53, 11270–11274. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhan, Y.; Zheng, Q.; Zheng, Y.; Chen, C.; She, Y.; Lin, X.; Wei, K. Water-Gas Shift Reaction over CuO/CeO2 Catalysts: Effect of the Thermal Stability and Oxygen Vacancies of CeO2 Supports previously prepared by different methods. Catal. Lett. 2009, 130, 532–540. [Google Scholar] [CrossRef]
- Duarte de Farias, A.M.; Nguyen-Thanh, D.; Fraga, M.A. Discussing the use of modified ceria as support for Pt catalysts on water–gas shift reaction. Appl. Catal. B Environ. 2010, 93, 250–258. [Google Scholar] [CrossRef]
- Hwang, K.R.; Park, J.S.; Ihm, S.K. Si-modified Pt/CeO2 catalyst for a single-stage water-gas shift reaction. Int. J. Hydrogen Energy 2011, 36, 9685–9693. [Google Scholar] [CrossRef]
- Montini, T.; Melchionna, M.; Monai, M.; Fornasiero, P. Fundamentals and Catalytic Applications of CeO2-Based Materials. Chem. Rev. 2016, 116, 5987–6041. [Google Scholar] [CrossRef] [PubMed]
- González-Castaño, M.; Ivanova, S.; Ioannides, T.; Centeno, M.A.; Odriozola, J.A. Deep insight into Zr/Fe combination for successful Pt/CeO2/Al2O3 WGS catalyst doping. Catal. Sci. Technol. 2017, 7, 1556–1564. [Google Scholar] [CrossRef]
- Bobrova, L.; Andreev, D.; Ivanov, E.; Mezentseva, N.; Simonov, M.; Makarshin, L.; Cribovskii, A.; Sadykov, V. Water-gas shift reaction over Ni/CeO2 catalysts. Catalysts 2017, 7, 310. [Google Scholar] [CrossRef]
- Wyvratt, B.M.; Gaudet, J.R.; Thompson, L.T. Effects of passivation on synthesis, structure and composition of molybdenum carbide supported platinum water–gas shift catalysts. J. Catal. 2015, 330, 280–287. [Google Scholar] [CrossRef]
- Posada-Pérez, S.; Gutiérez, R.A.; Zuo, Z.; Ramírez, P.J.; Viñes, F.; Liu, P.; Illas, F.; Rodriguez, J.A. Highly active Au/δ-MoC and Au/β-Mo2C catalysts for the low-temperature water gas shift reaction: Effects of the carbide metal/carbon ratio on the catalyst performance. Catal. Sci. Technol. 2017, 7, 5332–5342. [Google Scholar] [CrossRef]
- Schweitzer, N.M.; Schaidle, J.A.; Ezekoye, O.K.; Pan, X.; Linic, S.; Thompson, L.T. High Activity Carbide Supported Catalysts for Water Gas Shift. J. Am. Chem. Soc. 2011, 133, 2378–2381. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Wang, A.; Qiao, B.; Liu, X.; Yang, X.; Wang, X.; Liang, J.; Li, J.; Liu, J.; Zhang, T. Remarkable performance of Ir1/FeOx single-atom catalyst in water-gas shift reaction. J. Am. Chem. Soc. 2013, 135, 15314–15317. [Google Scholar] [CrossRef] [PubMed]
- Dufour, J.; Martos, C.; Ruiz, A.; Ayuela, F.J. Effect of the precursor on the activity of high temperature water gas shift catalysts. Int. J. Hydrogen Energy 2013, 38, 7647–7653. [Google Scholar] [CrossRef]
- Soria, M.A.; Pérez, P.; Carabineiro, S.A.C.; Maldonado-Hódar, F.J.; Mendes, A.; Madeira, L.M. Effect of the preparation method on the catalytic activity and stability of Au/Fe2O3 catalysts in the low-temperature water–gas shift reaction. Appl. Catal. A Gen. 2014, 470, 45–55. [Google Scholar] [CrossRef]
- Kono, E.; Tamura, S.; Yamamuro, K.; Ogo, S.; Sekine, Y. Pd/K/Co-oxide catalyst for water gas shift. Appl. Catal. A Gen. 2015, 489, 247–254. [Google Scholar] [CrossRef]
- Cybulskis, V.J.; Wang, J.; Pazmiño, J.H.; Ribeiro, F.H.; Delgass, W.N. Isotopic transient studies of sodium promotion of Pt/Al2O3 for the water–gas shift reaction. J. Catal. 2016, 339, 163–172. [Google Scholar] [CrossRef]
- Lenite, B.A.; Galletti, C.; Specchia, S. Studies on Au catalysts for water gas shift reaction. Int. J. Hydrogen Energy 2011, 36, 7750–7758. [Google Scholar] [CrossRef]
- Tibiletti, D.; Meunier, F.C.; Goguet, A.; Reid, D.; Burch, R.; Boaro, M.; Vicario, M.; Trovarelli, A. An investigation of possible mechanisms for the water–gas shift reaction over a ZrO2-supported Pt catalyst. J. Catal. 2006, 244, 183–191. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhan, Y.; Chen, C.; Cao, Y.; Lin, X.; Zheng, Q. Highly efficient Au/ZrO2 catalysts for low-temperature water-gas shift reaction: Effect of pre-calcination temperature of ZrO2. Int. J. Hydrogen Energ. 2012, 37, 12292–12300. [Google Scholar] [CrossRef]
- Luo, S.; Barrio, L.; Nguyen-Phan, T.D.; Vovchok, D.; Johnston-Peck, A.C.; Xu, W.; Stach, E.A.; Rodriguez, J.A.; Senanayake, S.D. Importance of Low Dimensional CeOx Nanostructures in Pt/CeOx-TiO2 Catalysts for the Water-Gas Shift Reaction. J. Phys. Chem. C 2017, 121, 6635–6642. [Google Scholar] [CrossRef]
- Silva, L.P.C.; Terra, L.E.; Coutinho, A.C.; Passos, F.B. Sour water-gas shift reaction over Pt/CeZrO2 catalysts. J. Catal. 2016, 341, 1–12. [Google Scholar] [CrossRef]
- Daly, H.; Goguet, A.; Hardacre, C.; Meunier, F.C.; Pilasombat, R.; Thompsett, D. The effect of reaction conditions on the stability of Au/CeZrO4 catalysts in the low-temperature water–gas shift reaction. J. Catal. 2010, 273, 257–265. [Google Scholar] [CrossRef]
- Sun, Y.; Hla, S.S.; Duffy, G.J.; Cousins, A.J.; French, D.; Morpeth, L.D.; Edwards, J.H.; Roberts, D.G. A comparative study of CeO2-La2O3-based Cu catalysts for the production of hydrogen from simulated coal-derived syngas. Appl. Catal. A Gen. 2010, 390, 201–209. [Google Scholar] [CrossRef]
- Rodriguez, J.A. Gold-based catalysts for the water–gas shift reaction: Active sites and reaction mechanism. Catal. Today 2011, 160, 3–10. [Google Scholar] [CrossRef]
- Williams, W.D.; Greeley, J.P.; Delgass, W.N.; Ribeiro, F.H. Water activation and carbon monoxide coverage effects on maximum rates for low temperature water-gas shift catalysis. J. Catal. 2017, 347, 197–204. [Google Scholar] [CrossRef]
- Ammal, S.C.; Heyden, A. Water-Gas Shift Catalysis at Corner Atoms of Pt Clusters in Contact with a TiO2 (110) Support Surface. ACS Catal. 2014, 4, 3654–3662. [Google Scholar] [CrossRef]
- Lin, Y.; Wu, Z.; Wen, J.; Ding, K.; Yang, X.; Poeppelmeier, K.R.; Marks, L.D. Adhesion and Atomic Structures of Gold on Ceria Nanostructures: The Role of Surface Structure and Oxidation State of Ceria Supports. Nano Lett. 2015, 15, 5375–5381. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.A.; Graciani, J.; Evans, J.; Park, J.B.; Yang, F.; Stacchiola, D.; Senanayake, S.D.; Ma, S.; Pérez, M.; Liu, P.; et al. Water-Gas Shift Reaction on a Highly Active Inverse CeOx/Cu(111) Catalyst: Unique Role of Ceria Nanoparticles. Angew. Chem. 2009, 121, 8191–8194. [Google Scholar] [CrossRef]
- Graciani, J.; Sanz, J.F. Designing a new generation of catalysts: Water gas shift reaction example. Catal. Today 2015, 240, 214–219. [Google Scholar] [CrossRef]
- Rodríguez, J.A.; Evans, J.; Graciani, J.; Park, J.B.; Liu, P.; Hrbek, J.; Sanz, J.F. High Water−Gas Shift Activity in TiO2(110) Supported Cu and Au Nanoparticles: Role of the Oxide and Metal Particle Size. J. Phys. Chem. C 2009, 113, 7364–7370. [Google Scholar] [CrossRef]
- Goddeti, K.C.; Kim, S.M.; Lee, Y.K.; Kim, S.H.; Park, J.Y. Chemical Doping of TiO2 with Nitrogen and Fluorine and Its Support Effect on Catalytic Activity of CO Oxidation. Catal. Lett. 2014, 144, 1411–1417. [Google Scholar] [CrossRef]
- Chen, X.; Liu, L.; Peter, Y.Y.; Mao, S.S. Increasing Solar Absorption for Photocatalysis with Black Hydrogenated Titanium Dioxide Nanocrystals. Science 2011, 331, 746–750. [Google Scholar] [CrossRef] [PubMed]
- Panagiotopoulou, P.; Christodoulakis, A.; Kondarides, D.I.; Boghosian, S. Particle size effects on the reducibility of titanium dioxide and its relation to the water–gas shift activity of Pt/TiO2 catalysts. J. Catal. 2006, 240, 114–125. [Google Scholar] [CrossRef]
- Yang, C.; Wang, Z.; Lin, T.; Yin, H.; Lü, X.; Wan, D.; Xu, T.; Zheng, C.; Lin, J.; Huang, F.; et al. Core-Shell Nanostructured “Black” Rutile Titania as Excellent Catalyst for Hydrogen Production Enhanced by Sulfur Doping. J. Am. Chem. Soc. 2013, 135, 17831–17838. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Li, W.; Wang, J.; Qu, Y.; Yang, Y.; Xie, Y.; Zhang, K.; Wang, L.; Fu, H.; Zhao, D. Ordered Mesoporous Black TiO2 as Highly Efficient Hydrogen Evolution Photocatalyst. J. Am. Chem. Soc. 2014, 136, 9280–9283. [Google Scholar] [CrossRef] [PubMed]
- Naldoni, A.; Allieta, M.; Santangelo, S.; Marelli, M.; Fabbri, F.; Cappelli, S.; Bianchi, C.L.; Psaro, R.; Santo, V.D. Effect of Nature and Location of Defects on Bandgap Narrowing in Black TiO2 Nanoparticles. J. Am. Chem. Soc. 2012, 134, 7600–7603. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Huang, B.; Lu, J.; Wang, Z.; Qin, X.; Zhang, X.; Dai, Y.; Whangbo, M.H. Hydrogenated titania: Synergy of surface modification and morphology improvement for enhanced photocatalytic activity. Chem. Commun. 2012, 48, 5733–5735. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yang, C.; Lin, T.; Yin, H.; Chen, P.; Wan, D.; Xu, F.; Huang, F.; Lin, J.; Xie, X.; et al. Visible-light Photocatalytic, Solar Thermal and Photoelectrochemical Properties of Aluminium-reduced Black Titania. Energy Environ. Sci. 2013, 6, 3007–3014. [Google Scholar] [CrossRef]
- Lu, X.; Wang, G.; Zhai, T.; Yu, M.; Gan, J.; Tong, Y.; Li, Y. Hydrogenated TiO2 Nanotube Arrays for Supercapacitors. Nano Lett. 2012, 12, 1690–1696. [Google Scholar] [CrossRef] [PubMed]
- He, Y.J.; Peng, J.F.; Chu, W.; Li, Y.Z.; Tong, D.G. Black mesoporous anatase TiO2 nanoleaves: A high capacity and high rate anode for aqueous Al-ion batteries. J. Mater. Chem. A 2014, 2, 1721–1731. [Google Scholar] [CrossRef]
- Kim, S.M.; Lee, H.; Park, J.Y. Charge Transport in Metal–Oxide Interfaces: Genesis and Detection of Hot Electron Flow and Its Role in Heterogeneous Catalysis. Catal. Lett. 2015, 145, 299–308. [Google Scholar] [CrossRef]
- Park, J.Y.; Lee, H.; Renzas, J.R.; Zhang, Y.; Somorjai, G.A. Probing Hot Electron Flow Generated on Pt Nanoparticles with Au/TiO2 Schottky Diodes during Catalytic CO Oxidation. Nano Lett. 2008, 8, 2388–2392. [Google Scholar] [CrossRef] [PubMed]
- Furube, A.; Du, L.; Hara, K.; Katoh, R.; Tachiya, M. Ultrafast Plasmon-Induced Electron Transfer from Gold Nanodots into TiO2 Nanoparticles. J. Am. Chem. Soc. 2007, 129, 14852–14853. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Lu, G.; Zhou, R.; Mao, J.; Chen, Y.; Zheng, X. Studies of pore structure, temperature-programmed reduction performance, and micro-structure of CuO/CeO2 catalysts. Appl. Surf. Sci. 2001, 173, 208–220. [Google Scholar] [CrossRef]
- Li, L.; Song, L.; Chen, C.; Zhang, Y.; Zhan, Y.; Lin, X.; Zheng, Q.; Wang, H.; Ma, H.; Ding, L.; et al. Modified precipitation processes and optimized copper content of CuO-CeO2 catalysts for water-gas shift reaction. Int. J. Hydrogen Energy 2014, 39, 19570–19582. [Google Scholar] [CrossRef]
- Li, L.; Song, L.; Wang, H.; Chen, C.; She, Y.; Zhan, Y.; Lin, X.; Zheng, Q. Water-Gas Shift Reaction over CuO/CeO2 Catalysts: Effect of CeO2 Supports previously prepared by precipitation with different precipitants. Int. J. Hydrogen Energy 2011, 36, 8839–8849. [Google Scholar] [CrossRef]
- Beuvier, T.; Richard-Plouet, M.; Brohan, L. Accurate Methods for Quantifying the Relative Ratio of Anatase and TiO2(B) Nanoparticles. J. Phys. Chem. C 2009, 113, 13703–13706. [Google Scholar] [CrossRef]
- Hemmingson, S.L.; James, T.E.; Feeley, G.M.; Tilson, A.M.; Campbell, C.T. Adsorption and Adhesion of Au on Reduced CeO2(111) Surfaces at 300 and 100 K. J. Phys. Chem. C 2016, 120, 12113–12124. [Google Scholar] [CrossRef]
- Li, B.; Zhao, Z.; Zhou, Q.; Meng, B.; Meng, X.; Qiu, J. Highly Efficient Low-Temperature Plasma-Assisted Modification of TiO2 Nanosheets with Exposed {001} Facets for Enhanced Visible-Light Photocatalytic Activity. Chem. Eur. J. 2014, 20, 14763–14770. [Google Scholar] [CrossRef] [PubMed]
- Kruse, N.; Chenakin, S. XPS characterization of Au/TiO2 catalysts: Binding energy assessment and irradiation effects. Appl. Catal. A Gen. 2011, 391, 367–376. [Google Scholar] [CrossRef]
- Kast, P.; Kučerová, G.; Behm, R.J. On the nature of the active Au species: CO oxidation on cyanide leached Au/TiO2 catalysts. Catal. Today 2015, 244, 146–160. [Google Scholar] [CrossRef]
- Magadzu, T.; Yang, J.H.; Henao, J.D.; Kung, M.C.; Kung, H.H.; Scurrell, M.S. Low-Temperature Water-Gas Shift Reaction over Au Supported on Anatase in the Presence of Copper: EXAFS/XANES Analysis of Gold-Copper Ion Mixtures on TiO2. J. Phys. Chem. C 2017, 121, 8812–8823. [Google Scholar] [CrossRef]
- Wang, G.; Wang, H.; Ling, Y.; Tang, Y.; Yang, X.; Fitzmorris, R.C.; Wang, C.; Zhang, J.Z.; Li, Y. Hydrogen-Treated TiO2 Nanowire Arrays for Photoelectrochemical Water Splitting. Nano Lett. 2011, 11, 3026–3033. [Google Scholar] [CrossRef] [PubMed]
- Sastre, F.; Oteri, M.; Corma, A.; García, H. Photocatalytic water gas shift using visible or simulated solar light for the efficient, room-temperature hydrogen generation. Energy Environ. Sci. 2013, 6, 2211–2215. [Google Scholar] [CrossRef]
- Deáka, P.; Kullgren, J.; Aradi, B.; Frauenheim, T.; Kavan, L. Water splitting and the band edge positions of TiO2. Electrochim. Acta 2016, 199, 27–34. [Google Scholar] [CrossRef]
- Zheng, Y.; Zheng, L.; Zhan, Y.; Lin, X.; Zheng, Q.; Wei, K. Ag/ZnO Heterostructure Nanocrystals: Synthesis, Characterization, and Photocatalysis. Inorg. Chem. 2007, 46, 6980–6986. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Kong, X.; Yu, Y.; Zhang, H. Synthesis and Characterization of Water-Soluble and Bifunctional ZnO−Au Nanocomposites. J. Phys. Chem. C 2007, 111, 3836–3841. [Google Scholar] [CrossRef]
- Park, J.Y.; Baker, L.R.; Somorjai, G.A. Role of Hot Electrons and Metal–Oxide Interfaces in Surface Chemistry and Catalytic Reactions. Chem. Rev. 2015, 115, 2781–2817. [Google Scholar] [CrossRef] [PubMed]
- Nedrygailov, I.I.; Lee, C.; Moon, S.Y.; Lee, H.; Park, J.Y. Hot Electrons at Solid-Liquid Interfaces: A Large Chemoelectric Effect during the Catalytic Decomposition of Hydrogen Peroxide. Angew. Chem. Int. Ed. 2016, 55, 10859–10862. [Google Scholar] [CrossRef] [PubMed]
- Renzas, J.R.; Somorjai, G.A. Rh Thin-Film Nanocatalysts as Chemical Sensors—The Hot Electron Effect. J. Phys. Chem. C 2010, 114, 17660–17664. [Google Scholar] [CrossRef]
- Maximoff, S.N.; Head-Gordon, M.P. Chemistry of fast electrons. Proc. Natl. Acad. Sci. USA 2009, 106, 11460–11465. [Google Scholar] [CrossRef] [PubMed]
- Gergen, B.; Nienhaus, H.; Weinberg, W.H.; McFarland, E.W. Chemically Induced Electronic Excitations at Metal Surfaces. Science 2001, 294, 2521–2523. [Google Scholar] [CrossRef] [PubMed]
- Nienhaus, H.; Bergh, H.S.; Gergen, B.; Majumdar, A.; Weinberg, W.H.; McFarland, E.W. Electron-Hole Pair Creation at Ag and Cu Surfaces by Adsorption of Atomic Hydrogen and Deuterium. Phys. Rev. Lett. 1999, 82, 446–449. [Google Scholar] [CrossRef] [Green Version]
- Meier, D.C.; Goodman, D.W. The Influence of Metal Cluster Size on Adsorption Energies: CO Adsorbed on Au Clusters Supported on TiO2. J. Am. Chem. Soc. 2004, 126, 1892–1899. [Google Scholar] [CrossRef] [PubMed]
- Green, I.X.; Tang, W.; Neurock, M.; Yates, J.T., Jr. Spectroscopic Observation of Dual Catalytic Sites during Oxidation of CO on a Au/TiO2 Catalyst. Science 2011, 333, 736–739. [Google Scholar] [CrossRef] [PubMed]
- Hammer, N.; Kvande, I.; Van Beek, W.; Chen, D.; Rønning, M. Identification of valence shifts in Au during the water-gas shift reaction. Top. Catal. 2007, 45, 25–29. [Google Scholar] [CrossRef]
- Liu, Z.; Jenkins, S.J.; King, D.A. Origin and Activity of Oxidized Gold in Water-Gas-Shift Catalysis. Phys. Rev. Lett. 2005, 94, 196102–196104. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Zhou, J.; Wu, Y.; Tang, L.; Zhu, L.; Gu, L. Self-assembled, robust titanate nanoribbon membranes for highly efficient nanosolid capture and molecule discrimination. Nanoscale 2013, 5, 3486–3495. [Google Scholar] [CrossRef] [PubMed]
- Otto, T.; Zones, S.I.; Iglesia, E. Challenges and strategies in the encapsulation and stabilization of monodisperse Au clusters within zeolites. J. Catal. 2016, 339, 195–208. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Anatase | |||||||
|---|---|---|---|---|---|---|---|---|
| 2theta (°) | d-Spacing (Å) | FWHM (°) | Crystal Size (nm) | Microstrain (Δd/d) (%) | a (Å) | C (Å) | V (Å3) | |
| TiO2-N | 25.287 | 3.519 | 0.564 | 14.52 | 0.600 | 3.7847 | 9.5181 | 136.33 |
| TiO2−x-N | 25.365 | 3.508 | 0.597 | 14.63 | 0.617 | 3.7845 | 9.5146 | 136.27 |
| Au-TiO2-N | 25.244 | 3.525 | 0.602 | 14.85 | 0.615 | 3.7838 | 9.5058 | 136.09 |
| Au-TiO2−x-N | 25.274 | 3.521 | 0.609 | 14.89 | 0.625 | 3.7818 | 9.5087 | 135.99 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, L.; Lu, Z.; Zhang, Y.; Su, Q.; Li, L. Hydrogen-Etched TiO2−x as Efficient Support of Gold Catalysts for Water–Gas Shift Reaction. Catalysts 2018, 8, 26. https://doi.org/10.3390/catal8010026
Song L, Lu Z, Zhang Y, Su Q, Li L. Hydrogen-Etched TiO2−x as Efficient Support of Gold Catalysts for Water–Gas Shift Reaction. Catalysts. 2018; 8(1):26. https://doi.org/10.3390/catal8010026
Chicago/Turabian StyleSong, Li, Zhufeng Lu, Yuting Zhang, Qi Su, and Lei Li. 2018. "Hydrogen-Etched TiO2−x as Efficient Support of Gold Catalysts for Water–Gas Shift Reaction" Catalysts 8, no. 1: 26. https://doi.org/10.3390/catal8010026
APA StyleSong, L., Lu, Z., Zhang, Y., Su, Q., & Li, L. (2018). Hydrogen-Etched TiO2−x as Efficient Support of Gold Catalysts for Water–Gas Shift Reaction. Catalysts, 8(1), 26. https://doi.org/10.3390/catal8010026