3.1. Catalyst Characterization
Each catalyst is composed of three distinctively different phases of magnetic nanoparticles (first phase) incorporated within the carbonaceous matrix (second phase) representing together a magnetically separable support for Ru nanoparticles (third phase). X-Ray Diffraction (XRD) patterns (
Figure 1) of Ru/C-Fe
2O
3, Ru/C-Fe
2O
3-300, and Ru/C-Fe
2O
3-500 are composed only of reflections characteristic of magnetic iron oxide and a broad hump at low angles characteristic of amorphous material. Average crystallite size of the iron oxide nanoparticles was found to be approx. 14 nm. XRD pattern of the Ru/C-Fe
2O
3-600 shows an additional weak reflection, a characteristic of nonmagnetic iron oxide FeO and BCC Fe indicating partial reduction of iron oxide. The composition of the Ru/C-Fe
2O
3-750 is significantly different. The catalyst is composed of cohenite Fe
3C and nanocrystalline graphite as main phases and smaller amounts of BCC Fe and magnetic iron oxide. Reflections characteristic of Ru were not observed in any of the XRD patterns suggesting its amorphous state. We can conclude that until 500 °C was reached the iron oxide nanoparticles remain practically intact. At 600 °C, a slow reduction occurs, and at 750 °C, most of the iron oxide reduces to Fe which substantially transforms to Fe
3C. Reduction is most likely due to CO that forms during decomposition of carbonaceous matter. More detailed characterisation results for the Ru/C-Fe
2O
3-750 are available in our previous work [
17].
Transmission Electron Microscopy (TEM) imaging of the catalyst particles revealed large differences between them in many aspects, such as distribution of magnetic phases within the carbonaceous matrix, size, and dispersion of the Ru nanoparticles for example (
Figure 2). Iron-oxide nanoparticles are visible as dark approximately spherical particles incorporated within the grey matrix of uniform and low contrast (
Figure 2a,b). Ru nanoparticles are visible as smaller dark particles seen mostly at higher magnification (
Figure 2d,f,h,j). Particles of the catalyst Ru/C-Fe
2O
3 appear as branched and rounded. Ru nanoparticles are barely visible and their number is relatively low in agreement with observed incomplete reduction of Ru
3+. Particles of catalyst Ru/C-Fe
2O
3-300 appear more compact and covered with Ru nanoparticles that are predominantly clustered and of relatively broad size distribution (
Figure 2c,d and
Figure 3,
Table 1). It should be mentioned that, also, in this case, the reduction of Ru
3+ was incomplete; however, it proceeded much further than in the case of Ru/C-Fe
2O
3. Particles of Ru/C-Fe
2O
3-500 are even more compact and completely covered with Ru nanoparticles of largest average size (
Figure 2e,f and
Figure 3,
Table 1). Particles of catalyst Ru/C-Fe
2O
3-600 appear similar but less densely covered with Ru nanoparticles (
Figure 2g,h). Ru nanoparticles are of two different sizes; smaller spherical and larger plate-like (
Figure 2h). In
Table 1 and
Figure 3 only the size distribution of smaller spherical ones is presented. Larger plate-like nanoparticles are predominantly oriented with the basal plane parallel to the electron beam making estimation of their size unreliable. However, a rough estimation of their thickness is 3 nm and the diameter of basal plane is 15 nm. TEM analysis of the catalyst Ru/C-Fe
2O
3-750 reveals rather irregular shape of particles (
Figure 2i). Larger Fe and Fe
3C nanoparticles of dark contrast are clearly visible within the matrix. Observation at higher magnification revealed that nanoparticles are enclosed with graphitic layer. Ru nanoparticles are homogeneously distributed over the support (
Figure 2j).
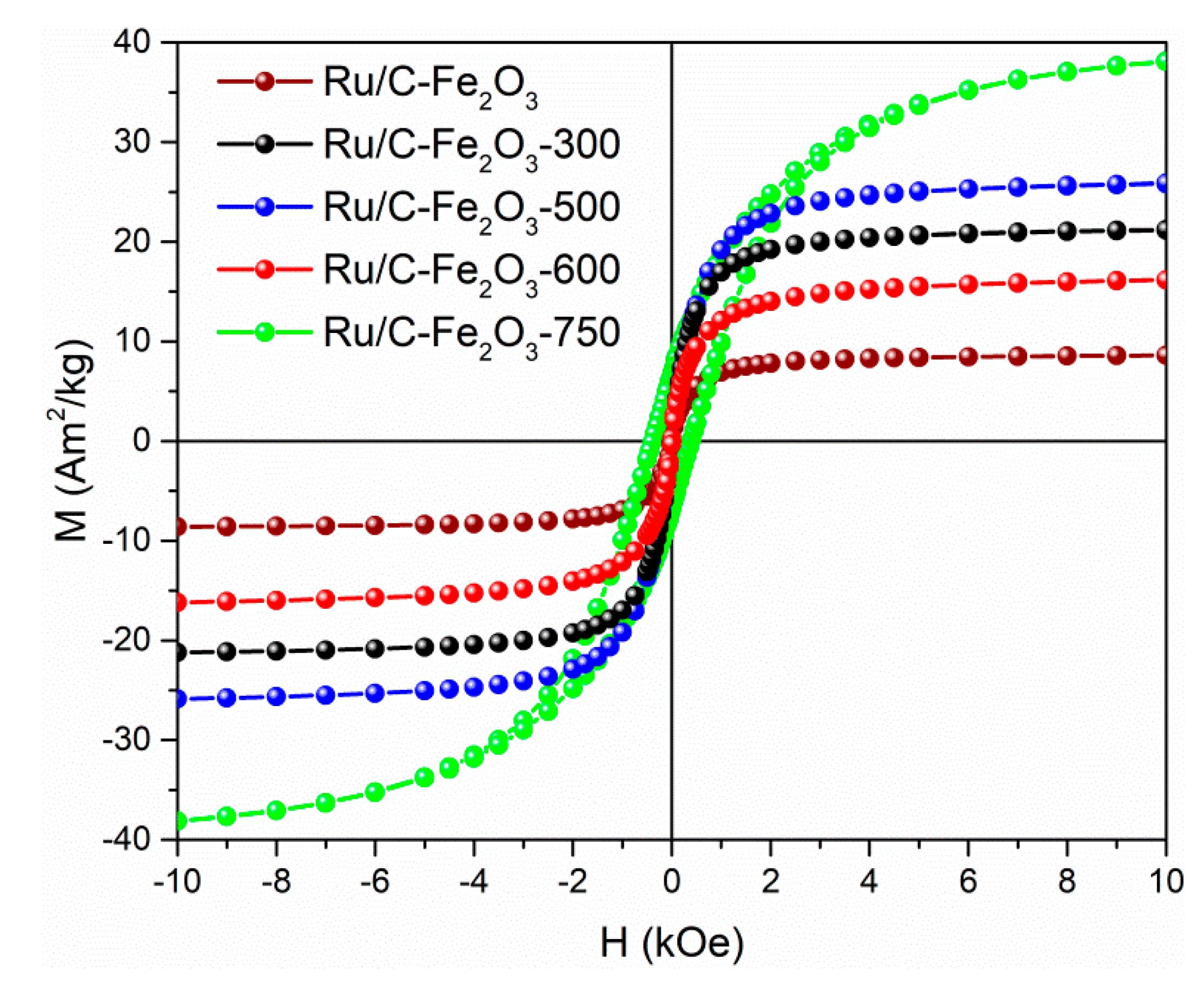
Room-temperature magnetisation curves of the catalysts, except Ru/C-Fe
2O
3-750, exhibited behaviour characteristic of the superparamagnetic state, namely showing zero coercivity and remanence (
Figure 4). Catalyst Ru/C-Fe
2O
3-750 exhibited ferromagnetic behaviour characteristic of Fe
3C [
18,
19]. Saturation magnetisation of the catalyst is proportional to the amount of incorporated magnetic nanoparticles. With increased temperature of annealing of the support the saturation magnetisation increased until 500 °C indicating partial loss of nonmagnetic carbonaceous matter (
Figure 4 and
Table 1). The drop in saturation magnetisation for the catalyst Ru/C-Fe
2O
3-600 is related to the formation of nonmagnetic FeO during annealing of the support at 600 °C (
Figure 1 and
Table 1). Substantial increase of saturation magnetisation of the catalyst Ru/C-Fe
2O
3-750 is related to formation of Fe and Fe
3C which both display much higher saturation magnetisations than magnetic iron oxide (
Figure 1 and
Table 1) [
18,
19]. Magnetic properties of the catalyst assured their rapid separation from reaction mixture using simple permanent magnet (
Figure S1).
The typical nitrogen adsorption–desorption isotherms of all the catalysts employed in the present study are presented in
Figure 5. Catalysts Ru/C-Fe
2O
3, Ru/C-Fe
2O
3-300, Ru/C-Fe
2O
3-500, and Ru/C-Fe
2O
3-600 all exhibit Type I isotherms represented by microporous solids, since they are composed of carbonaceous matrix, and thus having a relatively small external surface. This is also typical for activated carbons and zeolites (where the limiting uptake is governed by the accessible micropore volume rather than internal surface area). The result also corroborates well with the TEM micrographs of catalysts (
Figure 2a–h), where there is absence of obvious/substantial meso-to-macro porosity in the catalysts. The Ru/C-Fe
2O
3 catalyst exhibit the shallowest isotherm with the lowest nitrogen uptake, which is directly reflected in the lowest Brunauer-Emmett-Teller (BET) surface area of 8.4 m
2 g
−1 and pore volume of 0.089 cm
3 g
−1 (
Table 1), as a result of low number of barely visible Ru nanoparticles (
Figure 2a,b). The catalyst Ru/C-Fe
2O
3-300 exhibits an open-like, low pressure hysteresis, extending to the lowest attainable pressures (
Figure 5), which was unchanged even after prolonged equilibration time of the measurement (240 s instead of 60 s). The phenomenon can in principle be related to the swelling of a nonrigid porous structure, with the irreversible uptake of molecules in pores or an irreversible chemical interaction of the adsorbate with the adsorbent [
20]. However, considering the chemical properties of the catalyst Ru/Fe
2O
3-300, the first explanation seems most reasonable. The BET surface area and the pore volume of Ru/C-Fe
2O
3-300 in comparison to Ru/C-Fe
2O
3 catalysts were increased to 77 m
2 g
−1 and 0.176 cm
3 g
−1, respectively (
Table 1). The increase can be attributed to the formation of compact clusters of Ru nanoparticles of relatively broad size distribution. By further increase in the annealing temperature of the catalyst preparation to 500 and 600 °C, the microporous network channels were seemingly further evolved as evident from the surface properties (
Table 1). Namely, the BET surface area of Ru/C-Fe
2O
3-500 and Ru/C-Fe
2O
3-600 increased to 209 and 259 m
2 g
−1, while the pore volume increased to 0.165 to 0.398 cm
3 g
−1, respectively. The reason for such increase is twofold. Firstly, it can be associated with the compositional change of the catalysts, with the formation of partly reduced, nonmagnetic iron oxide FeO and BCC Fe phases (
Figure 1). Secondly, it can be also ascribed to the pronounced formation of Ru nanoparticles that are, in the case of Ru/C-Fe
2O
3-500, the largest in average size, more compact, and completely covering the catalyst (
Figure 2e,f and
Figure 3,
Table 1), while in the case of Ru/C-Fe
2O
3-600 (
Figure 2h), they are of two different sizes, i.e., smaller spherical and larger plate-like (
Figure 2h). The Type I isotherm of the catalyst Ru/C-Fe
2O
3-750 annealed at the highest temperatures was the only one to display a typical hysteresis loop at the highest p/p
0 pressures, which is typical for very narrow slit-like pores originating from the aggregates of loosely coherent particles. While on one hand, the formation of well-crystalline cohenite Fe
3C particles (and nanocrystalline graphite) (
Figure 1 and
Figure 2j) possibly attributed to the lowered BET surface area, i.e., 74 m
2 g
−1 (
Table 1), on the other hand, the decomposition of carbonaceous matter forming CO could well be responsible for “loosening” of the catalysts, providing an observed hysteresis loop (
Figure 5) and a relatively high pore volume of 0.236 cm
3 g
−1.
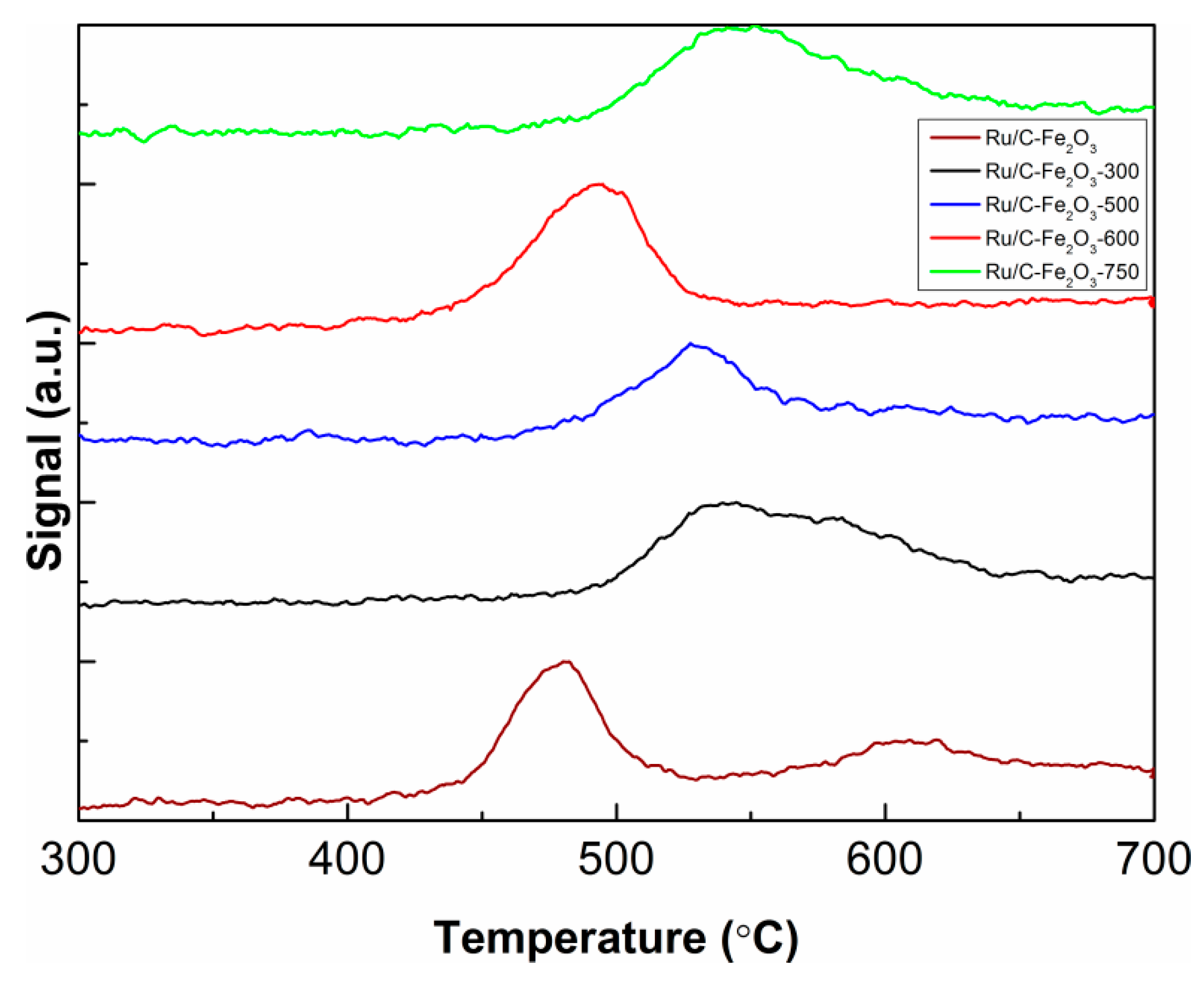
CO-TPD profiles are provided in
Figure 6, while the amount of metal active sites in
Table 1. Densities of active sites available for CO adsorption did not differ much among the catalysts; therefore, very significant differences in catalytic activities might be caused by having fewer active sites available for reactions. It might be worth mentioning that CO-TPD desorption profiles were differing in the number and the shape of peaks among the catalyst as
Figure 6 displays. Namely, CO desorption from nonannealed catalyst (wine line) resulted in two observable peaks, one strong at temperatures between 420 and 520 °C and other low and broad between temperatures 520 and 700 °C. This might indicate the presence of both weak and strong active sites, whereas weak are more dominant. Consequently the catalyst might catalyse various reactions with low activity. For the catalyst treated at 300 °C (black line), one broad peak, in the temperature range from 490 to 680 °C, was formed. According to the peak shape, one might say that the peak is composed of one sharp peak at 550 °C and a shoulder at 580 °C. Similar to the first, it could indicate the existence of moderately strong active sites and, therefore, moderate activity for both types of reaction (hydrogenation and deoxygenation). CO desorption from Ru/C-Fe
2O
3-500 (blue line) resulted in one strong and sharp peak in the range of 450 to 600 °C and one shoulder in between 600 and 700 °C. Such a CO-TPD profile could again indicate the presence of two types of active sites; moderately strong and dominant and another even stronger but present in small quantities. For the catalyst annealed at 600 °C (red line), one strong, sharp peak was observed ranging from 420 to 550 °C. Analogously to the previous cases, a single, intensive peak might point to the dominant presence of one type of moderate to strong active site, further indicating higher activity of the catalyst for one or more reactions. The Ru/C-Fe
2O
3-750 showed one broad, intensive peak ranging from 480 to 680 °C (green line). The amount of acid active sites, estimated by NH
3-TPD (
Figure S2), varied within experimental error making it difficult to draw a conclusive conclusion.
3.2. Hydrotreatment Results
The prepared catalysts differed in support annealing temperatures; specifically at 300, 500, 600, and 750 °C, thus catalysts were labelled as: Ru/C-Fe
2O
3-300, Ru/C-Fe
2O
3-500, Ru/C-Fe
2O
3-600, and Ru/C-Fe
2O
3-750. The Ru/C-Fe
2O
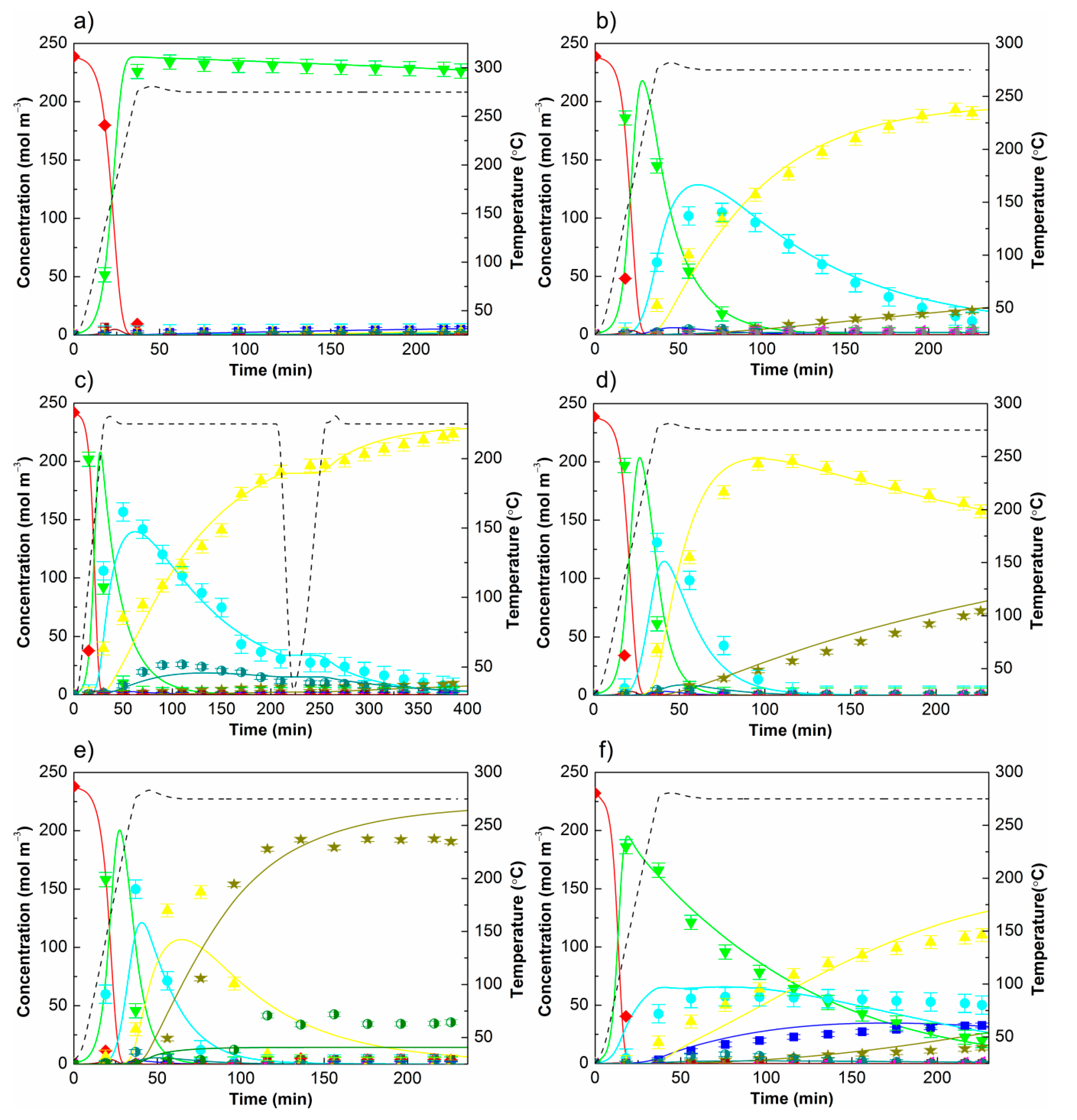
3 label refers to a nonannealed sample. Therefore hypothesis that annealing temperature influenced catalyst activity and selectivity for the hydrotreatment of eugenol was investigated. Detected products in the liquid phase and their abbreviations: eugenol (HMAB), 2-methoxy-4-propylphenol (HMPB), 2-methoxy-4-propycyclohexanol (HMPC), 4-propycyclohexane-1,2-diol (HHPC), 4-propyphenol (HPB), 4-propylcylohexanol (HPC), propylbenzene (PB), propylcyclohexane (PC), propylcyclopentane (PCP), 4-propycyclohexanone (KPC), and isoeugenol (IHMAB). Results are summarized in
Figure 7. Nonannealed catalyst showed a very low activity, as it can be seen in
Figure 7a. HMAB was fully, but mostly homogeneously (noncatalytically), converted to HMPB within the heating period of the experiment according to the previous results [
15] obtained without the catalyst. Further catalytic conversion of HMPB was negligible, since only approximately 2 mol% of HPB was observed in the final product, while the mole fraction of other components (HMPC, HHPC, KPC, HPC, and PC) was less than 1 mol% each. On the other hand, support annealed at the temperature of 300 °C significantly increased the final catalyst’s activity and selectivity (
Figure 7b). Hydrotreatment of HMAB over Ru/C-Fe
2O
3-300 after 3 h resulted in approximately 80 mol% of HPC, 9 mol% of PC, 5 mol% of HMPC, 2 mol% of PCP and KPC, and 1 mol% of PB or HPB. With further annealing temperature increase to 500 °C, catalytic activity was even higher, resulting in enhanced deoxygenation reactions and a higher yield of completely deoxygenated and hydrogenated product PC. 66 mol% of HPC, 30 mol% of PC, 2.5 mol% of PCP, and a remaining 1.5 mol% of others were detected in the final product at 275 °C (
Figure 7d). Higher Ru/C-Fe
2O
3-500 selectivity towards HPC was accomplished by lowering reaction temperature to 225 °C (
Figure 7c) as a result of the less promoted dehydroxylation reaction. The latter was also noticed based on a notable amount of HHPC in the system (approx. 10 mol%) during a wide reaction time interval. A fraction of this dihydroxyl intermediate was always lower than 4 mol% in all other runs. Deoxygenation has been reported as a high activation energy reaction; also confirmed in the present study (see
Section 4.3), thus being unfavourable at lower temperatures resulting in low conversion of HPC at 225 °C and therefore low high final yield. The reaction mixture was cooled after 216 min and again heated up to 225 °C, with the aim to test the ability of correct model response and test eventual catalyst deactivation during the cooling-down and reheating back to reaction temperature. The catalyst treated at 600 °C has exhibited a significantly enhanced degree of deoxygenation since 80 mol% of PC was detected in the final product, 15 mol% of PCP, and the remaining 5 mol% belonged to others (mostly HPC) (
Figure 7e). However, a notable amount of PCP takes side of disadvantage since ring contraction leads to unwanted carbon losses. When the catalyst’s support was treated at 750 °C (
Figure 7f), catalytic activity, as well as selectivity, was significantly reduced. Approximately 8.5 mol% of HMPB was still presented in the final product which was fully consumed within the first 100 min of the reaction by other prepared catalysts or Ru/C (5 wt% Ru, Sigma Aldrich, St. Louis, MO, USA, reference number 206180). Besides 8.5 mol% of HMPB, hydrotreatment of HMAB over Ru/C-Fe
2O
3-750 resulted in approximately 22 mol% of HMPC, 14 mol% of HPB, 47 mol% of HPC, 6 mol% of PC, and 2.5 mol% of others. A work by Whiffen and Smith also showed that the catalyst annealing temperature affected the product distribution [
21] of 4-methyphenol HDO. Namely, the Ni
2P catalyst annealed at 550 °C and displayed the lowest selectivity towards deoxygenated products, while when annealed at 700 °C displayed the highest. The same group, one year before, published a study again showing an influence of annealing temperature on the product distribution of 4-methylphenol HDO [
22] over the MoP-CA (CA-citric acid) catalyst. The MoP-CA has been annealed in a range of temperatures from 500 to 700 °C. The highest conversion was achieved on the catalyst treated at 550 °C (71%), obtaining the lowest yield of toluene. A slightly lower conversion of 4-methylphenol of 58%, but the highest selectivity towards toluene, was accomplished for the catalyst annealed at 500 °C.
The reaction network proposed in the previous work [
15] for HDO of HMAB over Ru/C has been shown to be valid for magnetic Ru catalysts according to the product evolution and distribution over the reaction time (
Figure 8). Several studies proposed reaction mechanisms of lignin model compounds (phenol, m-cresol) HDO over oxophilic metals (e.g., Fe-based catalysts) to describe the observed product distribution [
23,
24] ruling out direct Csp
2–O bond scission. Namely, it has been reported that (substituted) phenols might be in an equilibrium with an unstable ketone intermediate (3,5-cyclohexadienol) which can be hydrogenated over an oxophilic catalyst into unsaturated cycloalcohol (3,5-cyclohexadienol). The latter can be readily dehydrated (driven by aromatic stabilization) to (substituted) benzene. However, we have not observed a significant amount of deoxygenated aromatics in contrast to reports on Fe-based catalysts [
24,
25,
26,
27,
28]. Further comparing our results to those obtained over Ru/C (5 wt% Ru, Sigma Aldrich, St. Louis, MO, USA, reference number 206180) tested in the previous study (
Figure S3) [
15], one may say that our catalysts performed similar to Ru/C.
In general, an increase in annealing temperature up to 600 °C resulted in increasing HDO activity. Although further increase in the annealing temperature to 750 °C has not provided any improvement in the catalyst activity or selectivity, ring hydrogenation was of lower rate relative to other catalysts causing HPB detection of 14 mol% in the final product. Considering significant deviation from the results with Fe-based catalysts, one may say that Ru is a dominant active phase in our case where ring hydrogenation primarily took place as more favourable due to the planar adsorption of HMAB and HMPB on the Ru surface via ring [
29,
30,
31,
32]. Enhanced deoxygenation activity of catalysts annealed at higher temperature can be correlated to the dispersion and structure of Ru nanoparticles. Namely, an increase in the number of Ru nanoparticles (caused by greater reduction of Ru
3+) and their larger dispersity (according to the XRD and TEM results) is most likely responsible for the increased HDO activity of catalysts annealed at higher temperatures. Nonannealed catalyst possesses a small amount of Ru nanoparticles that are poorly dispersed on the support surface being almost inactive. The number of Ru nanoparticles increased when the catalyst was annealed at 300 °C showing significantly higher activity compared to nonannealed. Particles were mostly clustered due to the low surface area available for impregnation causing lower activity of this catalyst accompanied by their lower amount compared to those annealed at 500 and 600 °C. When the support was annealed at 500 °C, the number of Ru nanoparticles increased further, as well as the surface area and thus dispersity. Ru reduction proceeded even further when the support was annealed at 600 °C resulting in a larger number of Ru nanoparticles with even better dispersity on the support surface (due to even higher surface area), resulting in excellent HDO activity of this catalyst. Two types of particle shape were observed as possibly contributing to such catalytic behaviour, given that different plane might be exposed. A further increase of annealing temperature caused significant structural changes of Ru/C-Fe
2O
3-750 and, therefore, quite different activity. Considering CO TPD peak’s position, it seems that the strength of active sites is mostly moderate for all catalysts, indicating no clear connection between their strength and observed activity. However, the shape could be linked to activity since the most active catalysts (annealed at 500 and 600 °C) showed sharp, strong, and narrow peak indicating the dominant existence of one active site type, which might be appropriate for both hydrogenation and deoxygenation. Fe’s contribution to enhanced HDO activity is not expected or could be described as minor, as the Fe-phase in our catalysts represents a core coated by a carbon layer. Detailed TEM investigation of the support materials (prior deposition of Ru) showed that a small amount of Fe-containing nanoparticles might not be completely coated by a carbon layer when support was annealed at 600, and particularly, at 750 °C. Ru nanoparticles could be also deposited on these surfaces thus making intimate contact with Fe which can potentially cause a significant detection of HPB over Ru/C-Fe
2O
3-750. Yet this cannot be certainly said as a very thin carbon layer can be still present over the Fe core but not visible on TEM images.
Results over Ru/C are shown in
supplementary information as
Figure S3 (detailed results are available in the previous study) [
15]. In both cases, magnetic and commercial Ru/C, Ru phase is responsible (or at least mainly) for the observed catalytic activity and is most likely making the difference between them via, for example, the degree of dispersity or the type of Ru active sites exposed. Additionally, the potential impact of Fe might not be ruled out according to the TEM results and estimated acidity of the catalysts. Namely, somewhat higher acidity has been estimated for our catalysts relative to the Ru/C, which might be expected for Fe-containing catalyst being usually correlated to the grater HDO activity [
33,
34,
35]. Nevertheless, if there is a Fe contribution, it is probably rather low, as pointed out above.
3.3. Modelling Results
Adsorption–desorption equilibrium constants for hydrogen and organic components are estimated to be 3.17 × 10
−2 and 3.15 × 10
−3 m
3 mol
−1, respectively. Rate constants of observed reactions and activation energies accompanied are summarized in
Table 2 and
Table 3. It might be worth highlighting that estimated reaction rate constants are already normalized on the concentration of active sites obtained from CO-TPD results being thus a direct indication of catalyst activity. Considering the provided model results, it can be noted that the catalyst’s ability to hydrogenate a benzene ring increases with annealing temperature from 0 to 600 °C by two orders of magnitude between Ru/C-Fe
2O
3 and Ru/C-Fe
2O
3-300, 2.5-fold between being annealed at 300 and 500 °C, and 1.3-fold between being annealed at 500 and 600 °C, decreasing thereafter, as the catalyst annealed at 750 °C exhibited lower hydrogenation activity compared to other active catalysts, i.e. being between nonactive and annealed at 300 °C. Similarly, the increase of annealing temperature up to 600 °C facilitated deoxygenation of unsaturated and saturated intermediates (Ar–aromatics, Al–alkyl):
| Ar–OCH3: (Ru/C-Fe2O3) × 21 (Ru/C-Fe2O3-300) × 1.2 (Ru/C-Fe2O3-500) × 1.1 (Ru/C-Fe2O3-600); |
| Ar–OH: (Ru/C-Fe2O3) × 21 (Ru/C-Fe2O3-300) × 1.8 (Ru/C-Fe2O3-500) × 0.6 (Ru/C-Fe2O3-600); |
| Al–OCH3: (Ru/C-Fe2O3) × 19 (Ru/C-Fe2O3-300) × 3.5 (Ru/C-Fe2O3-500) × 1.1 (Ru/C-Fe2O3-600); |
| Al–OH: (Ru/C-Fe2O3) × 3.2 (Ru/C-Fe2O3-300) × 2.8 (Ru/C-Fe2O3-500) × 11 (Ru/C-Fe2O3-600). |
Generally, deoxygenation of saturated compounds was more favoured compared to the unsaturated shown also by Goncalves and coworkers [
36]. Further, hydrogenation is more favoured than deoxygenation over all tested catalysts being in an agreement with other kinetic studies [
37,
38]. Similar to the results of Massoth et al. [
38] and Shafaghat and coworkers [
39], we have also observed faster hydrogenation of less substituted benzene. In fact, Massoth et al. reported a hindered hydrogenation of methyl-substituted phenols if the number of methyl groups on the benzene ring was increasing. By investigating the hydrogenation of phenol, cresol, and guaiacol over Pd/C and zeolite solid acids, Shafaghat et al. observed a beneficial effect of methyl and methoxy groups in cresol and guaiacol on direct HDO mechanism at the expense of hydrogenation. Removal of oxygen-containing groups takes place to a larger extent via C–OCH
3 bond scission then by C–OH cleavage regardless of whether it appears on saturated or unsaturated components. Such behaviour has been also observed by performing DFT calculations of guaiacol HDO over a Ru (0001) plane [
30,
40].
HMAB transformation into HMPB is a fast reaction over all catalysts tested. The low hydrogenation constant of HMPB for a nonannealed catalyst clearly indicates the low activity of the catalyst. Almost negligible conversion of the first intermediate slows down or disables the downstream reactions. Other tested catalysts have shown good performance in catalysing the ring hydrogenation reaction. A significant amount of HPC in the system with Ru/C-Fe2O3-300 was a direct consequence of 15-fold intensive formation than disappearance. This ratio was even larger in the case of Ru/C-Fe2O3-500 (19-fold) resulting in a sharper increase of HPC concentration which started decreasing after 100 min due to no precursor being formed and a significantly high (higher than for Ru/C-Fe2O3-300) rate constant of the disappearance reaction producing a notable amount of oxygen-free component PC. A further increase in the catalyst annealing temperature up to 600 °C significantly improved catalyst activity in the dehydroxylation of saturated intermediates resulting in a quantitative production of PC already at 125 min of the reaction. Only deoxygenation of unsaturated HPB and formation of HHPC were of lower rate for Ru/C-Fe2O3-600 relative to the Ru/C-Fe2O3-500, all other reaction rates were the highest estimated in this group of catalysts. Catalyst annealed at 750 °C expressed the lowest hydrogenation activity among all other tested (active). Notable lower activity of Ru/C-Fe2O3-750 to hydrogenate HPB (over two orders of magnitude lower compared to other active catalysts) accompanied by moderate activity to remove the OCH3 group from HMPB which caused an appreciable detection of HPB in the system. The ratio between HMPB hydrogenation and demethoxylation clearly indicates the formation of PC via the hydrogenation route for all catalysts.
Hydrogenation of HMPB was a slightly more promoted reaction on Ru/C-Fe
2O
3-600, while demethoxylation of HMPC and dehydrohylation of HPC were 47 and 10 times faster reactions, respectively, compared to the Ru/C [
15]. Demethoxylation of HMPC was also more promoted over Ru/C-Fe
2O
3-750 in comparison to Ru/C (almost five times). Ru/C is, however, more active considering all other reactions and the other three tested catalysts in general.
Activation energies of hydrogenation reactions are significantly lower than for deoxygenation reactions (
Table 3). Besides, methoxy group removal is a less energy demanding reaction than the removal of hydroxyl groups, regardless of whether it occurs on a saturated or unsaturated species. Lu et al. [
40], in their DFT study, reported an approximately 11 kJ mol
−1 lower activation energy for guaiacol demethoxylation over the Ru catalyst, and 20 kJ mol
−1 higher for hydroxyl group removal than estimated in this work. Similarly, 30 kJ mol
−1 higher activation energy has been observed for m-cresol dehydroxylation (120 kJ mol
−1) by Tan et al. [
9] compared to our estimation for HPB, while it was significantly higher (90 kJ mol
−1) for its hydrogenation. Higher activation energy of benzene ring hydrogenation (79 kJ mol
−1) has been also proposed by He et al. but over the Pd/C catalyst [
41].
Product distribution has been affected by temperature as shown in
Figure 7c,d, refer to results obtained at 225 and 275 °C by the same order. Concentration profiles of formed components up to 50 min of the reaction (basically of hydrogenated products) are similar at these two temperatures, indicating lower activation energies of ring hydrogenation reactions (confirmed by the model results) and, thus, their lower sensitivity to temperature changes. More significant transformations of HMPC into HPC and further into PC at higher temperature, on the other hand, suggests higher activation energies of deoxygenation reactions also predicted by the model. The observation that deoxygenation reactions become more highly promoted at higher temperatures has been reported in numerous studies [
12,
28].
Catalyst coverage by each component has been estimated for the Ru/C-Fe
2O
3-300 catalyst and is shown here as
Figure 9 to illustrate the model abilities. Subplot a is related to the log scale of the time axis showing actual surface concentrations, while b represents the catalyst coverage by components at three reaction times. According to
Figure 9a, the dominant surface components, which take up to 10 min of the reaction, are HMAB and the solvent (HD). Ring hydrogenation mostly takes place within this period causing relatively constant and low hydrogen surface concentration, while it increases thereafter. Hydrogen concentration in the liquid phase was calculated according to the actual pressure in the reactor. HPC has become the dominant surface component after 100 min of the reaction.


 ,
, 









 HMAB,
HMAB,  HMPB,
HMPB,  HPB,
HPB,  HMPC,
HMPC,  PB,
PB,  HPC,
HPC,  PC,
PC,  IHMAB,
IHMAB,  PCP,
PCP,  HHPC,—temperature.
HHPC,—temperature.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}