Advances in Enantioselective C–H Activation/Mizoroki-Heck Reaction and Suzuki Reaction



Abstract
:1. Introduction
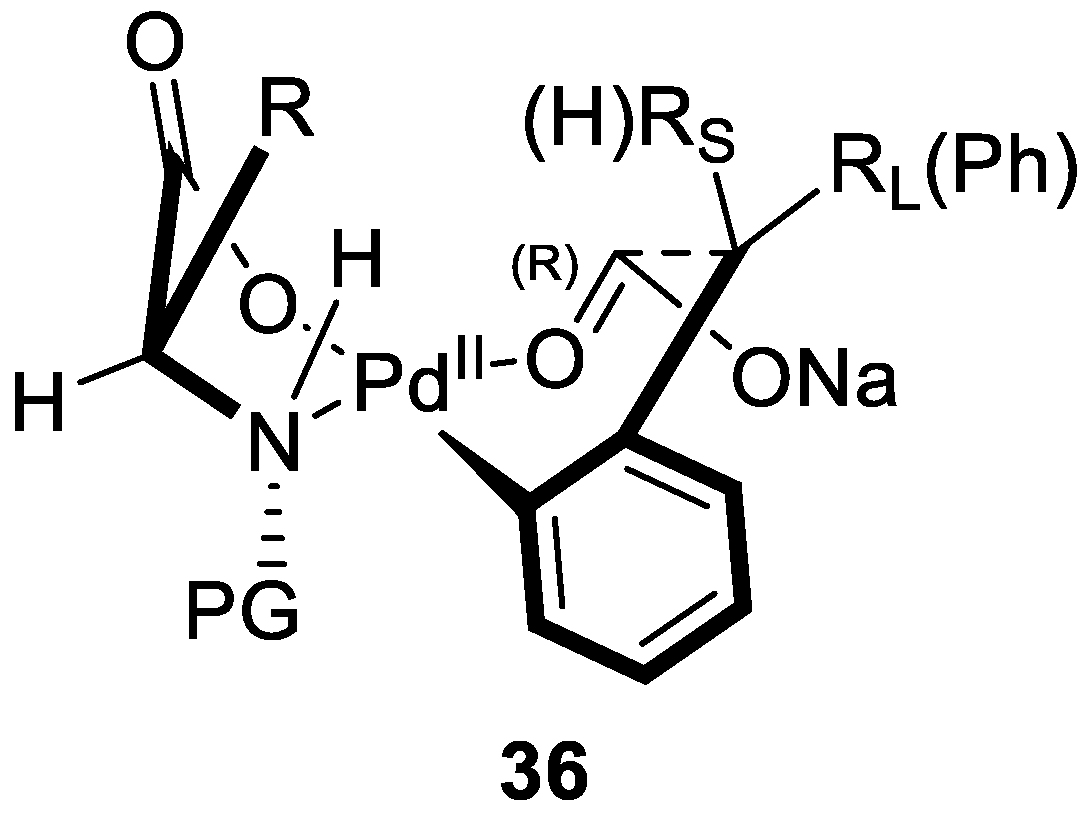
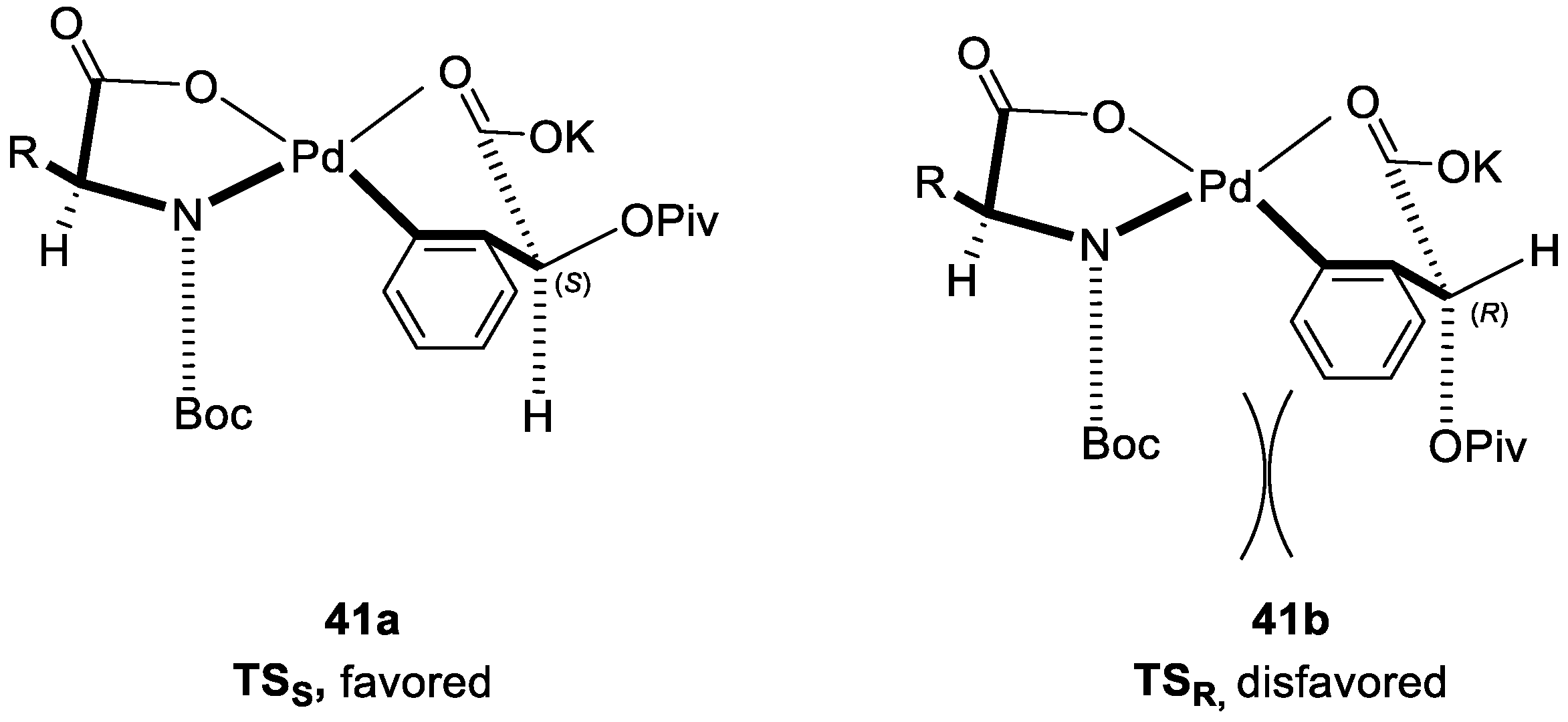
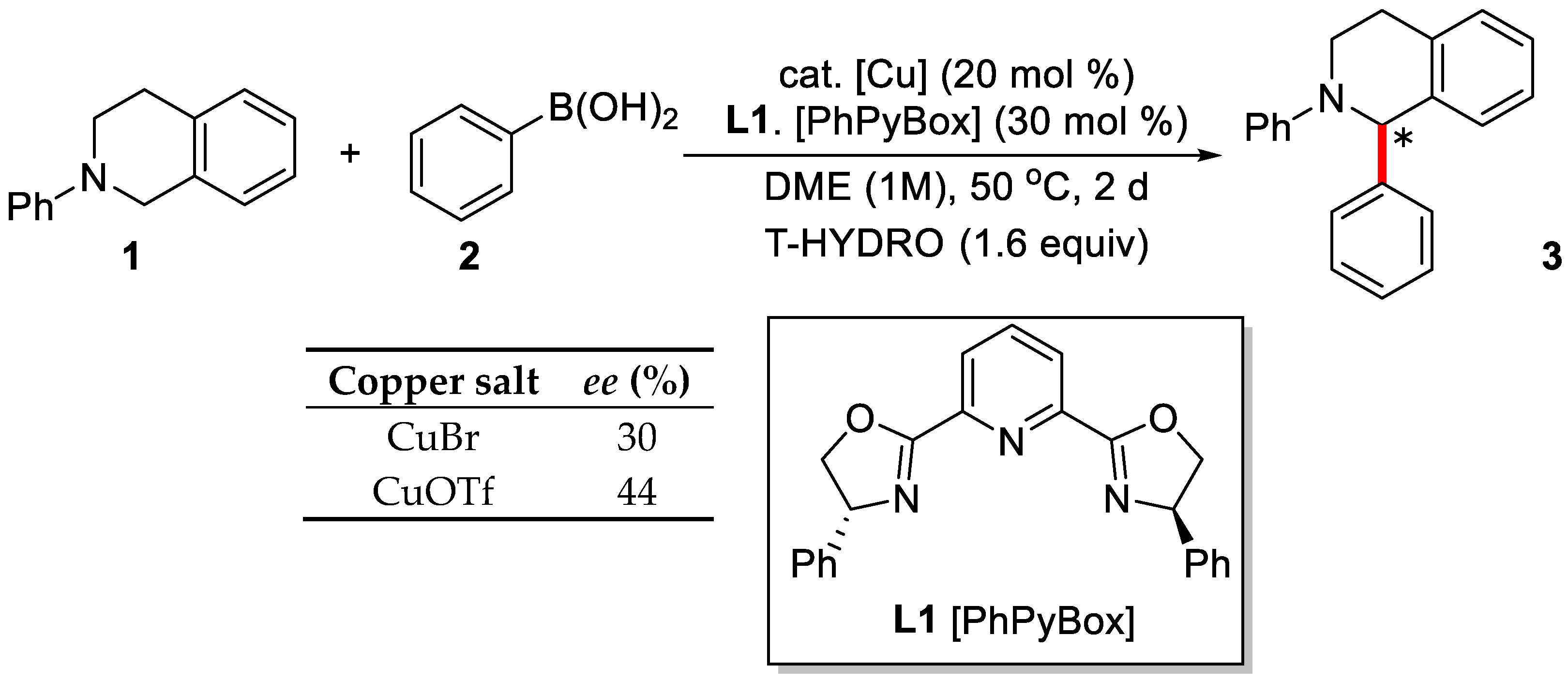
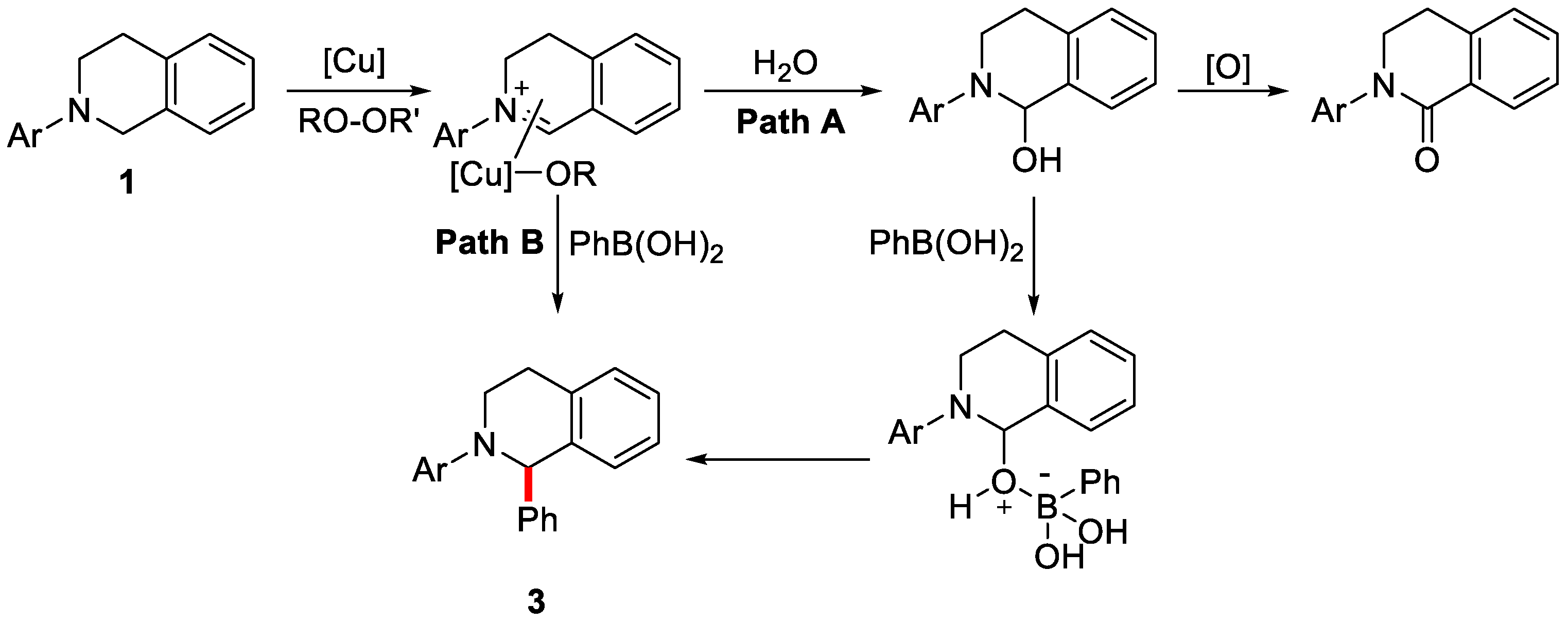
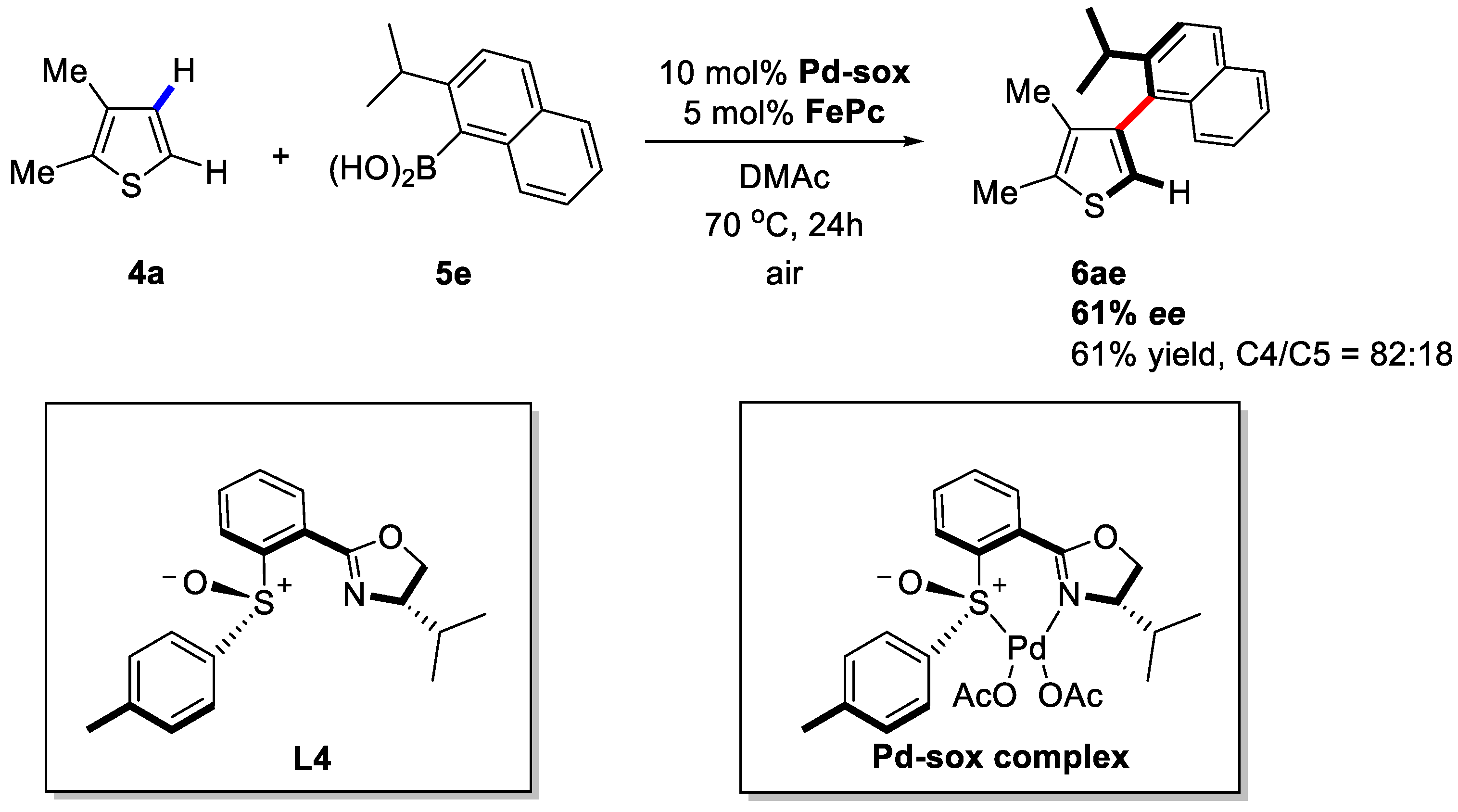
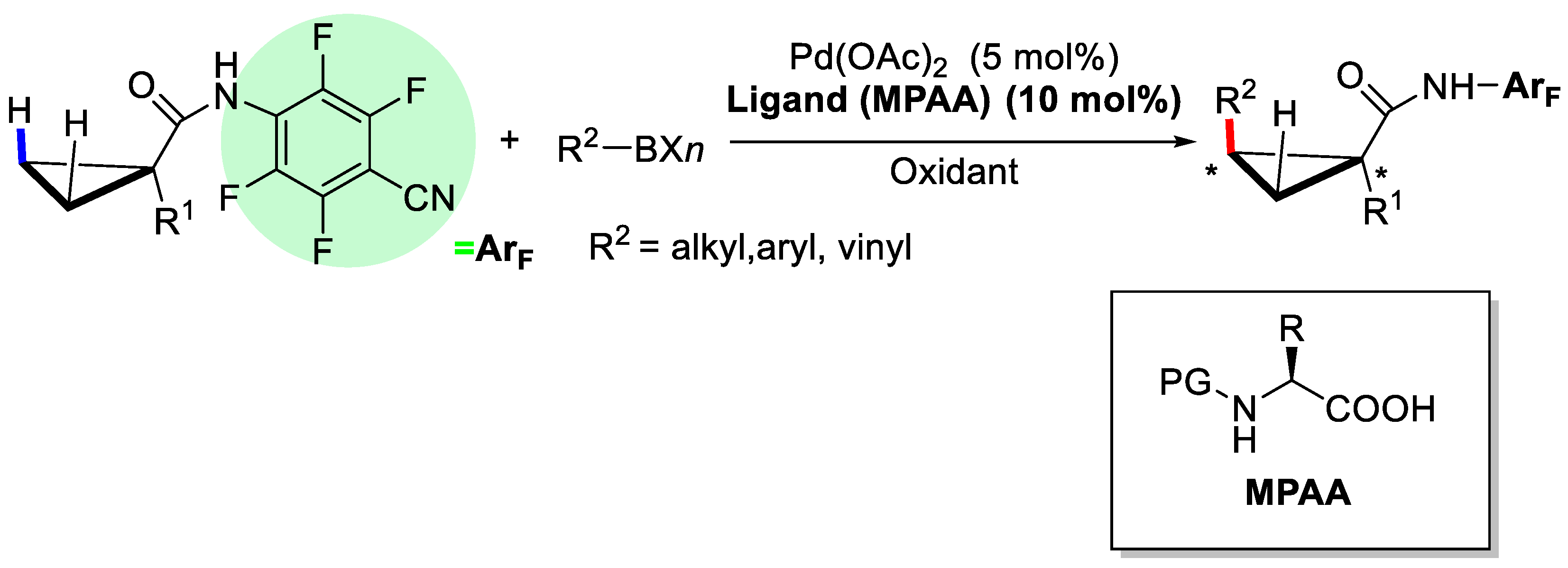
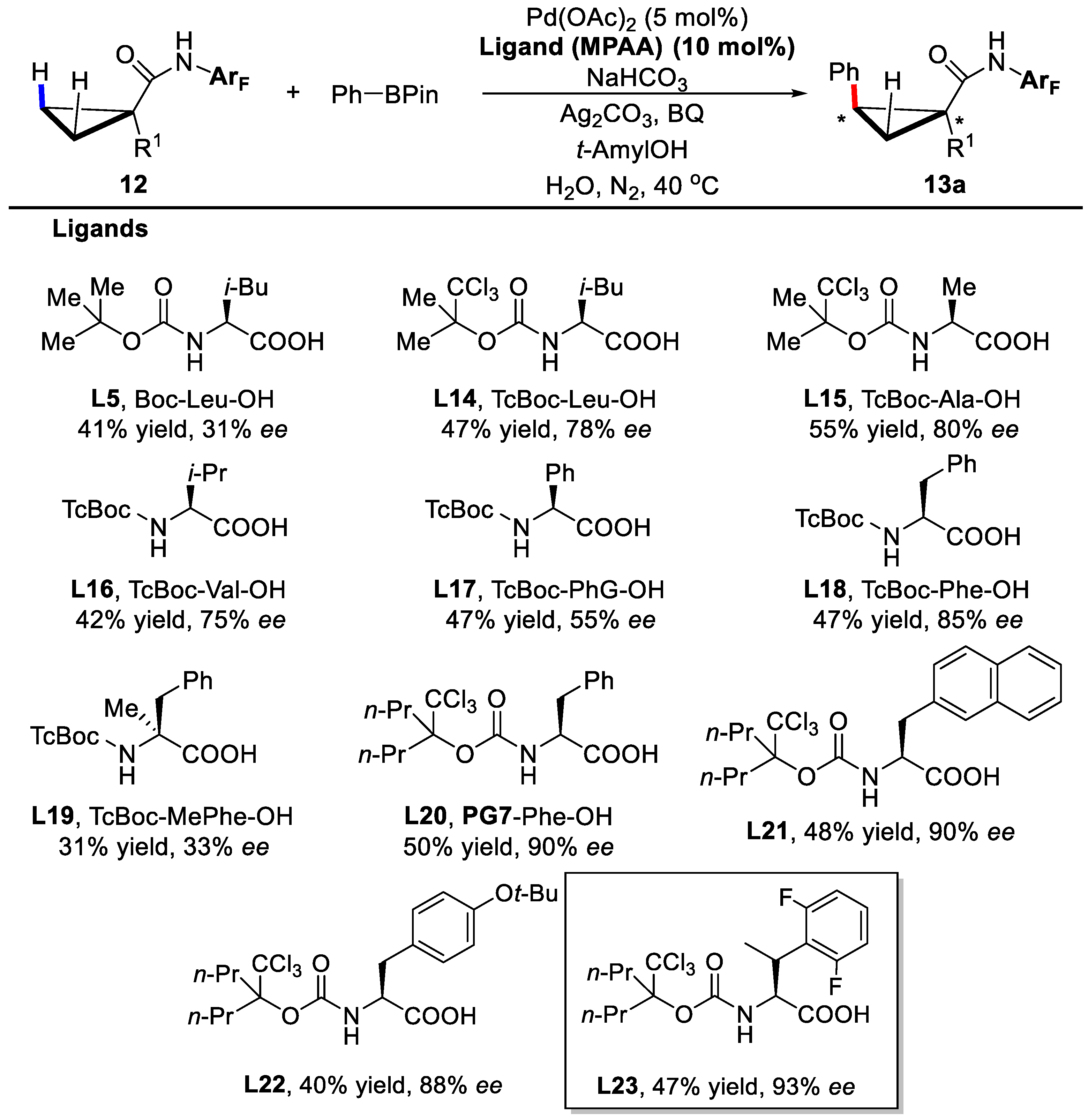
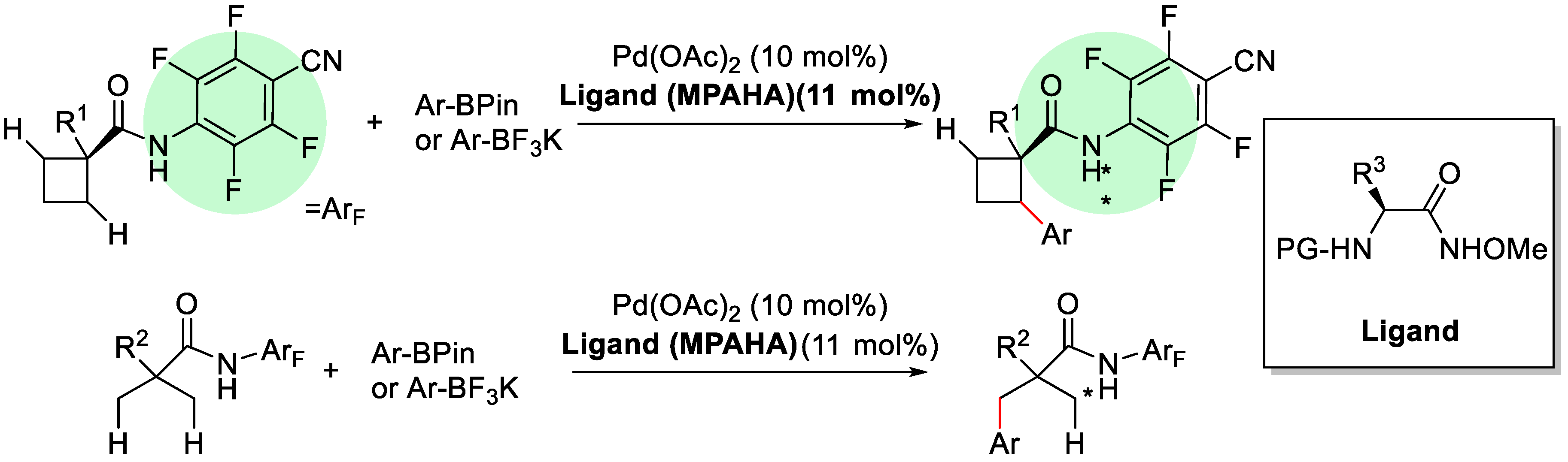
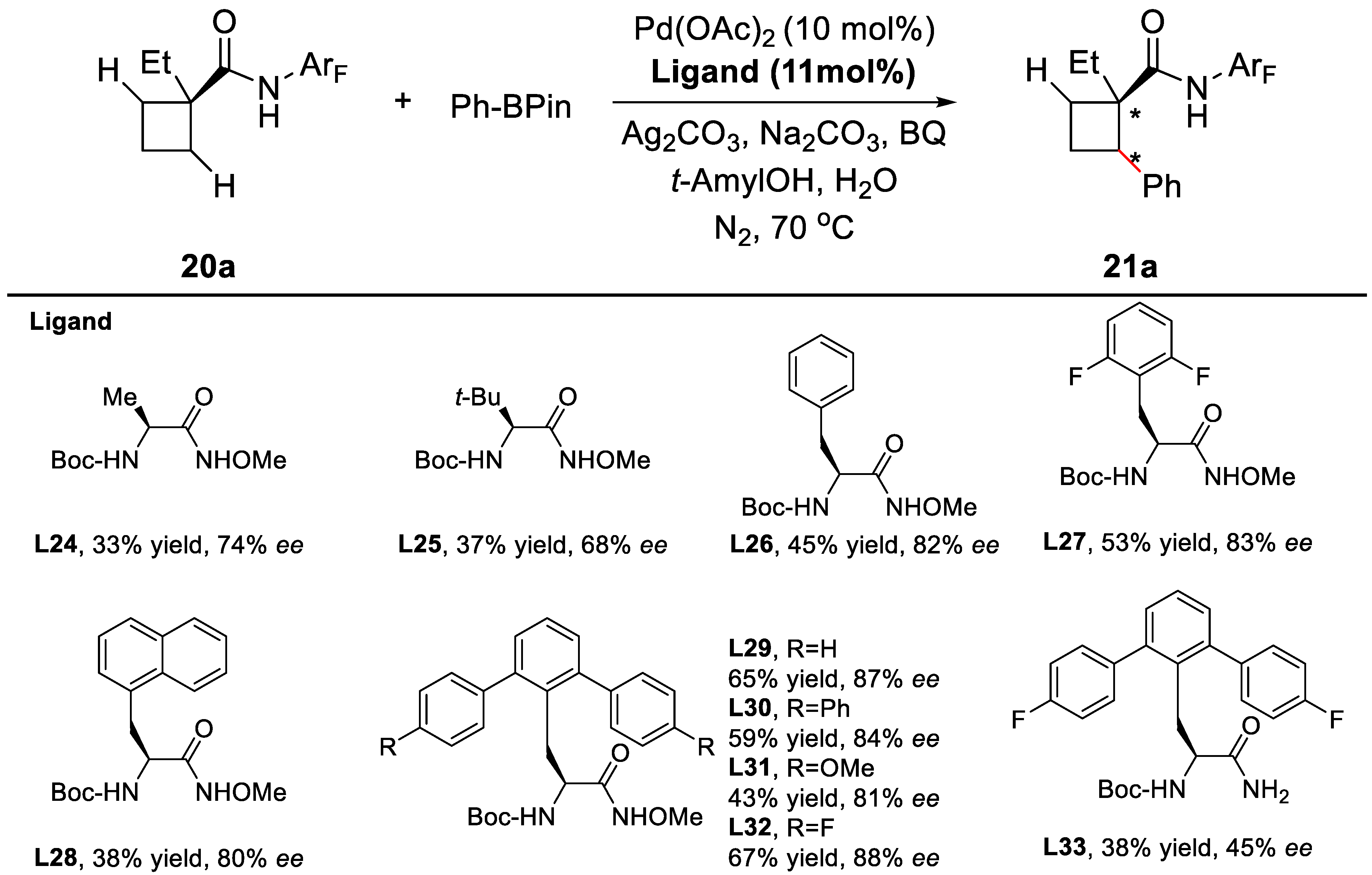
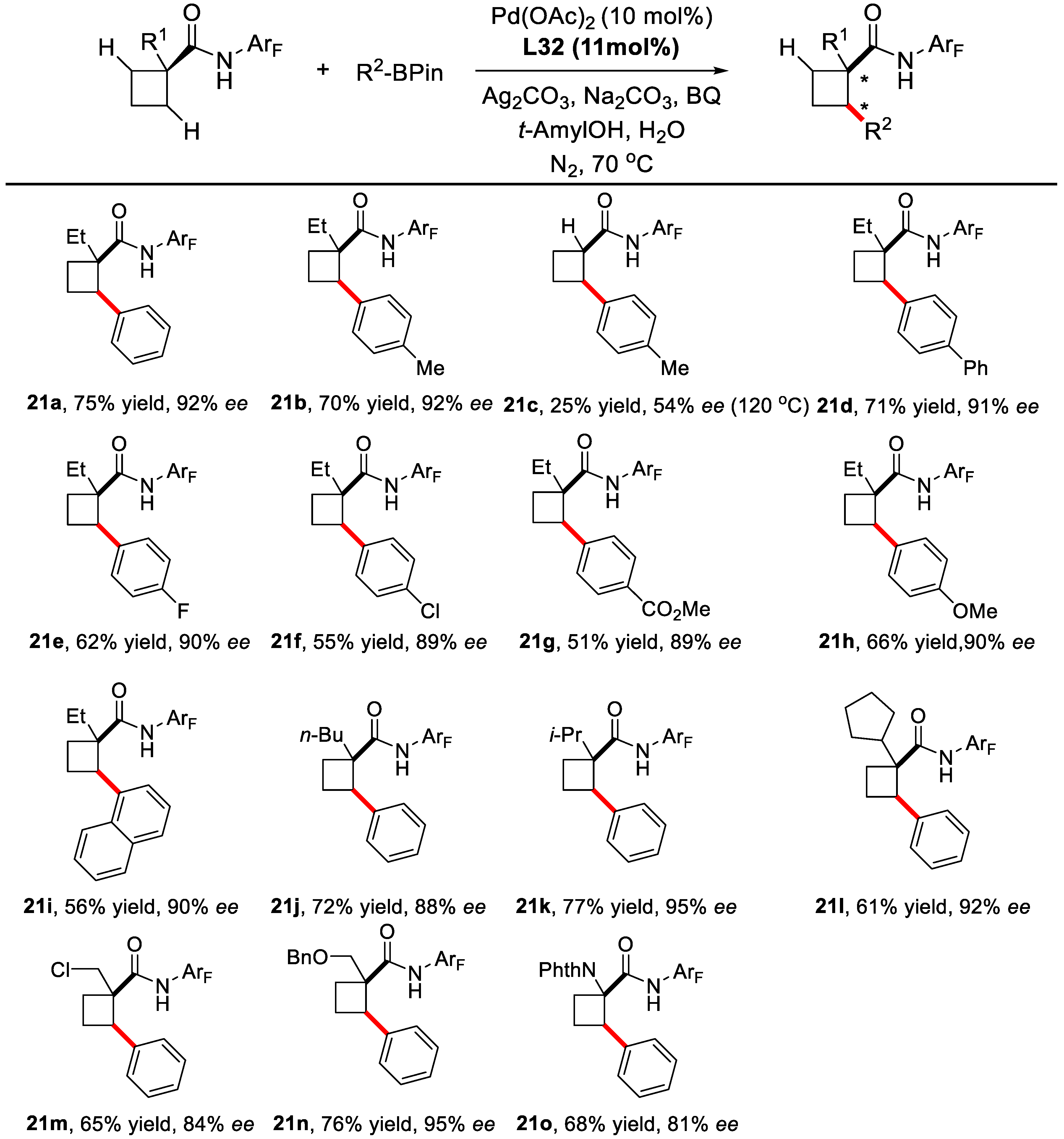
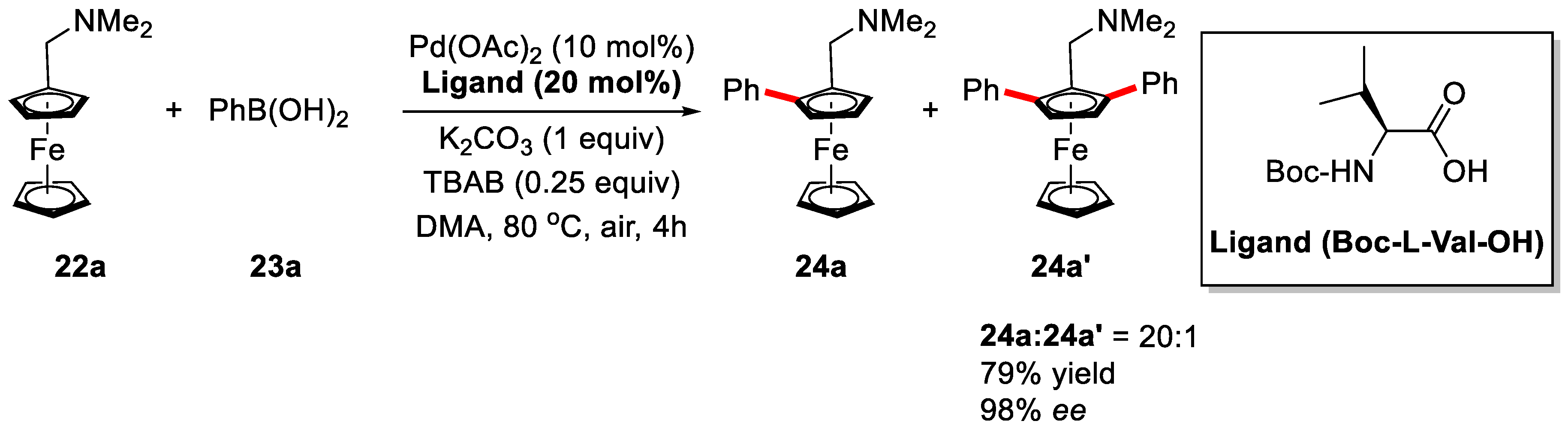
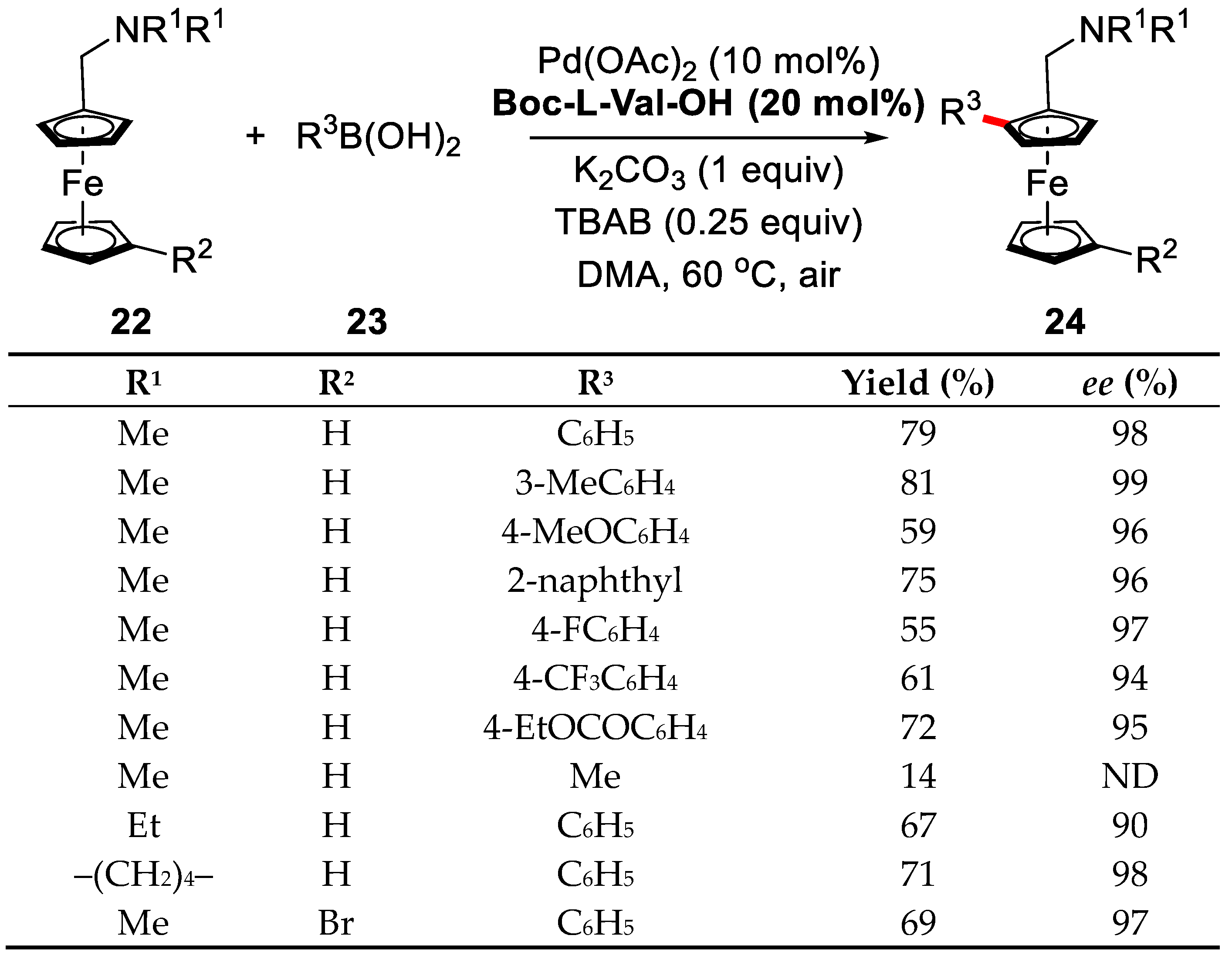
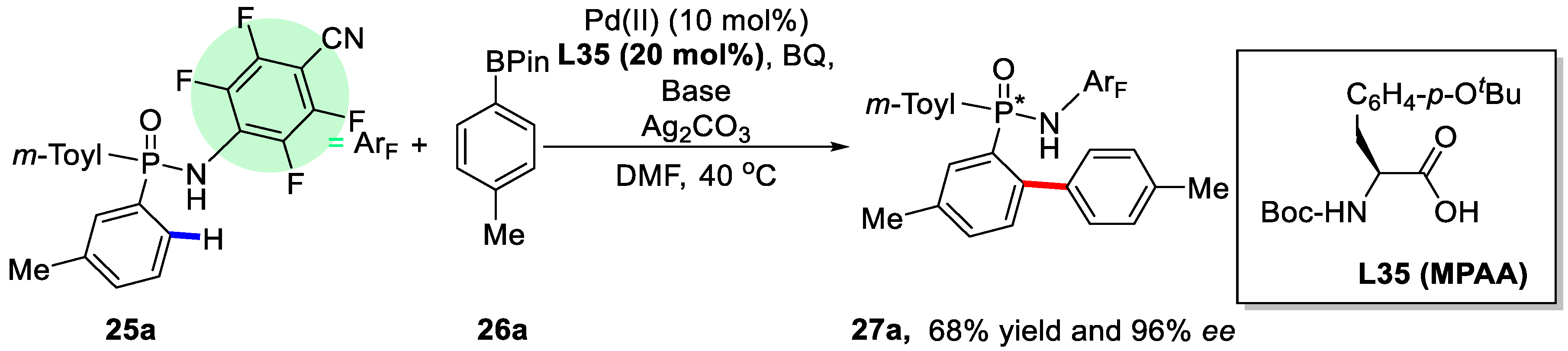
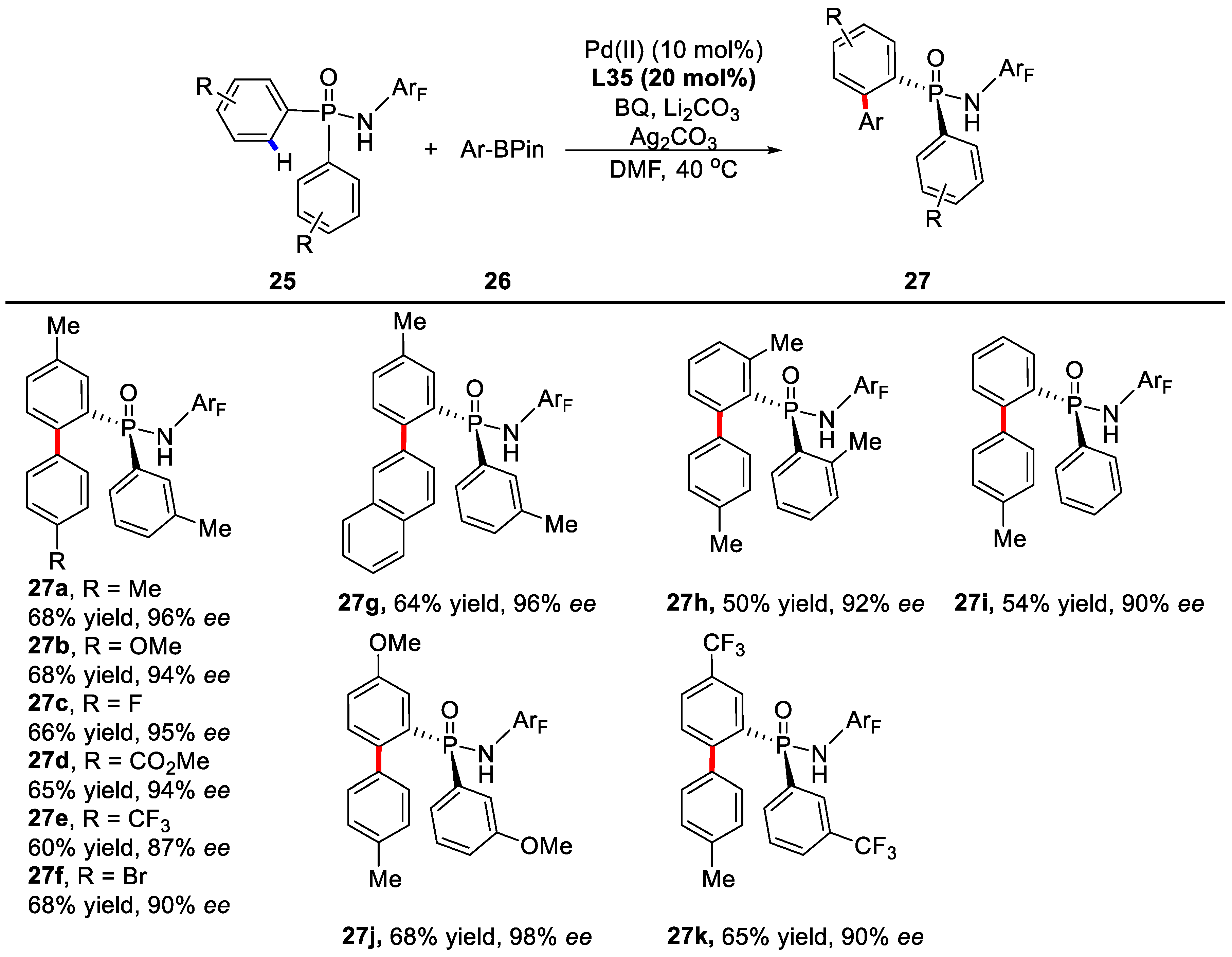
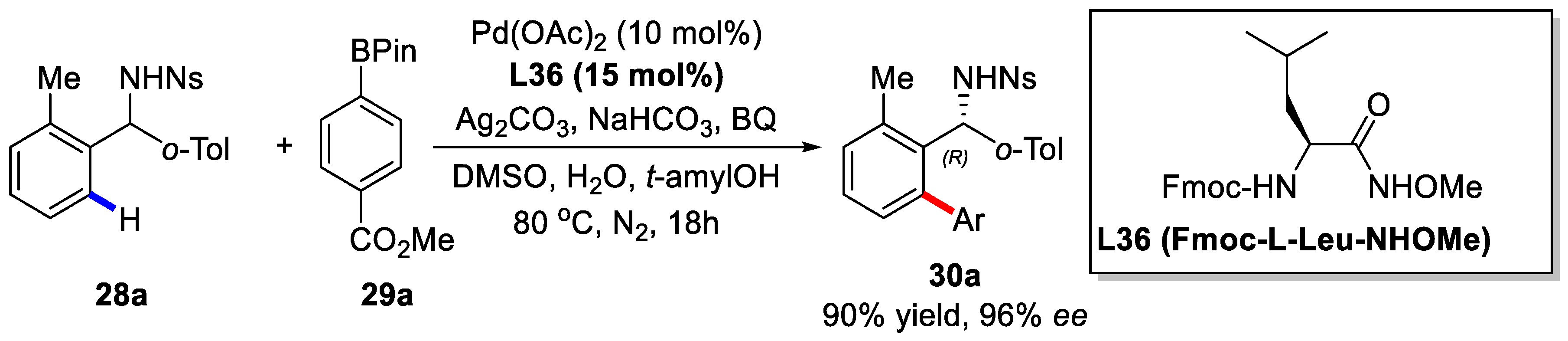
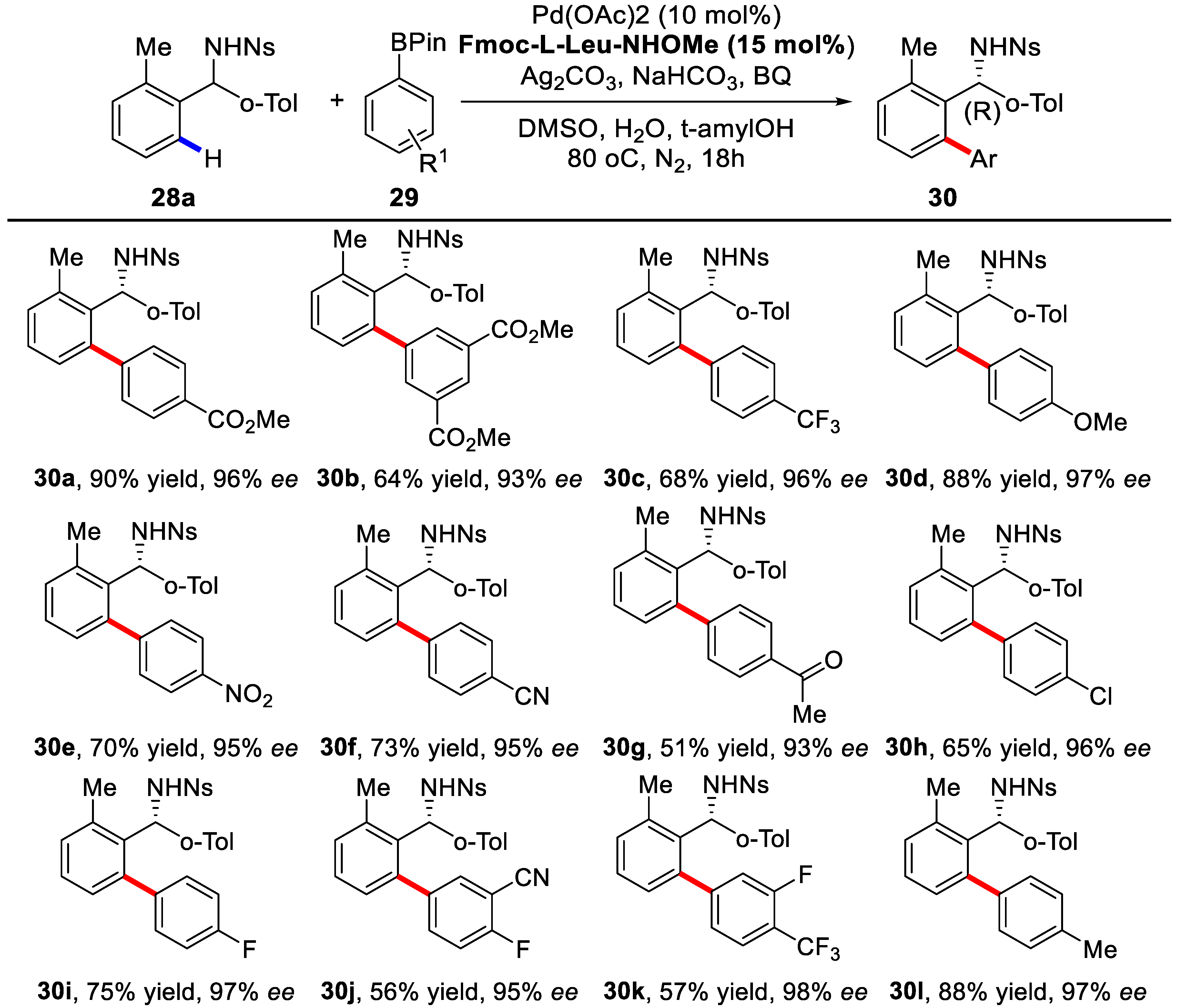
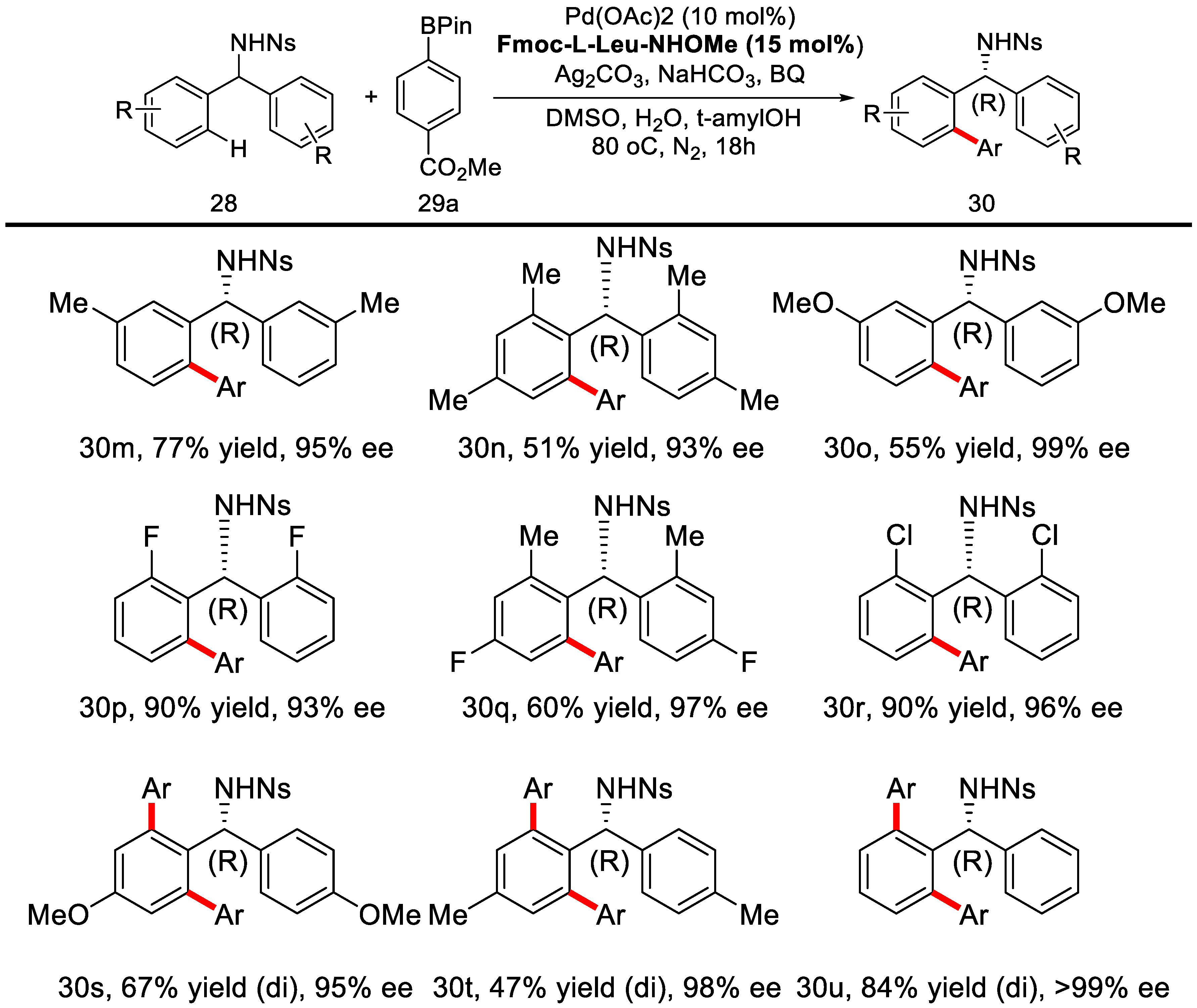
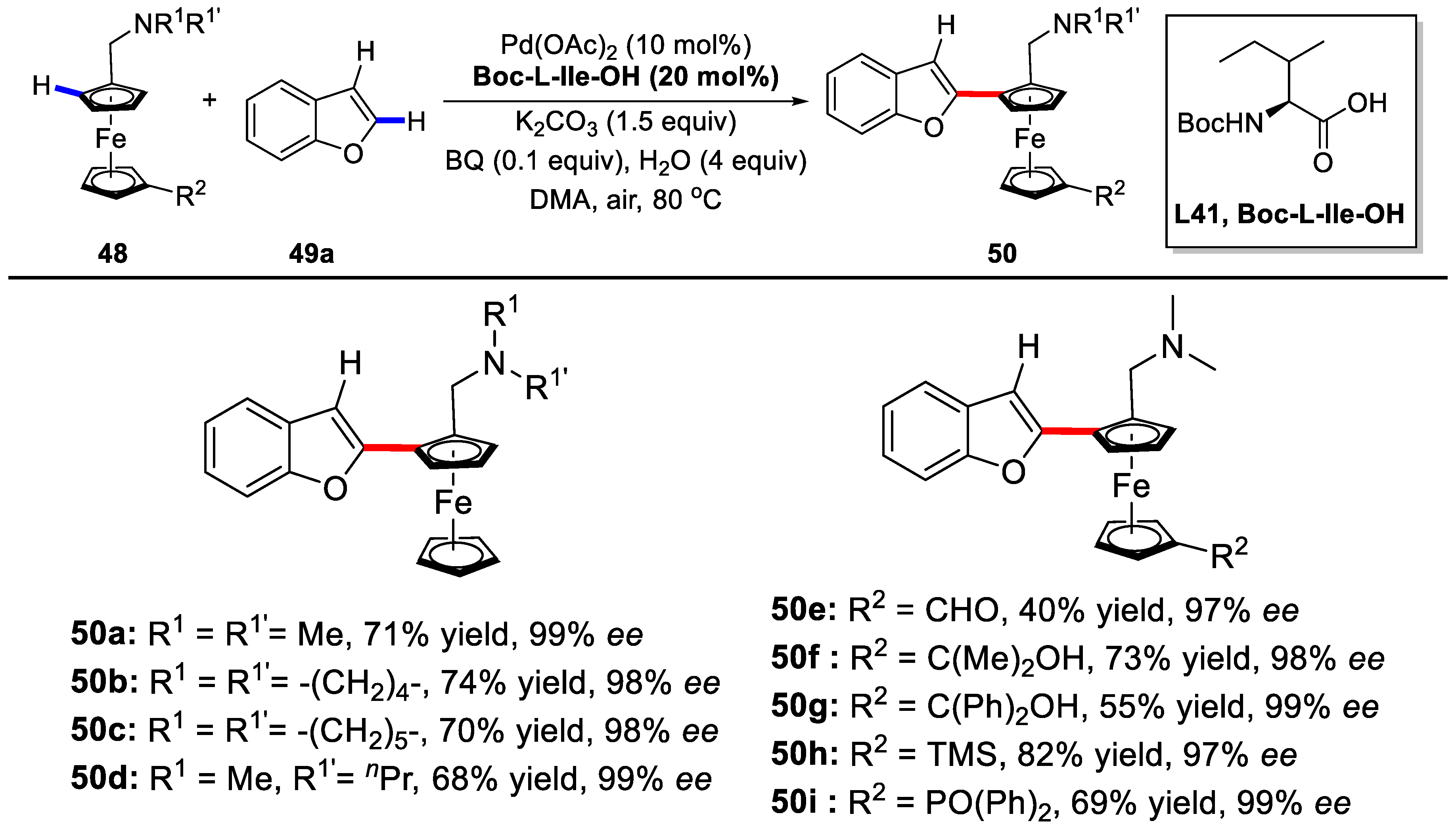
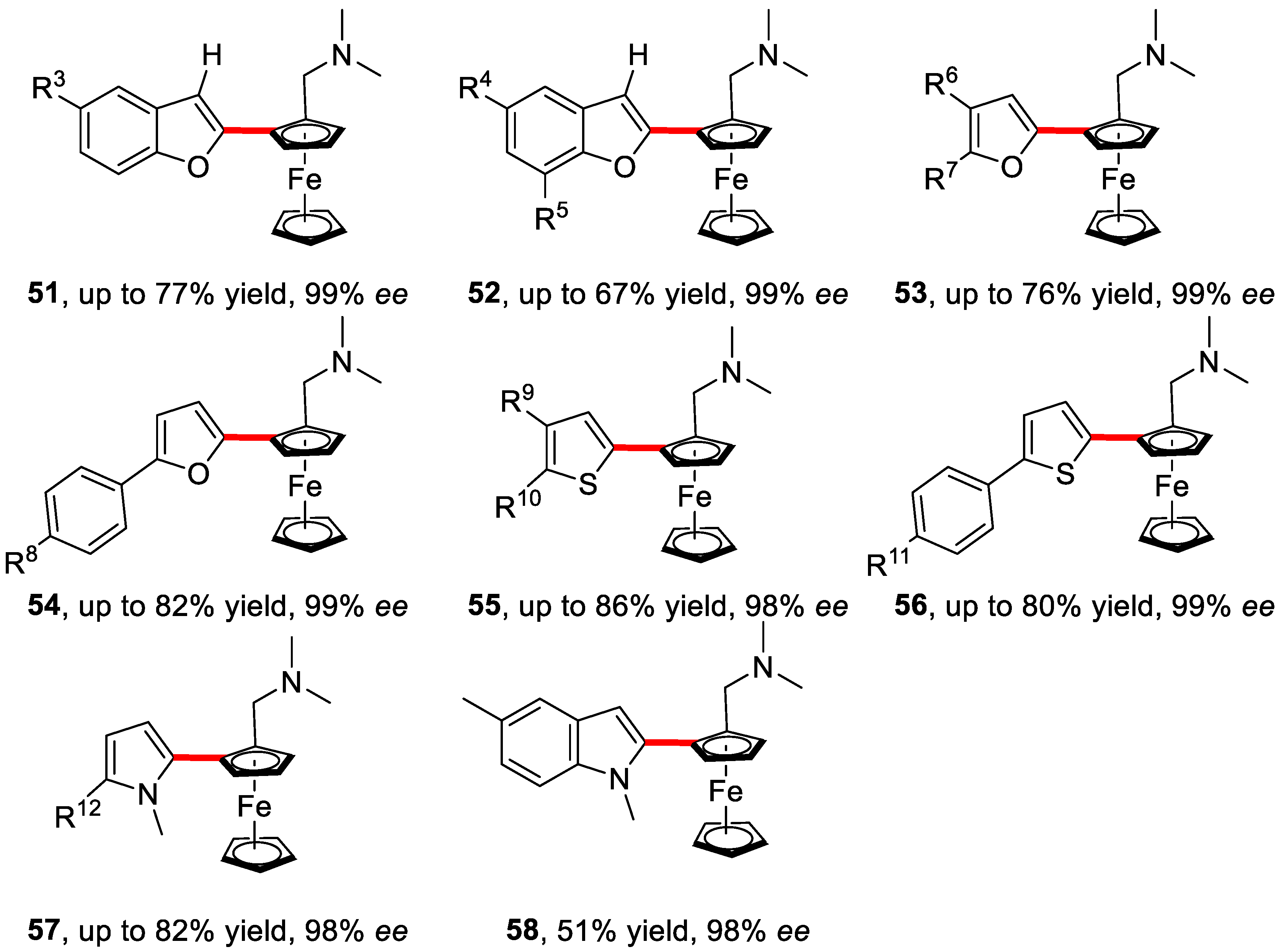
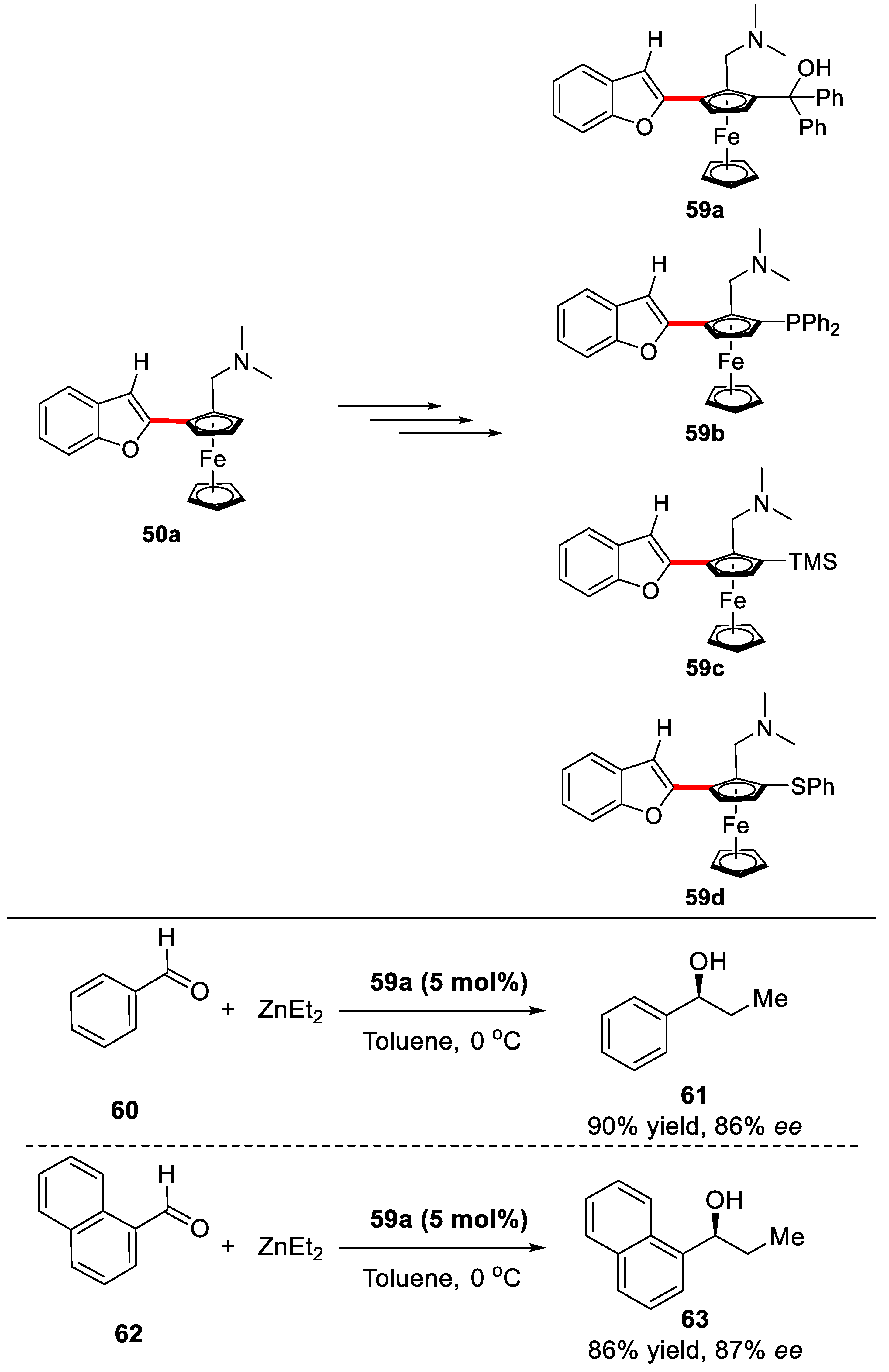
2. Enantioselective C–H Activation/Suzuki Reaction
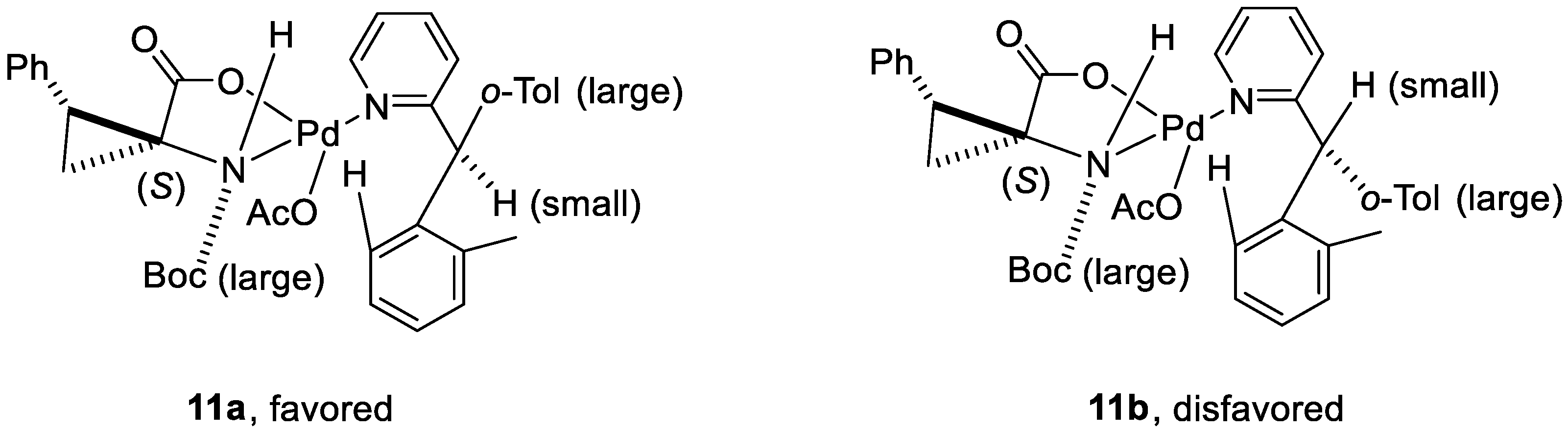
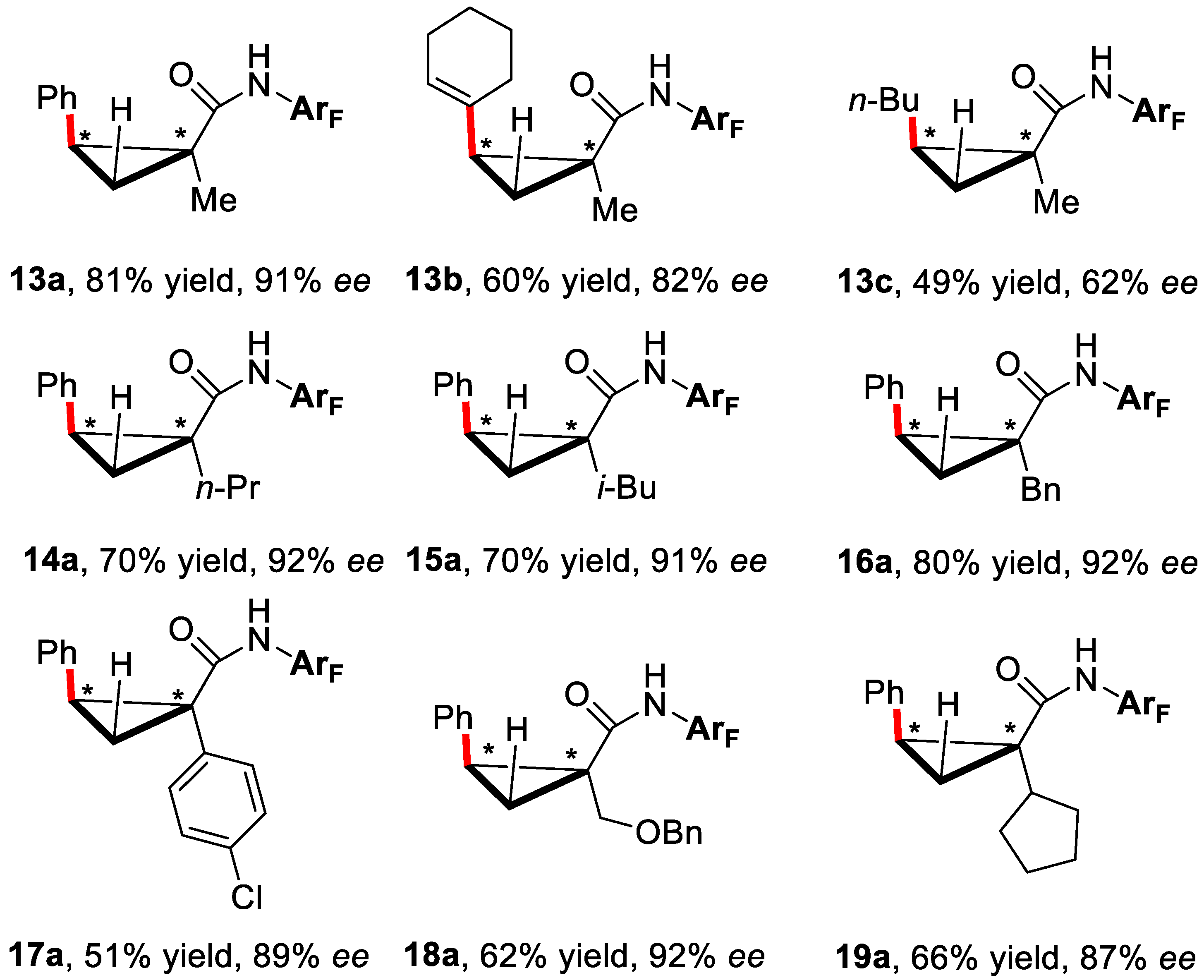
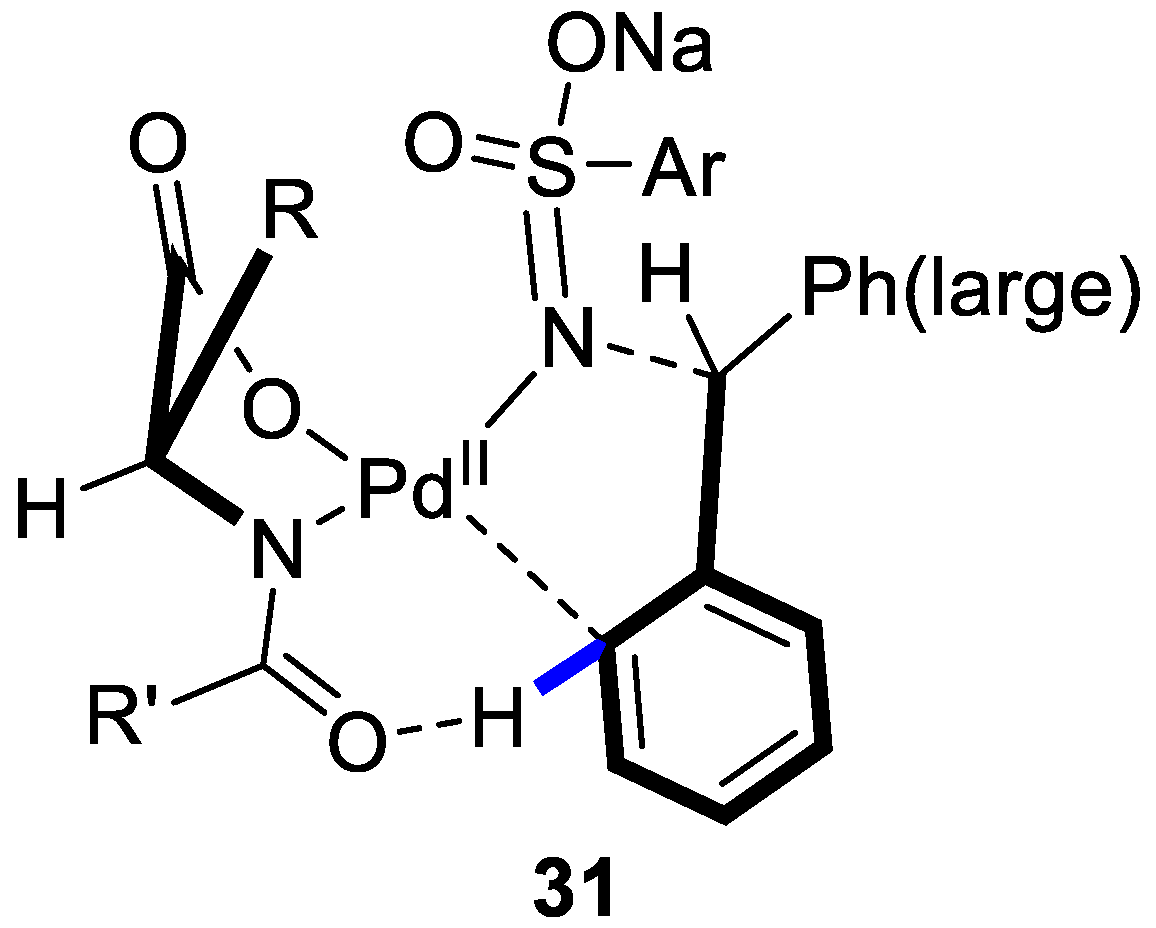
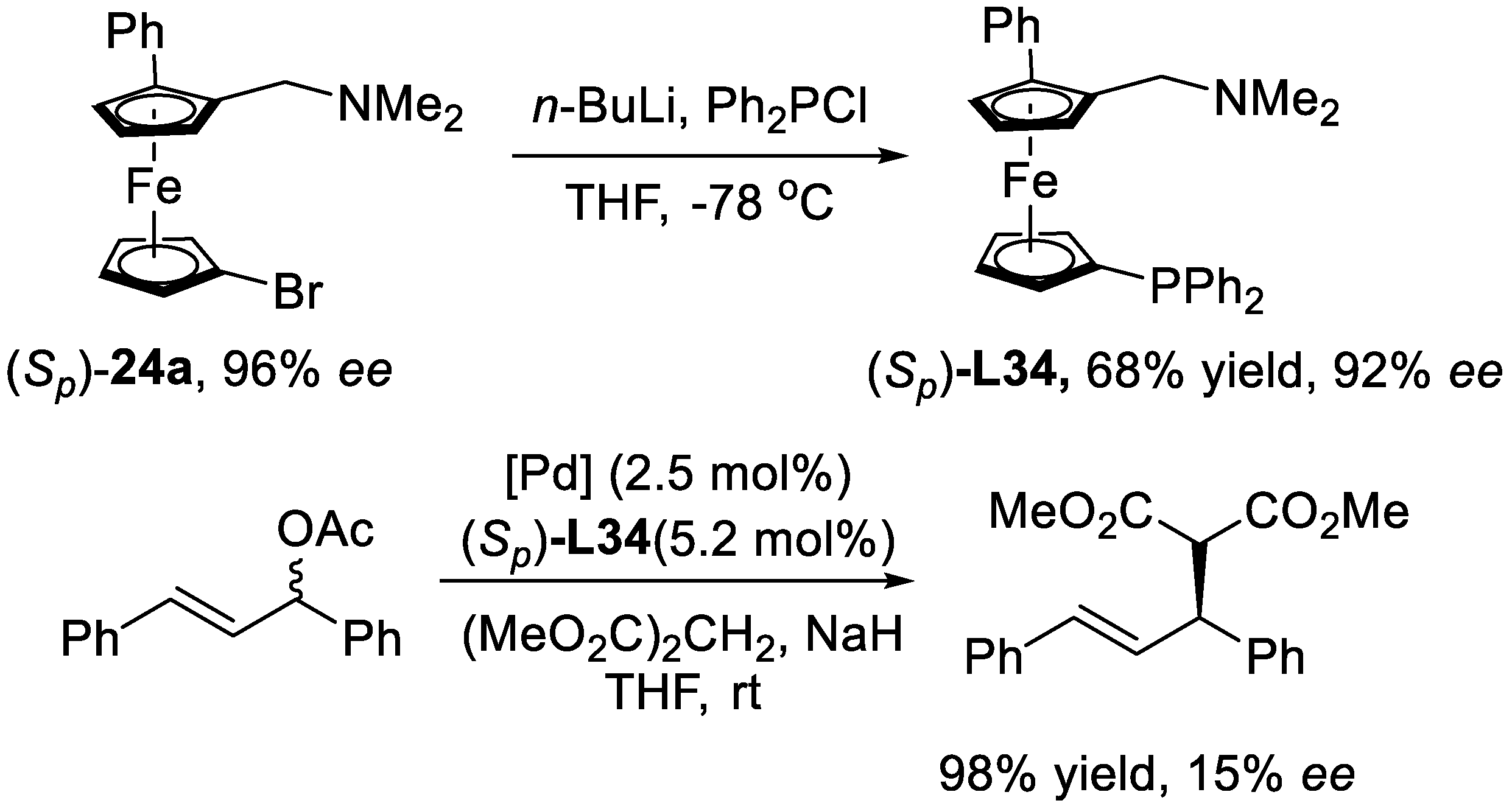
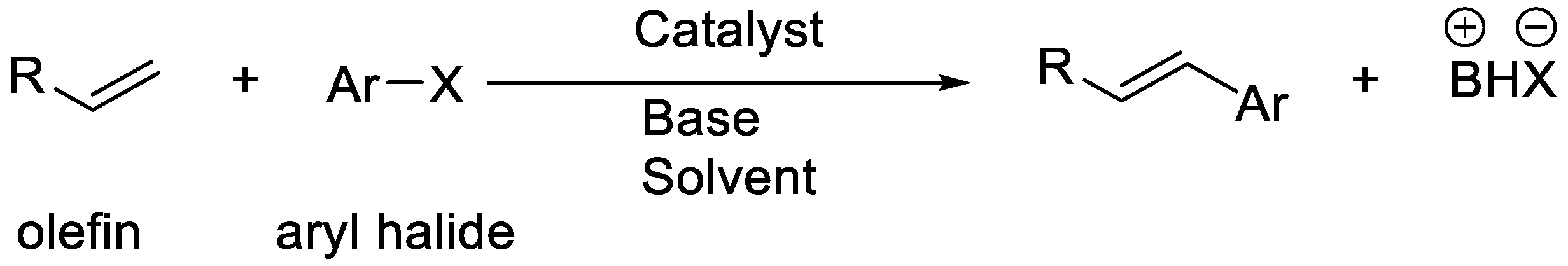
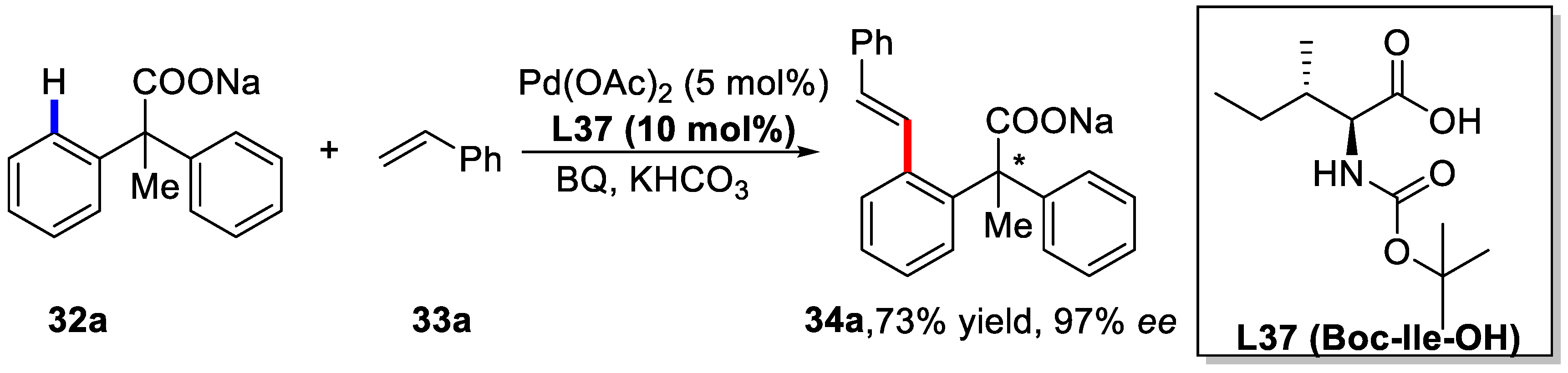
3. Enantioselective C–H Activation/Mizoroki-Heck Type Reaction
4. Summary
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mo, J.; Wang, L.; Liu, Y.; Cui, X. Transition-metal-catalyzed direct C–H functionalization under external-oxidant-free conditions. Synthesis 2015, 47, 439–459. [Google Scholar] [CrossRef]
- Li, S.-S.; Qin, L.; Dong, L. Rhodium-catalyzed C–C coupling reactions via double C–H activation. Org. Biomol. Chem. 2016, 14, 4554–4570. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.-S.; Ji, P.; Zhou, B.; Cheng, J.-P. The Essential Role of Bond Energetics in C–H Activation/Functionalization. Chem. Rev. 2017, 117, 8622–8648. [Google Scholar] [CrossRef] [PubMed]
- Gensch, T.; Hopkinson, M.; Glorius, F.; Wencel-Delord, J. Mild metal-catalyzed C–H activation: Examples and concepts. Chem. Soc. Rev. 2016, 45, 2900–2936. [Google Scholar] [CrossRef] [PubMed]
- Musaev, D.G.; Figg, T.M.; Kaledin, A.L. Versatile reactivity of Pd-catalysts: Mechanistic features of the mono-N-protected amino acid ligand and cesium-halide base in Pd-catalyzed C–H bond functionalization. Chem. Soc. Rev. 2014, 43, 5009–5031. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Lan, J.; You, J. Oxidative C–H/C–H Coupling Reactions between Two (Hetero) arenes. Chem. Rev. 2017, 117, 8787–8863. [Google Scholar] [CrossRef] [PubMed]
- Labinger, J.A. Platinum-Catalyzed C–H Functionalization. Chem. Rev. 2017, 117, 8483–8496. [Google Scholar] [CrossRef] [PubMed]
- Wencel-Delord, J.; Glorius, F. CH bond activation enables the rapid construction and late-stage diversification of functional molecules. Nat. Chem. 2013, 5, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Neufeldt, S.R.; Sanford, M.S. Controlling site selectivity in palladium-catalyzed C–H bond functionalization. Acc. Chem. Res. 2012, 45, 936–946. [Google Scholar] [CrossRef] [PubMed]
- Engle, K.M.; Mei, T.-S.; Wasa, M.; Yu, J.-Q. Weak coordination as a powerful means for developing broadly useful C–H functionalization reactions. Acc. Chem. Res. 2012, 45, 788–802. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Wang, F.; Li, X. C–C, C–O and C–N bond formation via rhodium (iii)-catalyzed oxidative C–H activation. Chem. Soc. Rev. 2012, 41, 3651–3678. [Google Scholar] [CrossRef] [PubMed]
- Hickman, A.J.; Sanford, M.S. High-valent organometallic copper and palladium in catalysis. Nature 2012, 484, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Patureau, F.W.; Wencel-Delord, J.; Glorius, F. Cp* Rh-Catalyzed C—H Activations. Versatile Dehydrogenative Cross-Couplings of Csp2 C—H Positions with Olefins, Alkynes, and Arenes. ChemInform 2013, 44. [Google Scholar] [CrossRef]
- Arockiam, P.B.; Bruneau, C.; Dixneuf, P.H. Ruthenium (II)-catalyzed C–H bond activation and functionalization. Chem. Rev. 2012, 112, 5879–5918. [Google Scholar] [CrossRef] [PubMed]
- Yeung, C.S.; Dong, V.M. Catalytic dehydrogenative cross-coupling: Forming carbon-carbon bonds by oxidizing two carbon-hydrogen bonds. Chem. Rev. 2011, 111, 1215–1292. [Google Scholar] [CrossRef] [PubMed]
- Wencel-Delord, J.; Dröge, T.; Liu, F.; Glorius, F. Towards mild metal-catalyzed C–H bond activation. Chem. Soc. Rev. 2011, 40, 4740–4761. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.H.; Kim, J.Y.; Kwak, J.; Chang, S. Recent advances in the transition metal-catalyzed twofold oxidative C–H bond activation strategy for C–C and C–N bond formation. Chem. Soc. Rev. 2011, 40, 5068–5083. [Google Scholar] [CrossRef] [PubMed]
- Baudoin, O. Transition metal-catalyzed arylation of unactivated C(sp3)–H bonds. Chem. Soc. Rev. 2011, 40, 4902–4911. [Google Scholar] [CrossRef] [PubMed]
- Rech, J.C.; Yato, M.; Duckett, D.; Ember, B.; LoGrasso, P.V.; Bergman, R.G.; Ellman, J.A. Synthesis of Potent Bicyclic Bisarylimidazole c-Jun N-Terminal Kinase Inhibitors by Catalytic C−H Bond Activation. J. Am. Chem. Soc. 2007, 129, 490–491. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, S.J.; Tan, K.L.; Watzke, A.; Bergman, R.G.; Ellman, J.A. Total synthesis of (+)-Lithospermic acid by asymmetric intramolecular alkylation via catalytic C−H bond activation. J. Am. Chem. Soc. 2005, 127, 13496–13497. [Google Scholar] [CrossRef] [PubMed]
- Hinman, A.; Du Bois, J. A stereoselective synthesis of (−)-tetrodotoxin. J. Am. Chem. Soc. 2003, 125, 11510–11511. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Wan, Z.; Kerr, R.G.; Davies, H.M. Synthetic and isolation studies related to the marine natural products (+)-elisabethadione and (+)-elisabethamine. J. Org. Chem. 2007, 72, 1895–1900. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M. Atom Economy-A Challenge for Organic Synthesis: Homogeneous Catalysis Leads the Way. Angew. Chem. Int. Ed. Engl. 1995, 34, 259–281. [Google Scholar] [CrossRef]
- Wehn, P.M.; Du Bois, J. A Stereoselective Synthesis of the Bromopyrrole Natural Product (−)-Agelastatin A. Angew. Chem. 2009, 121, 3860–3863. [Google Scholar] [CrossRef]
- Chen, K.; Baran, P.S. Total synthesis of eudesmane terpenes by site-selective C–H oxidations. Nature 2009, 459, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Tsai, A.S.; Bergman, R.G.; Ellman, J.A. Asymmetric Synthesis of (−)-Incarvillateine Employing an Intramolecular Alkylation via Rh-Catalyzed Olefinic C−H Bond Activation. J. Am. Chem. Soc. 2008, 130, 6316–6317. [Google Scholar] [CrossRef] [PubMed]
- Bowie, A.L.; Hughes, C.C.; Trauner, D. Concise synthesis of (±)-rhazinilam through direct coupling. Org. Lett. 2005, 7, 5207–5209. [Google Scholar] [CrossRef] [PubMed]
- Baran, P.S.; Corey, E. A short synthetic route to (+)-austamide, (+)-deoxyisoaustamide, and (+)-hydratoaustamide from a common precursor by a novel palladium-mediated indole → dihydroindoloazocine cyclization. J. Am. Chem. Soc. 2002, 124, 7904–7905. [Google Scholar] [CrossRef] [PubMed]
- Miyaura, N.; Yamada, K.; Suzuki, A. A new stereospecific cross-coupling by the palladium-catalyzed reaction of 1-alkenylboranes with 1-alkenyl or 1-alkynyl halides. Tetrahedron Lett. 1979, 20, 3437–3440. [Google Scholar] [CrossRef]
- Jagtap, S. Heck Reaction—State of the Art. Catalysts 2017, 7, 267. [Google Scholar] [CrossRef]
- Sonogashira, K.; Tohda, Y.; Hagihara, N. A convenient synthesis of acetylenes: Catalytic substitutions of acetylenic hydrogen with bromoalkenes, iodoarenes and bromopyridines. Tetrahedron Lett. 1975, 16, 4467–4470. [Google Scholar] [CrossRef]
- King, A.O.; Okukado, N.; Negishi, E.-I. Highly general stereo-, regio-, and chemo-selective synthesis of terminal and internal conjugated enynes by the Pd-catalysed reaction of alkynylzinc reagents with alkenyl halides. J. Chem. Soc. Chem. Commun. 1977, 683–684. [Google Scholar] [CrossRef]
- Milstein, D.; Stille, J. A general, selective, and facile method for ketone synthesis from acid chlorides and organotin compounds catalyzed by palladium. J. Am. Chem. Soc. 1978, 100, 3636–3638. [Google Scholar] [CrossRef]
- Tamao, K.; Sumitani, K.; Kumada, M. Selective carbon-carbon bond formation by cross-coupling of Grignard reagents with organic halides. Catalysis by nickel-phosphine complexes. J. Am. Chem. Soc. 1972, 94, 4374–4376. [Google Scholar] [CrossRef]
- Heck, R.F.; Negishi, E.-I.; Suzuki, A. Nobel Prizes 2010. Angew. Chem. Int. Ed. 2010, 49, 8300. [Google Scholar]
- Wencel-Delord, J.; Colobert, F. Asymmetric C(sp2)–H Activation. Chem. A Eur. J. 2013, 19, 14010–14017. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.C. Organocatalytic C–H activation reactions. Beilstein J. Org. Chem. 2012, 8, 1374. [Google Scholar] [CrossRef] [PubMed]
- Newhouse, T.; Baran, P.S. If C–H Bonds Could Talk: Selective C–H Bond Oxidation. Angew. Chem. Int. Ed. 2011, 50, 3362–3374. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.-M.; Dai, L.-X.; You, S.-L. Enantioselective Palladium-Catalyzed Direct Alkylation and Olefination Reaction of Simple Arenes. Angew. Chem. Int. Ed. 2010, 49, 5826–5828. [Google Scholar] [CrossRef] [PubMed]
- Giri, R.; Shi, B.-F.; Engle, K.M.; Maugel, N.; Yu, J.-Q. Transition metal-catalyzed C–H activation reactions: Diastereoselectivity and enantioselectivity. Chem. Soc. Rev. 2009, 38, 3242–3272. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.-L.; Wang, Y.; Luo, Y.-C.; Fu, X.-Z.; Xu, P.-F. Asymmetric C–H functionalization involving organocatalysis. Tetrahedron Lett. 2015, 56, 3703–3714. [Google Scholar] [CrossRef]
- Musaev, D.G.; Kaledin, A.; Shi, B.-F.; Yu, J.-Q. Key mechanistic features of enantioselective C–H bond activation reactions catalyzed by [(chiral mono-N-protected amino acid)–Pd (II)] complexes. J. Am. Chem. Soc. 2012, 134, 1690–1698. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Zhang, Y.; Wang, R. Catalytic Asymmetric Activation of a Csp3–H Bond Adjacent to a Nitrogen Atom: A Versatile Approach to Optically Active α-Alkyl α-Amino Acids and C1-Alkylated Tetrahydroisoquinoline Derivatives. Angew. Chem. Int. Ed. 2011, 50, 10429–10432. [Google Scholar] [CrossRef] [PubMed]
- Wasa, M.; Engle, K.M.; Lin, D.-W.; Yoo, E.J.; Yu, J.-Q. Pd (II)-catalyzed enantioselective C–H activation of cyclopropanes. J. Am. Chem. Soc. 2011, 133, 19598–19601. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Yu, Z.-X. Enantioselective Rhodium-Catalyzed Allylic C–H Activation for the Addition to Conjugated Dienes. Angew. Chem. 2011, 123, 2192–2195. [Google Scholar] [CrossRef]
- Davies, H.M.; Manning, J.R. Catalytic C–H functionalization by metal carbenoid and nitrenoid insertion. Nature 2008, 451, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Haines, B.E.; Yu, J.-Q.; Musaev, D.G. Enantioselectivity Model for Pd-Catalyzed C–H Functionalization Mediated by the Mono-N-protected Amino Acid (MPAA) Family of Ligands. ACS Catal. 2017, 7, 4344–4354. [Google Scholar] [CrossRef]
- Motevalli, S.; Sokeirik, Y.; Ghanem, A. Rhodium-Catalysed Enantioselective C–H Functionalization in Asymmetric Synthesis. Eur. J. Org. Chem. 2016, 2016, 1459–1475. [Google Scholar] [CrossRef]
- Loxq, P.; Manoury, E.; Poli, R.; Deydier, E.; Labande, A. Synthesis of axially chiral biaryl compounds by asymmetric catalytic reactions with transition metals. Coord. Chem. Rev. 2016, 308, 131–190. [Google Scholar] [CrossRef]
- Qin, Y.; Lv, J.; Luo, S. Catalytic asymmetric α-C(sp3)–H functionalization of amines. Tetrahedron Lett. 2014, 55, 551–558. [Google Scholar] [CrossRef]
- Yang, L.; Huang, H. Asymmetric catalytic carbon–carbon coupling reactions via C–H bond activation. Catal. Sci. Technol. 2012, 2, 1099. [Google Scholar] [CrossRef]
- Wasa, M.; Engle, K.M.; Yu, J.-Q. Cross-Coupling of C(sp3)–H Bonds with Organometallic Reagents via Pd(II)/Pd(0) Catalysis. Isr. J. Chem. 2010, 50, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.; Davies, H.M. High symmetry dirhodium (II) paddlewheel complexes as chiral catalysts. Coord. Chem. Rev. 2008, 252, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Enders, D.; Balensiefer, T. Nucleophilic carbenes in asymmetric organocatalysis. Acc. Chem. Res 2004, 37, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.K.; Kaval, N.; Tomar, S.; Eycken, E.V.; Parmar, V.S. Transition metal-catalyzed carbon-carbon bond formation Suzuki, Heck, and Sonogashira reactions using microwave and microtechnology. Org. Process Res. Dev. 2008, 12, 468–474. [Google Scholar] [CrossRef]
- Cantillo, D.; Kappe, C.O. Immobilized Transition Metals as Catalysts for Cross-Couplings in Continuous Flow—A Critical Assessment of the Reaction Mechanism and Metal Leaching. ChemCatChem 2014, 6, 3286–3305. [Google Scholar] [CrossRef]
- Shu, W.; Pellegatti, L.; Oberli, M.A.; Buchwald, S.L. Continuous-Flow Synthesis of Biaryls Enabled by Multistep Solid-Handling in a Lithiation/Borylation/Suzuki-Miyaura Cross-Coupling Sequence. Angew. Chem. 2011, 123, 10853–10857. [Google Scholar] [CrossRef]
- Basle, O.; Li, C.-J. Copper-Catalyzed Oxidative sp3 C–H Bond Arylation with Aryl Boronic Acids. Org. Lett. 2008, 10, 3661–3663. [Google Scholar] [CrossRef] [PubMed]
- Kirchberg, S.; Tani, S.; Ueda, K.; Yamaguchi, J.; Studer, A.; Itami, K. Oxidative Biaryl Coupling of Thiophenes and Thiazoles with Arylboronic Acids through Palladium Catalysis: Otherwise Difficult C4-Selective C–H Arylation Enabled by Boronic Acids. Angew. Chem. Int. Ed. 2011, 50, 2387–2391. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Yamaguchi, J.; Studer, A.; Itami, K. Hindered biaryls by C–H coupling: Bisoxazoline-Pd catalysis leading to enantioselective C–H coupling. Chem. Sci. 2012, 3, 2165. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Kondo, H.; Yamaguchi, J.; Itami, K. Aromatic C–H coupling with hindered arylboronic acids by Pd/Fe dual catalysts. Chem. Sci. 2013, 4, 3753. [Google Scholar] [CrossRef]
- Shi, B.-F.; Maugel, N.; Zhang, Y.-H.; Yu, J.-Q. Pd(II)-catalyzed enantioselective activation of C(sp2)–H and C(sp3)–H bonds using monoprotected amino acids as chiral ligands. Angew. Chem. Int. Ed. Engl. 2008, 47, 4882–4886. [Google Scholar] [CrossRef] [PubMed]
- Xiao, K.-J.; Lin, D.-W.; Miura, M.; Zhu, R.-Y.; Gong, W.; Wasa, M.; Yu, J.-Q. Palladium(II)-catalyzed enantioselective C(sp(3))–H activation using a chiral hydroxamic acid ligand. J. Am. Chem. Soc. 2014, 136, 8138–8142. [Google Scholar] [CrossRef] [PubMed]
- Dembitsky, V.M. Bioactive cyclobutane-containing alkaloids. J. Nat. Med. 2008, 62, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Kurosawa, K.; Takahashi, K.; Tsuda, E. SNF4435C and D, Novel Immunosuppressants Produced by a Strain of Streptomyces spectabilis. J. Antibiot. 2001, 54, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Fu, G.C. Applications of planar-chiral heterocycles as ligands in asymmetric catalysis. Acc. Chem. Res. 2006, 39, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Arae, S.; Ogasawara, M. Catalytic asymmetric synthesis of planar-chiral transition-metal complexes. Tetrahedron Lett. 2015, 56, 1751–1761. [Google Scholar] [CrossRef]
- Gómez Arrayás, R.; Adrio, J.; Carretero, J.C. Recent applications of chiral ferrocene ligands in asymmetric catalysis. Angew. Chem. Int. Ed. 2006, 45, 7674–7715. [Google Scholar] [CrossRef] [PubMed]
- Fu, G.C. Asymmetric catalysis with “planar-chiral” derivatives of 4-(dimethylamino) pyridine. Acc. Chem. Res. 2004, 37, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Colacot, T.J. A concise update on the applications of chiral ferrocenyl phosphines in homogeneous catalysis leading to organic synthesis. Chem. Rev. 2003, 103, 3101–3118. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.-X.; Tu, T.; You, S.-L.; Deng, W.-P.; Hou, X.-L. Asymmetric catalysis with chiral ferrocene ligands. Acc. Chem. Res. 2003, 36, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.-W.; Shi, Y.-C.; Gu, Q.; Zhao, Z.-L.; You, S.-L. Enantioselective synthesis of planar chiral ferrocenes via palladium-catalyzed direct coupling with arylboronic acids. J. Am. Chem. Soc. 2013, 135, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Parmar, D.; Sugiono, E.; Raja, S.; Rueping, M. Complete field guide to asymmetric BINOL-phosphate derived Brønsted acid and metal catalysis: History and classification by mode of activation; Brønsted acidity, hydrogen bonding, ion pairing, and metal phosphates. Chem. Rev. 2014, 114, 9047–9153. [Google Scholar] [CrossRef] [PubMed]
- Brak, K.; Jacobsen, E.N. Asymmetric Ion-Pairing Catalysis. Angew. Chem. Int. Ed. 2013, 52, 534–561. [Google Scholar] [CrossRef] [PubMed]
- Mahlau, M.; List, B. Asymmetric Counteranion-Directed Catalysis: Concept, Definition, and Applications. Angew. Chem. Int. Ed. 2013, 52, 518–533. [Google Scholar] [CrossRef] [PubMed]
- You, S.-L.; Cai, Q.; Zeng, M. Chiral Brønsted acid catalyzed Friedel–Crafts alkylation reactions. Chem. Soc. Rev. 2009, 38, 2190–2201. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.-J.; Guan, J.; Wu, G.-J.; Xu, P.; Gao, L.-X.; Han, F.-S. Pd(II)-catalyzed enantioselective synthesis of P-stereogenic phosphinamides via desymmetric C–H arylation. J. Am. Chem. Soc. 2015, 137, 632–635. [Google Scholar] [CrossRef] [PubMed]
- Laforteza, B.N.; Chan, K.S.; Yu, J.-Q. Enantioselective ortho-C–H cross-coupling of diarylmethylamines with organoborons. Angew Chem Int Ed Engl 2015, 54, 11143–11146. [Google Scholar] [CrossRef] [PubMed]
- Mizoroki, T.; Mori, K.; Ozaki, A. Arylation of olefin with aryl iodide catalyzed by palladium. Bull. Chem. Soc. Jpn. 1971, 44, 581. [Google Scholar] [CrossRef]
- Heck, R.F.; Nolley, J., Jr. Palladium-catalyzed vinylic hydrogen substitution reactions with aryl, benzyl, and styryl halides. J. Org. Chem. 1972, 37, 2320–2322. [Google Scholar] [CrossRef]
- Shi, B.-F.; Zhang, Y.-H.; Lam, J.K.; Wang, D.-H.; Yu, J.-Q. Pd(II)-catalyzed enantioselective C–H olefination of diphenylacetic acids. J. Am. Chem. Soc. 2010, 132, 460–461. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.; Xiao, K.-J.; Yu, J.-Q. Room-temperature enantioselective C–H iodination via kinetic resolution. Science 2014, 346, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.-W.; Gu, Q.; You, S.-L. Pd (II)-catalyzed intermolecular direct C–H bond Iodination: An efficient approach toward the synthesis of axially chiral compounds via kinetic resolution. ACS Catal. 2014, 4, 2741–2745. [Google Scholar] [CrossRef]
- Xiao, K.-J.; Chu, L.; Yu, J.-Q. Enantioselective C–H Olefination of alpha-Hydroxy and alpha-Amino Phenylacetic Acids by Kinetic Resolution. Angew. Chem. Int. Ed. Engl. 2016, 55, 2856–2860. [Google Scholar] [CrossRef] [PubMed]
- Kagan, H.; Fiaud, J. Kinetic resolution. Top. Stereochem 1988, 18, 21. [Google Scholar]
- Yao, Q.-J.; Zhang, S.; Zhan, B.-B.; Shi, B.-F. Atroposelective Synthesis of Axially Chiral Biaryls by Palladium-Catalyzed Asymmetric C–H Olefination Enabled by a Transient Chiral Auxiliary. Angew. Chem. Int. Ed. Engl. 2017, 56, 6617–6621. [Google Scholar] [CrossRef] [PubMed]
- Pi, C.; Li, Y.; Cui, X.; Zhang, H.; Han, Y.; Wu, Y. Redox of ferrocene controlled asymmetric dehydrogenative Heck reaction via palladium-catalyzed dual C–H bond activation. Chem. Sci. 2013, 4, 2675. [Google Scholar] [CrossRef]
- Gao, D.-W.; Gu, Q.; You, S.-L. An Enantioselective Oxidative C–H/C–H Cross-Coupling Reaction: Highly Efficient Method to Prepare Planar Chiral Ferrocenes. J. Am. Chem. Soc. 2016, 138, 2544–2547. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; You, S.-L. Construction of axial chirality by rhodium-catalyzed asymmetric dehydrogenative Heck coupling of biaryl compounds with alkenes. Angew. Chem. Int. Ed. Engl. 2014, 53, 13244–13247. [Google Scholar] [CrossRef] [PubMed]
- Egorova, K.S.; Ananikov, V.P. Which Metals are Green for Catalysis? Comparison of the Toxicities of Ni, Cu, Fe, Pd, Pt, Rh, and Au Salts. Angew. Chem. Int. Ed. Engl. 2016, 55, 12150–12162. [Google Scholar] [CrossRef] [PubMed]


















































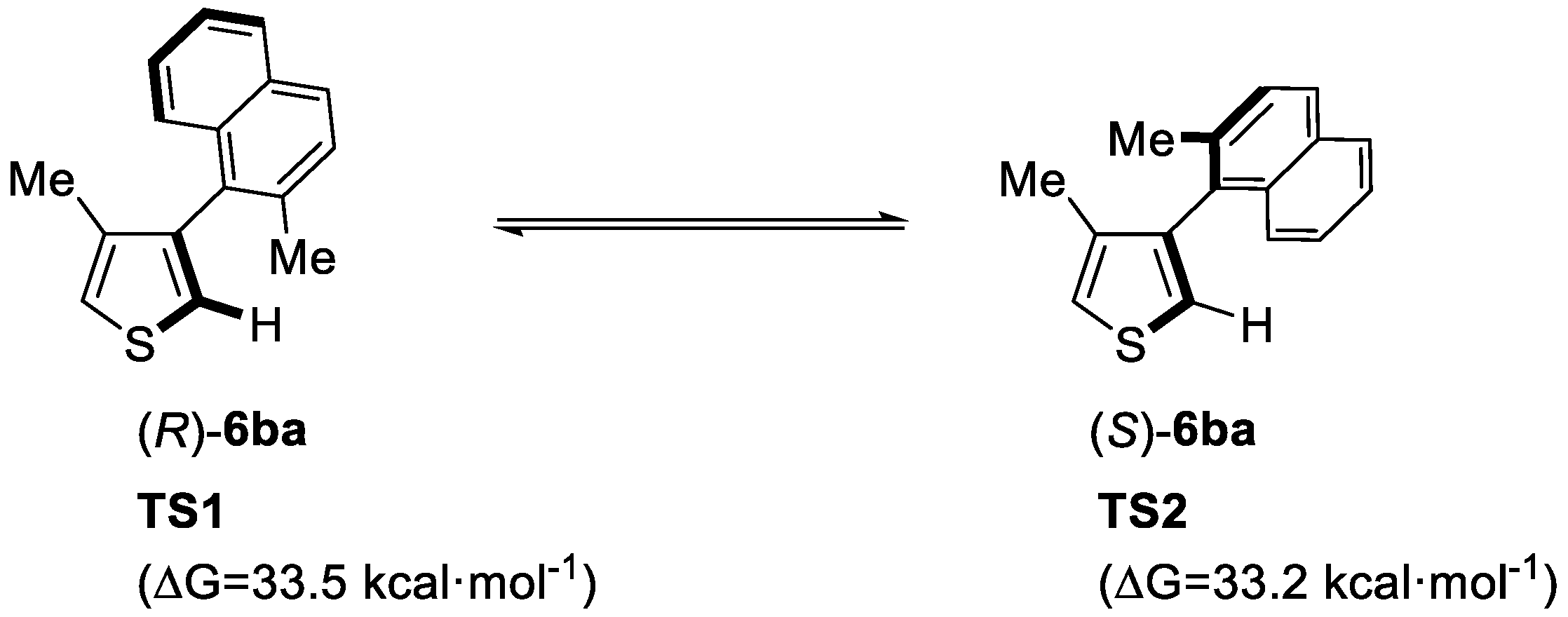{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
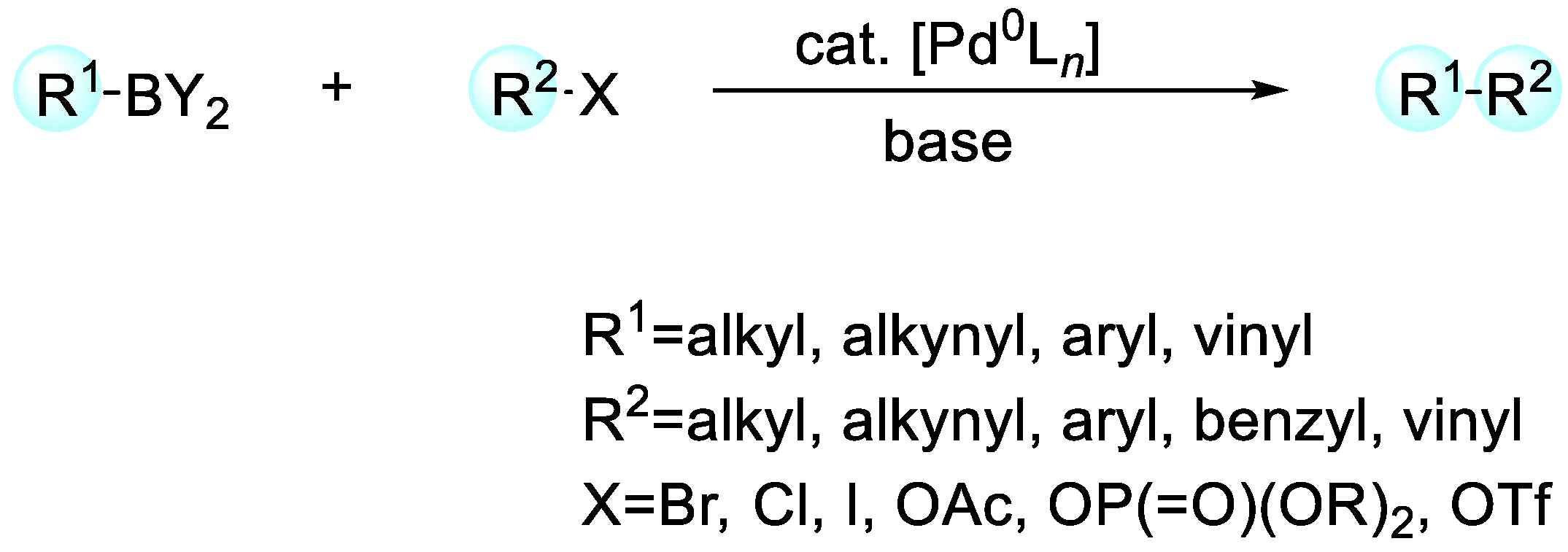{kind=link}
{kind=link}
{kind=link}
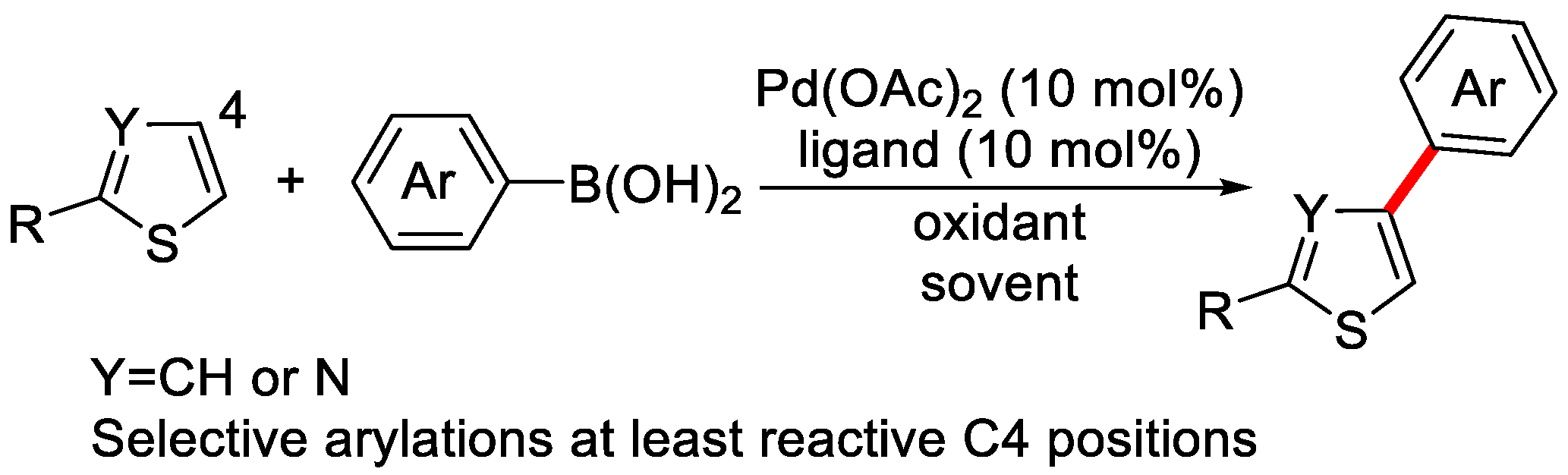{kind=link}
{kind=link}
{kind=link}
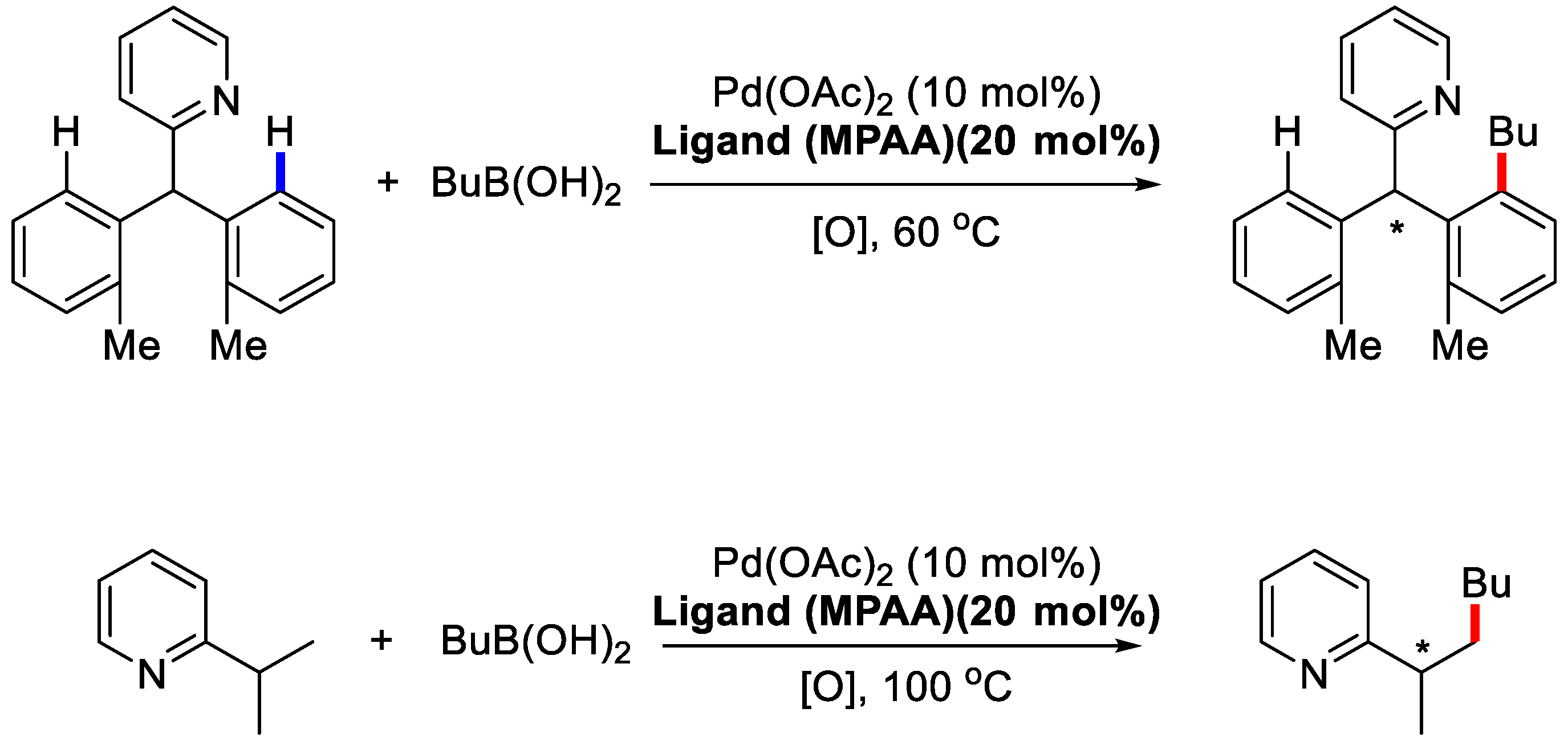{kind=link}
{kind=link}
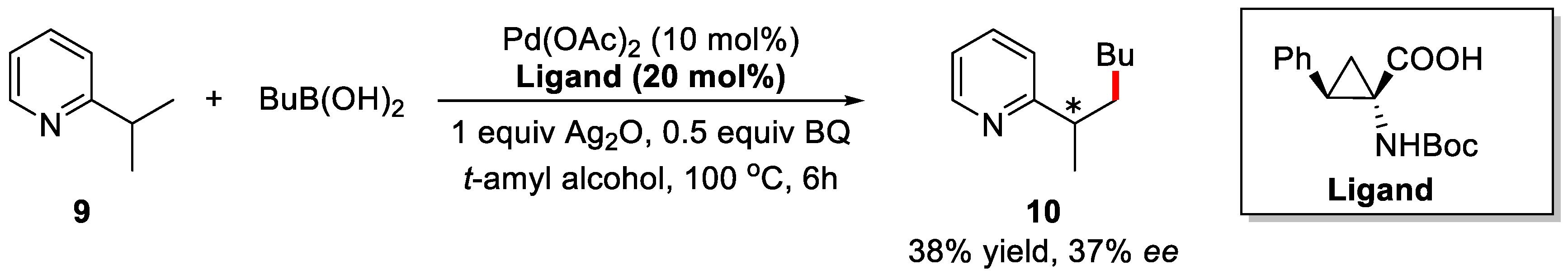{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
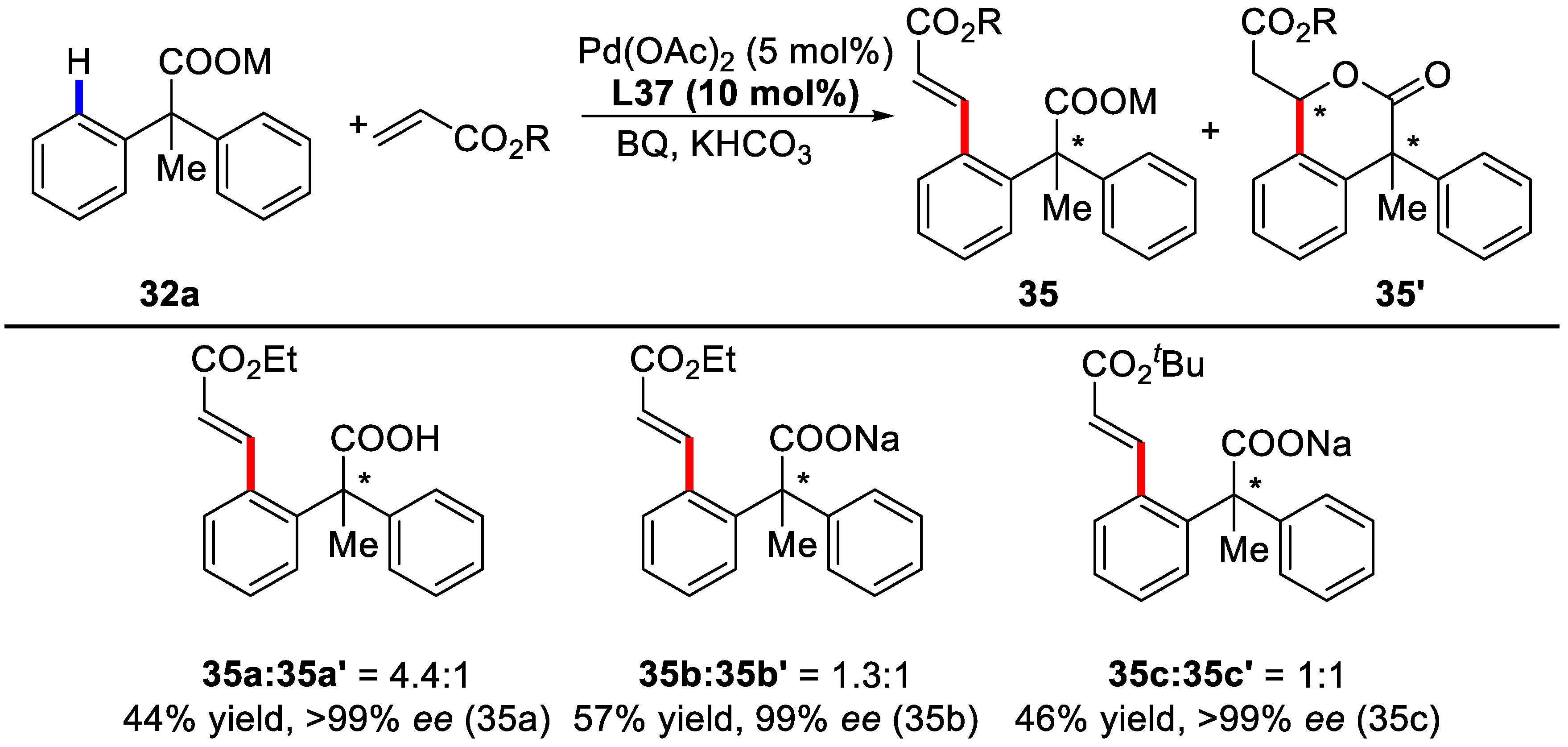{kind=link}
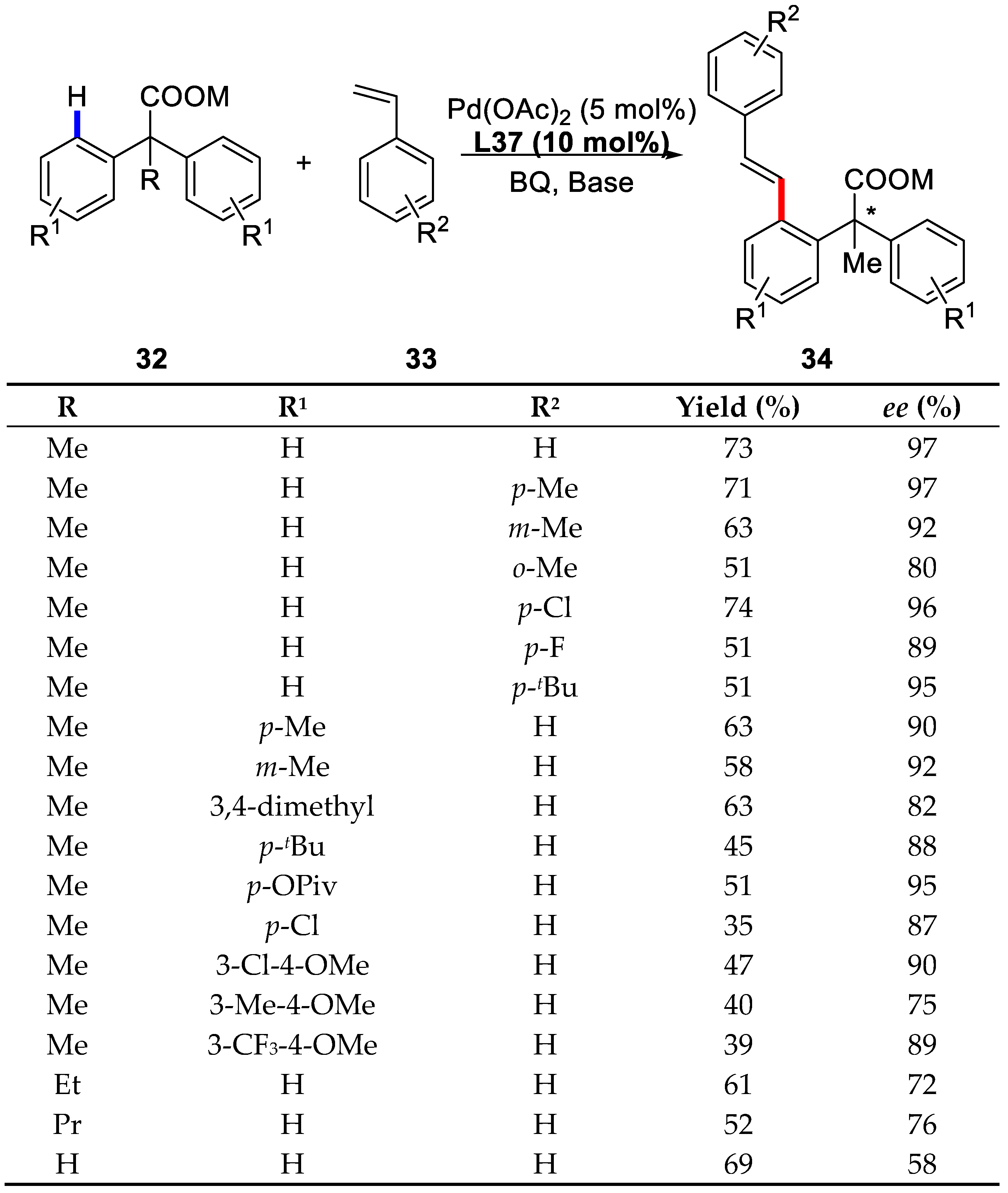{kind=link}
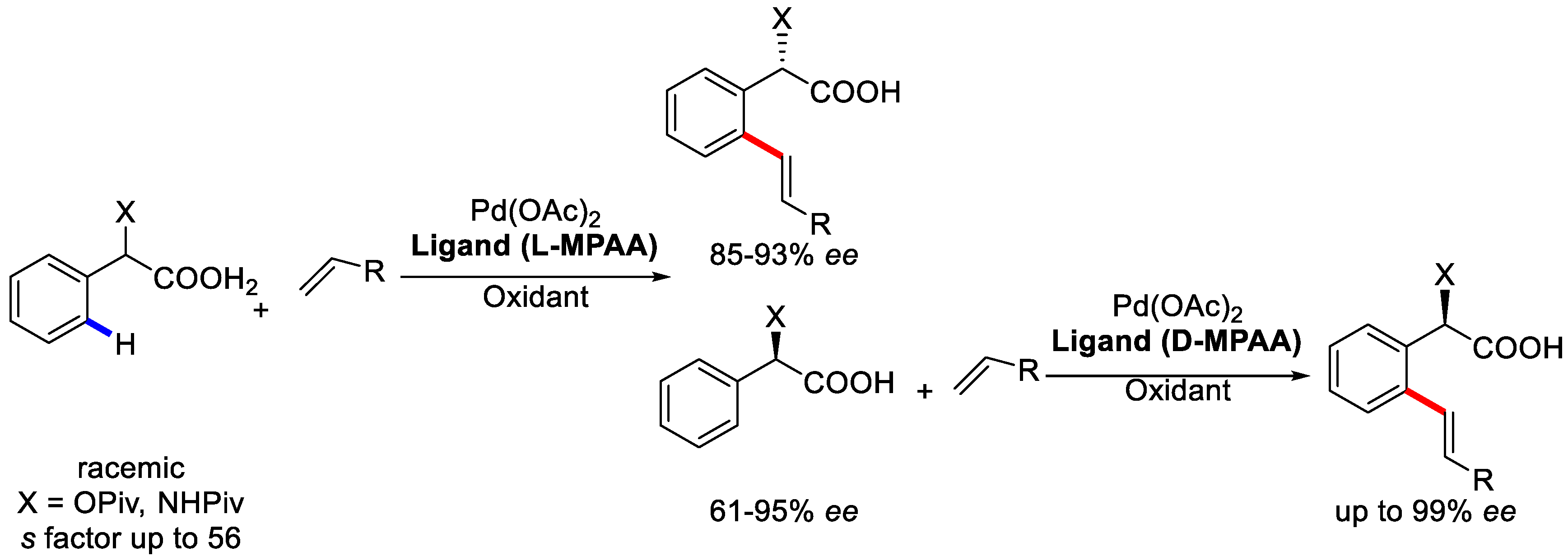{kind=link}
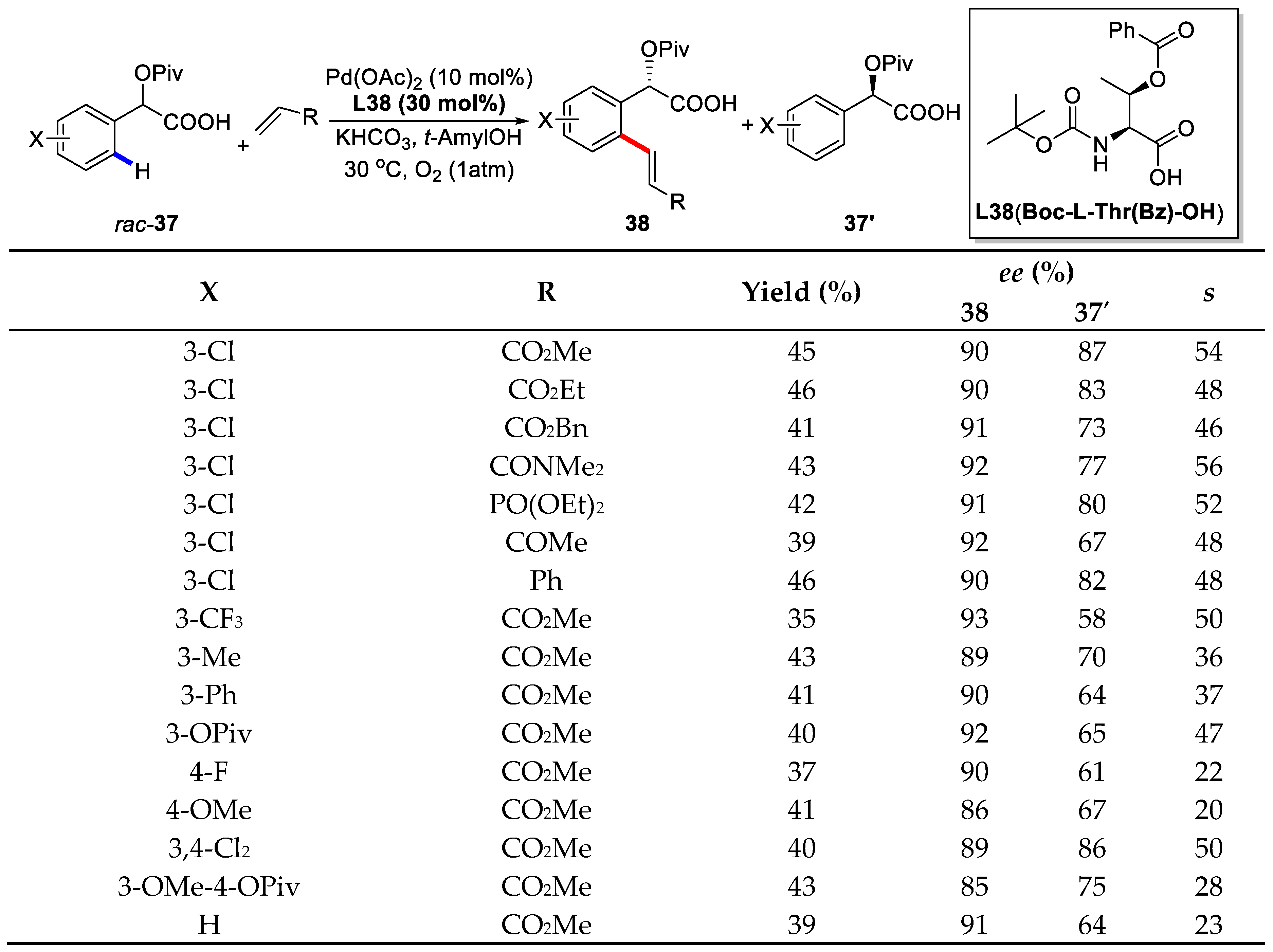{kind=link}
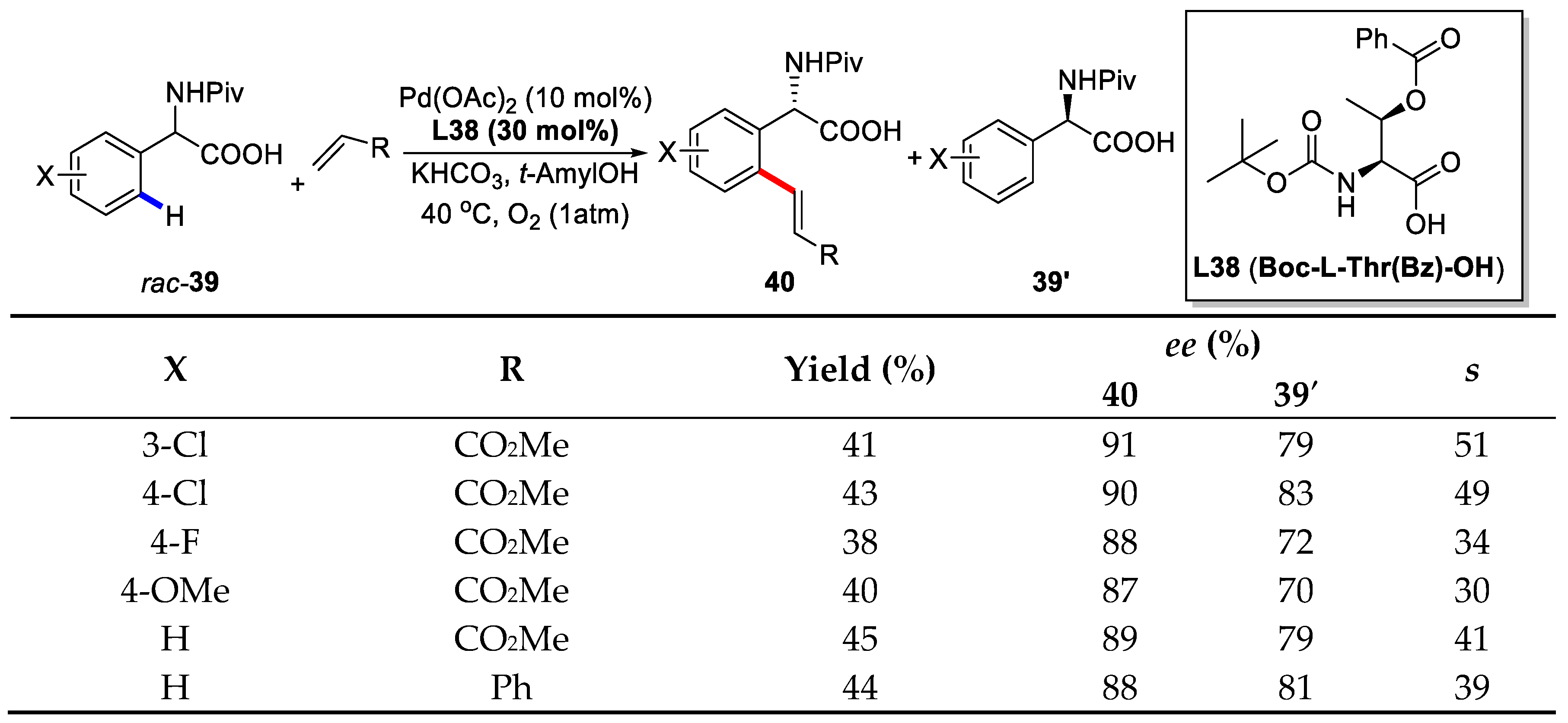{kind=link}
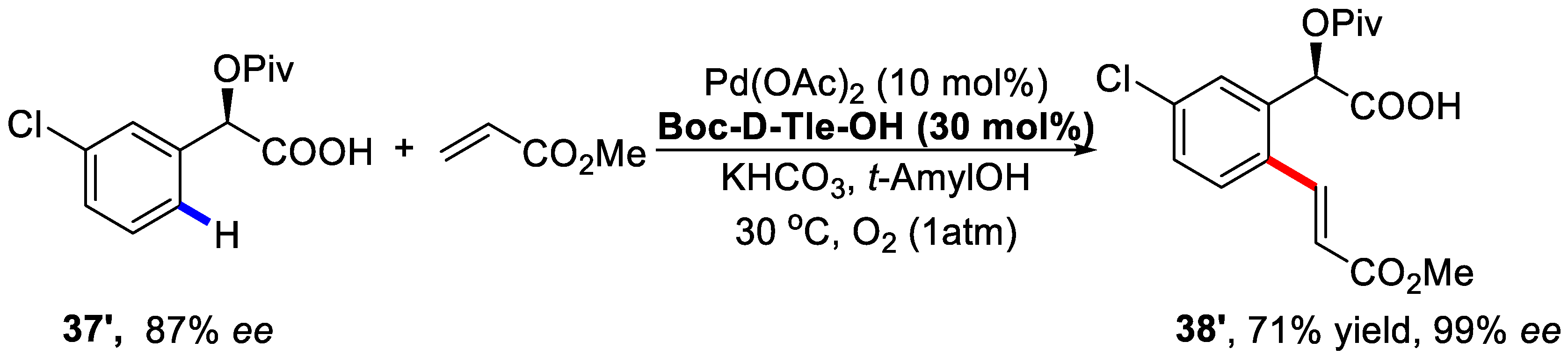{kind=link}
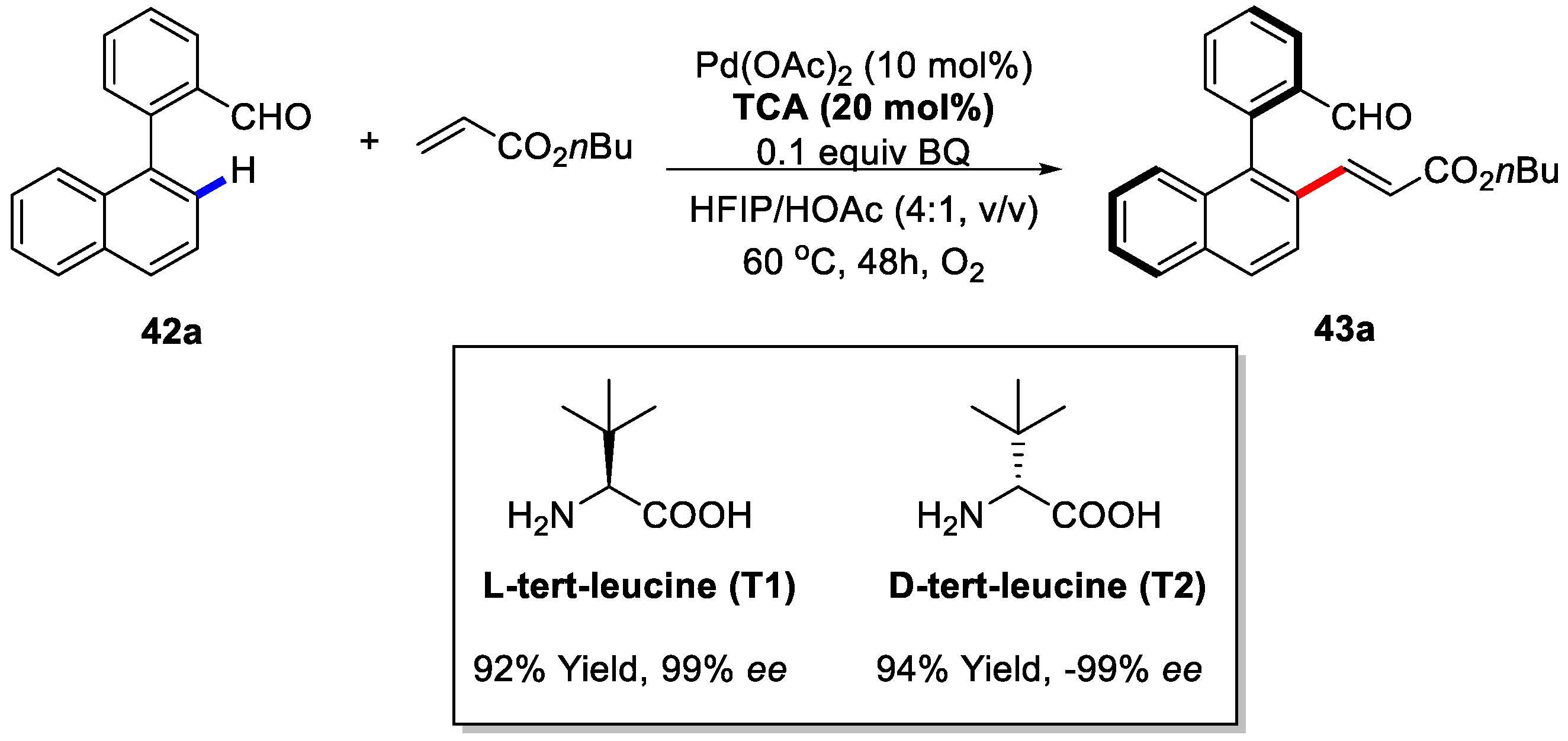{kind=link}
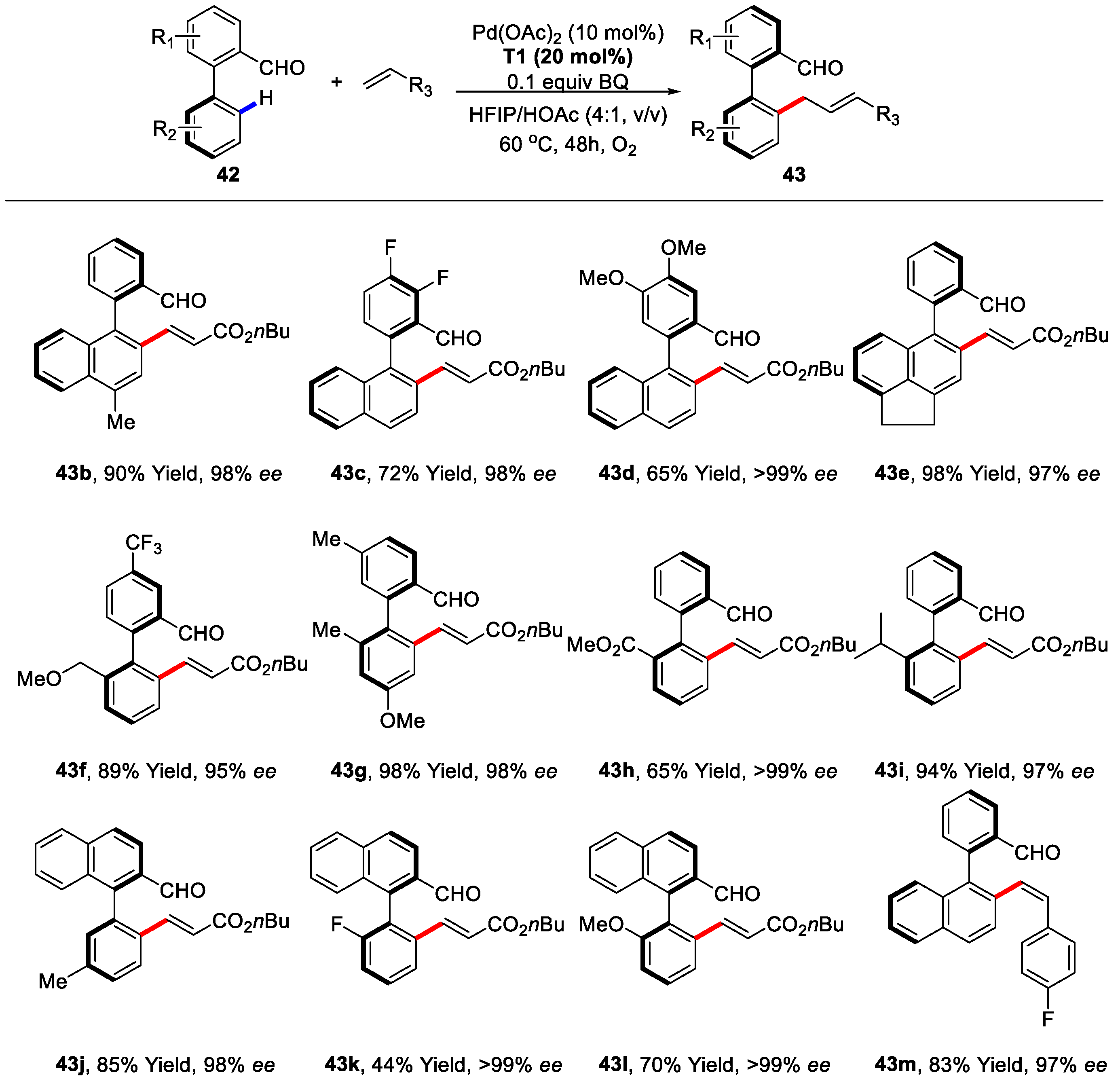{kind=link}
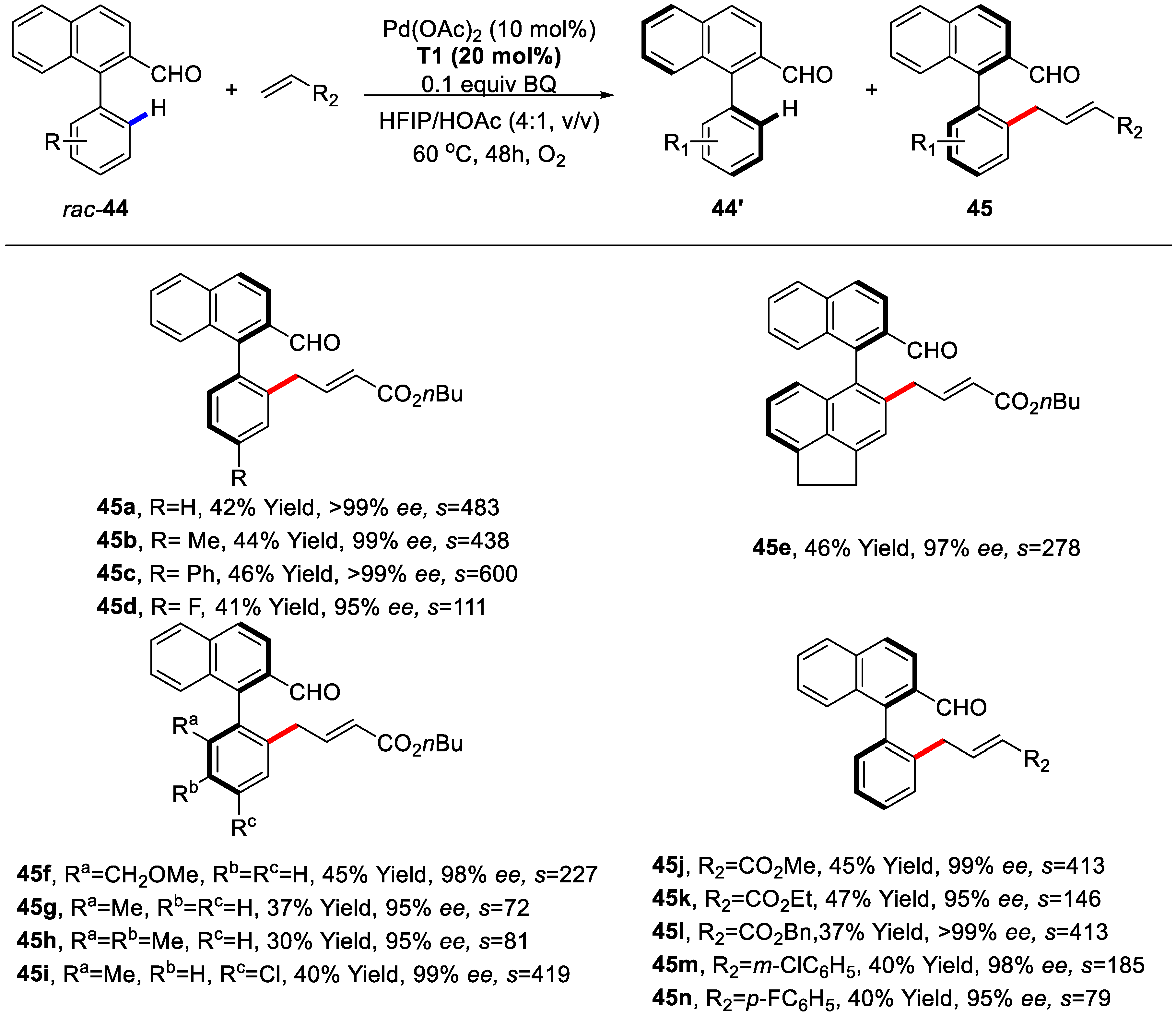{kind=link}
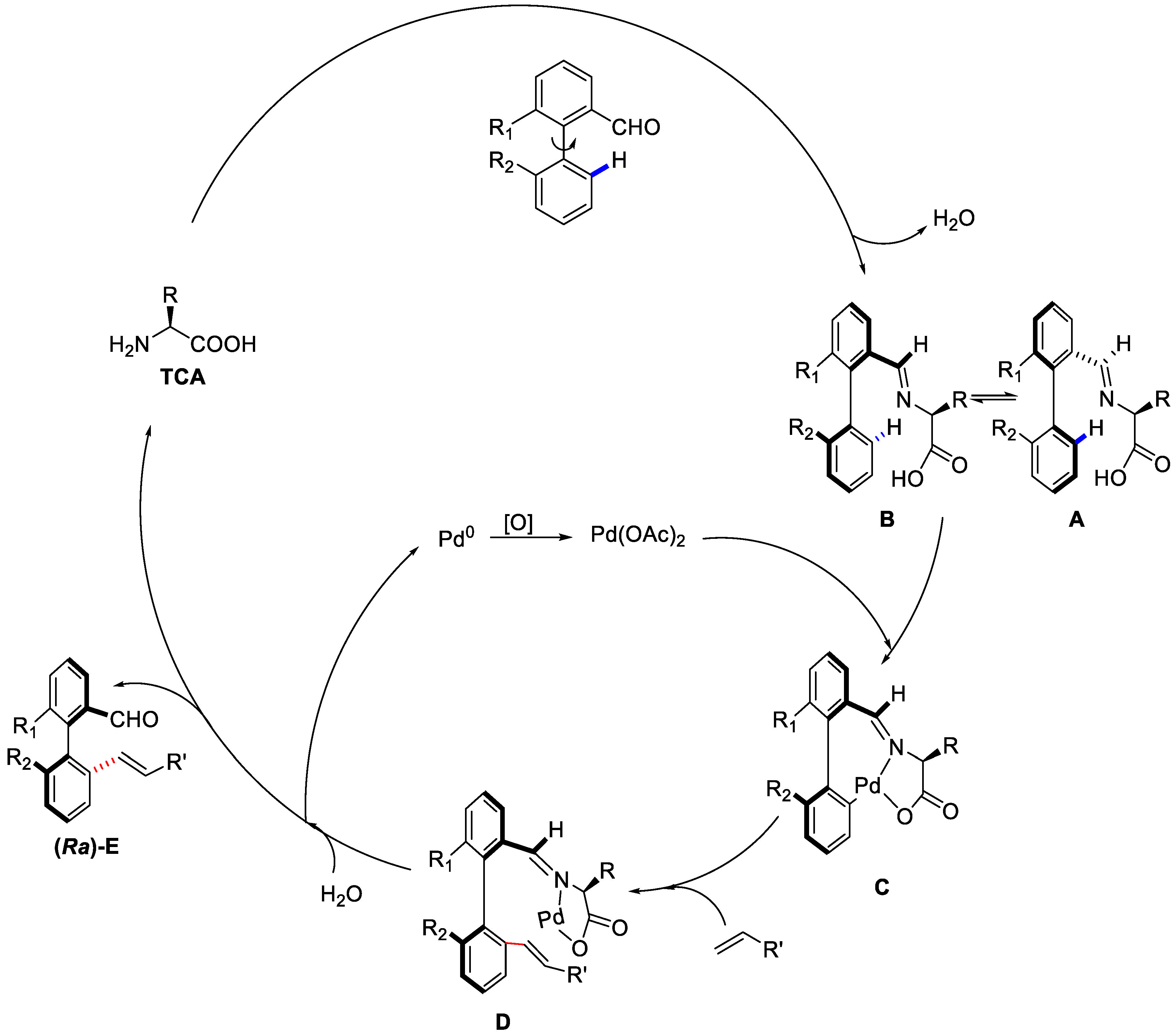{kind=link}
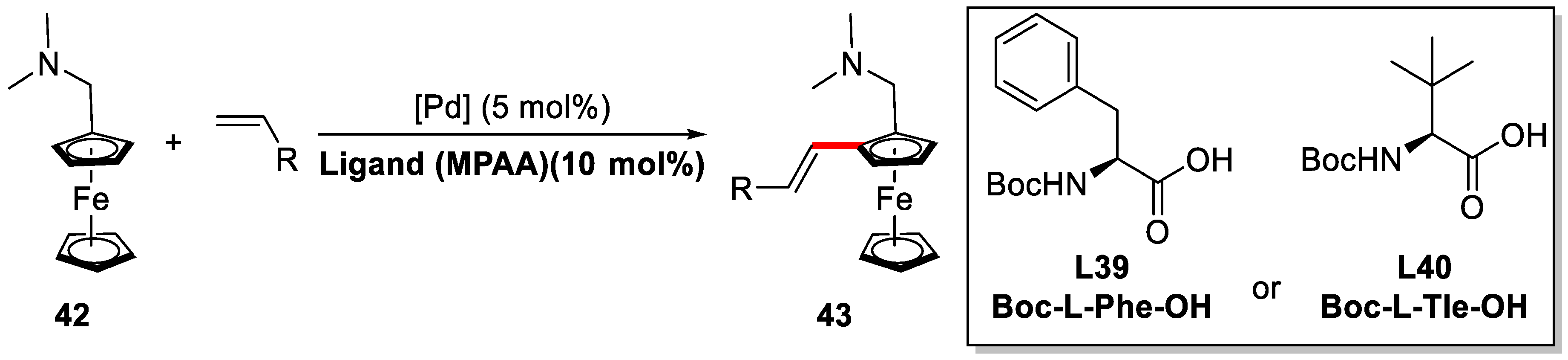{kind=link}
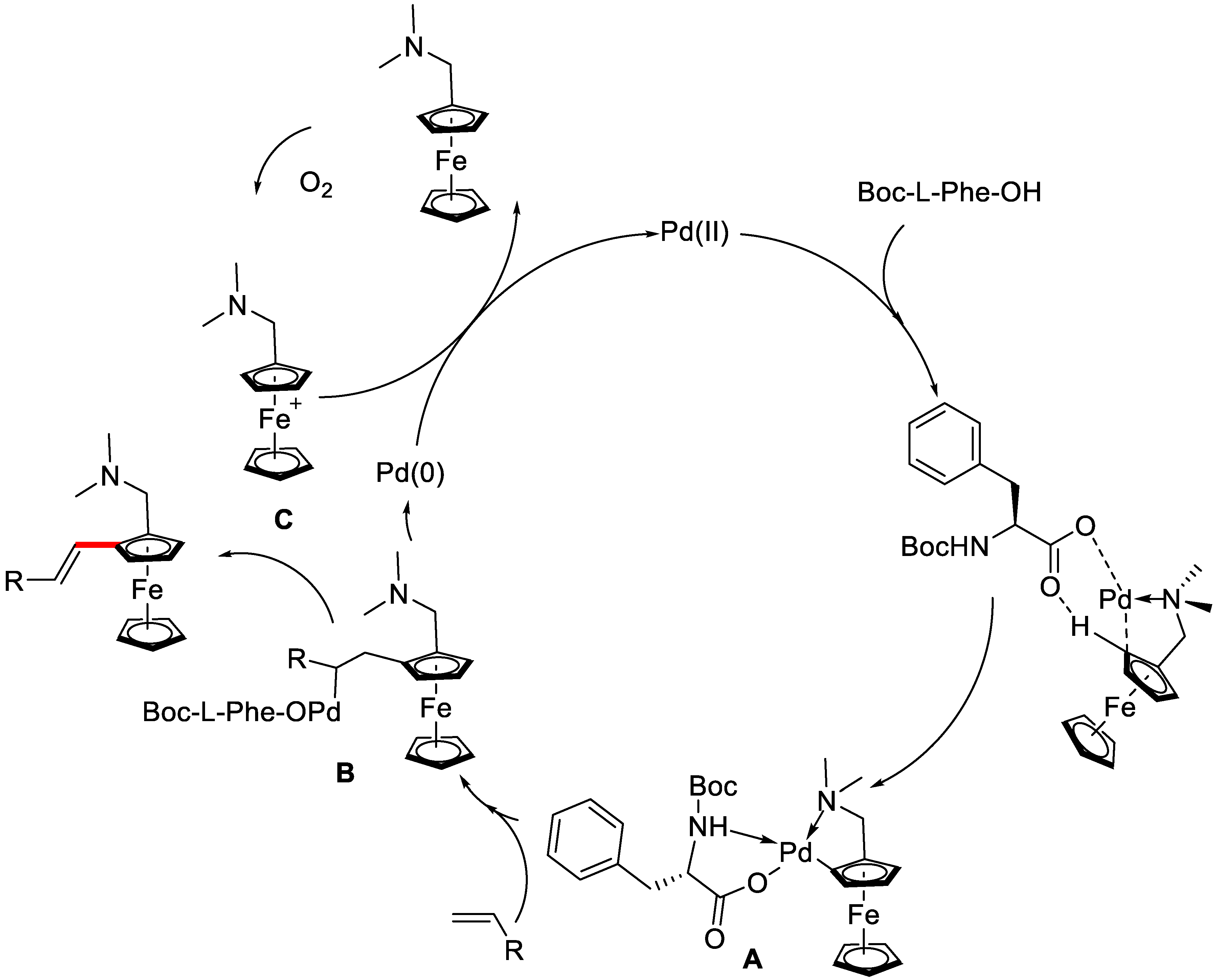{kind=link}
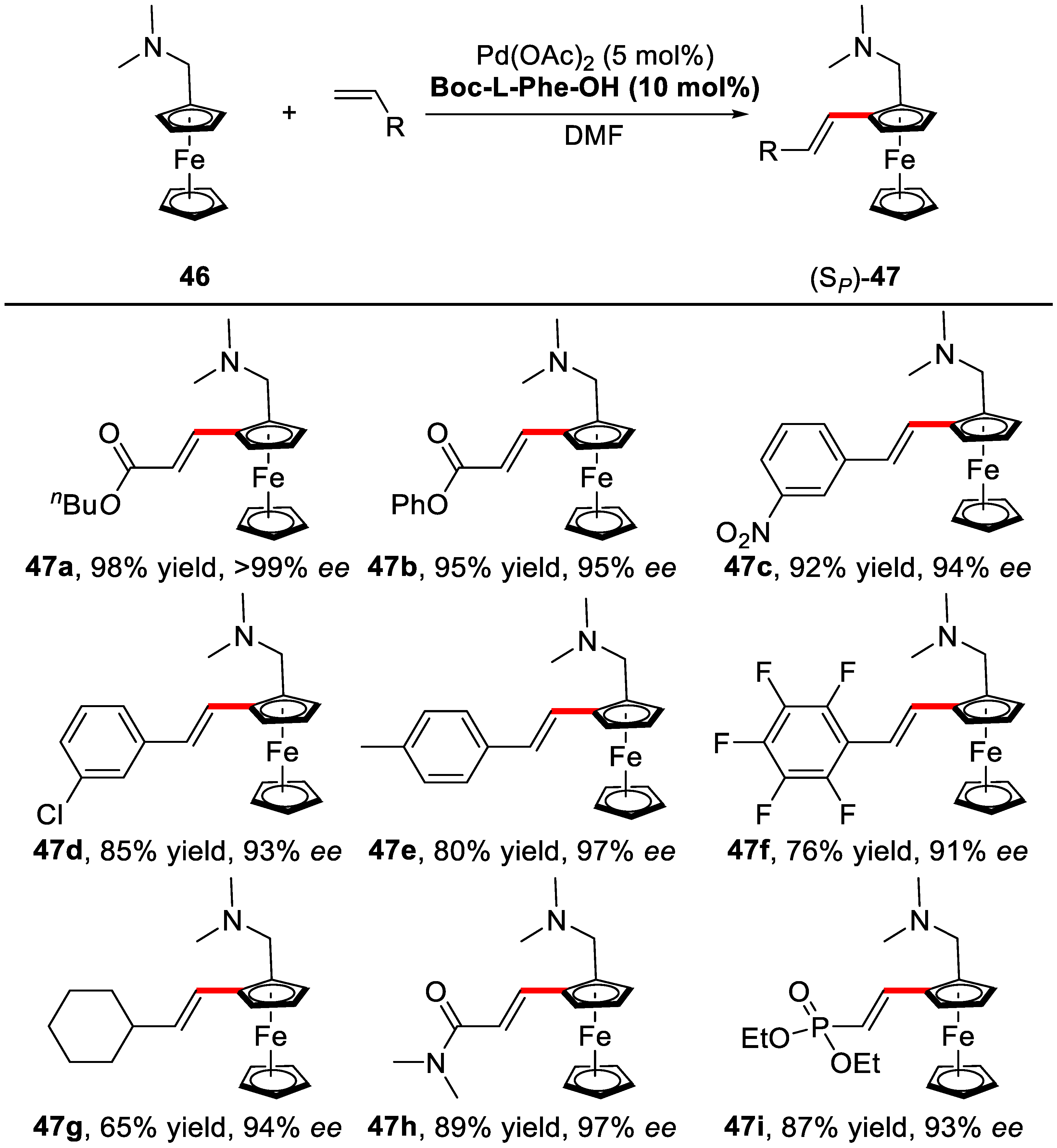{kind=link}
{kind=link}
{kind=link}
{kind=link}
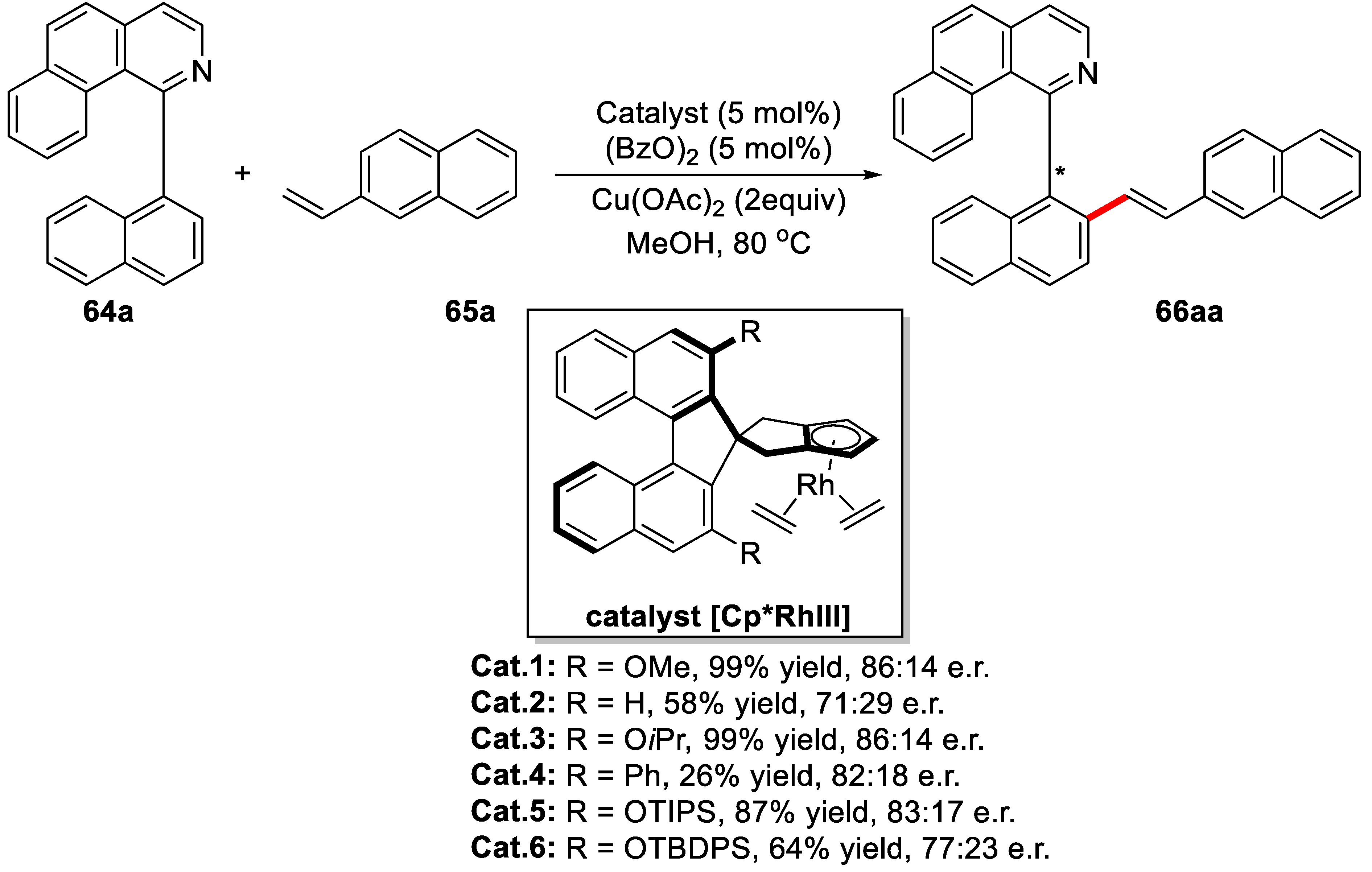{kind=link}
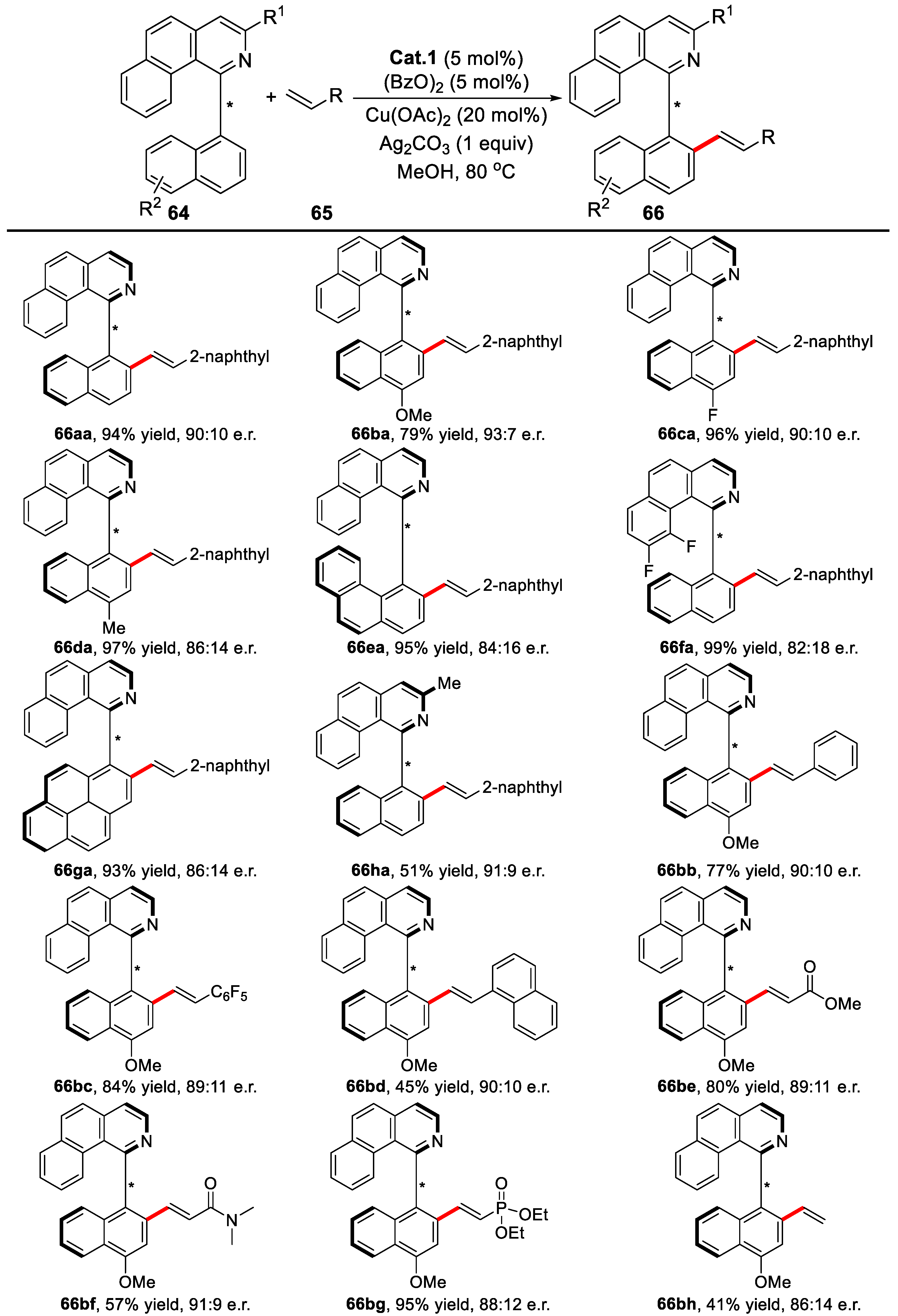{kind=link}
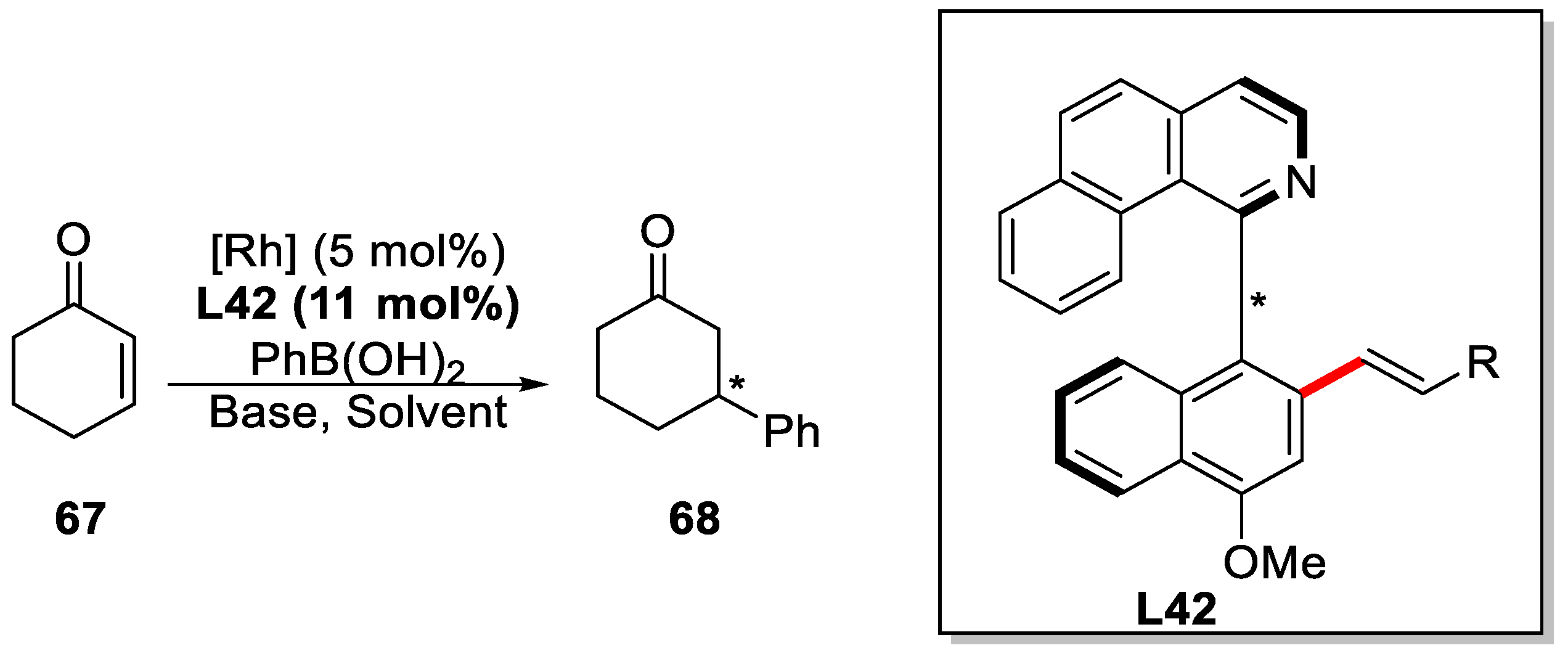{kind=link}

| R1 | R | T [°C] | L [mol %] | Yield (%) | ee (%) |
|---|---|---|---|---|---|
| o-Me | n-Bu | 50 | 20 | 50 | 95 |
| o-Me | n-Bu | 60 | 10 | 96 | 88 |
| H | n-Bu | 80 | 20 | 47 | 79 |
| o-Me | Et | 60 | 10 | 81 | 84 |
| o-Me | Cy | 60 | 10 | 61 | 89 |
| m-Me | n-Bu | 60 | 10 | 58 | 84 |
| m-OMe | n-Bu | 80 | 10 | 55 | 54 |
| m-OAc | n-Bu | 80 | 10 | 43 | 72 |
| p-Me | n-Bu | 80 | 10 | 61 | 78 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, S.; Nawaz, K.S.; Zaman, M.K.; Sun, Z. Advances in Enantioselective C–H Activation/Mizoroki-Heck Reaction and Suzuki Reaction. Catalysts 2018, 8, 90. https://doi.org/10.3390/catal8020090
Shi S, Nawaz KS, Zaman MK, Sun Z. Advances in Enantioselective C–H Activation/Mizoroki-Heck Reaction and Suzuki Reaction. Catalysts. 2018; 8(2):90. https://doi.org/10.3390/catal8020090
Chicago/Turabian StyleShi, Shuai, Khan Shah Nawaz, Muhammad Kashif Zaman, and Zhankui Sun. 2018. "Advances in Enantioselective C–H Activation/Mizoroki-Heck Reaction and Suzuki Reaction" Catalysts 8, no. 2: 90. https://doi.org/10.3390/catal8020090
APA StyleShi, S., Nawaz, K. S., Zaman, M. K., & Sun, Z. (2018). Advances in Enantioselective C–H Activation/Mizoroki-Heck Reaction and Suzuki Reaction. Catalysts, 8(2), 90. https://doi.org/10.3390/catal8020090